

Summary
TraR is a LuxR-type quorum sensing protein encoded by the Ti plasmid of Agrobacterium tumefaciens. TraR requires the pheromone 3-oxooctanoylhomoserine lactone (OOHL) for biological activity, and is dimeric both in solution and when bound to DNA. Dimerization is mediated primarily by two alpha helices, one in the N-terminal OOHL binding domain, and the other in the C-terminal DNA binding domain. Each of these helices forms a parallel coiled coil with the identical helix of the opposite subunit. We have previously shown that OOHL is essential for resistance to proteolysis, and here we asked whether dimerization is also required for protease resistance. We constructed a series of site-directed mutations at the dimer interface, and tested these mutants for activity in vivo. Alteration of residues A149, A150, A153, A222 and I229 completely abolished activity, while alteration of three other residues also caused significant defects. All mutants were tested for dimerization as well as for specific DNA binding. The cellular abundance of these proteins in A. tumefaciens was measured using western immunoblots and OOHL-sequestration, while the half-life was measured by pulse-chase radiolabelling. We found a correlation between defects in in vivo activity, in vitro dimerization, DNA binding, protein halflife. We conclude that dimerization of TraR enhances resistance to cellular proteases.
Introduction
The TraR protein of Agrobacterium tumefaciens is a member of the LuxR family of transcription factors, which are intracellular receptors of N-acylhomoserine lactones (AHLs), that act as pheromones in intercellular communication {Waters, 2005 #14}. AHLs are generally synthesized by a cognate LuxI-type protein (Pappas et al., 2004). These pheromones accumulate in a population density-dependent manner and form complexes with their receptors when they reach a threshold concentration. LuxR/LuxI-type regulatory systems therefore allow bacteria to monitor their cell population densities, and to coordinate their behavior in a process referred to as quorum sensing. This phenomenon was first discovered in the marine bacterium Vibrio fischeri, whose LuxR and LuxI proteins regulate genes required for bioluminescence. Homologous systems have also been identified in many proteobacteria where they regulate diverse activities, including pathogenesis, biofilm formation, horizontal DNA transfer and the production of secondary metabolites (Waters and Bassler, 2005).
TraR regulates genes that are required for vegetative replication (rep genes) and conjugal transfer (tra and trb genes) of the tumour-inducing (Ti) plasmid (Fuqua and Winans, 1994; Li and Farrand, 2000; Pappas and Winans, 2003). TraR activity requires N-3-oxooctanoyl-L-homoserine lactone (OOHL), which is synthesized by the Ti plasmid-encoded TraI protein. TraR subunits form dimers that bind to dyad-symmetrical DNA sequences. It has two domains, an N-terminal OOHL-binding domain and a C-terminal DNA binding domain (Pappas et al., 2004). OOHL is required for TraR to become resistant to proteolysis in vivo, and is thought to act as a scaffold for TraR folding (Zhu and Winans, 1999, 2001).
The structure of TraR bound to OOHL and tra box DNA has been solved by X-ray crystallography (Vannini et al., 2002; Zhang et al., 2002). The N-terminal and C-terminal domains both contribute to protein dimerization. The N-terminal domain dimerizes chiefly through ∀-helix 9 of each subunit (the longest ∀-helix in the protein), forming a coiled-coil (Figure 1A). The C-terminal domain dimerizes through ∀-helix 13 of each subunit, which form a second coiled-coil (Fig. 1A). Additional contacts include ionic interactions between the C-terminal carboxylate of each subunit and Arg230 of the opposite subunit.
Fig. 1. Dimerization interface of TraR.

A. Two alpha helices in the N-terminal domain form a coiled coil, and two additional alpha helices in the C-terminal domain form a second coiled coil. These two coiled coils provide the majority of the dimerization interface.
B. A monomer of TraR with the amino acid residues that were mutated in this study.
C. Helical wheel projection of alpha helix 9 and alpha helix 13. Residues depicted in black were altered by site-directed mutagenesis in this study. Positions a to g of the helical wheel projection are encircled and labeled with lower case letters.
Previous studies have shown that OOHL-mediated folding is essential for resistance to cytoplasmic proteases (Zhu and Winans, 1999, 2001). However, it was not known whether fully folded TraR monomers were protease resistant, or whether dimerizaton was also required. Cellular proteases are generally thought to detect denatured proteins by virtue of hydrophobic residues that would lie within the hydrophobic core of a properly folded protein (Wickner et al., 1999). The interface between TraR monomers is quite hydrophobic, and dimers are stabilized in large part by hydrophobic interactions. TraR monomers would therefore probably present a large hydrophobic surface, which might be recognized by proteases, and lead to proteolysis. Dimerization would sequester these hydrophobic patches, and could thereby block proteolysis. It therefore seemed plausible that protection from proteolysis might require not only binding of OOHL, but also dimerization of TraR monomers. If so, then mutations that block dimerization would be predicted to be more susceptible to proteolysis. In this study, TraR alleles were isolated that block biological activity, protein dimerization and DNA binding in vitro. These mutants showed an increased rate of turnover compared to wild type TraR.
Results
Structure-informed mutagenesis of the TraR dimerization interface
The goals of this study were to test amino acid residues that lie at the dimerization interface for their role in dimer formation and to determine whether defects in dimerization affect the rate at which these proteins are eliminated from the cells by proteolysis. Residues facing the dimerization interface and potentially mediating hydrophobic interactions were chosen for site-directed mutagenesis (Fig. 1B). In addition, we altered residues K119, and N122 because they interact with residues D6 and D10 of the opposite monomer (Zhang et al., 2002) and could potentially aid in dimerization. Residues I155 and S160 were chosen based on a previous study that showed their contribution to dimerization (Luo et al., 2003). Three other mutations were included as controls that were unlikely to have a role in dimerization. Of these, alteration of residue R206 confers a DNA binding defective phenotype (White and Winans, 2007), while alteration of residue G123 confers a positive control phenotype (Luo and Farrand, 1999), and alteration of residue L5 is phenotypically silent (this work). Wherever possible, mutations were made that preserve interactions with neighboring amino acid residues.
Transcriptional activation
Each TraR mutant was tested for its ability to activate a PtraI-lacZ fusion in vivo using a broad range of OOHL concentrations (Table 1). Mutations A149E, A149V, A150E, A150V, G153E, Q154E, A222D, and I229Y caused strong defects in TraR activation, with transcriptional levels less than 15% of the wild type for all OOHL concentrations tested. Mutation of other residues had weaker effects on the transcription activity, especially at higher concentrations of OOHL (Table 1). Residues A149 and A150, which are located on the alpha-helix 9, were especially critical for activity, as even conservative mutations strongly impaired transcriptional activity.
Table 1.
In vivo activity at PtraI-lacZ fusion of all point mutants relative to wild-type TraR at different concentrations of OOHL.
| OOHL concentration | 0.1 nM | 1nM | 10nM | 100nM |
|---|---|---|---|---|
| Wild type TraR | 100.0 (15.0) | 100.0 (15.6) | 100.0 (15.9) | 100.0 (9.9) |
| Vector Control | 0.1 (<0.1) | 0.1 (<0.1) | 0.1 (<0.1) | 0.1 (<0.1) |
| L5M | NT | 100.0 (4.8) | 100.0 (1.2) | 100.0 (0.2) |
| K119A | 28.6 (7.1) | 56.7 (11.9) | 40.2 (7.1) | 61.1 (28.9) |
| N122A | 0.1 (<0.1) | 2.9 (2.5) | 21.9 (8.2) | 29.3 (9.9) |
| G123R | NT | 0.1 (<0.1) | NT | 0.1 (<0.1) |
| V146A | 45.2 (9.5) | 41.3 (12.4) | 39.1 (14.6) | 33.9 (5.4) |
| V146E | 5.9 (2.1) | 56.4 (6.1) | 60.5 (10.7) | 75.7 (16.8) |
| A149E | 0.2 (<0.1) | 0.1 (<0.1) | 0.1 (<0.1) | 0.1 (<0.1) |
| A149V | 0.1 (<0.1) | 0.3 (0.2) | 1.1 (0.9) | 1.6 (0.8) |
| A150E | 0.1 (<0.1) | 1.3 (0.8) | 6.2 (2.4) | 8.0 (2.3) |
| A150V | 0.2 (<0.1) | 0.1 (<0.1) | 0.5 (0.4) | 1.5 (0.2) |
| G153A | 17.1 (3.1) | 84.8 (20.9) | 105.1 (12.2) | 90.0 (17.8) |
| G153E | 0.1 (<0.1) | 0.1 (<0.1) | 0.1 (<0.1) | 0.1 (<0.1) |
| Q154A | 0.6 (0.4) | 7.9 (3.4) | 56.0 (14.6) | 40.3 (31.8) |
| Q154E | 0.2 (<0.1) | 2.1 (1.8) | 6.9 (4.4) | 14.3 (10.1) |
| I155F | NT | 13.5 (1.7) | 60.7 (12.2) | 94.4 (5.1) |
| A157E | 10.6 (6.0) | 30.2 (5.7) | 56.7 (12.4) | 68.7 (12.0) |
| A157V | 9.2 (0.6) | 44.3 (8.9) | 55.7 (15.7) | 74.6 (8.6) |
| S160A | NT | 92.0 (12.1) | 99.0 (8.0) | 89.0 (15.5) |
| S160F | NT | 11.5 (1.4) | 62.4 (14.2) | 99.3 (2.0) |
| F161A | 5.5 (2.0) | 17.8 (6.1) | 42.8 (19.9) | 37.7 (18.5) |
| F161K | 0.8 (0.3) | 8.7 (1.7) | 33.5 (5.3) | 52.4 (13.9) |
| R206Q | NT | 0.1 (<0.1) | NT | NT |
| A222D | 0.2 (0.1) | 0.3 (0.1) | 0.8 (0.2) | 0.8 (0.4) |
| A222S | 29.9 (17.0) | 59.9 (4.4) | 52.5 (13.1) | 83.5 (11.1) |
| T225S | 1.0 (0.5) | 10.1 (3.4) | 17.5 (5.0) | 22.8 (3.1) |
| I229A | NT | 35.0 (5.1) | NT | NT |
| I229Y | 0.3 (0.1) | 0.1 (<0.1) | 0.1 (<0.1) | 0.1 (<0.1) |
Activity was measured at PtraI-lacZ fusion in NTL4 (pCEW260) with vector control (pPZP201), wild-type traR (pYC335) or each point mutant. Data represent the average of at least 3 repetitions compared to wild type TraR values that were set to 100%. Wild type TraR expression values were about 700, 2100, 2600 and 3000 units of∃-galactosidase activity at 0.1 nM, 1 nM, 10 nM and 100 nM of OOHL, respectively. NT - Not tested.
Assays for TraR dimerization in vitro
The assays described above measured the ability of TraR mutants to activate transcription in vivo, but did not directly address the role of the mutated residues in dimerization. We therefore developed assays that directly evaluate the ability of these mutant proteins to form dimers. These assays involved the construction of fusions between these mutants and the maltose binding protein (MBP) of E. coli. It was previously shown that native TraR can form heterodimers with MBP-TraR fusions (Zhu and Winans, 2001). Clarified lysates containing MBP-TraR fusion proteins were combined with lysates containing wild type TraR. After allowing sufficient time for heterodimer formation, MBP-TraR fusion proteins were purified using an amylose-agarose affinity resin. The bound fractions were eluted using maltose and examined by SDS-PAGE gels for the presence of native TraR. In these assays, only the fusion protein contains the point mutation of interest, while the native TraR has a wild type sequence. In contrast, in the in vivo assays described above, both subunits of TrarR dimers contained identical mutations.
As expected, a fusion protein containing wild-type TraR sequences efficiently bound native TraR protein (Fig. 2). As expected, native TraR was not detectably bound by MBP lacking TraR sequences (data not shown). In contrast, most of the fusion proteins containing TraR mutations were defective in binding native TraR (Fig. 2, Table 2). In many cases, a correlation was found between defects in in vivo activity and heterodimer formation (Table 2). Residues A149, A150, G153, Q154, and I229 were especially strongly defective in both assays. A few mutant proteins did not follow this pattern, in that they were strongly defective in activity, yet had only subtle defects in dimerization (see Table 1 and 2, mutants G153R, R206Q, and T225S). These mutants were judged to be defective in some other TraR property such as DNA binding or the ability to recruit RNA polymerase to the adjacent promoter.
Fig. 2.
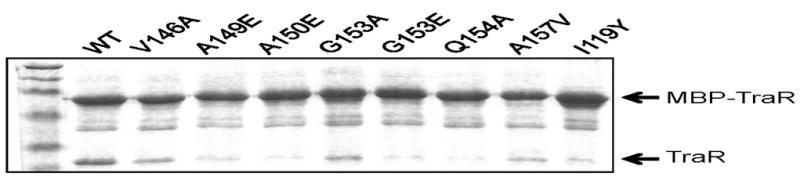
Dimerization of wild type TraR with MBP-TraR fusions containing point mutations at the subunit interface. Cell supernantants containing native TraR and various fusion proteins were combined and allowed to form heterodimers, then purified by amylose affinity chromatography. MBP-TraR fusions having wild type TraR sequence retain native TraR on the column, while some MBP-TraR fusions having mutant TraR sequences fail to retain TraR.
Table 2.
Correlation between Accumulation, DNA binding, Dimerization, and OOHL Retention.
| Accumulation1 | DNA binding2 | Heterodimer Formation1 | OOHL Retention1 | |
|---|---|---|---|---|
| Wild type TraR | 100 | Y | 100 | 100 |
| Vector Control | 0 | N | 0 | 0 |
| L5M | 100 (11) | Y | 99 (11) | 100 (5) |
| K119A | 82 (10) | Y | 94 (10) | 89 (15) |
| N122A | 28 (5) | Y | 28 (1) | 35 (16) |
| G123R | 51 (8) | Y | 49 (10) | 67 (10) |
| V146A | 110 (20) | Y | 61 (10) | 104 (19) |
| V146E | 65 (15) | Y | 52 (9) | 87 (11) |
| A149E | 44 (12) | N | 12 (6) | 38 (9) |
| A149V | 45 (15) | N | 19 (5) | 33 (6) |
| A150E | 60 (6) | N | 14 (1) | 55 (10) |
| A150V | 47 (13) | N | 15 (2) | 52 (6) |
| G153A | 63 (11) | Y | 61 (22) | 66 (10) |
| G153E | 33 (8) | N | 19 (4) | 39 (7) |
| Q154A | 45 (4) | N | 27 (7) | 45 (10) |
| Q154E | 36 (8) | N | 25 (5) | 28 (5) |
| I155F | 43 (7) | Y | 50 (7) | NT |
| A157E | 48 (6) | Y | 48 (4) | 65 (10) |
| A157V | 43 (8) | Y | 50 (5) | 75 (18) |
| S160A | 100 (10) | Y | 100 (6) | NT |
| S160F | 55 (5) | Y | 69 (4) | NT |
| F161A | 82 (7) | Y | 49 (7) | 73 (20) |
| F161K | 44 (6) | Y | 39 (6) | 57 (7) |
| R206Q | 105 (6) | N | 71 (14) | NT |
| A222D | 38 (10) | N | 29 (7) | 32 (14) |
| A222S | 66 (7) | Y | 53 (11) | 60 (14) |
| T225S | 39 (16) | N | 43 (6) | 35 (4) |
| I229A | NT | Y | NT | 33 (5) |
| I229Y | 41 (8) | N | 23 (7) | 28 (10) |
Values represent the average of at least 3 repetitions. Wild type levels were set to 100%. NT - not tested. Accumulation was measured by semi-quantitative western blots using strain NTL4 (pCEW260)(pYC335) or its derivatives carrying each traR mutant, dimerization was determined by the ability of a MBP-TraR fusion carrying each point mutation to form heterodimers with native TraR, and OOHL retention assays was tested as previously described (Chai and Winans, 2004).
DNA binding affinity of each TraR allele was evaluated by gel retardation assays using cleared cell lysates and tra box DNA repeated at least twice for each mutant. Y - TraR shifted DNA, N - TraR did not shift the DNA.
Assays for binding to DNA fragments containing TraR binding sites
It is well established that TraR heterodimers containing just one DNA binding domain fail to bind to tra box DNA sequences (Chai et al., 2001; Oger et al., 1998; Zhu and Winans, 1998). This indicates that TraR monomers do not bind DNA with high affinity and that dimerization is essential for high affinity DNA binding. To test whether defects in dimerization lead to defects in DNA binding, we conducted electrophoretic mobility shift assays (EMSA) using wild type or mutant proteins and a DNA fragment that contains a consensus tra box. As expected, virtually all mutants that had strong defects in transcription activation and dimerization were defective in DNA binding (Fig. 3, Table 2), while mutants with subtle phenotypes in the former assays were proficient in DNA binding. An exception to this pattern was found in mutant N122A, which was defective in in vivo activity, yet showed wild-type affinity for tra box DNA. We conclude that this mutant is defective in positive control. We have identified other residues that lie close to N122 that are also required for positive control (E. Costa, H. Cho, and S. C. Winans, manuscript submitted).
Fig. 3.
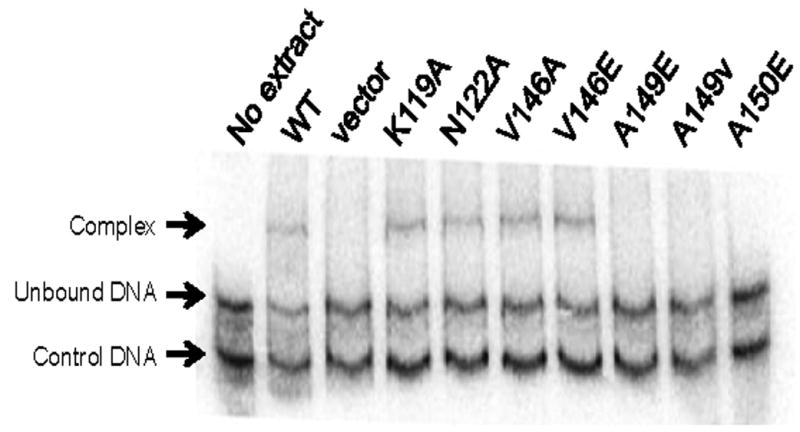
Electrophoretic mobility shift assays using clarified cell extracts containing wild type or mutant TraR proteins and a DNA fragment containing a consensus TraR binding site. Control DNA lacks a binding site for TraR. The amount of cell extracts added was normalized for TraR abundance using Western immunoblots.
In vivo accumulation and stability of dimerization mutants
The central question addressed in this study is whether dimerization of TraR is required for resistance to proteolysis. We addressed this question in three ways, two of which involve measurements of TraR abundance, and one of which directly measures the half-life of these mutant proteins. TraR abundance was assayed, first, by measuring the ability of whole A. tumefaciens cells expressing wild type or mutant protein to sequester exogenously provided OOHL. TraR mutants that are rapidly degraded or that cannot fold are defective in this assay (Chai and Winans, 2004). All dimerization mutants were proficient in binding OOHL, though the levels of OOHL binding were in many cases lower than with wild type TraR (Table 2).
We also assayed for the intracellular abundance of each mutant protein in A. tumefaciens by semi-quantitative western immunoblots. All mutant TraR proteins were readily detected immunologically, even those having very strong defects in transcription activation, dimerization, and DNA binding (Fig. 4, Table 2). These data, taken at face value, would tend to support the idea that dimerization does not play a critical role in resistance to proteolysis. However, subtle effects on protein stability could be hard to measure in this semi-quantitative assay of protein abundance.
Fig. 4.

Western immunoblots showing the abundance of wild type and mutant TraR proteins in clarified cell extracts. Cells were cultured in the presence of 10 nM OOHL. CRM: cross-reacting material, which was used to ensure that equivalent amounts of total protein was added to each lane. A strain carrying pPZP201 vector was used as a negative control while pYC335 served as a positive control for TraR.
We also used pulse-chase experiments in E. coli to evaluate the rate at which each mutant is degraded in vivo, as previously done using wild type protein (Zhu and Winans, 2001). As a negative control, a strain growing in the absence of OOHL showed very little accumulation of soluble TraR (Fig. 5), and the detectable TraR was rapidly degraded. The same strain cultivated in the presence of OOHL showed little or no TraR degradation. Mutant TraR proteins that have strong defects in dimerization, DNA binding, and activity were degraded more rapidly than the wild type protein in the presence of OOHL, but far more slowly than the wild type in the absence of OOHL. We conclude that blocks in dimerization lead to decreases in protease resistance, but that this effect is more subtle than the requirement of OOHL.
Fig. 5.
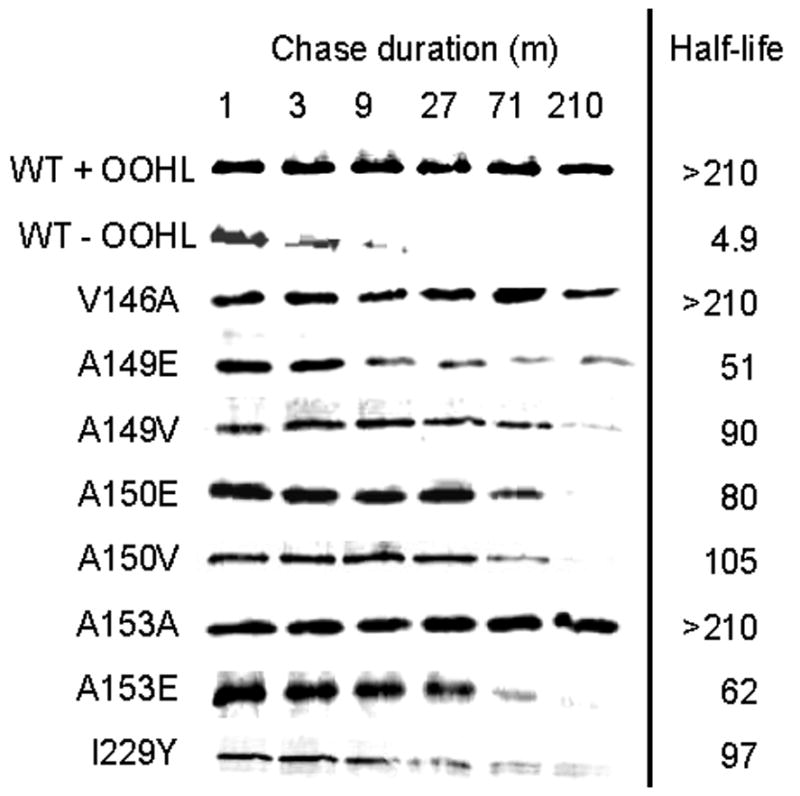
Pulse chase of TraR point mutants in E. coli. Cells expressing wild type or TraR point mutants from a phage T7 promoter were treated with rifampicin to block host transcription, then treated with [35S]methionine for 1 min, followed by addition of excess nonlabeled methionine. OOHL was provided prior to addition of radiolabel. At the indicated intervals after the addition of nonlabeled methionine, aliquots were frozen at −80°C to terminate proteolysis, then thawed and size-fractionated by SDS-PAGE. Radiolabel was quantitated using a Storm PhosphorImager. Calculated TraR half-lives are indicated at the right of each panel and represent the average of two independent experiments.
Discussion
The crystal structure of TraR-OOHL-DNA ternary complexes has enabled a series of studies about the roles of particular amino acid residues in binding of OOHL, in decoding of tra box DNA, in binding to RNA polymerase (Chai and Winans, 2004; White and Winans, 2005, 2007), and now in the dimerization of the TraR subunits. Each of these studies provides an ever-sharper picture of how TraR subunits interact with their molecular environment. The results obtained in this work corroborate a model (Fig. 6) in which OOHL binds to and stabilizes TraR, allowing time for monomers to form dimers. Dimerization further enhances resistance to degradation.
Fig. 6.
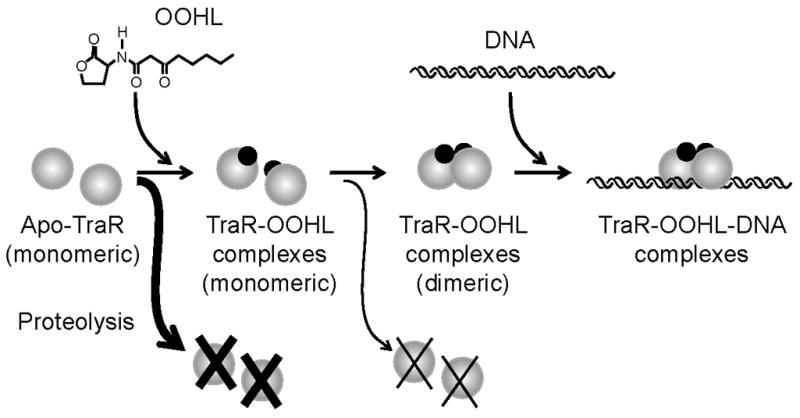
TraR folding and dimerization model. TraR monomers that fail to bind OOHL after synthesis are immediately targeted for proteolysis (Zhu and Winans, 1999, 2001), while TraR-OOHL complexes that fail to dimerize are also targeted for proteolysis, though not as rapidly as apo-TraR. TraR-OOHL dimers are competent to bind tra box DNA with high affinity and specificity.
By altering all residues at the subunit interface, we were able to evaluate the contribution of each to dimer formation and overall function. Residues A149, A150, and G153, all from helix 9, were critical for dimerization, as the mutations A149V, A149E, A150V, A150E, and G153E caused very strong defects in dimer formation (Fig. 2, Table 2). This could be due to the larger size of the mutant residues, and to the lack of available unoccupied space in this region of the dimer interface. Elsewhere in helix 9, mutations V146A, V146E, A157V and A157E caused relatively mild defects, even though the wild type residues make direct contacts with the opposite subunit. The structure suggests sufficient space to accommodate these bulkier residues. Helix 13 must also play a role in dimerization, as mutations A222D and I229Y caused severe defects in activity, DNA binding, and heterodimer formation (Tables 1 and 2), though more conservative mutations had milder defects. Overall, these results indicate that positions a and d on the helical wheel projection (which corresponds to residues 149, 153, 222, 225, and 229 on TraR, Fig. 1) show specific defects in protein dimerization (Lupas, 1996). A study with the E. coli FNR transcription factor also showed that residues located at similar positions on the helical wheel projection were the main ones directly involved in FNR dimerization (Moore and Kiley, 2001).
Though this study focused on the use of TraR mutants, we hope to make inferences about the wild type protein. Does wild type TraR ever exist as a monomer, and if so, what are its properties? At the earliest stages of quorum sensing induction, some cells could have as few as one TraR molecule, obviously a monomer. Even in cells with more than one monomer, newly synthesized TraR monomers must require a short time interval to dimerize, and even then, dimers exist in a dynamic equilibrium with monomers (Chai et al., 2001; Oger et al., 1998; Zhu and Winans, 1998). Would such monomers be protease-sensitive? For several reasons, we believe that the mutants we have described are a good approximation of wild type monomers. First, wherever possible, mutations were chosen that preserve interactions with neighboring amino acid residues and change only the surface of the protein. This was done to try to minimize defects in tertiary structure. Second, most of the mutations described here did not cause large decreases in the sequestration of OOHL in vivo, suggesting that their overall tertiary structure was intact. Third, a general pattern was observed, in which mutants with severe defects in dimerization tended to have severe defects in protein instability, and conversely (Fig. 2 and Fig. 5). These data strongly suggest that blocks in dimerization lead directly to decreased half-life, and that these mutants are informative about the properties of wild type monomeric protein.
TraR monomers would likely have a considerable hydrophobic surface exposed to solvent, which could in principle target the protein for proteolysis. It is conceivable however, that the hydrophobic patches of the two domains could interact with each other, possibly forming an antiparallel coiled coil. If so, this might shield both domains from proteases, and possibly increase the solubility of the protein. If that is true, the formation of a TraR dimer would require the dissociation of the N-terminal and C-terminal domains of each monomer before they could interact with their counterparts of the opposite subunit.
Dimerization of transcription factors is often mediated by parallel or antiparallel helices (Lupas, 1996). For instance, the catabolite activator protein (CAP) of E. coli dimerizes via two parallel ∀-helices that extend from residues 108 to 137 of each subunit (Joung et al., 1995; Schultz et al., 1991). The E. coli protein FNR (a CAP homolog), has been mutagenized in the corresponding dimerization interface, and the resulting mutants invariably showed defects in dimerization and in DNA binding (Moore and Kiley, 2001). FNR dimerization occurs only in anaerobic conditions, in which a (4Fe-4S)2+ cluster in each subunit is reduced to a (2Fe-2S)2+ form (Kiley and Beinert, 1998). The former is thought to distort the dimerization helix, thereby blocking dimer formation (Kiley and Beinert, 1998). The receiver domain of the E. coli PhoB protein has been resolved in the active and inactive conformations (Bachhawat et al., 2005). Dimerization of the inactive form is mediated primarily by ∀helix 1 of each subunit, while dimerization of the active form is mediated by ∀-helix 4, ∃-strand 5, and ∀-helix 5 of each subunit. In the former state, the two DNA binding domains of the dimer cannot associate, while in the latter state, they are brought into close proximity, allowing them to bind DNA. The transcription factor BmrR of Bacillus subtilis consists of an N-terminal DNA binding domain and a C-terminal ligand binding domain that are connected by a long ∀helix (residues 77–119). This helix forms a long antiparallel coiled coil with its counterpart from the opposite subunit (Heldwein and Brennan, 2001). This structure is thought to be conserved among other members of the MerR family. Two dimers of E. coli Lac repressor associate via four C-terminal ∀-helices, each 20 amino acids in length, with two antiparallel helices contributed from each dimer (Lewis et al., 1996). Each of these helices is connected to the rest of the subunit by a flexible linker, allowing considerable conformational freedom between the two dimers. The LysR-type regulator CbnR of Ralstonia eutropha consists of a N-terminal DNA binding domain fused to a C-terminal ligand binding domain via a long ∀ helix that forms a parallel coiled coil with the opposite subunit (Muraoka et al., 2003). In contrast, some transcription factors, including Lrp of E. coli and PrgX of Enterococcus facaelis, dimerize by other sorts of secondary structures (de los Rios and Perona, 2007; Shi et al., 2005). We have not seen other reports showing that multimerization of any of these proteins plays a role in their stability.
Experimental Procedures
Bacterial strains and plasmids
Bacterial strains and plasmids used in this study are listed in Table 3. E. coli strains were cultured in Luria broth (LB) or solid medium at 37°C (Miller, 1972). A. tumefaciens strains were cultured in AT minimal medium at 28°C (Cangelosi et al., 1991). Synthetic OOHL was provided by Dr. Anatol Eberhard (Cornell University). Antibiotics were added at the following concentrations: 100 μg ml−1 spectinomycin; 100 μg ml−1 kanamycin, and 100 μg ml−1 ampicillin. IPTG was added at 500 μM.
Table 3.
Strains and plasmids.
| Strains | Relevant features | References |
|---|---|---|
| DH5∀ | E. coli, ∀-complementation | Stratagene |
| NTL4 | A. tumefaciens C58, pTi less | (Luo et al., 2001) |
| BL21/DE3 | E. coli B Plac-gene 1 of bacteriophage T7 | (Studier et al., 1990) |
| KY2347 | E. coli MG1655, (clpPX-lon) 1196::cat | (Herman et al., 1998) |
| Plasmids | ||
| pPZP201 | Broad-host-range cloning vector, SpcR | (Hajdukiewicz et al., 1994) |
| pMCSG9 | PT7-his6-MBP-TEV, AmpR | (Donnelly et al., 2006) |
| pJZ358 | PT7-traR, AmpR | (Zhu and Winans, 1999) |
| pYC335 | Plac-traR in pPZP201 | (Chai and Winans, 2004) |
| pCEW250 | pBluescriptSK+ with consensus tra box at PtraI, KmR | (White and Winans, 2007) |
| pCEW260 | Consensus PtraI lacZ fusion, KmR | (White and Winans, 2007) |
| pAFM02 | traR cloned into pMCSG9 | This study |
| pUP-5M to 229Y | traR point mutants cloned into pYC335 | This study |
| pUP2-5M to 229Y | traR point mutants cloned into pAFM02 | This study |
| pUP3-5M to 229Y | traR point mutants cloned into pJZ358 | This study |
DNA manipulations
Recombinant DNA techniques were performed using established procedures (Sambrook and Russel, 2001). Plasmid DNA was isolated using QIAprep spin miniprep kits (Qiagen). DNA fragments generated by PCR or restriction digestion were gel purified using QIAquick Gel Extraction Kit (Qiagen). Restriction endonucleases were obtained from New England Biolabs and used according to methods described by the manufacturers. Plasmid DNA was introduced into E. coli and A. tumefaciens by electroporation (Cangelosi et al., 1991).
Site-directed mutagenesis of traR and cloning into different vectors
Site-directed mutagenesis of traR was performed by using a synthetic overlap extension PCR and two complementary mutagenic primers (Sambrook and Russel, 2001). For mutations on the N-terminal domain of TraR, a 978 bp fragment of pYC335 (Chai and Winans, 2004) was amplified using Pfx DNA Polymerase (Invitrogen) to include an unique EcoRI site (located upstream of traR) and a native SacII site (near codon 168 of traR). For mutations on the C-terminal domain of TraR, a 303 bp fragment of pYC335 was amplified as above to include a native SacII site (near codon 168 of TraR) and a unique MfeI site (just downstream of the stop codon of TraR). All oligonucleotides used in this study are listed in Table 4 and were obtained from Integrated DNA Technologies (Coralville, IA). The flanking primers were used in separate reactions with two different mutagenic primers that overlap at the mutation, using pYC335 as the template. These two PCR products were then combined and used as the template in a second round of PCR with the same flanking primers to generate the complete fragment. The second set of PCR products was digested with EcoRI and SacII (for N-terminal domain mutations) or SacII and MfeI (for C-terminal domain mutations), and ligated to pYC335 digested with the same enzymes. Mutant sequences were confirmed by automated DNA sequencing.
Table 4.
Oligonucleotide primers used in this study.
| Oligonucleotide Name | DNA Sequence |
|---|---|
| Flanking primers - N-terminal region | |
| PT1 | 5′ - CTCACTCATTAGGCACCCCAG - 3′ |
| TraRb | 5′ - GTACAACGTGTAGGGCAACGC- 3′ |
| Flanking primers - C-terminal region | |
| TraRa | 5′ - CATTCCTTCGCACCACCCC - 3′ |
| TraRb | 5′ - GTACAACGTGTAGGGCAACGC - 3′ |
| Mutagenic primers | |
| L5M-F | 5′ - CAGCACTGGATGGACAAGCTG - 3′ |
| L5M-R | 5′-CAGCTTGTCCATCCAGTGCTG - 3′ |
| K119A-F | 5′ - AATACCCATCGCGACCGCCAACG - 3′ |
| K119A-R | 5′ - CGTTGGCGGTCGCGATGGGTATT - 3′ |
| N122A-F | 5′ - CAAGACCGCCGCCGGCTTTATGT - 3′ |
| N122A-R | 5′ - ACATAAAGCCGGCGGCGGTCTTG - 3′ |
| V146A-F | 5′ - GATCGATGCAGCCGCAGCCGCTG - 3′ |
| V146A-R | 5′ - CAGCGGCTGCGGCTGCATCGATC - 3′ |
| V146E-F | 5′ - GATCGATGCAGAAGCAGCCGCTG - 3′ |
| V146E-R | 5′ - CAGCGGCTGCTTCTGCATCGATC - 3′ |
| A149E-F | 5′ - AGTCGCAGCCGAGGCAACCATCG - 3′ |
| A149E-R | 5′ - CGATGGTTGCCTCGGCTGCGACT - 3′ |
| A149V-F | 5′ - AGTCGCAGCCGTTGCAACCATCG - 3′ |
| A149V-R | 5′ - CGATGGTTGCAACGGCTGCGACT - 3′ |
| A150E-F | 5′ - CGCAGCCGCTGAAACCATCGGGC - 3′ |
| A150E-R | 5′ - GCCCGATGGTTTCAGCGGCTGCG - 3′ |
| A150V-F | 5′ - CGCAGCCGCTGTAACCATCGGGC - 3′ |
| A150V-R | 5′ - GCCCGATGGTTACAGCGGCTGCG - 3′ |
| G153A-F | 5′ - TGCAACCATCGCGCAGATCCATG - 3′ |
| G153A-R | 5′ - CATGGATCTGCGCGATGGTTGCA - 3′ |
| G153E-F | 5′ - TGCAACCATCGAGCAGATCCATG - 3′ |
| G153E-R | 5′ - CATGGATCTGCTCGATGGTTGCA - 3′ |
| Q154A-F | 5′ - AACCATCGGGGCGATCCATGCCC - 3′ |
| Q154A-R | 5′ - GGGCATGGATCGCCCCGATGGTT - 3′ |
| Q154E-F | 5′ - AACCATCGGGGAGATCCATGCCC - 3′ |
| Q154E-R | 5′ - GGGCATGGATCTCCCCGATGGTT - 3′ |
| I155F-F | 5′ - ATCGGGCAGTTCCATGCCCGC - 3′ |
| I155F-R | 5′ - GCGGGCATGGAACTGCCCGAT - 3′ |
| A157E-F | 5′ - GCAGATCCATGAGCGCATCTCAT - 3′ |
| A157E-R | 5′ - ATGAGATGCGCTCATGGATCTGC - 3′ |
| A157V-F | 5′ - GCAGATCCATGTCCGCATCTCAT - 3′ |
| A157V-R | 5′ - ATGAGATGCGGACATGGATCTGC - 3′ |
| S160A-F | 5′ - CCGCATCGCATTCCTTCGCA - 3′ |
| S160A-R | 5′ - TGCGAAGGAATGCGATGCGG - 3′ |
| S160F-F | 5′ - GCCCGCATCTTCTTCCTTCGC - 3′ |
| S160F-R | 5′ - GCGAAGGAAGAAGATGCGGGC - 3′ |
| F161A-F | 5′ - CCGCATCTCAGCCCTTCGCACCA - 3′ |
| F161A-R | 5′ - TGGTGCGAAGGGCTGAGATGCGG - 3′ |
| F161K-F | 5′ - CCGCATCTCAAAACTTCGCACCA - 3′ |
| F161K-R | 5′ - TGGTGCGAAGTTTTGAGATGCGG - 3′ |
| A222D-F | 5′ - GCAGCAAGGACCATCTTACC - 3′ |
| A222D-R | 5′ - GGTAAGATGGTCCTTGCTGC - 3′ |
| A222S-F | 5′ - GCAGCAAGTCCCATCTTACC - 3′ |
| A222S-R | 5′ - GGTAAGATGGGACTTGCTGC - 3′ |
| T225S-F | 5′ - CCCATCTTTCCGCGCTCGCC - 3′ |
| T225S-R | 5′ - GGCGAGCGCGGAAAGATGGG - 3′ |
| I229Y-F | 5′ - GCTCGCCTACCGGCGGAAAC - 3′ |
| I229Y-R | 5′ - GTTTCCGCCGGTAGGCGAGC - 3′ |
| traR cloning into pMCSG9 | |
| traR-fusion-F | 5′ - TACTTCCAATCCAATATGCAGCACTGGCT - 3′ |
| traR-fusion-R | 5′ - TTATCCACTTCCAATTCTCAGATGAGTTTCCG - 3′ |
| To check correct cloning into pMCSG9 | |
| pMCSG9-F | 5′ - ACGAGGAAGAGTTGGCGAAAGATC - 3′ |
| pMCSG9-R | 5′ - TTAGAGGCCCCAAGGGGTTATGCTA - 3′ |
| Oligonucleotides used in the gel shift assays | |
| Ptra-box For | 5′ - GAATTCTATGTGCAGATCTGCACATAGC - 3′ |
| Ptra-box Rev | 5′ - GGATCAATACGACGAGCTCGAGGATCCAGC - 3′ |
| Pcontrol gel shift For | 5′ - CCGCTACAGGGCGCGTCC - 3′ |
| Pcontrol gel shift rev | 5′ - CCAATTCGCCCTATAGTG - 3′ |
The traR point mutants were also cloned into vectors pAFM02 and pJZ358. For the former plasmid, mutants that lie between codons 85 and 168 were cloned by digesting pYC335 derivatives with BstB1 and SacII enzymes. The digestion of pYC335 derivatives with the above enzymes generated four fragments of distinct sizes. The one of 306 bp contained the mutated codons that were ligated in pAFM02 digested with the same enzymes. For other mutants located downstream of the codon 168, the unique sites, in both plasmids, SacII and HindIII were used. For cloning into pJZ358, digestion was performed with HindIII and BbsI enzymes. A fragment of 867 bp originating from pYC335 derivatives starting at codon 24 (BbsI site) of traR and going 227 bp downstream of its stop codon (HindIII site) was used to replace a fragment of 922 bp from pJZ358 digested with the same enzymes.
Measurement of TraR activity in vivo
Bioassays of TraR activity were performed with each TraR mutant using NTL4(pCEW260)(pYC335) or derivatives of pYC335 carrying each of the traR mutants. Strains were cultured in AT minimal medium at 28°C to an OD600 of 0.3–0.4. Each culture was then diluted 20-fold into AT medium containing concentrations of 0.1, 1, 10 or 100 nM OOHL, incubated with vigorous aeration at 28°C for 8 h, and assayed for∃-galactosidase activity (Miller, 1972). Experiments were performed in triplicate using three different isolates for each strain.
Heterodimer formation assay
To measure the ability of TraR to form heterodimers with MBP-TraR fusions, clarified extracts containing MBP-TraR fusions expressed from E. coli BL21/DE3(pAFM02) or pAFM02 derivatives carrying each TraR mutant were combined with equal volumes of clarified extracts containing native TraR protein expressed from BL21/DE3(pJZ358). Combined supernatants were incubated for 2 h at 28≅C to allow subunit exchange, and were applied to an amylose affinity chromatography column (New England Biolabs). Proteins were step eluted with buffer containing 10 mM maltose. Purified proteins were size-fractionated using 12% SDS-PAGE gels and band intensities were quantified using ImageJ software (http://rsbweb.nih.gov/ij/). Cells expressing only TraR or only MBP-TraR respectively were used as negative and positive controls.
Electrophoretic mobility shift assays
To perform gel shift assays, clarified cell extracts were prepared from E. coli strain KY2347 (a clp, lon mutant) carrying pYC335 or its derivatives with each of the traR point mutants. Strains were cultured in LB broth containing 100 μg mL−1 of spectinomycin in the presence of 200 nM OOHL at 28− C. When cultures reached OD600 of 0.2, IPTG was added to a final concentration of 500 μM to induce TraR expression, and incubation was followed by an additional 6 h. TraR abundance in the soluble fraction was estimated using Western immunoblots as previously described (White and Winans, 2005). A 247 nucleotide PCR fragment containing a consensus tra box was obtained from plasmid pCEW250 using primers Ptra-box For and Ptra-box Rev (Table 4). A negative control fragment of 211 nucleotides in length was PCR amplified from plasmid pCEW250 with primers Pcontrol-gel-shift-For and Pcontrol-gel-shift-Rev. Both fragments were end-labeled with [(-32P]-dATP (Pelkin Elmer) using T4 polynucleotide kinase (New England Biolabs). Binding reactions were performed as previously described (White and Winans, 2005), with equivalent amounts of soluble TraR in each reaction. Gel shift assays were performed with independent clarified lysates in duplicate for each strain, and data were analyzed using a Storm B840 Phosphorimager.
Measurements of TraR abundance in A. tumefaciens
The abundance of each TraR allele was determined using strain NTL4(pCEW160)(pYC335) or derivatives of pYC335 carrying each of the traR mutants. Strains were cultured in AT at 28− C with 10 nM OOHL. Cells were harvested at mid-log phase, and westerns were performed as described previously (White and Winans, 2005). Westerns were prepared with independent cell lysates at least twice for each strain. Data were analyzed using IMAGEJ and normalized against cross-reacting material in each lane.
OOHL sequestration assay by TraR in whole cells
A. tumefaciens strain NTL4(pCEW260)(pYC335) or derivatives of pYC335 carrying each TraR mutant allele were used for OOHL sequestration assays as previously described (Chai et al., 2001). All assays were performed twice with independent cultures.
TraR stability in E. coli
The measurement of TraR turnover for key TraR point mutants was determined using strain BL21/DE3(pJZ358) (Zhu and Winans, 1999) or derivatives of pJZ358 carrying traR mutants. The experiments were performed as described previously (Zhu and Winans, 2001) except that OOHL was added 10 min before the addition of radiolabel.
Acknowledgments
The authors gratefully acknowledge Dr Anatol Eberhard (Cornell University) for the synthesis and purification of OOHL used in this study, and Ana Lidia Flores-Mireles for construction of pAFM02 plasmid. We also thank all members of our laboratory for helpful discussions and critical review of the manuscript. Funding for this study was provided by the National Institute of General Medical Sciences (GM41892). UMP acknowledges the financial support of the Brazilian government through a fellowship grant from the Coordenacao de Aperfeicoamento de Pessoal de Nivel Superior (Capes).
References
- Bachhawat P, Swapna GV, Montelione GT, Stock AM. Mechanism of activation for transcription factor PhoB suggested by different modes of dimerization in the inactive and active states. Structure. 2005;13:1353–1363. doi: 10.1016/j.str.2005.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Zhu J, Winans SC. TrlR, a defective TraR-like protein of Agrobacterium tumefaciens, blocks TraR function in vitro by forming inactive TrlR:TraR dimers. Mol Microbiol. 2001;40:414–421. doi: 10.1046/j.1365-2958.2001.02385.x. [DOI] [PubMed] [Google Scholar]
- Chai Y, Winans SC. Site-directed mutagenesis of a LuxR-type quorum-sensing transcription factor: alteration of autoinducer specificity. Mol Microbiol. 2004;51:765–776. doi: 10.1046/j.1365-2958.2003.03857.x. [DOI] [PubMed] [Google Scholar]
- de los Rios S, Perona JJ. Structure of the Escherichia coli leucine-responsive regulatory protein Lrp reveals a novel octameric assembly. J Mol Biol. 2007;366:1589–1602. doi: 10.1016/j.jmb.2006.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly MI, Zhou M, Millard CS, Clancy S, Stols L, Eschenfeldt WH, Collart FR, Joachimiak A. An expression vector tailored for large-scale, high-throughput purification of recombinant proteins. Protein Expr Purif. 2006;47:446–454. doi: 10.1016/j.pep.2005.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhard A. Inhibition and activation of bacterial luciferase synthesis. J Bacteriol. 1972;109:1101–1105. doi: 10.1128/jb.109.3.1101-1105.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuqua WC, Winans SC. A LuxR-LuxI type regulatory system activates Agrobacterium Ti plasmid conjugal transfer in the presence of a plant tumor metabolite. J Bacteriol. 1994;176:2796–2806. doi: 10.1128/jb.176.10.2796-2806.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajdukiewicz P, Svab Z, Maliga P. The small, versatile pPZP family of Agrobacterium binary vectors for plant transformation. Plant Mol Biol. 1994;25:989–994. doi: 10.1007/BF00014672. [DOI] [PubMed] [Google Scholar]
- Heldwein EE, Brennan RG. Crystal structure of the transcription activator BmrR bound to DNA and a drug. Nature. 2001;409:378–382. doi: 10.1038/35053138. [DOI] [PubMed] [Google Scholar]
- Herman C, Thevenet D, Bouloc P, Walker GC, D’Ari R. Degradation of carboxy-terminal-tagged cytoplasmic proteins by the Escherichia coli protease HflB (FtsH) Genes Dev. 1998;12:1348–1355. doi: 10.1101/gad.12.9.1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung JK, Chung EH, King G, Yu C, Hirsh AS, Hochschild A. Genetic strategy for analyzing specificity of dimer formation: Escherichia coli cyclic AMP receptor protein mutant altered in its dimerization specificity. Genes Dev. 1995;9:2986–2996. doi: 10.1101/gad.9.23.2986. [DOI] [PubMed] [Google Scholar]
- Kiley PJ, Beinert H. Oxygen sensing by the global regulator, FNR: the role of the iron-sulfur cluster. FEMS Microbiol Rev. 1998;22:341–352. doi: 10.1111/j.1574-6976.1998.tb00375.x. [DOI] [PubMed] [Google Scholar]
- Lewis M, Chang G, Horton NC, Kercher MA, Pace HC, Schumacher MA, Brennan RG, Lu P. Crystal structure of the lactose operon repressor and its complexes with DNA and inducer. Science. 1996;271:1247–1254. doi: 10.1126/science.271.5253.1247. [DOI] [PubMed] [Google Scholar]
- Li PL, Farrand SK. The replicator of the nopaline-type Ti plasmid pTiC58 is a member of the repABC family and is influenced by the TraR-dependent quorum-sensing regulatory system. J Bacteriol. 2000;182:179–188. doi: 10.1128/jb.182.1.179-188.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZQ, Farrand SK. Signal-dependent DNA binding and functional domains of the quorum-sensing activator TraR as identified by repressor activity. Proc Natl Acad Sci U S A. 1999;96:9009–9014. doi: 10.1073/pnas.96.16.9009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZQ, Clemente TE, Farrand SK. Construction of a derivative of Agrobacterium tumefaciens C58 that does not mutate to tetracycline resistance. Mol Plant Microbe Interact. 2001;14:98–103. doi: 10.1094/MPMI.2001.14.1.98. [DOI] [PubMed] [Google Scholar]
- Luo ZQ, Smyth AJ, Gao P, Qin Y, Farrand SK. Mutational analysis of TraR. Correlating function with molecular structure of a quorum-sensing transcriptional activator. J Biol Chem. 2003;278:13173–13182. doi: 10.1074/jbc.M210035200. [DOI] [PubMed] [Google Scholar]
- Lupas A. Coiled coils: new structures and new functions. Trends Biochem Sci. 1996;21 :375–382. [PubMed] [Google Scholar]
- Moore LJ, Kiley PJ. Characterization of the dimerization domain in the FNR transcription factor. J Biol Chem. 2001;276:45744–45750. doi: 10.1074/jbc.M106569200. [DOI] [PubMed] [Google Scholar]
- Muraoka S, Okumura R, Ogawa N, Nonaka T, Miyashita K, Senda T. Crystal structure of a full-length LysR-type transcriptional regulator, CbnR: unusual combination of two subunit forms and molecular bases for causing and changing DNA bend. J Mol Biol. 2003;328:555–566. doi: 10.1016/s0022-2836(03)00312-7. [DOI] [PubMed] [Google Scholar]
- Oger P, Kim KS, Sackett RL, Piper KR, Farrand SK. Octopine-type Ti plasmids code for a mannopine-inducible dominant-negative allele of traR, the quorum-sensing activator that regulates Ti plasmid conjugal transfer. Mol Microbiol. 1998;27:277–288. doi: 10.1046/j.1365-2958.1998.00671.x. [DOI] [PubMed] [Google Scholar]
- Pappas KM, Winans SC. A LuxR-type regulator from Agrobacterium tumefaciens elevates Ti plasmid copy number by activating transcription of plasmid replication genes. Mol Microbiol. 2003;48:1059–1073. doi: 10.1046/j.1365-2958.2003.03488.x. [DOI] [PubMed] [Google Scholar]
- Pappas KM, Weingart CL, Winans SC. Chemical communication in proteobacteria: biochemical and structural studies of signal synthases and receptors required for intercellular signalling. Mol Microbiol. 2004;53:755–769. doi: 10.1111/j.1365-2958.2004.04212.x. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russel DW. Molecular Cloning - A laboratory manual. New York: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Schultz SC, Shields GC, Steitz TA. Crystal structure of a CAP-DNA complex: the DNA is bent by 90 degrees. Science. 1991;253:1001–1007. doi: 10.1126/science.1653449. [DOI] [PubMed] [Google Scholar]
- Shi K, Brown CK, Gu ZY, Kozlowicz BK, Dunny GM, Ohlendorf DH, Earhart CA. Structure of peptide sex pheromone receptor PrgX and PrgX/pheromone complexes and regulation of conjugation in Enterococcus faecalis. Proc Natl Acad Sci U S A. 2005;102:18596–18601. doi: 10.1073/pnas.0506163102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- Vannini A, Volpari C, Gargioli C, Muraglia E, Cortese R, De Francesco R, Neddermann P, Marco SD. The crystal structure of the quorum sensing protein TraR bound to its autoinducer and target DNA. Embo J. 2002;21:4393–4401. doi: 10.1093/emboj/cdf459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters CM, Bassler BL. Quorum sensing: cell-to-cell communication in bacteria. Annu Rev Cell Dev Biol. 2005;21:319–346. doi: 10.1146/annurev.cellbio.21.012704.131001. [DOI] [PubMed] [Google Scholar]
- White CE, Winans SC. Identification of amino acid residues of the Agrobacterium tumefaciens quorum-sensing regulator TraR that are critical for positive control of transcription. Mol Microbiol. 2005;55:1473–1486. doi: 10.1111/j.1365-2958.2004.04482.x. [DOI] [PubMed] [Google Scholar]
- White CE, Winans SC. The quorum-sensing transcription factor TraR decodes its DNA binding site by direct contacts with DNA bases and by detection of DNA flexibility. Mol Microbiol. 2007;64:245–256. doi: 10.1111/j.1365-2958.2007.05647.x. [DOI] [PubMed] [Google Scholar]
- Wickner S, Maurizi MR, Gottesman S. Posttranslational quality control: folding, refolding, and degrading proteins. Science. 1999;286:1888–1893. doi: 10.1126/science.286.5446.1888. [DOI] [PubMed] [Google Scholar]
- Zhang RG, Pappas T, Brace JL, Miller PC, Oulmassov T, Molyneaux JM, Anderson JC, Bashkin JK, Winans SC, Joachimiak A. Structure of a bacterial quorum-sensing transcription factor complexed with pheromone and DNA. Nature. 2002;417:971–974. doi: 10.1038/nature00833. [DOI] [PubMed] [Google Scholar]
- Zhu J, Winans SC. Activity of the quorum-sensing regulator TraR of Agrobacterium tumefaciens is inhibited by a truncated, dominant defective TraR-like protein. Mol Microbiol. 1998;27:289–297. doi: 10.1046/j.1365-2958.1998.00672.x. [DOI] [PubMed] [Google Scholar]
- Zhu J, Winans SC. Autoinducer binding by the quorum-sensing regulator TraR increases affinity for target promoters in vitro and decreases TraR turnover rates in whole cells. Proc Natl Acad Sci U S A. 1999;96:4832–4837. doi: 10.1073/pnas.96.9.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Winans SC. The quorum-sensing transcriptional regulator TraR requires its cognate signaling ligand for protein folding, protease resistance, and dimerization. Proc Natl Acad Sci U S A. 2001;98:1507–1512. doi: 10.1073/pnas.98.4.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]