

Abstract
Calcium (Ca2+) signaling plays a major role in a wide range of physiological functions including control and regulation of cardiac and skeletal muscle performance and vascular tone [1, 2]. As all Ca2+ signals require proteins to relay intracellular Ca2+ oscillations downstream to different signaling networks, a specific toolkit of Ca2+-sensor proteins involving members of the EF-hand S100 Ca2+ binding protein superfamily maintains the integrity of the Ca2+ signaling in a variety of cardiac and vascular cells, transmitting the message with great precision and in a temporally and spatially coordinated manner [3–6]. Indeed, the possibility that S100 proteins might contribute to heart and vascular diseases was first suggested by the discovery of distinctive patterns of S100 expression in healthy and diseased hearts and vasculature from humans and animal heart failure (HF) models [7–18]. Based on more elaborate genetic studies in mice and strategies to manipulate S100 protein expression in human cardiac, skeletal muscle and vascular cells, it is now apparent that the integrity of distinct S100 protein isoforms in striated muscle and vascular cells such as S100A1, S100A4, S100A6, S100A8/A9 or S100B is a basic requirement for normal cardiovascular and muscular development and function; loss of integrity would naturally lead to profound deregulation of the implicated Ca2+ signaling systems with detrimental consequences to cardiac, skeletal muscle, and vascular function [7–20]. The brief debate and discussion here are confined by design to the biological actions and pathophysiological relevance of the EF-hand Ca2+-sensor protein S100A1 in the heart, vasculature and skeletal muscle with a particular focus on current translational therapeutic strategies [4, 21, 22]. By virtue of its ability to modulate the activity of numerous key effector proteins that are essentially involved in the control of Ca2+- and NO-homeostasis in cardiac, sketelal muscle and vascular cells, S100A1 has been proven to play a critical role both in cardiac performance, blood pressure regulation and skeletal muscle function [4, 21, 23]. Given that deregulated S100A1 expression in cardiomyocytes and endothelial cells has recently been linked to heart failure and hypertension [4, 21, 23], it is arguably a molecular target of considerable clinical interest as S100A1 targeted therapies have already been successfully investigated in preclinical translational studies.
Keywords: S100, S100A1, Calcium, Nitric oxide, Cardiomyocyte, Endothelial Cell
1. S100A1 and the S100 gene family: genomic organization and molecular structure
1.1 S100A1 genomic organization
S100A1 is a member of the multigenic S100 protein family constituting by far the largest subgroup within the EF-hand Ca2+ binding protein superfamily [6, 24]. These proteins were first discovered in 1965 by B.W. Moore and characterized as “S100”, accounting for their solubility in a 100%-saturated solution with ammonium sulphate [24]. Since then S100 proteins have emerged as regulators of fundamental cellular and molecular functions, including cell proliferation, differentiation, survival and motility as well as protein phosphorylation, enzyme activity, cytoskeleton dynamics, transcriptional activity, and NO homeostasis and Ca2+ dynamics.
At present, 25 individual human S100 genes have been identified, most of which are tightly clustered in a 2.05 Mbp segment of the human chromosome 1q21 (2q34 in rat and 3F1–F2 in mouse) including S100A1 (Figure 1, red box) [6]. The organization of this gene cluster is highly conserved in evolution and the adopted nomenclature designates clustered genes as S100 followed by arabic numbers (S100A1, S100A2, S100A3, etc.) (Figure 1) [6]. S100A1, alike other S100 proteins, does not have apparent intrinsic catalytic activity and is considered to function as an intracellular Ca2+ sensor [25, 26]. Likewise calmodulin (CaM) or troponin C, S100A1 undergoes a conformational change (Figure 2A and B) [27, 28] enabling Ca2+ dependent interaction with a specific subset of target proteins in cardiac and vascular cells and eventually modulation of their activity.
Figure 1. Comparative map of the conserved S100 gene cluster on human chromosome 1q21 and mouse chromosome 3F1–3F2.
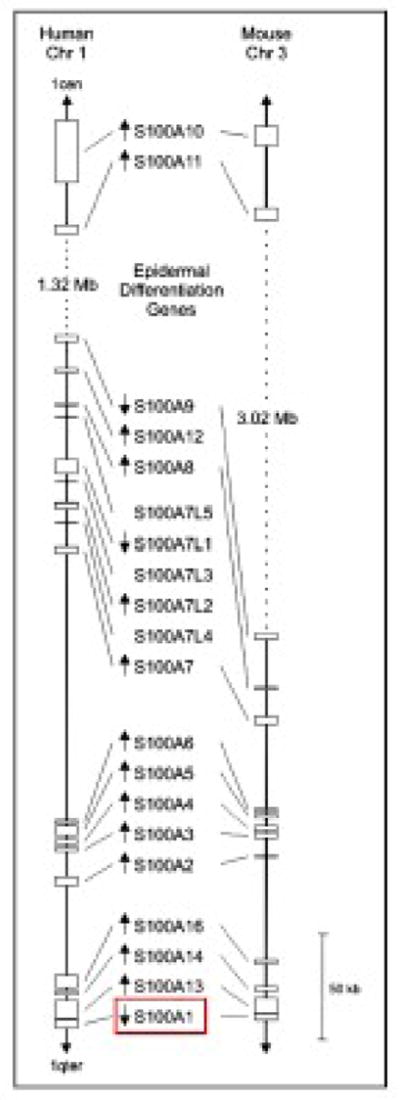
Position and size of each gene corresponds to the NCBI genome sequences of human and mouse (as of May 2009). In both species, a cluster of genes involved in epidermal differentiation is located between S100A9 and S100A11. The transcriptional direction of each gene is indicated by arrow. Position of S100A1 within the S100 gene cluster is highlighted in red. Reproduced with modifications from Marenholz et al. [6].
Figure 2. Three dimensional (3D) solution structure of S100A1 determined by NMR spectroscopy.

A,3D S100A1 structure in the apo state: S100A1 is composed of two identical subunits (blue/red) each composed of a repetitive helix-loop-helix calcium binding motif that is connected by a short linear linker (hinge) region. Dimerization occurs in an antiparallel manner between helix (H) 1 and 1′ stabilized by hydrophobic bonds. Hydrophobic residues in the hinge region and H4 in the blue coloured dimer are shown in yellow. This 3D structure reflects the characteristic molecular backbone of S100 proteins. B, 3D S100A1 structure in the calcium bound state: Ca2+ binding both to the N- and C- terminal EF hand motif mainly results in altered orientation of H3 and H4 as well as the hinge region while the residual structure of the dimer remains nearly unaffected. As indicated by arrows, the N-terminal part of H3/3′ and the C-terminal part of H4/4′ in both dimers swing apart exposing hydrophobic residues of both helices and the hinge region to the surface facilitating their Ca2+ dependent interaction with target molecules. S100A1ct is indicated by the dashed red box. Reproduced with modifications from Wright et al. [27].
1.2 S100A1 molecular structure
Within cells, the structural and functional unit of S100A1 has been described as a symmetric, antiparallel dimer of two monomeric subunits (Figure 2A) [27, 28]. The dimeric structure is favored even at picomolecular monomer concentrations and reflects the common intracellular architecture typical for all S100 proteins. The monomer itself with an apparent molecular weight of 10.4 kDa consists of two EF-hand Ca2+ binding motifs interconnected by an intermediate region often referred to as the hinge region (Figure 3). In each EF hand, a Ca2+ binding loop is flanked by α-helices, resulting in a helix E-loop-F-helix arrangement as prototypically described for parvalbumin. Thus as depicted in Figure 3, helices I and II flank the N-terminal loop (loop I), whereas helices III and IV flank the C-terminal loop (loop II) and stabilization of the dimeric structure actually occurs independent of Ca2+ binding through hydrophobic bonds between helices I of each monomer (Figure 2A) [27, 28].
Figure 3. Schematic representation of the secondary structure of an S100 protein.

The monomeric structure consists of a repetitive α-helix-loop-α-helix EF-hand Ca2+ binding motif. The non-canonical N-terminal and canonical C-terminal EF-hand is connected by a linker region (hinge region). The hinge region and C-terminal extension (boxed in red) display the least amount of sequence homology among S100 paralogs. Reproduced with modifications from Donato et al. [77].
Upon Ca2+ binding to both EF-hands, S100A1 alike other S100 proteins undergoes a major conformational change in the C-terminal EF-hand leading to the exposure of a concave hydrophobic pocket in each monomer (Figure 2A and B) [27, 28]. The pocket, which is mainly composed of residues of the hinge region, helix III and the C-terminus (helix IV), is considered to be critically involved in Ca2+-dependent recognition of S100A1 target proteins. Given the fact that the pocket forming domains, including both hinge and C-terminal residues, exhibit the highest sequence variation, they are further viewed to convey isoform-specific target recognition and functional specificity of individual S100 proteins [5, 29].
As Ca2+ binding to individual EF-hands in the S100A1 dimer has been estimated to occur with a Kd of ≈ 200–500 μM at the N-terminal and a Kd ≈ 10–50 μM at the C-terminal site, it is relevant to point out that S100A1 Ca2+ affinity is apparently tightly regulated by redox- and NO-dependent posttranslational modifications [30–32]. S-nitrosylation or S-gluthionylation of a single cysteine residue in the C-terminal region of the S100A1 monomer dramatically enhances Ca2+ affinity of both EF-hands thereby facilitating Ca2+ binding even at nanomolar free Ca2+ concentrations ([Ca2+]) [30–32]. In other words, redox- and NO-related posttranslational modification of a hydrophobically compartmentalized single cysteine might enable S100A1 to sense spatially defined short-term as well as long-term global Ca2+ oscillations in cardiac and vascular cells over a relatively wide concentration range.
Therefore, it is tempting to speculate that the biological function of S100A1 as a calcium signal transmitter is probably regulated by the delicately balanced NO/redox equilibrium within cardiac and vascular cells [33]. However, whether the Ca2+ sensor can indeed activate and modulate target proteins on a millisecond time scale with a short-lived or even persistent activation after intracellular [Ca2+] has returned to low resting levels due to S100A1 Ca2+ on- and off-rates remains to be determined.
2. Cardiovascular S100A1 expression pattern, molecular targets and physiological functions
2.1 S100A1 expression in cardiomyocytes
S100A1 is preferentially expressed in human cardiac muscle but also found in skeletal muscle, brain and kidney (most recent expression profiles are accessible through http://www.genecards.org/cgi-bin/carddisp.pl?gene=S100A1&search=S100A1), albeit at much lower concentrations. Comparative analyses revealed a similar S100A1 expression pattern in rodents and larger mammals [34–38]. Heizmann and co-workers showed that S100A1 mRNA and protein levels steadily increase in the developing mouse heart during embryonic development and reach a plateau in ventricular myocardium in the postnatal state [36], where it predominantly resides within cardiomyocytes [38].
Although vascular S100A1 expression in mice has not been detected in the fetal state, S100A1 mRNA and protein expression have most recently been described in adult rodent and human capillary, aortic and femoral artery endothelial cells (ECs) [23, 39, 40]. Comparative protein expression analyses revealed an approximately 50-fold lower S100A1 protein level in adult ECs than ventricular cardiomyocytes (Patrick Most, unpublished results). It is therefore possible that S100A1 is either not expressed in maturing ECs and vascular structures until a relatively late developmental stage or vascular S100A1 expression in fetal vascular structures has just been below the detection threshold of the methods employed.
Analysis of the murine S100A1 promoter sequence by Kiewitz et al. recently identified various transcription factor (TF) consensus sites (i.e. homeobox protein NK-2 homolog E, myocyte enhancer factor-2 and GATA4 among others) [36] that drive cardiac-specific expression of cardiac troponin C or calsequestrin. Although these TFs might account for predominant cardiac and cardiomyocyte expression of S100A1, it is currently unclear which factors actually control S100A1 expression in vascular ECs.
2.2 S100A1 subcellular location and molecular targets in cardiomyocytes
Sarcoplasmic reticulum
In ventricular cardiomyocytes, S100A1 mostly displays a striated-like pattern and resides both at the junctional and longitudinal sarcoplasmic reticulum (SR) [13, 34, 41] that functions as the major calcium source and sink during the contraction-relaxation cycle. The EF-hand Ca2+ sensor co-localizes and interacts both with the Ca2+ ATPase (SERCA2)/phospholamban (PLB) complex (representative images are shown in Figure 4A–C) [13, 42–44] and the cardiac SR Ca2+ release channel/ryanodine receptor (RyR2) [13, 34, 41, 42, 44–46] (Figure 4D). Current evidence indicates that S100A1 binding to the RyR2 is indeed Ca2+ dependent and mapping studies disclosed amino acid residues 75–94 within the α-helical S100A1 C-terminal domain as a crucial interface for the interaction [13, 45–48]. Although, recent studies revealed that S100A1 binds to the calmodulin binding domain in the cytoplasmic portion of the skeletal muscle ryanodine receptor isoform (RyR1) [47, 48] - which is conserved between RyR1 and RyR2 - its interaction sites and binding stoichiometry with the macromolecular RyR2 complex have yet to be characterized.
Figure 4. Mechanisms conveying S100A1 inotropic actions in ventricular cardiomyocytes.

A–C, representative confocal fluorescent images highlight subcellular S100A1 distribution in a rat ventricular cardiomyocyte exhibiting a striated-like pattern. Note that S100A1 (red) partially co-localizes (violett) with SERCA2 (blue) as illustrated in the magnified inlets (images taken from Most et al. [4]). D, Normal excitation contraction (ec) coupling in a non-failing cardiomyocyte requires (1) action potential dependent systolic opening of the L-type calcium channel (LLC) enabling a transsarcolemmal Ca2+ entry (2) triggering sarcoplasmic reticulum (SR) Ca2+ release via the ryanodine receptor 2 (RyR2) that in turn (3) activates myofilament cross-bridge cycling and force development. During diastole, (4) SR Ca2+ resequestration occurs through the SR Ca2+-ATPase (SERCA2) allowing Ca2+ dissociation from myofilaments and relaxation to occur. (5) Under steady-state conditions, the sodium-calcium exchanger (NCX) exerts balanced extrusion of LLC-mediated Ca2+ entry. S100A1 interacts both with RyR2 and the SERCA2a/PLB complex and is present at myofilaments and in mitochondria. E, Proposed mechanistic model for S100A1 actions in cardiomyocytes: Increased cardiomyocyte S100A1 protein levels result (a) in enhanced ec coupling gain and systolic SR Ca2+ release through enhanced RyR2 but not LLC activity. (b) Myofilament compliance is increased via S100A1/titin interaction accompanied by (c) facilitated diastolic Ca2+ dissociation independent of PKA. Augmented systolic SR Ca2+ release is balanced by (d) enhanced diastolic SERCA2a activity independent of PKA and CaMKII. Enhanced SERCA2a activity together with (e) diminished diastolic RyR2 activity result in increased SR Ca2+ resequestration and prevents SR Ca2+ leak whereas S100A1 does not affect diastolic Ca2+ extrusion through the NCX. Finally, S100A1/F1-ATPase and S100A1/ANT interaction is associated with (f) enhanced generation and cytoplasmic content of ATP in cardiomyocytes. Inlets illustrate the net effect of enhanced cardiomyocyte S100A1 protein levels evoking augmented cytosolic Ca2+ transients and force generation. Reproduced with modifications from Most et al. [4].
Despite these unresolved questions, numerous studies show that S100A1 modulates both systolic and diastolic RyR2 function and cardiomyocte SR Ca2+ release, respectively [13, 42, 45, 46, 49]. At nanomolar [Ca2+], S100A1 was found to decrease [3H]-ryanodine binding to the cardiac SR Ca2+ release channel [13, 42]. In line with these results, Voelkers et al. showed that S100A1 can reduce Ca2+ spark frequency and SR Ca2+ leak in quiescent cardiomyocytes [44] and cardiac SR vesicle preparations [13, 46, 49] indicating that S100A1 promotes diastolic RyR2 closure during cardiomyocyte relaxation. However, at micromolar [Ca2+], S100A1 resulted in enhanced [3H]-ryanodine binding to the RyR2. In accord, Kettlewell et al. [41] demonstrated increased SR fractional Ca2+ release in voltage-clamped cardiomyocytes and SR vesicular preparations and [42, 45, 49] suggesting that S100A1 can also augment RyR2 opening during systole. Given that these effects occur in the presence of unchanged sarcolemmal Ca2+ fluxes via L-type Ca2+ channel (ICa) and sodium-calcium exchanger (NCX) [42], S100A1 protein levels seem to modulate and enhance the excitation-contraction coupling gain in cardiomyocytes.
The differential effects on RyR2 activity rather predict several than a single S100A1 binding site at the RyR2. This notion is further supported by previous mapping studies that have identified at least three distinct S100A1 binding domains along with different binding affinities within the cytoplasmic portion of the RyR1 [50]. Although previous studies demonstrated that the Ca2+ dependent interaction between S100A1 and the RyR2 neither alter FKBP12.6 nor sorcin stoichiometry with the SR Ca2+ release channel at nanomolar [Ca2+] [46], it is tempting to speculate that inhibitory and stimulatory binding sites for S100A1 might co-exist at the RyR2. Finally S100A1, in concert with other accessory modulatory proteins such as FKBP12.6 and sorcin, might eventually account for normal diastolic and systolic function of the macromolecular SR Ca2+ release channel.
The presence of S100A1 at the junctional SR prompted further studies with the goal of identifying target proteins at the highly specialized SR site that mediates rapid diastolic Ca2+ resequestration. Several studies reported co-localization of S100A1 with the SERCA2/PLB complex in cardiomyocytes (Figure 4) and demonstrated a Ca2+ dependent interaction between the EF-hand Ca2+ sensor and the SR Ca2+ pump as well as with its inhibitory peptide PLB [13, 42–44]. Functional analyses disclosed that S100A1 stimulates SERCA2 activity in cardiomyocytes eventually resulting in improved SR Ca2+ uptake and enhanced SR Ca2+ load [13, 42, 45, 49, 51]. Similar effects in isolated SR cardiac vesicles were obtained with a synthetic peptide consisting of the S100A1 C-terminal amino acid residues 75–94 [13], which indicates a crucial role of this domain for the Ca2+ dependent functional interaction between the EF-hand Ca2+ sensor protein and SERCA2a as well. Interestingly, S100A1 binding to the SERCA2/PLB complex neither changed PLB/SERCA2 stoichiometry nor PLB phosphorylation at serine 16 or threonine 17 [51]. As the former indicates that S100A1 might interact both with PLB and SERCA2 outside their predicted interaction domain, the latter also suggests that S100A1 neither limits protein kinase A (PKA) and calmodulin kinase II (CaMKII) access to their PLB phosphorylation sites.
Mitochondria
More recently, several studies reported on a mitochondrial location of S100A1 in cardiomyocytes (Figure 4) combining immunoelectron and confocal microscopy [52, 53] and its mitochondrial presence supported a role for the EF-hand Ca2+ sensor protein in cardiac energy metabolism. In line with this notion, several mitochondrial S100A1 target proteins have been identified. Boerries et al. were the first to describe a Ca2+ dependent interaction between S100A1 and the F1 portion (α/β-chain) of the mitochondrial ATP synthase [52]; an enzyme of crucial importance in almost all organisms, because ATP as the result of mitochondrial oxidative phosphorylation is the common ”energy currency” of cells [54]. The same study provided further evidence that S100A1 can directly stimulate ATP synthase activity in a Ca2+ dependent manner resulting in enhanced ATP content in cardiomyocytes [52], which further supports a crucial role of the calcium sensor in cardiac energy homeostasis.
Given that Boerries et al. went on to characterize the adenine nucleotide translocator (ANT) as mitochondrial S100A1 target [52], it is tempting to speculate that the interaction of S100A1 with ANT might facilitate ADP and ATP exchange between the mitochondrial matrix and the cytoplasm, and in addition, might be involved in the control of cardiomyocyte apoptosis [54]. In addition, the same study identified hydroxyl-coenzyme A dehydrogenase and isocitrate dehydrogenase (IDH) as S100A1 targets [52]; both key regulators in cardiac fatty acid metabolism and citric acid cycle [54]. Due to the fact that S100A1 is apparently able to increase mitochondrial Ca2+ uptake (Melanie Boerries, personal communication), additional studies are warranted to address the impact of S100A1 on the interplay between SR and mitochondrial Ca2+ handling in greater detail and to elucidate S100A1 impact on mitochondrial citric acid cycle and fatty acid metabolism in cardiac muscle.
Sarcomere
S100A1 has further been detected at different sites within the cardiac sarcomer [34, 41, 55, 56] (Figure 4), indicating potential roles in myofilament function. Employing immunoelectron and confocal microscopy, S100A1 has been described at the Z-line, at the periphery of the sarcomeric M-lines, and in the myofibrillar I-bands and A-bands [34, 41, 55, 56]. Within the I-band, Granzier and co-workers provided comprehensive evidence for S100A1 as a binding partner for the giant myofilament protein titin [55]. Within titin’s force generating region, they identified three S100A1 interaction sites including the force-generating PEVK domain and the N2B element. Granzier’s group could show that Ca2+ dependent S100A1-PEVK interaction reduces the force that arises as F-actin slides relative to the PEVK domain [55], and speculated that S100A1 may provide a mechanism to free the thin filament from titin, potentially reducing titin-based tension before active contraction. In addition, S100A1 has previously been shown to decrease Ca2+ sensitivity and Ca2+ cooperativeness of cardiac myofilaments without changing Ca2+ dependent maximal force development [51] that, in concert with attenuated passive pre-systolic sarcomeric tension, might facilitate diastolic cardiomyocte function. It is currently unclear whether S100A1 has additional sarcomeric target proteins but given the critical role played by Z-disc adjacent proteins in cardiomyocyte function and growth [57], further studies are clearly needed to determine putative S100A1 interaction sites at the Z-disc and the thin and thick myofilament.
2.3 S100A1 impact on cardiomyocyte Ca2+ handling and contractile performance
A compelling body of evidence, accomplished by in vitro and in vivo genetic, biochemical and viral-based manipulation of cardiomyocyte S100A1 protein levels over the last years, has disclosed a role for S100A1 as a critical regulator of cardiomyocyte Ca2+ cycling and contractile performance [13, 38, 39, 44–46, 49, 51, 52, 58–60]. Employing viral-based S100A1 gene delivery, numerous studies demonstrated that enhanced S100A1 protein levels can increase intracellular Ca2+ transients, SR Ca2+ load and contractile performance in field-stimulated isolated rat and rabbit ventricular cardiomyocytes in vitro [13, 44, 51, 52, 61] (Figure 4). Enhancing cardiomyocyte S100A1 protein levels by intracellular application via the patch pipette of recombinant S100A1 protein augmented intracellular Ca2+ transients just as well and amplified excitation-contraction coupling gain and systolic SR Ca2+ release in voltage-clamped isolated rabbit ventricular cardiomyocytes [42]. In line with these results, transgenic cardiomyocyte-specific S100A1 overexpression in mouse hearts lead to chronically heightened cardiac performance without heart rate changes or abnormal cardiac growth [45]. The apparent independence of S100A1 inotropic actions from the β-adrenergic receptor (βAR) signaling pathway is of particular interest to cardiovascular physiology. S100A1-mediated inotropy unfolds independent and on top of βAR-stimulated contractility with unchanged βAR downstream signaling and expression of related molecular components in S100A1 genetically manipulated hearts and S100A1 overexpressing cardiomyocytes [13, 45, 49, 51, 52, 58].
The phenotypic changes in cardiomyocyte performance are consistent with the underlying S100A1 molecular framework as discussed above (Figure 4). S100A1-mediated enhancement of SERCA2 activity and tight closure of the RyR2 during diastole most likely accounts for increased SR Ca2+ load and accelerated relaxation seen in S100A1 overexpressing cardiomyocytes [45, 49, 51]. In addition, facilitated dissociation of Ca2+ from myofilaments in combination with decreased sarcomeric passive tension [51, 55] might further contribute to improved diastolic function in S100A1 overexpressing myocardium. On the other hand, a S100A1-mediated increase in SR Ca2+ load that co-incides with the ability for augmented fractional SR Ca2+ release and systolic RyR2 opening [13, 42, 44, 45, 49, 51], respectively, is an evident powerful molecular mechanism to augment Ca2+ transients and contractile performance. Finally, enhanced mitochondrial ATP generation and cytoplasmic availability [52] might complement S100A1 molecular actions enabling the cardiomyocyte to match energetic demands caused by enhanced SR and myofilament ATPase activity.
Although these mechanisms reflect a growing awareness that S100A1 modulates several critical nodal points, our understanding of how S100A1 modulates cardiac performance is still incomplete and warrants further investigation. In particular, its impact on endothelial NO-homeostasis [23, 39] (see detailed discussion below) and NO-dependent regulation of S100A1 activity [30–32] might suggest a link between S100A1 and cardiomyocyte NO homeostasis. First evidence supporting this notion stems from data showing that S100A1 can prevent nitration of the SR Ca2+ ATPase in diseased myocardium [49]. To the extent NO governs key processes in the heart, it is clear that future studies will have to address this critical issue in greater detail.
Thus, although chronic inotropic molecular interventions have traditionally been viewed with strong reservations due to devastating consequences of direct and indirect sympathomimetics in chronic HF [62], S100A1-dependent signaling apparently assembles an alternative molecular toolkit eventually resulting in a chronically heightened cardiac state without detrimental consequences on cardiac development, cardiac growth and survival.
2.4 S100A1 molecular targets and physiological function in endothelial cells
The most recent and unexpected finding of S100A1’s dual physiological role in vascular tone and arterial blood pressure regulation [23, 39] prompted immediate interest in S100A1 vascular biology. Employing immunofluorescent microscopy, several laboratories have described S100A1 both in endothelial (EC) [23, 39, 40] and smooth muscle cells (SMC) [63]. With regard to vascular endothelium, S100A1 is present both in capillary, coronary, aortic and femoral artery endothelial cells from rodent and human vasculature. Within cultured aortic ECs, S100A1 is mainly dispersed in a granular, punctate pattern throughout the cytoplasm (Figure 5A) but also shows a zonular enrichment around the nucleus [23]. In line with a presence at the endoplasmatic reticulum (ER), confocal immunofluorescent microscopy disclosed S100A1 colocalization with the sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA) (representative images are shown in Figure 5A–C) as well as with the inositol-3-phosphate receptor (IP3R) (Figure 5D) [23].
Figure 5. Subcellular S100A1 location and mechanisms conveying S100A1-mediated NO production in endothelial cells.

A–C, representative confocal fluorescent images highlight partial co-localization (yellow) of S100A1 (red) with SERCA (green) in an aortic mouse EC at the endoplasmic reticulum and nuclear envelope as illustrated by the magnified inlets (images taken from Pleger et al. [23]). D, Gq-protein-coupled muscarinergic (M3) and B2-kininergic receptor Ca2+ dependent endothelial NO production (1) requires IP3-mediated Ca2+ release from the ER (2) resulting in a cytosolic [Ca2+] elevation that in turn activates calmodulin (CaM) binding to eNOS (3) and enhances NO generation (4). Lack of S100A1 in ECs impairs M3 and B2-kininergic receptor mediated intracellular [Ca2+] rise and subsequent endothelial NO production. In contrast, enhanced EC S100A1 protein levels enhance a) both M3 and B2-kininergic receptor mediated intracellular [Ca2+] rise and b) Ca2+ dependent endothelial NO production. E, Subcellular distribution of S100A1 protein (red) in an isolated aortic mouse EC. Note the granular, punctate pattern (arrows) throughout the cytoplasm with a S100A1-free nuclear cavity (asterisk). S100A1 is present at the ER interacting both with the IP3R and SERCA as provided in schematic figure D. Reproduced with modifications from Pleger et al [4].
Due to their function as major effectors of ER Ca2+ uptake and release in ECs, this pattern reveals a striking similarity with cardiomyocytes where S100A1 targets both the SR Ca2+ ATPase (SERCA2) and Ca2+ release channel (RyR2). Indeed, Pleger et al. were the first to demonstrate abnormal Ca2+ handling in aortic ECs isolated from S100A1 knock out (SKO−/−) mice [22]. These cells exhibited blunted muscarinergic and kininergic receptor stimulated cytosolic Ca2+ release and transients, respectively [23, 39], pointing towards a critical role for S100A1 in IP3-regulated Ca2+ signaling in ECs (Figure 5C). In support of a specific S100A1 related signaling defect, the same study showed that S100A1 siRNA treated ECs with a 3-fold decrease in S100A1 protein levels display similar abnormalities in Ca2+ homesostasis [23]. Accordingly, S100A1 gene delivery to SKO−/− ECs restored attenuated bradykinin (BK) and acetylcholine (ACh) induced Ca2+ transients. Although it is tempting to speculate that S100A1 might function as a Ca2+ dependent regulator of IP3R and SERCA activity in ECs, it remains to be established whether S100A1 effects on intracellular EC Ca2+ fluxes might solely rely on IP3-activated emptying of Ca2+ stores encompassing store-dependent or capacitative Ca2+ entry, or even include alternative pathways such as non capacitative Ca2+ entry.
In addition, S100A1 effects on Ca2+ regulated NO homeostasis in ECs seems to be of equal importance given the critical role of NO in vascular biology. Pleger et al. and Desjardins et al. showed that SKO−/− ECs and vessels exhibit impaired baseline NO production and as well as in response to vasodilating agents such as BK, ACh or thrombin despite unchanged eNOS expression [23, 39]. In addition, similar results were obtained in S100A1 siRNA treated ECs providing corroborating evidence for a functional significance of S100A1 in EC NO homeostasis [22]. Enhanced baseline and potentiated acetycholine-induced NO production after S100A1 gene delivery to human coronary artery endothelial cells on the other hand actually adds on first therapeutic weight to S100A1 vascular effects [23]. Given unchanged eNOS expression, complete lack or decrease of EC S100A1 protein presumably interrupts normal muscarinergic and kininergic receptor signaling to eNOS at the level of IP3-triggered ER Ca2+ release and potentially interferes with Ca2+ activated calmodulin binding to eNOS. The former notion is further supported by experimental evidence showing that the IP3R inhibitor 2-ABP abolishes S100A1-mediated potentiation of ACh-mediated NO production in S100A1 overexpressing human coronary artery ECs [23].
2.5 S100A1 impact on vascular tone and arterial blood pressure
Evidence for physiological significance eventually emerged from experiments demonstrating that preconstricted ringed segments of aorta and mesenteric vessels from SKO−/− mice when tested ex vivo lack endothelium-dependent relaxation in response to ACh [23, 39]. Of note, SMC-mediated and endothelium-independent relaxation to sodium nitroprusside and isoproterenol was preserved intact [23]. Hence, the abnormal vascular phenotype in SKO−/− mice can most likely be linked back to molecular defects in muscarinergic receptor downstream Ca2+ and NO signaling in SKO−/− ECs. Although previous studies described S100A1 expression in SMCs and suggested a regulatory role by reversing caldesmon-induced inhibition of actomyosin ATPase activity in vitro [64], unchanged SMC-mediated vessel relexation in SKO−/− mice in response to NO and isoproterenol argues against a relevant role of S100A1 in SMC contractile function. Most importantly, SKO−/− mice eventually develop arterial hypertension and completely lacked in vivo blood pressure responsiveness to BK that induced a marked drop in arterial blood pressure in control mice at equimolar doses [23, 39]. Interestingly, Parker and co-workers most recently disclosed evidence for a gender-dependent aggravated hypertensive phenotype and post-MI mortality in male SKO−/− mice [39] suggesting a modulatory role for estrogens in the SKO−/− phenotype.
Thus, deficient vascular endothelial NO production due to a lack of S100A1 results in abnormal vascular relaxation with arterial hypertension that finally rises the question whether S100A1-related vascular abnormalities might also extend to abnormal pulmonary vessel and blood pressure regulation.
2.6 S100A1 role in skeletal muscle Ca2+ handling and contractile performance
Although skeletal muscle expresses S100A1 at significantly lower levels than cardiac muscle [65], numerous studies support a likewise important role for S100A1 in skeletal muscle EC coupling [47, 48, 50, 65]. First evidence for this notion came from the observation by Treves et al. [50] showing that S100A1 binds the RyR1 and potentiates its open probability in lipid bilayer experiments. The same study further identified three different S100A1 binding sites at the RyR1 of which one overlapped with the Ca2+ dependent CaM binding domain. Subsequent studies that employed saponin skinned murine skeletal muscle fibers with functionally intact SR and myofilaments demonstrated that exogenously added S100A1 protein can enhance SR Ca2+ release and isometric force development both in fast- and slow-twitch skeletal muscle [65]. This effect was found to be dose-dependent with a maximal effect at 100 nM S100A1 protein. However, in this model S100A1 neither activated RyR1 in its closed state nor initiated SR Ca2+ release at diastolic Ca2+ and physiological Mg2+ concentrations. Instead, S100A1 increases the activation state of the opening RyR1 and ongoing SR Ca2+ release, respectively, following initial stimulation of the channel, which leads to greater force generation and isometric contraction of the fibers[65]. However, despite these striking similarity with its effect on EC coupling gain in cardiomyocytes, S100A1 neither enhanced SR Ca2+ resequestration nor altered Ca2+ sensitivity in skinned murine skeletal muscle fibers [65]. These findings indicate that S100A1 effects in skeletal muscle EC coupling might be limited to SR Ca2+ release sites and potentially mitochondria.
Taking advantage of SKO−/− skeletal muscle fibers, most recent work from the groups of Martin Schneider and David Weber provided ultimate proof of concept for a significant physiological role of S100A1 in skeletal muscle EC coupling [47, 48]. They were able to show that voltage-induced SR Ca2+ release (VICR) in intact SKO−/− skeletal muscle fibers is decreased eventually resulting in diminished global Ca2+ transients and contractile force development [47, 48]. Importantly, viral-based S100A1 gene delivery to SKO−/− skeletal muscle restoring S100A1 protein expression rescued the in vivo phenotype suggesting that the loss of S100A1 but not adaptive changes account for the loss-in-function phenotype [47]. Weber and co-workers further revealed that S100A1 and CaM compete for the same binding site at the RyR1 in a Ca2+ dependent manner and characterized the hydrophobic RyR1 residues 3416–3427 as the common binding domain [48]. Importantly, these residues interact with the hydrophobic pocket of S100A1 that is formed upon Ca2+ binding of helix 3 and helix 4 [27]. Given that a synthetic helix 4 S100A1 peptide, which consists of S100A1 hydrophobic residues 75–94, completely mimicked the inotropic effect of native S100A1 in skinned skeletal muscle fibers [65], the most recent findings significantly advance our molecular understanding of the molecular basis underlying S100A1 regulation of RyR function. Given that the S100A1 binding domain, which is conserved between RyR1 and RyR2, is involved in intersubunit interactions as well as close in space to distal regions of the RyR [66], the authors proposed an exciting model where one dimeric S100A1 protein is involved in linking together two subunits of the RyR tetramer [48].
Overall, these data suggest that, likewise cardiac muscle, S100A1 plays a significant role in skeletal muscle EC coupling, primarily through regulation of RyR1-mediated SR Ca2+ release but not through modulation of SR Ca2+ load and myofilament function. Given the importance of NO for normal RyR1 function [33], further studies are warranted to determine whether S100A1 might regulate RyR1 also through modulation of skeletal muscle NO-homeostasis. In light of the clinical importance of EC coupling defects in several congenital skeletal myopathies and dysfunctional skeletal muscle in HF, further molecular and in vivo studies are needed to determine both therapeutic and pathophysiolocal impact of S100A1 in skeletal muscle disorders.
3. S100A1 in cardiovascular pathology and therapeutic potential
3.1. S100A1 in cardiac disease
Chronically dysfunctional human myocardium is characterized by progressively diminished S100A1 mRNA and protein levels that inversely correlate with the severity of the disease [67]. Given the same pathological plasticity in chronically diseased rodent, rabbit and pig hearts [13, 49, 58–60, 68], decreased S100A1 expression seems to be a common pathognomonic molecular signature of failing myocardium. Abnormal in vivo cardiac S100A1 expression has been recapitulated in vitro by chronic stimulation of cardiomyocytes with various hypertrophic stimuli including Gq-protein coupled receptor [49], receptor serine/threonine as well as receptor tyrosine kinase agonists. Stimulation with endothelin-1 and phenylephrine [49] as well as transforming growth factor β (TGF-β) and acidic fibroblast growth factor (FGF-1) (Patrick Most unpublished results) caused a progressive decrease in cultured cardiomyocyte S100A1 mRNA and protein in a time- and concentration dependent manner indicating a transcriptional inhibitory mechanism in the course of maladaptive growth. AP-1 (Fos/Jun) binding sites located within the S100A1 promoter sequence known to act downstream of activated Gq-protein coupled receptors might confer S100A1 transcriptional inhibition. In light of the low abundance of S100A1 in the developing fetal heart [36], downregulation of S100A1 in hypertrophied cardiomyocytes could be considered a part of the reprogrammed fetal gene pattern.
Pathophysiological relevance of deregulated S100A1 expression in hearts finally stems from the observation that SKO−/− mice exhibited enhanced susceptibility and accelerated functional deterioration in response to chronic cardiac stress and ischemic damage [38, 39, 49]. SKO−/− mice subjected to transaortic constriction (TAC) showed accelerated deterioration of cardiac performance and transition to failure while TAC control mice developed a functionally compensated state [38]. Further analysis showed equal cardiac remodeling and expression of hypertrophic markers in both groups. Interestingly, heterozygous SKO−/+ subjected to TAC increased cardiac S100A1 protein concentrations to levels seen in control mice eventually enabling them to achieve and maintain a functionally compensated state [38]. Thus, normal left ventricular S100A1 expression levels are apparently required to cope with chronically elevated afterload and similar observations have been made with right ventricular S100A1 expression levels in a pig model of pulmonary hypertension [69]. Alike TAC, SKO−/− hearts exhibit enhanced susceptibility to ischemic damage [39, 49]. Myocardial infarction (MI) in SKO−/− mice resulted in accelerated deterioration of left ventricular function and transition to failure together with exaggerated cardiac remodeling and cardiomyocyte apoptosis, abrogated β-AR responsiveness and enhanced overall mortality [39, 49]. The latter could either be due to pump failure or a most recently reported enhanced pro-arrhythmic susceptibility of SKO−/− mice in response to sympathetic stimulation [70]. As predicted by S100A1 molecular effects on SR function, infarcted SKO−/− mice showed early signs of SR dysfunction including enhanced SR Ca2+ leakage as well as decreased SR Ca2+ load and release, respectively [49], potentially providing the substrate for Ca2+ triggered afterdepolarizations and tachyarrhthmias.
On the contrary, hypercontractile S100A1 transgenic hearts subjected to MI maintained almost normal left ventricular function, exhibited only minimal signs of cardiac hypertrophy and programmed cell death together with improved post-MI survival [49]. In line with S100A1 molecular actions, remote myocardium from infarcted S100A1-overexpressing hearts showed superior SR Ca2+ fluxes and storage capabilities compared to control mice that exhibit a progressive loss of cardiac S100A1 protein levels after ischemic injury [49]. Interestingly, previous studies demonstrated significant extracellular S100A1 protein release from infarcted human hearts [17]. Given that S100A1, alike other S100 proteins, can exhibit extracellular functions and has been shown to protect ventricular cardiomyocytes from apoptosis in vitro [71], it is tempting to speculate that damage-released S100A1 protein might actually exert a cardioprotective effect and mitigate cardiomyocte apoptosis after ischemic damage. Vice versa, lack of S100A1 release in damaged myocardium might result in less paracrine cardioprotection and contribute to augmented cardiomyocyte death in infarcted SKO−/− hearts [49].
3.2. S100A1 therapy of diseased myocardium
Together, these results provided a strong rationale to propose S100A1 as a novel therapeutic target for acute and chronic cardiac dysfunction. Indeed, viral-based S100A1 gene delivery to isolated failing ventricular rat cardiomyocytes provided first proof of concept for the therapeutic potential of S100A1 gene therapy [13]. Adenoviral-based S100A1 gene transfer normalized S100A1 protein expression in failing cardiomyocytes and, in turn, restored normal contractile function and cellular Ca2+ handling [13]. Detailed analysis of SR Ca2+ handling in S100A1-treated failing cardiomyocytes disclosed normalized SR Ca2+ load and improved SERCA2 activity together with decreased SR Ca2+ leakage and normalized diastolic [Ca2+]. Interestingly, restored S100A1 protein levels also normalized elevated cytosolic free sodium concentrations ([Na+]) [13]; an effect which is directly linked to the balance between SERCA2 and Na/Ca exchanger (NCX) activity [72]. Given that S100A1 increased SERCA2 activity together with a decrease in NCX expression, lowered [Na+] most likely reflects the favorable shift towards improved diastolic SR Ca2+ uptake and diminished sarcolemmal Ca2+ extrusion. Most recently, another study showed that S100A1 gene delivery can actually prevent β-AR triggered proarrhythmic SR Ca2+ leakage and diastolic Ca2+ waves in cardiomyocytes [73]; an effect that could contribute to previously reported improved post-MI survival in S100A1 transgenic mice [49].
In vivo proof of concept for the therapeutic effectiveness and long-term beneficial effects of cardiac S100A1 gene therapy stems from studies employing intracoronary viral-based S100A1 gene delivery to acutely and chronically failing hearts [13, 58, 60]. Both adenoviral and adeno-associated (AAV) based S100A1 gene delivery rescued contractile performance of failing rat hearts through restored Ca2+ cycling [13, 58, 60]. In addition, S100A1 gene therapy exerted remarkable antihypertrophic effects in vivo reversing increased heart-to-body weight and fetal gene expression, and prevented progressive left chamber dilatation [13, 58, 60]. Moreover, restored S100A1 expression in failing myocardium normalized the PCr/ATP ratio indicating improved cardiomyocyte energy homeostasis [13], a fact that correlates well with clinical survival. Although, no direct effects of S100A1 on hypertrophic signaling has been demonstrated yet, restored cardiac output in S100A1-treated hearts most likely curtails neurohumoral activation and abrogates diastolic wall stress in diseased hearts. Thus, S100A1 seems to directly reverse the pathophysiological sequence that leads to sympathetic overdrive and load-induced prohypertrophic activation in HF.
Most recently, data showing long-term rescue of HF by AAV9-S100A1 gene delivery in an experimental post-MI HF model in domestic pigs provided proof of therapeutic effectiveness and feasibility for S100A1 HF gene therapy in a near-clinical setting [68]. Employed cardiac gene delivery modalities, applied gene dosage and post-treatment analyses are directly applicable to humans and might pave the way for first-in-human clinical S100A1 gene therapy saftey trails. The S100A1-AAV9 virus delivered via the anterior coronary vein 2 weeks post-MI protected from progressive hemodynamic deterioration and improved cardiac performance to near-normal levels [68]. In addition, S100A1 gene therapy reversed the enhanced heart-to-body weight ratio and BNP mRNA expression reflecting abrogated in vivo pathological remodeling. Moreover, S100A1-treated infarcted animals had normalized heart rate post-MI clearly reflecting curtailed neurohumoral activation most likely due to sufficient global cardiac output [68].
As highlighted on the US Department of Agriculture website on Animal Models in Biomedical Research (http://www.nal.usda.gov/awic/pubs/swine/swine.htm), pigs share striking anatomic and physiologic characteristics with humans making them a unique and viable model for biomedical research and, more importantly, develop an infarction pattern and post-MI HF like humans. This study therefore actually closes the gap between basic research, which started almost a decade ago aiming to determine S100A1 physiological and pathophysiological cardiovascular significance, and the urgent need to translate these findings into novel clinical treatments for HF. With the large-animal HF model at hand, further studies are warranted to determine the therapeutic impact and disclose protective molecular effects of S100A1 on cardiac energetics, arrhythmias and even compatibility with established pharmacological and antiarrhythmic clinical device therapy.
However, the use of S100A1 as a therapeutic target is actually not restricted to cardiac gene therapy but could potentially be employed in the emerging fields of cardiac tissue engineering and cell-based therapy. Engineered heart tissue from rat cardiomyocytes showed enhanced contractile performance at baseline and in response to βAR stimulation after S100A1 gene addition [74] indicating that S100A1-based genetic manipulation might be a promising strategy to strengthen therapeutic effectiveness of arteficial cardiac tissue or even artificial hearts. In addition, it is reasonable to assume that either bone-marrow or cardiac derived progenitor cells with the ability to regenerate cardiomyocytes [75, 76] could be subject to S100A1 gene addition prior to their therapeutic use. Such strategies could potentially result in newly formed cardiac cells with superior contractile properties and enhanced resistance against apoptosis, hypertrophic and proarrhythmic alterations.
3.3. S100A1 in vascular disease and therapy
The hitherto unknown role of S100A1 in endothelial Ca2+ signaling and NO homeostasis disclosed by Pleger et al. [22] has broad pathophysiological implications. The fact that vascular S100A1 deficiency impairs function of the endothelial cell layer eventually resulting in hampered vascular relaxation and elevated in vivo arterial blood pressure [23, 39] might offer new perspectives on mechanisms of actions underlying vasoconstriction in hypertension and heart failure. However, besides control of vascular tone, NO plays a vital role in vascular protection from atherosclerosis including coronary and peripheral artery disease, prothrombotic platelet activation, diabetes-related vascular dysfunction as well as in (neo)angiogenesis and endothelial progenitor cell function [33].
Decreased endothelial S100A1 expression in HF [23] may therefore limit vascular NO availability and further exacerbate the NO/redox disequilibrium resulting from increased oxidase activity in the vascular wall [32]. On the other hand, endothelin-1 and angiotensin II both stimulating vascular reactive oxygen species production have been shown to diminish endothelial S100A1 abundance [23], which might indeed further contribute to vasoconstriction in patients with hypertension and heart failure. From this perspective, S100A1 can be seen as a novel target amenable to therapeutic modulation of vascular function through restoration of the NO/redox balance. This novel concept is supported by data showing that S100A1 gene delivery to human coronary artery endothelial cells enhanced baseline and potentiated ACh-induced NO production in vitro [23]. However, it requires a systematic further evaluation of S100A1 expression in various human vascular disorders and corresponding animal models to fully establish a clinical relevance of these findings and sound understanding of S100A1 vascular biology.
4. Conclusions and future perspective
In recent years, S100A1 has clearly emerged as a critical regulator of Ca2+ handling and NO homeostasis in cardiac and vascular cells reflected by numerous reports highlighting its physiological and pathophysiological relevance in the cardiovascular system (reviewed in [4, 21]). However, S100A1 appears to have a likewise significant role in skeletal muscle function although its pathophysiological and therapeutic relevance in congential and HF-associated skeletal muscle disorders have not been fully explored yet. Nevertheless, in light of most recent advances demonstrating the unique and beneficial long-term therapeutic effects of the Ca2+ sensor in small and large postischemic HF animal models [58, 68], cardiac-targeted S100A1 HF gene therapy steadily moves forward towards first clinical safety trials. It is therefore exciting to speculate that manipulation of endothelial S100A1 expression might also hold therapeutic potential as a novel vascular therapeutic strategy against clinically common disorders including hypertension, atherosclerosis or peripheral artery disease. Given the tremendous health and economic burden of chronic cardiac and vascular diseases, it is mandatory to pursue S100A1-based translational strategies with the ultimate goal of closing the wide open gap between basic research and clinical therapy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tran QK, Ohashi K, Watanabe H. Calcium signalling in endothelial cells. Cardiovasc Research. 2000 Oct;48(1):13–22. doi: 10.1016/s0008-6363(00)00172-3. [DOI] [PubMed] [Google Scholar]
- 2.Bers DM. Calcium cycling and signaling in cardiac myocytes. Ann Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 3.Donato R. Intracellular and extracellular roles of S100 proteins. Microsc Res Tech. 2003;60(6):540–51. doi: 10.1002/jemt.10296. [DOI] [PubMed] [Google Scholar]
- 4.Most P, Remppis A, Pleger ST, Katus HA, Koch WJ. S100A1: a novel inotropic regulator of cardiac performance. Transition from molecular physiology to pathophysiological relevance. Am J Physiol. 2007;293(2):R568–77. doi: 10.1152/ajpregu.00075.2007. [DOI] [PubMed] [Google Scholar]
- 5.Santamaria-Kisiel L, Rintala-Dempsey AC, Shaw GS. Calcium-dependent and -independent interactions of the S100 protein family. Biochem J. 2006;396(2):201–14. doi: 10.1042/BJ20060195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marenholz I, Heizmann CW, Fritz G. S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature) Biochem Biophys Res Commun. 2004;322(4):1111–22. doi: 10.1016/j.bbrc.2004.07.096. [DOI] [PubMed] [Google Scholar]
- 7.Boyd JH, Kan B, Roberts H, Wang Y, Walley KR. S100A8 and S100A9 mediate endotoxin-induced cardiomyocyte dysfunction via the receptor for advanced glycation end products. Circ Res. 2008;102(10):1239–46. doi: 10.1161/CIRCRESAHA.107.167544. [DOI] [PubMed] [Google Scholar]
- 8.Ehlermann P, Eggers K, Bierhaus A, Most P, Weichenhan D, Greten J, et al. Increased proinflammatory endothelial response to S100A8/A9 after preactivation through advanced glycation end products. Cardiovasc Diabet. 2006;5:6. doi: 10.1186/1475-2840-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foell D, Ichida F, Vogl T, Yu X, Chen R, Miyawaki T, et al. S100A12 (ENRAGE) in monitoring Kawasaki diesease. Lancet. 2003;361:1270–2. doi: 10.1016/S0140-6736(03)12986-8. [DOI] [PubMed] [Google Scholar]
- 10.Inamoto S, Murao S, Yokoyama M, Kitazawa S, Maeda S. Isoproterenol-induced myocardial injury resulting in altered S100A4 and S100A11 protein expression in the rat. Pathol Internat. 2000 Jun;50(6):480–5. doi: 10.1046/j.1440-1827.2000.01069.x. [DOI] [PubMed] [Google Scholar]
- 11.Mazzini GS, Schaf DV, Oliveira AR, Goncalves CA, Bello-Klein A, Bordignon S, et al. The ischemic rat heart releases S100B. Life Sciences. 2005;77(8):882–9. doi: 10.1016/j.lfs.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 12.Miyamoto S, Ueda M, Ikemoto M, Naruko T, Itoh A, Tamaki S, et al. Increased serum levels and expression of S100A8/A9 complex in infiltrated neutrophils in atherosclerotic plaque of unstable angina. Heart (British Cardiac Society) 2008;94(8):1002–7. doi: 10.1136/hrt.2007.121640. [DOI] [PubMed] [Google Scholar]
- 13.Most P, Pleger ST, Völkers M, Heidt B, Boerries M, Weichenhan D, et al. Cardiac adenoviral S100A1 gene transfer rescues failing myocardium. J Clin Invest. 2004;114:1550–63. doi: 10.1172/JCI21454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider M, Kostin S, Strom CC, Aplin M, Lyngbaek S, Theilade J, et al. S100A4 is upregulated in injured myocardium and promotes growth and survival of cardiac myocytes. Cardiovasc Res. 2007;75(1):40–50. doi: 10.1016/j.cardiores.2007.03.027. [DOI] [PubMed] [Google Scholar]
- 15.Tsoporis JN, Marks A, Haddad A, Dawood F, Liu PP, Parker TG. S100B expression modulates left ventricular remodeling after myocardial infarction in mice. Circulation. 2005;111:598–606. doi: 10.1161/01.CIR.0000154554.65287.F5. [DOI] [PubMed] [Google Scholar]
- 16.Tsoporis JN, Marks A, Van Eldik LJ, O’Hanlon D, Parker TG. Regulation of the S100B gene by alpha 1-adrenergic stimulation in cardiac myocytes. Am J Physiol. 2003;284(1):H193–203. doi: 10.1152/ajpheart.00161.2002. [DOI] [PubMed] [Google Scholar]
- 17.Usui A, Kato K, Sasa H, Minaguchi K, Abe T, Murase M, et al. S-100ao protein in serum during acute myocardial infarction. Clin Chem. 1990;36(4):639–41. [PubMed] [Google Scholar]
- 18.Viemann D, Strey A, Janning A, Jurk K, Klimmek K, Vogl T, et al. Myeloid-related proteins 8 and 14 induce a specific inflammatory response in human microvascular endothelial cells. Blood. 2005;105(7):2955–62. doi: 10.1182/blood-2004-07-2520. [DOI] [PubMed] [Google Scholar]
- 19.Tsoporis JN, Izhar S, Parker TG. Expression of S100A6 in cardiac myocytes limits apoptosis induced by tumor necrosis factor-alpha. J Biol Chem. 2008;283(44):30174–30183. doi: 10.1074/jbc.M805318200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsoporis JN, Marks A, Haddad A, O’Hanlon D, Jolly S, Parker TG. S100A6 is a negative regulator of the induction of cardiac genes by trophic stimuli in cultured rat myocytes. Exp Cell Res. 2005;303(2):471–81. doi: 10.1016/j.yexcr.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 21.Most P, Koch WJ. S100A1: a calcium-modulating inotropic prototype for future clinical heart failure therapy. Future Cardiol. 2007;3:5–11. doi: 10.2217/14796678.3.1.5. [DOI] [PubMed] [Google Scholar]
- 22.Pleger ST, Boucher M, Most P, Koch WJ. Targeting myocardial beta-adrenergic receptor signaling and calcium cycling for heart failure gene therapy. J Card Failure. 2007;13(5):401–14. doi: 10.1016/j.cardfail.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 23.Pleger ST, Harris DM, Shan C, Vinge LE, Chuprun JK, Berzins B, et al. Endothelial S100A1 modulates vascular function via nitric oxide. Circ Res. 2008;102(7):786–94. doi: 10.1161/CIRCRESAHA.108.172031. [DOI] [PubMed] [Google Scholar]
- 24.Moore BW. A soluble protein characteristic of the nervous system. Biochem Biophys Res Com. 1965;19(6):739–44. doi: 10.1016/0006-291x(65)90320-7. [DOI] [PubMed] [Google Scholar]
- 25.Donato R. Functional roles of S100 proteins, calcium-binding proteins of the EF- hand type. Biochim Biophys Acta. 1999;1450(3):191–231. doi: 10.1016/s0167-4889(99)00058-0. [DOI] [PubMed] [Google Scholar]
- 26.Zimmer DB, Wright SP, Weber DJ. Molecular mechanisms of S100-target protein interactions. Microsc Res Tech. 2003;60:552–9. doi: 10.1002/jemt.10297. [DOI] [PubMed] [Google Scholar]
- 27.Wright NT, Varney KM, Karen CE, Markowitz J, Rossitza KG, Zimmer DB, et al. The three-dimensional solution structure of Ca2+-bound S100A1 as determined by NMR Spectroscopy. J Mol Biol. 2005;353:410–26. doi: 10.1016/j.jmb.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 28.Rustandi RR, Baldisseri DM, Inman KG, Nizner P, Hamilton SM, Landar A, et al. Three-dimensional solution structure of the calcium-signaling protein apo-S100A1 as determined by NMR. Biochem. 2002;41(3):788–96. doi: 10.1021/bi0118308. [DOI] [PubMed] [Google Scholar]
- 29.Osterloh D, Ivanenkov VV, Gerke V. Hydrophobic residues in the C-terminal region of S100A1 are essential for target protein binding but not for dimerization. Cell Calcium. 1998;24:137–51. doi: 10.1016/s0143-4160(98)90081-1. [DOI] [PubMed] [Google Scholar]
- 30.Zhukova L, Zhukova I, Bal W, Wyslouch-Cieszynska A. Redox modifications of the C-terminal cysteine residue cause structural changes in S100A1 and S100B proteins. Biochem Biophys Acta. 2004;1742:191–201. doi: 10.1016/j.bbamcr.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 31.Goch G, Vdovenko S, Kozlowska H, Bierzynski A. Affinity of S100A1 protein for calcium increases dramatically upon glutathionylation. Febs J. 2005;272(10):2557–65. doi: 10.1111/j.1742-4658.2005.04680.x. [DOI] [PubMed] [Google Scholar]
- 32.Zhukov I, Ejchart A, Bierzynski A. Structural and motional changes induced in apo-S100A1 protein by the disulfide formation between its Cys 85 residue and beta-mercaptoethanol. Biochem. 2008;47(2):640–50. doi: 10.1021/bi701762v. [DOI] [PubMed] [Google Scholar]
- 33.Hare JM, Stamler JS. NO/redox disequilibrium in the failing heart and cardiovascular system. J Clin Invest. 2005;115(3):509–17. doi: 10.1172/JCI200524459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haimoto H, Kato K. S100a0 (alpha alpha) protein in cardiac muscle. Isolation from human cardiac muscle and ultrastructural localization. Eur J Biochem. 1988;171(1–2):409–15. doi: 10.1111/j.1432-1033.1988.tb13805.x. [DOI] [PubMed] [Google Scholar]
- 35.Kato K, Kimura S, Haimoto H, Suzuki F. S100a0 (alpha alpha) protein: distribution in muscle tissues of various animals and purification from human pectoral muscle. J Neurochem. 1986;46(5):1555–60. doi: 10.1111/j.1471-4159.1986.tb01776.x. [DOI] [PubMed] [Google Scholar]
- 36.Kiewitz R, Lyons GE, Schafer BW, Heizmann CW. Transcriptional regulation of S100A1 and expression during mouse heart development. Biochim Biophys Acta. 2000;1498(2–3):207–19. doi: 10.1016/s0167-4889(00)00097-5. [DOI] [PubMed] [Google Scholar]
- 37.Zimmer DB, Song W, Zimmer WE. Isolation of a rat S100 alpha cDNA and distribution of its mRNA in rat tissues. Brain Res Bull. 1991;27(2):157–62. doi: 10.1016/0361-9230(91)90061-n. [DOI] [PubMed] [Google Scholar]
- 38.Du XJ, Cole TJ, Tenis N, Gao XM, Kontgen F, Kemp BE, et al. Impaired cardiac contractility response to hemodynamic stress in S100A1-deficient mice. Mol Cell Biol. 2002;22(8):2821–9. doi: 10.1128/MCB.22.8.2821-2829.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Desjardins JF, Pourdjabbar A, Quan A, Leong-Poi H, Teichert-Kuliszewska K, Verma S, et al. Lack of S100A1 in mice confers a gender-dependent hypertensive phenotype and increased mortality following myocardial infarction. Am J Physiol. 2009;296(5):H1457–1465. doi: 10.1152/ajpheart.00088.2008. [DOI] [PubMed] [Google Scholar]
- 40.Lefranc F, Decaestecker C, Brotchi J, Heizmann CW, Dewitte O, Kiss R, et al. Co-expression/co-location of S100 proteins (S100B, S100A1 and S100A2) and protein kinase C (PKC-beta, -eta and -zeta) in a rat model of cerebral basilar artery vasospasm. Neuropath Appl Neurobiol. 2005;31(6):649–60. doi: 10.1111/j.1365-2990.2005.00682.x. [DOI] [PubMed] [Google Scholar]
- 41.Kato K, Kimura S. S100ao (alpha alpha) protein is mainly located in the heart and striated muscles. Biochim Biophys Acta. 1985;842(2–3):146–50. doi: 10.1016/0304-4165(85)90196-5. [DOI] [PubMed] [Google Scholar]
- 42.Kettlewell S, Most P, Currie S, Koch H, Smith GL. S100A1 increases the gain of excitation-contraction coupling in isolated rabbit ventricular cardiomyocytes. J Mol Cell Cardiol. 2005;39:900–10. doi: 10.1016/j.yjmcc.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 43.Kiewitz R, Acklin C, Schafer BW, Maco B, Uhri KB, Wuytack F, et al. Ca(2+)-dependent interaction of S100A1 with the sarcoplasmic reticulum Ca(2+)-ATPase2a and phospholamban in the human heart. Biochem Biophys Res Commun. 2003;306(2):550–7. doi: 10.1016/s0006-291x(03)00987-2. [DOI] [PubMed] [Google Scholar]
- 44.Most P, Eicher C, Börries M, Schweda C, Völkers M, Wedel T, et al. Distinct subcellular location of the Ca2+-binding protein S100A1 differentially modulates Ca2+-cycling in ventricular rat cardiomyocytes. J Cell Sci. 2005;118:421–31. doi: 10.1242/jcs.01614. [DOI] [PubMed] [Google Scholar]
- 45.Most P, Remppis A, Pleger ST, Löffler E, Ehlermann P, Bernotat J, et al. Transgenic overexpression of the Ca2+ binding protein S100A1 in the heart leads to increased in vivo myocardial contractile performance. J Biol Chem. 2003;278(5):33809–17. doi: 10.1074/jbc.M301788200. [DOI] [PubMed] [Google Scholar]
- 46.Volkers M, Loughrey CM, Macquaide N, Remppis A, DeGeorge BR, Jr, Wegner FV, et al. S100A1 decreases calcium spark frequency and alters their spatial characteristics in permeabilized adult ventricular cardiomyocytes. Cell Calcium. 2007;41(2):135–43. doi: 10.1016/j.ceca.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 47.Prosser BL, Wright NT, Hernandez-Ochoa EO, Varney KM, Liu Y, Olojo RO, et al. S100A1 binds to the calmodulin-binding site of ryanodine receptor and modulates skeletal muscle excitation-contraction coupling. J Biol Chem. 2008;283(8):5046–57. doi: 10.1074/jbc.M709231200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wright NT, Prosser BL, Varney KM, Zimmer DB, Schneider MF, Weber DJ. S100A1 and calmodulin compete for the same binding site on ryanodine receptor. J Biol Chem. 2008;283(39):26676–83. doi: 10.1074/jbc.M804432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Most P, Seifert H, Gao E, Funakoshi H, Volkers M, Heierhorst J, et al. Cardiac S100A1 protein levels determine contractile performance and propensity toward heart failure after myocardial infarction. Circulation. 2006;114(12):1258–68. doi: 10.1161/CIRCULATIONAHA.106.622415. [DOI] [PubMed] [Google Scholar]
- 50.Treves S, Scutari E, Robert M, Groh S, Ottolia M, Prestipino G, et al. Interaction of S100A1 with the Ca2+ release channel (ryanodine receptor) of skeletal muscle. Biochem. 1997;36(38):11496–503. doi: 10.1021/bi970160w. [DOI] [PubMed] [Google Scholar]
- 51.Most P, Bernotat J, Ehlermann P, Pleger ST, Reppel M, Borries M, et al. S100A1: a regulator of myocardial contractility. Proc Nat Acad Sci USA. 2001;98(24):13889–94. doi: 10.1073/pnas.241393598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Most P, Boerries M, Gledhill JR, Walker JE, Katus HA, Koch WJ, et al. Ca2+-dependent interaction of S100A1 with F1-ATPase leads to an increased ATP content in cardiomyocytes. Mol Cell Biol. 2007;27(12):4365–73. doi: 10.1128/MCB.02045-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brezova A, Heizmann CW, Uhrik B. Immunocytochemical localization of S100A1 in mitochondria on cryosections of the rat heart. Gen Physiol Biophys. 2007;26(2):143–9. [PubMed] [Google Scholar]
- 54.Balaban RS. Cardiac Energy Metabolism Homeostasis: Role of Cytosolic Calcium. J Mol Cell Cardiol. 2002;34:1259–71. doi: 10.1006/jmcc.2002.2082. [DOI] [PubMed] [Google Scholar]
- 55.Yamasaki R, Berri M, Wu Y, Trombitas K, McNabb M, Kellermayer MS, et al. Titin-actin interaction in mouse myocardium: passive tension modulation and its regulation by calcium/s100a1. Biophys J. 2001;81(4):2297–313. doi: 10.1016/S0006-3495(01)75876-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maco B, Mandinova A, Durrenberger MB, Schafer BW, Uhrik KB, Heizmann CW. Ultrastructural distribution of the S100A1 Ca2+-binding protein in the human heart. Physiol Res. 2001;50:567–74. [PubMed] [Google Scholar]
- 57.Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly. Ann Rev Physiol. 2003;65:45–79. doi: 10.1146/annurev.physiol.65.092101.142243. [DOI] [PubMed] [Google Scholar]
- 58.Pleger ST, Most P, Boucher M, Soltys S, Chuprun JK, Pleger W, et al. Stable myocardial-specific AAV6-S100A1 gene therapy results in chronic functional heart failure rescue. Circulation. 2007;115(19):2506–15. doi: 10.1161/CIRCULATIONAHA.106.671701. [DOI] [PubMed] [Google Scholar]
- 59.Pleger ST, Most P, Heidt B, Voelkers M, Hata JA, Katus HA, et al. S100A1 gene transfer in myocardium. Europ Journal Med Res. 2006 Oct 27;11(10):418–22. [PubMed] [Google Scholar]
- 60.Pleger ST, Remppis A, Heidt B, Völkers M, Chaprun JK, Kuhn M, et al. S100A1 gene therapy preserves in vivo cardiac function after myocardial infarction. Mol Therapy. 2005;12:1120–9. doi: 10.1016/j.ymthe.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 61.Remppis A, Most P, Löffler E, Ehlermann P, Bernotat J, Pleger ST, et al. The small EF-hand Ca2+ binding protein S100A1 increases contractility and Ca2+ cycling in rat cardiac myocytes. Basic Res Cardiol. 2002;97(Suppl 1):I/56–I/62. doi: 10.1007/s003950200031. [DOI] [PubMed] [Google Scholar]
- 62.Overgaard CB, Dzavik V. Inotropes and vasopressors: review of physiology and clinical use in cardiovascular disease. Circulation. 2008;118(10):1047–56. doi: 10.1161/CIRCULATIONAHA.107.728840. [DOI] [PubMed] [Google Scholar]
- 63.Mandinova A, Atar D, Schafer BW, Spiess M, Aebi U, Heizmann CW. Distinct subcellular localization of calcium binding S100 proteins in human smooth muscle cells and their relocation in response to rises in intracellular calcium. J Cell Sci. 1998;111 (Pt 14):2043–54. doi: 10.1242/jcs.111.14.2043. [DOI] [PubMed] [Google Scholar]
- 64.Polyakov AA, Huber PA, Marston SB, Gusev NB. Interaction of isoforms of S100 protein with smooth muscle caldesmon. FEBS Let. 1998;422(2):235–9. doi: 10.1016/s0014-5793(98)00014-3. [DOI] [PubMed] [Google Scholar]
- 65.Most P, Remppis A, Weber C, Bernotat J, Ehlermann P, Pleger ST, et al. The C-terminus (aa 75–94) and the linker region (aa 42–54) of the Ca2+ binding protein S100A1 differentially enhance sarcoplasmic Ca2+ release in murine skinned skeletal muscle fibres. J Biol Chem. 2003;278(29):26356–64. doi: 10.1074/jbc.M303338200. [DOI] [PubMed] [Google Scholar]
- 66.Zhang H, Zhang JZ, Danila CI, Hamilton SL. A noncontiguous, intersubunit binding site for calmodulin on the skeletal muscle Ca2+ release channel. J Biol Chem. 2003;278(10):8348–55. doi: 10.1074/jbc.M209565200. [DOI] [PubMed] [Google Scholar]
- 67.Remppis A, Greten T, Schafer BW, Hunziker P, Erne P, Katus HA, et al. Altered expression of the Ca(2+)-binding protein S100A1 in human cardiomyopathy. Biochim Biophys Acta. 1996;1313(3):253–7. doi: 10.1016/0167-4889(96)00097-3. [DOI] [PubMed] [Google Scholar]
- 68.Pleger ST, Shan C, Kziencek J, Mueller O, Bekereddjian R, Remppis A, et al. Retroinfusion-facilitated inotropic AAV9-S100A1 gene therapy restores global cardiac function in a clinically relevant pig heart failure model. Circulation. 2008;118:S_792. [Google Scholar]
- 69.Ehlermann P, Remppis A, Guddat O, Weimann J, Schnabel PA, Motsch J, et al. Right ventricular upregulation of the Ca(2+) binding protein S100A1 in chronic pulmonary hypertension. Biochim Biophys Acta. 2000;1500(2):249–55. doi: 10.1016/s0925-4439(99)00106-4. [DOI] [PubMed] [Google Scholar]
- 70.Ackermann GE, Domenighetti AA, Deten A, Bonath I, Marenholz I, Pedrazzini T, et al. S100A1 deficiency results in prolonged ventricular repolarization in response to sympathetic activation. Gen Physiol Biophys. 2008 Jun;27(2):127–42. [PubMed] [Google Scholar]
- 71.Most P, Boerries M, Eicher C, Schweda C, Ehlermann P, Pleger ST, et al. Extracellular S100A1 protein inhibits apoptosis in ventricular cardiomyocytes via activation of the extracellular-regulated kinase (ERK1/2) pathway. J Biol Chem. 2003;278:48404–12. doi: 10.1074/jbc.M308587200. [DOI] [PubMed] [Google Scholar]
- 72.Pieske B, Houser SR. [Na+]i handling in the failing heart. Cardiovasc Res. 2003;57:874–86. doi: 10.1016/s0008-6363(02)00841-6. [DOI] [PubMed] [Google Scholar]
- 73.Voelkers M, Weidenhammer C, Herzog N, Friedrich O, Fink RHA, Koch WJ, et al. S100A1 prevents arrythmogenic diastolic sarcoplasmic reticulum calcium leak in ventricular cardiomyocytes. Circulation. 2008;118:S_527. [Google Scholar]
- 74.Remppis A, Pleger ST, Most P, Lindenkamp J, Ehlermann P, Löffler E, et al. S100A1 gene transfer: A strategy to strenghten engineered cardiac grafts. J Gen Med. 2004;6:387–94. doi: 10.1002/jgm.513. [DOI] [PubMed] [Google Scholar]
- 75.deGeorge B, Jr, Rosenberg M, Eckstein V, Gao E, Herzog N, Katus HA, et al. BMP-2 and FGF-2 synergistically facilitate adoption of a cardiac phenotype in somatic bone marrow c-kit+/sca-1+ stem cells. Clin Trans Sci. 2008;2:116–25. doi: 10.1111/j.1752-8062.2008.00034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dimmeler S, Zeiher AM, Schneider MD. Unchain my heart: the scientific foundations of cardiac repair. J Clin Invest. 2005 Mar;115(3):572–83. doi: 10.1172/JCI24283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Donato R. S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int J Biochem Cell Biol. 2001;33(7):637–68. doi: 10.1016/s1357-2725(01)00046-2. [DOI] [PubMed] [Google Scholar]