

Abstract
Endogenous protein quality control machinery has long been suspected of influencing the onset and progression of neurodegenerative diseases characterized by accumulation of misfolded proteins. Huntington's disease (HD) is a fatal neurodegenerative disorder caused by an expansion of a polyglutamine (polyQ) tract in the protein huntingtin (htt), which leads to its aggregation and accumulation in inclusion bodies. Here, we demonstrate in a mouse model of HD that deletion of the molecular chaperones Hsp70.1 and Hsp70.3 significantly exacerbated numerous physical, behavioral and neuropathological outcome measures, including survival, body weight, tremor, limb clasping and open field activities. Deletion of Hsp70.1 and Hsp70.3 significantly increased the size of inclusion bodies formed by mutant htt exon 1, but surprisingly did not affect the levels of fibrillar aggregates. Moreover, the lack of Hsp70s significantly decreased levels of the calcium regulated protein c-Fos, a marker for neuronal activity. In contrast, deletion of Hsp70s did not accelerate disease in a mouse model of infectious prion-mediated neurodegeneration, ruling out the possibility that the Hsp70.1/70.3 mice are nonspecifically sensitized to all protein misfolding disorders. Thus, endogenous Hsp70s are a critical component of the cellular defense against the toxic effects of misfolded htt protein in neurons, but buffer toxicity by mechanisms independent of the deposition of fibrillar aggregates.
Introduction
Many neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), prion disease and HD, are characterized by conformational changes in disease-causing proteins that result in misfolding and aggregation and have collectively been termed protein-conformational disorders. In contrast to AD, PD, and ALS, in which the vast majority of cases are idiopathic, HD is one of a number of inherited neurodegenerative disorders, collectively termed polyQ diseases, which are caused by an expansion of CAG repeats, coding for glutamine, in their respective disease proteins. The deposition of aggregation-prone proteins that contain expanded polyQ repeats in inclusion bodies is a neuropathological hallmark of the majority of these disorders.
The accumulation of misfolded proteins in cells triggers a protective stress response that includes the upregulation of heat shock proteins (Hsps) that function as molecular chaperones to help to restore cellular homeostasis (Lindquist, 1986). Postmitotic neurons, unable to dilute misfolded and/or aggregated proteins through cell division, are particularly vulnerable to the deleterious effects of misfolded proteins (Muchowski and Wacker, 2005). Accordingly, the endogenous protein quality control system is speculated to be critical in controlling the onset and severity of protein-conformational diseases that affect the brain.
The 70 kDa Hsps (Hsp70s) are abundantly expressed molecular chaperones that participate in a variety of fundamental cellular processes. Hsp70s promote the renaturation of misfolded and/or aggregated proteins through ATP-dependent cycles of binding and release and are likely to provide a first line of defense against aggregation-prone disease proteins in vivo (Hartl and Hayer-Hartl, 2002). Indeed, genetic screens and directed studies have shown that Hsp70 and its partners potently modulate the aggregation and/or suppresses the toxicity of mutant polyQ proteins in cell-, yeast-, worm- and fly-based models of polyQ aggregation and disease (Warrick et al., 1998; Chai et al., 1999; Warrick et al., 1999; Jana et al., 2000; Krobitsch and Lindquist, 2000; Muchowski et al., 2000; Kobayashi and Sobue, 2001; Zhou et al., 2001; Gunawardena et al., 2003; Nollen et al., 2004). Hsp70 overexpression also conferred a dose-dependent improvement in behavioral phenotypes of transgenic mouse models of Spinocerebellar ataxia-1 (SCA1) and Spinal and bulbar muscular atrophy (SBMA) (Cummings et al., 2001; Adachi et al., 2003). Conversely, overexpression of Hsp70 in the R6/2 mouse model of HD had only a marginal effect on weight loss and no effect on other behavioral and neuropathological features (Hansson et al., 2003; Hay et al., 2004).
The goal of this study was to determine whether endogenous Hsp70s can modulate the onset, progression and/or severity of pathogenesis in a mouse model of HD. We used the well characterized R6/2 transgenic model of HD, in which expression of htt exon 1 with ∼150 CAG repeats causes a progressive HD-like behavioral phenotype, including a robust decline in motor performance, alterations in activity level, weight loss and premature death (Mangiarini et al., 1996). R6/2 mice also accumulate mutant htt exon 1 in intranuclear and cytoplasmic inclusion bodies (Davies et al., 1997), a feature of HD brains (DiFiglia et al., 1997). To determine whether inducible Hsp70s play a protective role in the R6/2 model of HD, we crossed transgenic R6/2 mice with knock-out mice that lack both Hsp70.1 and Hsp70.3.
Materials and Methods
Animals and breeding strategy.
The University of Washington Animal Care and Use Committee, the University of California San Francisco IACUC Committee, or the Massachusetts Institute of Technology (MIT) Committee on Animal Care approved all experiments and procedures involving mice. Mice were maintained and bred in accordance with National Institutes of Health guidelines. Hemizygous transgenic R6/2tg/− male founder mice were kindly provided by Dr. James Olson (Fred Hutchinson Cancer Research Center, Seattle, WA). The R6/2tg/− male mice were backcrossed five times to C57BL/6 female mice to generate a colony of R6/2tg/− mice. The Hsp70.1/3 knock-out mice were originally generated by simultaneously targeting the Hsp70.1 and Hsp70.3 genes so that homologous recombination with the targeting construct resulted in a 12 kb deletion of both Hsp70.1 and Hsp70.3 coding regions as well as insertion of a neomycin-resistance gene (Hampton et al., 2003). A breeding pair of double knock-out Hsp70.1/3−/− mice (herein referred to as Hsp70−/− mice) were obtained with the permission of Dr. David Dix from Dr. Philip Mirke (University of Washington, Seattle, WA) and used to establish a colony of Hsp70−/− mice that was maintained on a C57BL/6 background for R6/2 studies and on 129Sv/Ev for prion studies. Hsp70−/− females were mated with R6/2tg/− males. Resulting R6/2tg/−;Hsp70−/+ males were mated with Hsp70−/− females to yield four genotypes: R6/2−/−;Hsp70−/+, R6/2tg/−;Hsp70−/+, R6/2−/−;Hsp70−/−, and R6/2tg/−;Hsp70−/−. Female mice of these four genotypes were analyzed alongside female R6/2tg/−;Hsp70+/+and R6/2−/−;Hsp70+/+ mice for a total of six genotypes. The number of mice in each cohort that was analyzed in the behavioral paradigms was as follows: R6/2tg/−;Hsp70+/+ (n = 21), R6/2tg/−;Hsp70+/+ (n = 18), R6/2−/−;Hsp70−/+ (n = 27), R6/2tg/−;Hsp70−/+ (n = 22), R6/2−/−;Hsp70−/−(n = 18), and R6/2tg/−;Hsp70−/− (n = 18). The experimenter was blind to the genotype during all testing paradigms. At 4 weeks of age the mice were weaned and housed randomly in groups of five. Mice were allowed access to water and food ad libitum and maintained on a 12 h light-dark cycle. At 10 weeks of age, mice were given powdered chow mixed with water (mash) to provide adequate nutrition and hydration.
Genotyping.
Mouse tail DNA was analyzed by PCR to determine the genotype. The R6/2 transgene was identified as described using the following primer sequences to identify the R6/2 transgene: forward-CGCAGGCTAGGGCTGTCAATCATGCT and reverse-TCATCAGCTTTTCCAGGGTCGCCAT (Hockly et al., 2003). Hsp70−/− and Hsp70−/+ mice were genotyped using a protocol established by the mutant mouse regional resource center at UC Davis (http://www.mmrrc.org/strains/372/0372.html). The primer sequences used to identify the targeted knock-out Hsp70.1/3-neo were: forward-GAACGGAGGATAAAGTTAGG and reverse-AGTACACAGTGCCAAGACG. The primer sequences used to identify the wild-type (WT) Hsp70.3 allele were: forward-GTACACTTTAAACTCCCTCC and reverse-CTGCTTCTCTTGTCTTCG.
We used GeneMapper techniques to determine the CAG repeat number by measuring the size of fluorescently labeled PCR products that cover the CAG repeat region in the exon 1 of HD gene. GeneMapper results showed that the R6/2tg/−;Hsp70+/+ mice used in the behavioral assays of this study had a CAG repeat length of ∼185. Genetic deletion of Hsp70.1/3 did not have a dramatic effect on CAG repeat length, which was ∼181 in the R6/2tg/−;Hsp70−/− mice used in the behavioral assays. R6/2tg/−;Hsp70+/+ mice used in the neuropathological assays of this study had a CAG repeat length of 115.
Prion studies. Hsp70.1/3 knock-out mice (Hsp70−/−) used for the prion studies were the same mice used for the R6/2 study, other than being maintained on a 129Sv/Ev pure background (Hampton et al., 2003). The Hsp70 overexpressing transgenic mouse was maintained on a hybrid C57BL/6-SJL background and expresses the rat inducible Hsp70 gene of a rat under a β-actin promoter (Marber et al., 1995). Hsp70−/− (n = 19) and Hsp70+/+ (n = 12) mice were injected intracranially with 30 μl of the Rocky Mountain Laboratory (“RML”) strain of murine prions, corresponding to a dose of ∼3.5 log LD50/30 μl. Hsp70 Tg+/− (n = 14) and Hsp70 Tg−/− (n = 10) mice were injected intracranially with 5.5 log LD50/30 μl. Hsp70−/−(n = 19) and Hsp70+/+ (n = 11) mice were also injected with “22L” strain of murine prions at a dose of ∼3.5 log LD50/30 μl. Hsp70 Tg+/− (n = 12) and Hsp70 Tg−/− (n = 11) mice were injected intraperitoneally with 100 μl of 4.5 log LC50 RML. Mice were monitored daily for typical prion symptoms, such as imbalance, priapism (males), and weight loss.
Survival.
For the R6/2 study (performed at the University of Washington) mice were observed twice daily, in the early morning and late afternoon. Survival was evaluated as the time to which the mice either died spontaneously, or exceeded a defined endpoint criterion. Motor performance, neurobehavioral and physical symptoms, weight, and ability to feed were closely monitored. Mice were killed when they had lost >20% of their maximal weight, and were no longer actively eating or drinking. For the prion studies (performed at MIT), mice were closely monitored and killed when they were unable to reach the food bin or water spout or regain posture after being placed on their side.
Rotarod experiments.
A Rotamex rotarod (Rotamex 4/8, Columbus Instruments International) was programmed to accelerate from 4 to 40 rpm over a period of 10 min and measure the latency to fall. Testing was performed every 2 weeks, starting at week 4. During the first week of testing (week 4) the mice performed three trials per day on four consecutive days. Data from day 1 of week 4 was excluded from the analysis as the mice were learning the task. During the subsequent weeks of testing (6, 8, 10, 12, and 14 weeks), the mice were tested on three consecutive days for three trials per day and all of the trial data were included in the analysis. For each week, the trials were pooled and used to calculate the average latency to fall for each mouse.
Weight loss.
Starting at 28 d, the mice were weighed twice weekly, at the same time of day, to the nearest 0.1 g.
Neurobehavioral and physical phenotype assessment.
Beginning week 6, mice were evaluated once a week, as described (Ditzler et al., 2003) to extensively characterize their neurobehavioral and physical phenotype. Each mouse was removed from its home cage and placed into a new, sterile cage where it was observed for 2 min. Briefly, to assess the neurobehavioral phenotype mice were scored for grooming, spontaneous activity, and locomotor activity. During the same 2 min period the physical phenotype of each mouse was scored for palpebral closure, coat appearance, body position and tail position. The scoring protocol for the neurobehavioral and physical assessment is detailed in supplemental Table 1, available at www.jneurosci.org as supplemental material. At the end of the 2 min period the mouse was removed and suspended by the tail ∼10 cm above the cage for 30 s to analyze pathogenic clasping behavior. Paw clasping behavior was scored from 0 to 2 points as described in supplemental Table 1, available at www.jneurosci.org as supplemental material.
Statistics.
All data are expressed at the mean ± SEM. For each outcome measure a two-way ANOVA was performed to determine whether there was a significant interaction between the R6/2 transgene and the Hsp70.1/3 genes. Specifically, the Mixed Models ANOVA in SPSS 13 was used with week as a repeated variable, mouse as a subject variable and the R6/2 transgene or Hsp70 deletion as factors. An unstructured repeated covariance was used to analyze weight, rotarod, clasping, tremor, body postion, tail position, grooming, locomotor activity, and spontaneous activity outcome measures. A compound asymmetry repeated covariance was used to evaluate eye closure and fur phenotypes. In cases in which differences between the various genotypes were examined at a single time-point, a one-way ANOVA in conjunction with the Bonferroni post hoc test was performed in GraphPad Prism. The Kaplan–Meier method was used to evaluate survival, followed by the log rank test to identify significant changes in GraphPad Prism.
Biochemical experiments.
At 14 weeks of age mice were killed with CO2. The brains were removed and homogenized with 5 μl/mg tissue RIPA buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mm β-mercaptoethanol, 1 mm PMSF, and a protease inhibitor cocktail (Roche Diagnostics) and centrifuged at 10,000 × g for 90 min at 4°C. A Bradford assay was used to determine protein concentration of the supernatant fraction. For Western blots, 3× SDS sample buffer was added, and the samples were heated at 95°C for 5 min. Equal amounts of protein (25 μg) were loaded in each well, separated by 4–20% SDS/PAGE, transferred to nitrocellulose membranes, blocked for 30 min at room temperature in 5% milk/TBST. After overnight incubation with primary antibody (made in 5% milk/TBST, blots were rinsed three times in TBST, incubated with secondary antibodies for 2 h at room temperature, rinsed three times in TBST and detected with enhanced chemiluminescence (GE Healthcare). Antibodies and concentrations were as follows: EM48 (1:500, a kind gift from Dr. Xiao-Jiang Li, Emory University), GAPDH (1:200, Millipore Bioscience Research Reagents), secondary antibodies (1:5000, Jackson ImmunoResearch). All chaperone antibodies were from Stressgen Biotechnologies, and dilutions were as follows: Hsp70 (1:1000), Hsp40 (1:10,000), Hsp27 (1:1000), Hsp25 (1:5000), Hsc70 (1:1000), Hsp90 (1:5000). To detect formic acid-sensitive monomer/oligomers, 100 μg of total protein lysate was incubated with 100 μl of formic acid at room temperature for 1 h. Treating the lysate with formic acid releases mutant htt species that migrate at the approximated molecular weight of a trimer/tetramer, although it is possible that this species is an aberrantly migrating monomer. Formic acid was removed in a speed vacuum and 30 μl of SDS loading buffer was added. The samples were neutralized with 2 μl of 5 m NaOH and heated at 95°C for 5 min. For the filter assay, 30 μl of 1× SDS sample buffer (4% SDS) were incubated with 100 μg of total protein lysate at 95°C for 5 min and then filtered onto a cellulose acetate membrane with a slot blot manifold. For densitometry films were scanned using ArcSoft PhotoStudio 5.5, and analyzed with ImageQuant V2005 (GE Healthcare).
Neuropathology.
At 14 weeks of age, mice were deeply anesthetized with halothane and perfused with 100 ml of phosphate buffer, followed by 100 ml of 4% paraformaldehyde in phosphate buffer, pH 7.4. Brains were removed, cryoprotected overnight in 30% sucrose and frozen in cooled isopentane. To investigate the effects of Hsp70.1/Hsp70.3 deletion on levels of mutant htt immunoreactivity in R6/2 mutant mice, the sections were immunolabeled overnight with a rabbit polyclonal antibody (EM48, Millipore Bioscience Research Reagents) against a glutathione S-transferase fusion protein containing the first 256 aa of htt lacking the polyQ and polyproline stretches. Sections were washed in PBS and then placed in biotinylated secondary antibody (1:100) (Vector Laboratories) for 2 h. Sections were placed in 20% diaminobenzidine (DAB) (Vector Laboratories), mounted, dried, and coverslipped with Entillin (Fisher). Three immunostained sections per mouse were imaged with an Olympus digital microscope. A total of 10 digital images per section and region of interest were analyzed with Image-Pro Plus (MediaCybernetics) to determine the optical density per field and the mean diameter and number of intranuclear inclusions. Individual values were averaged and expressed as mean value. To quantify microglial activation, microtome sections from R6/2 mice were immunostained with a mouse monoclonal antibody against Iba-1 (1:1000, DakoCytomation) followed by biotinylated secondary antibody, avidin coupled to horseradish peroxidase and reacted with DAB. Sections were analyzed and the numbers of Iba-1-positive microglia were averaged and expressed as total number per 0.1 mm3. Ten digital images per field were obtained and analyzed with Image-Pro Plus (MediaCybernetics) to determine the number of microglia per unit area. Similar immunohistochemical methods were performed to quantify astrocyte activation with a mouse monoclonal antibody against GFAP (1:1000, Abcam), c-Fos with a rabbit polyclonal antibody (1:500, Abcam) and synaptophysin with a mouse monoclonal antibody (1:200, Sigma). From each animal at least three blind-coded random sections were analyzed, and the results were averaged and expressed as mean value. Two sets of mice were used for the pathology experiments. For the first analysis, the mice that remained alive after the 14 week behavioral study were perfused and the brains were harvested. For the second analysis, mice were bred specifically for the biochemical and pathology experiments so that a more comprehensive analysis could be performed with a larger number of mice/genotype. Shown here are the results of the second analysis, but results were similar in both groups of animals.
Results
Deletion of Hsp70.1 and Hsp70.3 decreases survival in the R6/2 mouse, but not in prion-infected mice
To determine whether endogenous Hsp70s play an important role in combating the toxic effects induced by a mutant htt fragment, we crossed the R6/2 mouse model of HD to knock-out mice lacking the inducible Hsp70.1 and Hsp70.3 genes (herein referred to as Hsp70.1/3). We subsequently analyzed a number of physical and behavioral outcome measures in six genotypes of mice: R6/2−/−;Hsp70+/+, R6/2−/−;Hsp70−/+, R6/2−/−;Hsp70−/−, R6/2tg/−;Hsp70+/+, R6/2tg/−;Hsp70−/+, and R6/2tg/−;Hsp70−/−.
Kaplan–Meier survival analysis demonstrated that deletion of one copy of Hsp70.1/3 did not alter the lifespan of the R6/2 mouse (details of all statistical analyses used in this study can be found in Materials and Methods). The survival curves of the R6/2tg/−;Hsp70+/+ and the R6/2tg/−;Hsp70−/+ mice were indistinguishable, as were the endpoint survival rates of 83% and 82%, respectively (Fig. 1A). Strikingly, deletion of both copies of Hsp70.1/3 profoundly affected R6/2 survival: only 50% of the R6/2tg/−;Hsp70−/− mice were alive at the study endpoint. Survival analysis demonstrated a significant decrease in the lifespan of the R6/2tg/−;Hsp70−/− mice relative to the R6/2tg/−;Hsp70+/+ (p = 0.033, log rank test) and the R6/2tg/−;Hsp70−/+ mice (p = 0.026). An intact endogenous Hsp70 response, thus, appears to be critical for survival in the R6/2 mice.
Figure 1.

Deletion of Hsp70.1 and Hsp70.3 decreases survival in the R6/2 mouse, but not in mice infected with prions. A, Kaplan–Meier survival curve for the indicated genotypes [R6/2−/−;Hsp70+/+ (n = 21), R6/2tg/−;Hsp70+/+ (n = 18), R6/2−/−;Hsp70−/+ (n = 27), R6/2tg/−;Hsp70−/+ (n = 22), R6/2−/−;Hsp70−/−(n = 18), and R6/2tg/−;Hsp70−/− (n = 18)] demonstrates that the absence of Hsp70.1/3 significantly decreased survival of R6/2 mice (log rank: p = 0.033). No nontransgenic, Hsp70 heterozygous knock-out, or Hsp70 homozygous knock-out mice died during the 14 week time course. B, C, Kaplan–Meier survival curves for Hsp70+/+ (n = 11, and 12, respectively) and Hsp70−/− (n = 19) mice inoculated intracranially with 3.5 LC50 22L prion or 3.5 LC50 RML prion indicate that deletion of Hsp70.1/3 did not affect survival (log rank: p = 0.207 and 0.495, respectively).
Hsp70s are presumed to be broadly protective against the toxic effects of misfolded protein in the CNS. Therefore, we examined the effect of deleting endogenous Hsp70s on survival in two mouse models of prion disease. A dose corresponding to ∼3.5 log LD50 of the 22L strain of murine prions was injected intracranially into Hsp70+/+ and Hsp70−/− mice. Surprisingly, the absence of endogenous Hsp70s had no effect on lifespan (Fig. 1B). The survival curves of the Hsp70+/+ and Hsp70−/− mice were indistinguishable, and the median survival times of the Hsp70+/+ and Hsp70−/− mice injected with the 22L prion strain were 25.0 and 24.1 weeks, respectively. Similarly, the median survival times of the Hsp70+/+ and Hsp70−/− mice injected with the RML prion strain were identical at 26.4 weeks post-prion inoculation, and the two survival curves were indistinguishable (Fig. 1C). Moreover, transgenic overexpression of Hsp70 did not prolong survival of prion-infected mice (data not shown).
Deletion of Hsp70.1/3 worsens motor deficits in R6/2 mice
We used a panel of diverse outcome measures to systematically characterize the effect of deleting endogenous Hsp70s on the phenotypes of R6/2 mice. We first evaluated the effects of Hsp70 deletion on motor performance, as measured by rotarod analysis, which is widely used to characterize the progressive decline in motor performance of R6/2 mice (Carter et al., 1999; Hockly et al., 2002). We found that deletion of Hsp70.1/3 significantly enhanced (p < 0.05) the severity of rotarod deficits in R6/2 mice (Fig. 2A). As expected, at the early time point of 4 weeks the performances of wild-type and R6/2 mice were well matched. In contrast, the R6/2tg/−;Hsp70−/− mice were already significantly impaired at 4 weeks (p < 0.001), demonstrating that the absence of inducible Hsp70s decreases the age of onset of the R6/2 motor phenotype. The intermediate motor phenotype of the R6/2tg/−;Hsp70−/+ mice, when compared with the R6/2tg/−;Hsp70+/+ and the R6/2tg/−;Hsp70−/− mice, suggests that the relative expression levels of inducible Hsp70s modulate both the progression and severity of motor abnormalities in R6/2 mice.
Figure 2.

Deletion of Hsp70.1/3 worsens motor deficits in R6/2 mice. A–D, Deletion of Hsp70.1/3 decreases the latency to fall of R6/2 mice (two-way ANOVA: p < 0.05) (A), and increases severity of clasping (two-way ANOVA: p < 0.001) (B), tremor (two-way ANOVA: p < 0.001) (C), and grooming (two-way ANOVA: p < 0.03) (D). E, F, Deletion of Hsp70.1/3 decreases R6/2 spontaneous activity (two-way ANOVA: p < 0.001) but has only a moderate effect on locomotor activity. Error bars indicate SEM. Note that in the absence of the R6/2 transgene, the loss of one or both copies of Hsp70.1/3 does not influence any of the presented outcome measures.
Deletion of Hsp70.1 and Hsp70.3 exacerbates neurobehavioral phenotypes in R6/2 mice
To characterize the neurobehavioral and physical decline of our mice we used a modified SHIRPA assessment (Rogers et al., 1997). This behavioral protocol was recently refined to provide a rapid, reproducible and quantitative means of examining numerous outcome measures that clearly distinguish R6/2 transgenic mice from their wild-type littermates (Ditzler et al., 2003). The protocol includes a number of neurobehavioral (clasping, tremor, grooming, spontaneous and locomotor activities) and physical (weight, palpebral closure, coat appearance, body and tail position) outcome measures (supplemental Table 1, available at www.jneurosci.org as supplemental material).
Progressive clasping of the front and hind limbs that is triggered by a tail suspension test is a conserved motor abnormality observed in numerous mouse models of neurological disease and is widely used as a marker of neuronal dysfunction (Mangiarini et al., 1996; Carter et al., 1999; Stack et al., 2005). We analyzed clasping behavior once a week by suspending each mouse above its cage for 30 s and scoring 0 for no clasp, 1 for a mild clasp in which only the fore or hind-limbs press into the stomach, and 2 for a severe clasp in which both fore and hind-limbs touch and press into the stomach. Deletion of Hsp70.1/3 significantly worsened (p < 0.001) the average clasping score of the R6/2 mice (Fig. 2B). In contrast to the R6/2tg/−;Hsp70+/+mice, the R6/2tg/−;Hsp70−/+ and the R6/2tg/−;Hsp70−/− mice already displayed significant clasping by 6 weeks (p < 0.01). Moreover, the R6/2tg/−;Hsp70−/− mice consistently exhibited the most severe clasping score, followed by the R6/2tg/−;Hsp70−/+ mice and finally the R6/2tg/−;Hsp70+/+ mice, suggesting a gene dose-dependent effect on the onset, progression and severity of this R6/2 phenotype (Fig. 2B).
R6/2 mice develop a progressive, resting tremor in the limbs, trunk and head, which was scored as 0 (no tremor,) 1 (mild tremor), or 2 (severe tremor) (Mangiarini et al., 1996; Ditzler et al., 2003). Tremor analysis showed that deletion of Hsp70.1/3 significantly increased (p < 0.001) the score of the R6/2 mice (Fig. 2C). At 6 weeks, the R6/2tg/−;Hsp70+/+ mice had a negligible tremor score, whereas the R6/2tg/−;Hsp70−/+ and the R6/2tg/−;Hsp70−/− mice exhibited a significantly higher average score of 0.5 (p < 0.001), demonstrating that deletion of one or both alleles of Hsp70.1/3 decreased the age of tremor onset. The consistently intermediate score of the R6/2tg/−;Hsp70−/+ mice relative to the R6/2tg/−;Hsp70+/+ and R6/2tg/−;Hsp70−/− micesuggests a gene dose-dependent effect of Hsp70.1/3 on the R6/2 tremor phenotype.
As R6/2 mice become symptomatic, either a complete lack of grooming or a stereotypic, repetitive grooming behavior is often observed (Mangiarini et al., 1996; Carter et al., 1999). Repetitive hindlimb grooming is thought to mimic the choreiform movements displayed by HD patients (Mangiarini et al., 1996). Mice received a score of 1 for normal grooming and a score of 2 for abnormal grooming. Analysis of cumulative grooming scores revealed that the deletion of Hsp70.1/3 genes significantly worsened (p < 0.03) the abnormal grooming behavior of the R6/2 at later time points (Fig. 2D). In this case, however, the loss of both alleles of Hsp70.1/3 was required to enhance the progression and endpoint severity of the R6/2 grooming phenotype.
The progressive development of abnormalities in the activity level of R6/2 HD mice has been well characterized (Dunnett et al., 1998; Bolivar et al., 2003; Stack et al., 2005), and our modified SHIRPA protocol included two measures of activity. We first measured spontaneous activity by scoring the coverage of four delineated cage quadrants by each mouse during a 2 min testing period. A score of 1 denoted movement into all four quadrants, 2 denoted slow movement in three or less quadrants, 3 denoted no movement or stereotypic darting/circling movements. We found that deletion of Hsp70.1/3 significantly exacerbated (p < 0.001) the spontaneous activity phenotype of the R6/2 mouse, most noticeably after 8 weeks of age (Fig. 2E). The absence of both alleles of Hsp70.1/3 had a marked effect on the onset, progression and endpoint severity. Despite the fact that deletion of one allele of Hsp70.1/3 also had a more moderate effect, there was still a trend toward gene dose dependence for this outcome measure. We also performed the locomotor test as a second measure of activity by scoring the number of times that each mouse touched the side of the cage during a 2 min observation period. The locomotor activity test did not reveal a significant effect of the inducible Hsp70s on the R6/2 phenotype, although there was a trend toward a gene dose-dependent effect of Hsp70.1/3 deletion to enhance motor abnormalities (Fig. 2F). Thus, two distinct outcome measures showed that Hsp70.1/3 affects the development, progression and severity of activity deficits in the R6/2 mouse.
Deletion of Hsp70.1/3 exacerbates the physical phenotypes of R6/2 mice
To characterize decline in the physical phenotypes of the R6/2 mice, we measured weight and scored for coat appearance, body position, tail position and palpebral closure. Female R6/2 mice show a characteristic weight loss pattern: weight plateaus around week 8 and declines significantly at 12 weeks (Hockly et al., 2003). The weights of the R6/2−/−;Hsp70+/+, R6/2−/−;Hsp70−/+, and the R6/2−/−;Hsp70−/− mice were indistinguishable, demonstrating that the absence of inducible Hsp70s alone does not influence body weight (Fig. 3A). Analysis of the weight of the R6/2tg/−;Hsp70+/+ and R6/2tg/−;Hsp70−/− mice showed a significant interaction (p < 0.05) on this phenotype and a Hsp70.1/3 gene dose-dependent trend on weight loss was observed, suggesting that the inducible Hsp70s may modulate the onset, progression and endpoint severity of the R6/2 weight loss phenotype.
Figure 3.
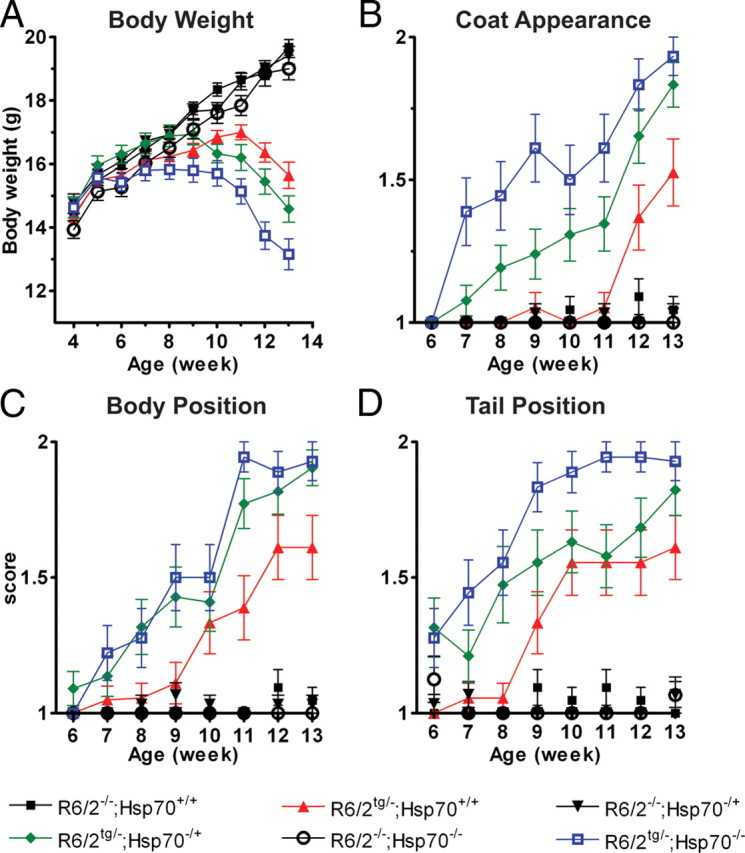
Deletion of Hsp70.1/3 exacerbates the physical phenotypes of R6/2 mice. The absence of Hsp70.1/3 significantly exacerbates the weight loss phenotype (two-way ANOVA: p < 0.05) (A), and worsens the coat appearance (two-way ANOVA: p < 0.001) (B), body position (two-way ANOVA: p < 0.02) (C), and tail position (two-way ANOVA p < 0.001) (D) of R6/2 mice. Error bars indicate SEM. Note that in the absence of the R6/2 transgene, the loss of one or both copies of Hsp70.1/3 does not influence any of the presented outcome measures.
The coat appearance of the R6/2 mice declines as the disease state progresses and is characterized by a score of 1 for a shiny, well groomed coat and a score of 2 for a scruffy and/or piloerected coat. Deletion of Hsp70.1/3 significantly worsened (p < 0.001) the R6/2 coat appearance phenotype (Fig. 3B). A decrease in the age of onset, enhanced progression and increase in endpoint severity were observed with a trend toward Hsp70.1/3 gene dose dependence. We also scored body position and tail position to further evaluate the effect of Hsp70.1/3 on the decline of the R6/2 physical phenotype. The R6/2 body position phenotype was scored as 1 for normal, and 2 for a hunched and rounded stature. Tail position was scored as 1 for normal or horizontally extended, and 2 for dragging/straub. Deletion of Hsp70.1/3 significantly enhanced the severity of the body position outcome measure (p < 0.02, Fig. 3C) and the tail position outcome measure (p < 0.001, Fig. 3D). In both cases a trend toward an Hsp70.1/3 gene-dose dependent enhancement of phenotypic severity was observed. The only component of the physical phenotype test that was unaffected by the deletion of Hsp70.1/3 was palpebral closure (data not shown). Importantly, all outcome measures included in the neurobehavioral and physical phenotype assessment showed that there were no significant differences between the wild-type nontransgenic mice and the Hsp70.1/3 heterozygous or homozygous knock-out mice.
Deletion of Hsp70.1/3 increases the size of inclusion bodies in R6/2 mice
To determine whether the exacerbated behavioral and physical phenotypes observed in R6/2 mice that lacked inducible Hsp70s correlated with changes in the density or size of inclusion bodies formed by mutant htt exon 1, we examined serial sections from the neocortex of 14-week-old mice with immunohistochemistry and the EM48 anti-htt antibody. As expected, the R6/2−/−;Hsp70+/+, R6/2−/−;Hsp70−/− and R6/2−/−;Hsp70−/+ mice did not display EM48-positive inclusion bodies (data not shown). Immunohistochemical analyses on cortical brain sections with EM48 suggested R6/2tg/−;Hsp70−/− had an increased density of inclusion bodies compared with R6/2tg/−;Hsp70+/+ mice (Fig. 4A). However, quantification of the number of inclusion bodies in a defined brain volume indicated this difference only showed a trend toward statistical significance (p = 0.086) (Fig. 4B).
Figure 4.

Deletion of Hsp70.1/3 increases the size of inclusion bodies in R6/2 mice. A, Representative images (600×) of inclusion bodies in the neocortex of R6/2 mice as detected with the anti-htt antibody EM48. B, Quantification of inclusion body number in the neocortex shows that R6/2tg/−;Hsp70−/− mice have an increase in the density of inclusion bodies compared with R6/2tg/−;Hsp70+/+ mice, although this difference only showed a trend toward statistical significance (p = 0.086). C, Representative images (1000×) illustrating the size of inclusion bodies in the neocortex of R6/2 mice as detected with the anti-htt antibody EM48. D, Quantification of inclusion body size shows that the average size of the inclusion bodies was significantly larger (p < 0.001) in R6/2tg/−;Hsp70−/− mice compared with R6/2tg/−;Hsp70+/+ mice. Statistical comparisons were performed by one-way ANOVA (n = 6–11 mice per group).
Analysis of average inclusion body diameter demonstrated that the R6/2tg/−;Hsp70+/+ (4.22 ± 0.55 μm) and R6/2tg/−;Hsp70−/+ (3.81 ± 0.34 μm) mice were indistinguishable (p > 0.05, Fig. 4D). In comparison, the R6/2tg/−;Hsp70−/− inclusion bodies (7.68 ± 0.44 μm) stained with EM48 were dramatically and significantly larger (p < 0.001) than in R6/2tg/−;Hsp70+/+ mice (Fig. 4C, D). The pixel intensity of EM48 staining in R6/2 mice lacking both alleles of Hsp70.1/3 also appeared greater than in R6/2 mice alone. In summary, these results indicate that the complete absence of inducible Hsp70s increased the size of inclusion bodies formed by mutant htt exon 1 in R6/2 mice, consistent with in vitro data indicating Hsp70 can directly modulate the misfolding and aggregation of mutant htt (Muchowski et al., 2000; Wacker et al., 2004).
Deletion of Hsp70.1/3 does not modulate levels of SDS-insoluble fibrillar protein aggregates formed by mutant Htt exon 1 in R6/2 mice
We next sought to determine whether the increased size of inclusion bodies in the absence of inducible Hsp70s in R6/2 mice was attributed to increased levels of aggregates formed by mutant htt exon 1. We used Western immunoblots, filter-trap assays and agarose gels to measure the relative levels of SDS-insoluble aggregates and formic acid–sensitive htt species in the brains of 14-week-old R6/2tg/−;Hsp70+/+ and R6/2tg/−;Hsp70−/− mice.
Western blots probed with the EM48 antibody on total brain homogenates from R6/2tg/−;Hsp70+/+ mice showed that all of the reactivity corresponded to large SDS-insoluble aggregates that were retained in the stacking portion of the gel (Fig. 5A) as described (Davies et al., 1997). An identical pattern of reactivity is observed on Western blots that contain purified mutant htt exon 1 with 53Q that has been aggregated into fibrillar protein assemblies (Wacker et al., 2004). Surprisingly, analysis of the pixel intensities relative to the GAPDH loading controls showed that the average EM48 reactivities for the R6/2tg/−;Hsp70+/+ and R6/2tg/−;Hsp70−/− mice were indistinguishable (Fig. 5A, B). Previous studies showed that HD brain homogenates treated with formic acid liberate a SDS-resistant oligomer as analyzed by Western immunoblots (Iuchi et al., 2003; Hoffner et al., 2005). Similar to the results observed in HD brain homogenates, we found that treatment of total brain homogenates from R6/2tg/−;Hsp70+/+ mice with formic acid released two bands that reacted with EM48 which migrated at an apparent molecular mass of 70–85 kDa, while concomitantly leading to nearly a complete loss of reactivity in the stacking gel (Fig. 5C). Based on their apparent molecular mass, these bands may reflect a low molecular mass SDS-resistant oligomer or aberrantly migrating monomers of mutant htt exon 1. The levels of formic acid-sensitive monomers/oligomers (normalized to GAPDH reactivity) appeared to increase in the absence of Hsp70.1/3, but did not reach statistical significance (Student's t test p = 0.15). (Fig. 5D). Identical findings were observed using the 3B5H10 antibody generated by the Finkbeiner laboratory (data not shown).
Figure 5.
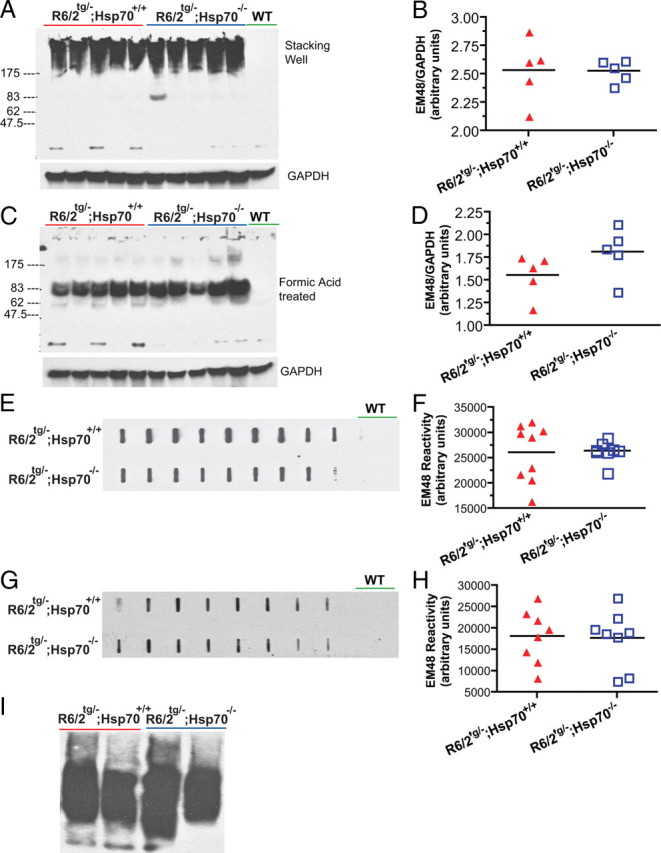
Deletion of Hsp70.1/3 does not modulate levels of SDS-insoluble fibrillar protein aggregates formed by mutant htt exon 1 in R6/2 mice. A, B, Deletion of Hsp70.1/3 does not alter the levels of EM48 reactive SDS-insoluble aggregates (normalized to GAPDH reactivity) measured with Western immunoblots in 14-week-old R6/2 brain homogenates (Student's t test p = 0.96). C, D, Treatment of brain homogenates with formic acid liberates an SDS-resistant monomeric/oligomeric mutant huntingtin exon 1 species. The levels of formic acid-sensitive monomers/oligomers (normalized to GAPDH reactivity) appeared to increase in the absence of Hsp70.1/3, but did not reach statistical significance (Student's t test p = 0.15). E, F, The levels of SDS-insoluble EM48-positive fibrillar aggregates in brain measured by a filter-trap assay do not change in the absence of Hsp70.1/3 (Student's t test p = 0.89). G, H, Formic acid-treated brain homogenates were subjected to the filter-trap assay, which showed no change in EM48 immunoreactivity in the absence of Hsp70.1/3 (Student's t test p = 0.90). I, A native agarose gel used to examine EM48 immunoreactive oligomeric species in R6/2 brain homogenates shows no apparent change in the absence of Hsp70.1/3.
We next used filter-trap assays as an independent method to evaluate total SDS-insoluble material formed by mutant htt exon 1 in R6/2 brains in the presence and absence of inducible Hsp70s. In this assay, total brain homogenates were boiled in SDS and filtered through a cellulose acetate membrane that contains 0.2 μm pores. Previous studies with brains from R6/2 mice showed that this method traps large (>0.2 μm) SDS-insoluble aggregates of fibrillar material (Scherzinger et al., 1997). Consistent with the Western immunoblots, we found that the levels of SDS-insoluble material detected by EM48 in filter-trap assays were not significantly different between R6/2tg/−;Hsp70+/+ and R6/2tg/−;Hsp70−/− mice (Fig. 5E, F). Interestingly, total brain homogenates treated with formic acid and SDS were still detected by EM48 in filter assays (Fig. 5G), suggesting that this treatment releases oligomeric species larger than 0.2 μm in size. However, as with the other assays, no significant differences were observed in brain homogenates analyzed in this manner between R6/2tg/−;Hsp70+/+ and R6/2tg/−;Hsp70−/− mice (Fig. 5G, H). Similar results were also obtained when agarose native gels were used to detect oligomeric species in total brain homogenates from R6/2tg/−;Hsp70+/+ and R6/2tg/−;Hsp70−/− mice (Fig. 5I). Thus, three independent approaches used to evaluate mutant htt exon 1 aggregates in 14-week-old R6/2 brain homogenates showed no significant differences in aggregate levels in the presence or absence of inducible Hsp70s. Similar results were obtained in 7-week-old R6/2 brain homogenates (data not shown).
Previous studies demonstrated changes in the relative levels of chaperone proteins in mouse models over the course of polyQ disease (Cummings et al., 2001; Hay et al., 2004). For example, expression of mutant ataxin-1 in a mouse model of SCA1 elicits an increase in Hsp70 expression (Cummings et al., 2001), whereas levels of Hsp70 and other chaperones decreased progressively in R6/2 mice (Hay et al., 2004). To test whether deletion of Hsp70.1/3 caused compensatory changes in the relative levels of other heat shock proteins, we performed Western immunoblots on brain homogenates from R6/2tg/−;Hsp70+/+, R6/2tg/−;Hsp70−/−, R6/2−/−;Hsp70+/+ and R6/2−/−;Hsp70−/− mice. At 14 weeks of age, no significant changes were detected in the levels of Hsp27 and Hsp90 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), or Hsp25, Hsp40 and Hsc70 (data not shown), relative to a GAPHD loading control. Thus, the lack of both Hsp70 alleles on a wild-type or R6/2 strain background did not appear to confer compensatory changes in levels of other major heat shock proteins.
Deletion of Hsp70.1/3 exacerbates the loss of c-Fos immunoreactivity and other neuropathological deficits in R6/2 mice
To determine the effect of Hsp70 deletion on neuronal loss in R6/2 mice we used immunohistochemistry and unbiased stereology with an antibody against the neuronal-specific protein NeuN in brain sections from 14-week-old mice. These analyses showed no significant change in NeuN immunoreactivity in the cortex or striatum of R6/2tg/−;Hsp70+/+ mice compared with WT animals, and no significant difference in NeuN levels between R6/2tg/−;Hsp70+/+ and R6/2tg/−;Hsp70−/− mice (data not shown). In an independent study, we recently found that immunoreactivity for the presynaptic protein synaptophysin and the calcium regulated immediate early gene product c-Fos, a surrogate marker for neuronal activity, were decreased in R6/2 mice relative to nontransgenic littermate controls, and that these changes were attenuated in R6/2 mice treated with a small-molecule inhibitor of kynurenine 3-monooxygenase in a manner that correlated with survival (P. Guidetti, W. Kwan, S.-Y. Huang, J. Lee, C. Patrick, F. Giorgini, T. Möller, C. S. Cheah, T. Wu, K. Scearce-Levie, J. M. Muchowski, E. Masliah, R. Schwarcz, and P. J. Muchowski, unpublished observations). In the current study immunohistochemical analysis of brain sections from R6/2tg/−;Hsp70+/+ mice showed a significant (p < 0.05) decrease in c-Fos immunoreactivity relative to WT mice in the cortex and striatum, and this loss was further and significantly exacerbated (p < 0.05) in R6/2tg/−;Hsp70−/− mice (Fig. 6A, B). Importantly, levels of c-Fos were not significantly different between R6/2−/−;Hsp70+/+ and R6/2−/−;Hsp70−/− mice. Levels of synaptophysin immunoreactivity also appeared to be decreased in the cortex and striatum of R6/2tg/−;Hsp70−/− mice compared with R6/2−/−;Hsp70−/− mice, although this decrease did not reach statistical significance possibly explained by the lack of statistical power due to the small numbers of mice analyzed (n = 4–6 per group) (Fig. 6C, D). A recent study showed increased levels of immunoreactivity for the microglia-specific protein Iba1 in R6/2 mice (Simmons et al., 2007). We observed a significant (p = 0.0297) increase in immunoreactivity for Iba1, and a trend toward an increase in the astrocyte-specific marker GFAP in the cortex of R6/2tg/−;Hsp70−/− mice compared with R6/2−/−;Hsp70+/− and −/− mice (Fig. 6E, F). Insufficient brain material unfortunately precluded the analysis of other genotypes in these studies. Our results demonstrate that endogenous Hsp70s protect against the loss of c-Fos in a highly significant manner, and suggest these chaperones are critical regulators of neuronal activity and inflammatory responses in R6/2 mice.
Figure 6.
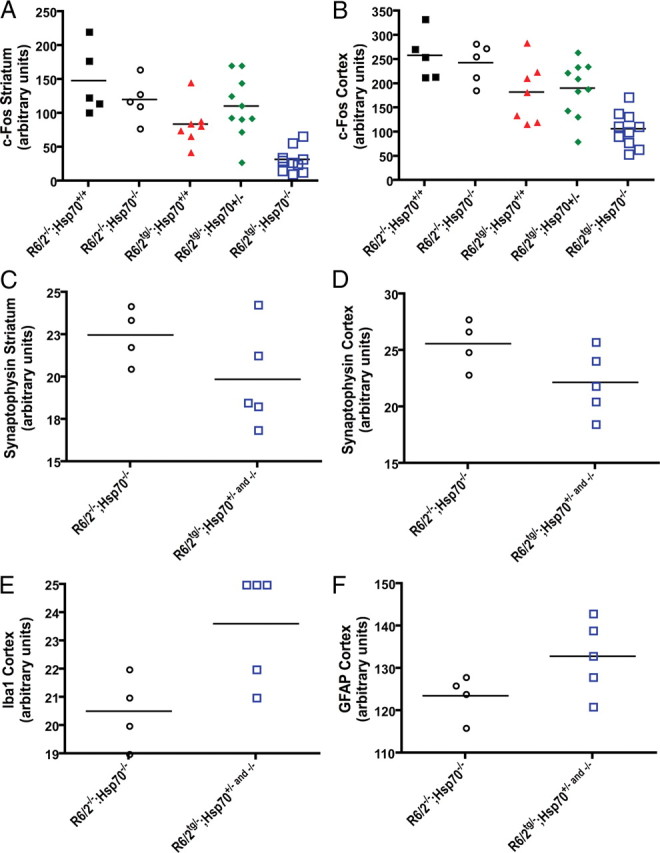
Deletion of Hsp70.1/3 exacerbates the loss of c-Fos immunoreactivity and other neuropathological deficits in R6/2 mice. A, B, Quantification of c-Fos immunohistochemistry in the neocortex from 14-week-old mice shows that R6/2tg/−;Hsp70−/− mice have a significant decrease (p < 0.05) in c-Fos levels compared with R6/2tg/−;Hsp70+/+ mice. C, D, Quantification of synaptophysin immunohistochemistry in the neocortex from 14-week-old mice shows that R6/2tg/−;Hsp70+/− and −/− mice have a significant decrease (p < 0.05) in synaptophysin levels compared with R6/2−/−;Hsp70−/− mice. E, F, Quantification of Iba1 and GFAP immunohistochemistry in the neocortex from 14-week-old mice shows that R6/2tg/−;Hsp70+/− and −/− mice have a significant increase (p = 0.0297) in Iba1 levels, and a trend toward increased GFAP levels (p = 0.1048), respectively, compared with R6/2−/−;Hsp70−/− mice.
Discussion
Here we showed that endogenous Hsp70s critically regulate the toxicity of a disease-causing misfolded protein in a mouse model of HD. The absence of even one allele of the Hsp70.1/Hsp70.3 genes significantly exacerbated the severity of a number of outcome measures for the R6/2 mouse model of HD (rotarod, clasping, activity, weight, coat appearance, and body position). The absence of both alleles of Hsp70.1/3 profoundly enhanced the onset, severity and progression of behavioral phenotypes in R6/2 mice, including a significant decrease in median lifespan. R6/2 mice completely lacking inducible Hsp70s showed an increase in the number and size of inclusion bodies, although these findings did not correlate with a biochemical changes in the relative levels of SDS-insoluble fibrillar aggregates as measured by multiple independent approaches. Finally, we found that deletion of Hsp70.1/3 exacerbated the loss of c-Fos, a surrogate marker for neuronal activity, in a highly significant manner. These findings indicate that the absence of inducible Hsp70s increased neuronal sensitivity to mutant htt exon 1 in the R6/2 mouse model of HD, without affecting htt expression or its accumulation into SDS-insoluble aggregates.
Deletion of Hsp70.1/3 had no significant effect on lifespan in two mouse models of transmissible prion disease. This is not simply because prion diseases are so extreme that they can not be modified. Indeed, deletion of HSF1, a master regulator of homeostatic stress responses, has a pronounced effect on the course of these same prion models (Steele et al., 2008). Thus, the striking effect of the absence of inducible Hsp70s on R6/2 mice indicates that a specific genetic interaction occurs between the inducible Hsp70s and the mutant htt fragment in vivo. Although the inducible Hsp70s may play a pivotal role in prion propagation in yeast (Tutar et al., 2006; Loovers et al., 2007), our results suggest that the inducible Hsp70s do not influence toxicity in mice infected with established strains of prions. Importantly, unlike HD, which is an autosomal dominant inherited neurodegenerative disorder, prion disease encompasses diverse etiologies in addition to acute infection (Kingsbury et al., 1983). The inducible Hsp70s may possibly play a role in suppressing toxicity in other mouse models of spontaneous and/or genetically derived prion disease.
The loss of one copy of Hsp70.1/3 did not decrease the lifespan of R6/2 mice, suggesting a potent activity of endogenous Hsp70 chaperones, even when present at half of their normal concentration, to mitigate pathogenic cascades and modulate disease onset, progression and severity in vivo. However, the decrease in the age of onset observed for the majority of behavioral and physical testing parameters in both the R6/2tg/−;Hsp70−/+ and the R6/2tg/−;Hsp70−/− mice demonstrates that an intact inducible Hsp70s response is required to limit mutant htt toxicity at the earliest stages of pathogenesis in R6/2 mice.
The absence of both alleles of Hsp70.1/3 significantly increased the average size and appeared to increase the number of inclusion bodies in R6/2 brains, yet paradoxically did not alter the total load of fibrillar aggregates detected biochemically. What mutant htt species detected by EM48 in brain sections can account for the increased size and abundance of inclusion bodies? Our biochemical studies excluded the possibility that the increased size and abundance of inclusion bodies were due to any significant changes in fibrillar and/or large oligomeric species that are insoluble in SDS. In addition, formic acid-treated R6/2 brain lysates had similar levels of mutant htt monomers and oligomers in the presence or absence of Hsp70s. Our previous in vitro studies used atomic force microscopy and biochemical approaches to demonstrate that the cooperative activity of Hsp70 and Hsp40 stabilized a monomeric conformation of a mutant htt fragment (HD53Q), while concomitantly suppressing the accumulation of annular and spherical oligomeric assemblies (Wacker et al., 2004). However, a recent study indicated Hsp70 and Hsp40 can also partition onto SDS-soluble mutant htt oligomers in an ATP-dependent manner (G. P. Lotz, J. Legleiter, E. Mitchell, S.-Y. Huang, C.-P. Ng, C. Glabe, L. M. Thompson, and P. J. Muchowski, unpublished observations). Therefore we speculate that, in the absence of inducible Hsp70s in R6/2 mice, small, SDS-soluble mutant htt exon 1 assemblies that accumulate may account for the increase in inclusion body density and size in the R6/2tg/−;Hsp70−/− mice. Consistent with this interpretation, deletion of C terminus of Hsp70 interacting protein (CHIP) in a mouse model of Spinocerebellar Ataxia Type 3 (SCA3) markedly increased levels of ataxin-3 microaggregates in a manner that correlated with exacerbated behavioral phenotypes in these mice (Williams et al., 2009). We hypothesize that inducible Hsp70s buffer toxicity by binding monomeric and/or low molecular mass SDS-soluble oligomers that are likely off-pathway to fibril formation, but may be potentially pathogenic. However, based on the multifunctional nature of Hsp70 it is very likely that this chaperone can also suppress protein misfolding toxicity by multiple mechanisms independent of its direct effects on misfolded protein (see below).
Although larger inclusion bodies were observed in R6/2 mice in the absence of Hsp70s, this does not necessarily suggest that inclusion bodies are toxic entities. Indeed, in direct contrast to the current study, we recently observed a strong positive correlation between survival and inclusion body size in mice treated with an inhibitor of the mitochondrial enzyme kynurenine 3-monooxygenase (P. Guidetti, W. Kwan, S.-Y. Huang, J. Lee, C. Patrick, F. Giorgini, T. Möller, C. S. Cheah, T. Wu, K. Scearce-Levie, J. M. Muchowski, E. Masliah, R. Schwarcz, and P. J. Muchowski, unpublished observations). Furthermore, systematic analysis of the effects of genetic enhancers (Willingham et al., 2003) or suppressors (Giorgini et al., 2005) of mutant htt exon 1 toxicity in yeast showed no correlations between toxicity and inclusion bodies. These experiments underscore the inherent limitations of quantifying inclusion body size and abundance in mouse brain sections using immunohistochemistry to draw meaningful deductions of the role of these abnormal brain deposits on in vivo pathogenesis. We propose that the molecular composition of SDS-soluble conformers that may exist in a diffuse fraction or in inclusion bodies, be they monomers or small oligomers, will be the key to understanding which structures mediate pathogenesis. Thus, tools to identify and track such structures in situ, such as antibodies, will be required before unequivocal experiments can determine which are the toxic species of mutant htt in mouse models of HD.
Although a primary function of Hsp70s in animal models of polyQ disease may be to counteract the assembly process that leads to the accumulation of toxic monomers/oligomers, the inducible Hsp70s may also buffer the toxicity of mutant htt monomers/oligomers by masking surfaces that promote pathogenic interactions with essential cellular proteins. For example, in one study, a mutant htt monomer underwent an intramolecular transition that facilitated an interaction with the Tata binding protein (TBP) and ultimately resulted in the functional inactivation of this important transcription factor (Schaffar et al., 2004). Addition of Hsp70 to the in vitro system prevented the conformational rearrangement of mutant htt and thus inhibited the pathogenic interaction with TBP, suggesting that the activity of Hsp70 to bind and hold mutant htt monomers can prevent aberrant protein-protein interactions that lead to neuronal dysfunction. Mutant htt, in addition to causing transcriptional repression, has also been shown to upregulate p53 associated transcriptional events in neuronal cultures (Bae et al., 2005). p53 is a strong suppressor of Hsp70 expression in specific neuronal subtypes that are affected in HD (Tagawa et al., 2007), and, moreover, genetic deletion of p53 ameliorates behavioral abnormalities in the N171-82Q mouse model of HD (Schilling et al., 1999; Bae et al., 2005). Thus, it is tempting to speculate that the effect of p53 on HD pathogenesis may be at least partially mediated by changes in the expression of inducible Hsp70s.
The exacerbation of R6/2 phenotypes in mice lacking Hsp70s may also be due to an overall disruption in the protein homeostasis network, as suggested from studies in Caenorhabditis elegans by Morimoto and colleagues (Gidalevitz et al., 2006). Consistent with this scenario, we observed that levels of the calcium regulated immediate early gene c-Fos and the presynaptic protein synaptophysin were decreased in R6/2 mice lacking Hsp70s relative to controls, whereas levels of protein markers for inflammatory responses (Iba1 and GFAP) were increased. Although the functional significance of these changes in R6/2 mice has not yet been investigated, the levels of c-Fos, which is used a surrogate marker for neuronal activity, are tightly linked to cognitive deficits in mouse models of AD (Palop et al., 2003). The apparent loss of synaptophysin in R6/2 mice lacking Hsp70 is consistent with previous studies in R6/2 mice (Cepeda et al., 2003) and, more broadly, with studies that suggest synaptic loss may be important for pathogenesis in HD (Li et al., 2003). Interestingly, as mutant htt inhibits the acetyltransferase activity of CREB-binding protein (CBP) (Steffan et al., 2000; Steffan et al., 2001), which itself controls c-Fos expression (Yuan et al., 2009), it is possible that aberrant protein interactions between mutant htt and CBP, and suppression of these interactions by Hsp70 (Schaffar et al., 2004), mediate in part the effects we observed on c-Fos expression in R6/2 mice. Our results also indicate endogenous Hsp70s may influence inflammatory responses, consistent with previous reports (Van Molle et al., 2002; Hampton et al., 2003; Singleton and Wischmeyer, 2006; Mycko et al., 2008). Collectively these studies strongly suggest that Hsp70s may modulate pathogenesis of protein misfolding diseases in vivo by direct and indirect effects in multiple cell types that may only be dissected by modulating levels of these proteins and their interacting proteins in specific cell types in vivo.
The majority of the behavioral outcome measures that we examined in R6/2 mice showed a trend toward Hsp70.1/3 gene dose dependence, demonstrating that the relative levels of inducible Hsp70s can dramatically alter pathogenesis in vivo. A recent study used RNA interference (RNAi) to show that the expression levels of Hsp70 dictate the susceptibility of primary neurons to mutant htt toxicity (Tagawa et al., 2007). Thus, even a modest increase in the levels of molecular chaperones may suffice to decrease the severity of protein-conformational disorders. Indeed, treatment with arimoclomol, a compound that acts to amplify the endogenous heat shock response to the accumulation of misfolded disease-causing proteins, significantly delayed disease progression in a mouse model of ALS (Kieran et al., 2004) and is being evaluated in a clinical trial in ALS patients. In addition, geranylgeranylacetone, which acts to increase the levels of heat shock proteins in vivo, significantly decreased the severity of the neuromuscular phenotype in a mouse model of SBMA (Katsuno et al., 2005). Pharmacological strategies aimed at enhancing the production or activity of molecular chaperones, such as Hsp70, may prove beneficial in the treatment of protein-conformational disorders.
The demonstration in this study that the endogenous Hsp70.1 and Hsp70.3 chaperones are an integral component of the physiological response to an aggregation-prone disease protein in vivo highlights the importance of investigating genetic modifiers of disease pathogenesis as potential therapeutic targets. The multifunctional activity of Hsp70 in vivo contributes to its attraction as a potential therapeutic target for diseases associated with protein misfolding and aggregation. Modification of the levels and/or activity of Hsp70 can potentially impact a number of important cellular pathways that influence HD pathogenesis, and likely plays similar role in modulating pathogenic cascades in diverse protein-conformational disorders.
Footnotes
Support for this study was provided by National Institute of Neurological Disease Grants NS47237 and NS054753 (P.J.M.) and National Institute of Aging Grant AG022074 (P.J.M., E.M.). We thank S. Ordway and G. Howard for editorial assistance and Artur Topolszki for assistance with mouse colony management for PrP studies. S.L. is an investigator of the Howard Hughes Medical Institute.
References
- Adachi H, Katsuno M, Minamiyama M, Sang C, Pagoulatos G, Angelidis C, Kusakabe M, Yoshiki A, Kobayashi Y, Doyu M, Sobue G. Heat shock protein 70 chaperone overexpression ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model by reducing nuclear-localized mutant androgen receptor protein. J Neurosci. 2003;23:2203–2211. doi: 10.1523/JNEUROSCI.23-06-02203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, Hayward SD, Moran TH, Montell C, Ross CA, Snyder SH, Sawa A. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron. 2005;47:29–41. doi: 10.1016/j.neuron.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Bolivar VJ, Manley K, Messer A. Exploratory activity and fear conditioning abnormalities develop early in R6/2 Huntington's disease transgenic mice. Behav Neurosci. 2003;117:1233–1242. doi: 10.1037/0735-7044.117.6.1233. [DOI] [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Hurst RS, Calvert CR, Hernandez-Echeagaray E, Nguyen OK, Jocoy E, Christian LJ, Ariano MA, Levine MS. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington's disease. J Neurosci. 2003;23:961–969. doi: 10.1523/JNEUROSCI.23-03-00961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Koppenhafer SL, Bonini NM, Paulson HL. Analysis of the role of heat shock protein (Hsp) molecular chaperones in polyglutamine disease. J Neurosci. 1999;19:10338–10347. doi: 10.1523/JNEUROSCI.19-23-10338.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings CJ, Sun Y, Opal P, Antalffy B, Mestril R, Orr HT, Dillmann WH, Zoghbi HY. Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet. 2001;10:1511–1518. doi: 10.1093/hmg/10.14.1511. [DOI] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Ditzler S, LeBlanc M, Kooperberg C, Hansen S, Coppin L, Olson JM. A rapid neurobehavioral assessment reveals that FK506 delays symptom onset in R6/2 Huntington's disease mice. Preclinica. 2003;1:115–126. [Google Scholar]
- Dunnett SB, Carter RJ, Watts C, Torres EM, Mahal A, Mangiarini L, Bates G, Morton AJ. Striatal transplantation in a transgenic mouse model of Huntington's disease. Exp Neurol. 1998;154:31–40. doi: 10.1006/exnr.1998.6926. [DOI] [PubMed] [Google Scholar]
- Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–1474. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- Giorgini F, Guidetti P, Nguyen Q, Bennett SC, Muchowski PJ. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat Genet. 2005;37:526–531. doi: 10.1038/ng1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- Hampton CR, Shimamoto A, Rothnie CL, Griscavage-Ennis J, Chong A, Dix DJ, Verrier ED, Pohlman TH. HSP70.1 and -70.3 are required for late-phase protection induced by ischemic preconditioning of mouse hearts. Am J Physiol Heart Circ Physiol. 2003;285:H866–H874. doi: 10.1152/ajpheart.00596.2002. [DOI] [PubMed] [Google Scholar]
- Hansson O, Nylandsted J, Castilho RF, Leist M, Jaattela M, Brundin P. Overexpression of heat shock protein 70 in R6/2 Huntington's disease mice has only modest effects on disease progression. Brain Res. 2003;970:47–57. doi: 10.1016/s0006-8993(02)04275-0. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- Hay DG, Sathasivam K, Tobaben S, Stahl B, Marber M, Mestril R, Mahal A, Smith DL, Woodman B, Bates GP. Progressive decrease in chaperone protein levels in a mouse model of Huntington's disease and induction of stress proteins as a therapeutic approach. Hum Mol Genet. 2004;13:1389–1405. doi: 10.1093/hmg/ddh144. [DOI] [PubMed] [Google Scholar]
- Hockly E, Cordery PM, Woodman B, Mahal A, van Dellen A, Blakemore C, Lewis CM, Hannan AJ, Bates GP. Environmental enrichment slows disease progression in R6/2 Huntington's disease mice. Ann Neurol. 2002;51:235–242. doi: 10.1002/ana.10094. [DOI] [PubMed] [Google Scholar]
- Hockly E, Woodman B, Mahal A, Lewis CM, Bates G. Standardization and statistical approaches to therapeutic trials in the R6/2 mouse. Brain Res Bull. 2003;61:469–479. doi: 10.1016/s0361-9230(03)00185-0. [DOI] [PubMed] [Google Scholar]
- Hoffner G, Island ML, Djian P. Purification of neuronal inclusions of patients with Huntington's disease reveals a broad range of N-terminal fragments of expanded huntingtin and insoluble polymers. J Neurochem. 2005;95:125–136. doi: 10.1111/j.1471-4159.2005.03348.x. [DOI] [PubMed] [Google Scholar]
- Iuchi S, Hoffner G, Verbeke P, Djian P, Green H. Oligomeric and polymeric aggregates formed by proteins containing expanded polyglutamine. Proc Natl Acad Sci U S A. 2003;100:2409–2414. doi: 10.1073/pnas.0437660100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana NR, Tanaka M, Wang G, Nukina N. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum Mol Genet. 2000;9:2009–2018. doi: 10.1093/hmg/9.13.2009. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Sang C, Adachi H, Minamiyama M, Waza M, Tanaka F, Doyu M, Sobue G. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci U S A. 2005;102:16801–16806. doi: 10.1073/pnas.0506249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med. 2004;10:402–405. doi: 10.1038/nm1021. [DOI] [PubMed] [Google Scholar]
- Kingsbury DT, Kasper KC, Stites DP, Watson JD, Hogan RN, Prusiner SB. Genetic control of scrapie and Creutzfeldt-Jakob disease in mice. J Immunol. 1983;131:491–496. [PubMed] [Google Scholar]
- Kobayashi Y, Sobue G. Protective effect of chaperones on polyglutamine diseases. Brain Res Bull. 2001;56:165–168. doi: 10.1016/s0361-9230(01)00593-7. [DOI] [PubMed] [Google Scholar]
- Krobitsch S, Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc Natl Acad Sci U S A. 2000;97:1589–1594. doi: 10.1073/pnas.97.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, Plomann M, Brundin P. Huntington's disease: a synaptopathy? Trends Mol Med. 2003;9:414–420. doi: 10.1016/j.molmed.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Lindquist S. The heat-shock response. Annu Rev Biochem. 1986;55:1151–1191. doi: 10.1146/annurev.bi.55.070186.005443. [DOI] [PubMed] [Google Scholar]
- Loovers HM, Guinan E, Jones GW. Importance of the Hsp70 ATPase domain in yeast prion propagation. Genetics. 2007;175:621–630. doi: 10.1534/genetics.106.066019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Mahal A, Mott R, Seller M, Bates GP. Instability of highly expanded CAG repeats in mice transgenic for the Huntington's disease mutation. Nat Genet. 1997;15:197–200. doi: 10.1038/ng0297-197. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Marber MS, Mestril R, Chi SH, Sayen MR, Yellon DM, Dillmann WH. Overexpression of the rat inducible 70-kD heat stress protein in a transgenic mouse increases the resistance of the heart to ischemic injury. J Clin Invest. 1995;95:1446–1456. doi: 10.1172/JCI117815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- Muchowski PJ, Schaffar G, Sittler A, Wanker EE, Hayer-Hartl MK, Hartl FU. Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc Natl Acad Sci U S A. 2000;97:7841–7846. doi: 10.1073/pnas.140202897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mycko MP, Cwiklinska H, Walczak A, Libert C, Raine CS, Selmaj KW. A heat shock protein gene (Hsp70.1) is critically involved in the generation of the immune response to myelin antigen. Eur J Immunol. 2008;38:1999–2013. doi: 10.1002/eji.200737661. [DOI] [PubMed] [Google Scholar]
- Nollen EA, Garcia SM, van Haaften G, Kim S, Chavez A, Morimoto RI, Plasterk RH. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc Natl Acad Sci U S A. 2004;101:6403–6408. doi: 10.1073/pnas.0307697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc Natl Acad Sci U S A. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome. 1997;8:711–713. doi: 10.1007/s003359900551. [DOI] [PubMed] [Google Scholar]
- Schaffar G, Breuer P, Boteva R, Behrends C, Tzvetkov N, Strippel N, Sakahira H, Siegers K, Hayer-Hartl M, Hartl FU. Cellular toxicity of polyglutamine expansion proteins; mechanism of transcription factor deactivation. Mol Cell. 2004;15:95–105. doi: 10.1016/j.molcel.2004.06.029. [DOI] [PubMed] [Google Scholar]
- Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, Copeland NG, Price DL, Ross CA, Borchelt DR. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- Simmons DA, Casale M, Alcon B, Pham N, Narayan N, Lynch G. Ferritin accumulation in dystrophic microglia may be an early event in the development of Huntington's disease. Glia. 2007;55:1074–1084. doi: 10.1002/glia.20526. [DOI] [PubMed] [Google Scholar]
- Singleton KD, Wischmeyer PE. Effects of HSP70.1/3 gene knock-out on acute respiratory distress syndrome and the inflammatory response following sepsis. Am J Physiol Lung Cell Mol Physiol. 2006;290:L956–L961. doi: 10.1152/ajplung.00466.2005. [DOI] [PubMed] [Google Scholar]
- Stack EC, Kubilus JK, Smith K, Cormier K, Del Signore SJ, Guelin E, Ryu H, Hersch SM, Ferrante RJ. Chronology of behavioral symptoms and neuropathological sequela in R6/2 Huntington's disease transgenic mice. J Comp Neurol. 2005;490:354–370. doi: 10.1002/cne.20680. [DOI] [PubMed] [Google Scholar]
- Steele AD, Hutter G, Jackson WS, Heppner FL, Borkowski AW, King OD, Raymond GJ, Aguzzi A, Lindquist S. Heat shock factor 1 regulates lifespan as distinct from disease onset in prion disease. Proc Natl Acad Sci U S A. 2008;105:13626–13631. doi: 10.1073/pnas.0806319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagawa K, Marubuchi S, Qi ML, Enokido Y, Tamura T, Inagaki R, Murata M, Kanazawa I, Wanker EE, Okazawa H. The induction levels of heat shock protein 70 differentiate the vulnerabilities to mutant huntingtin among neuronal subtypes. J Neurosci. 2007;27:868–880. doi: 10.1523/JNEUROSCI.4522-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tutar Y, Song Y, Masison DC. Primate chaperones Hsc70 (constitutive) and Hsp70 (induced) differ functionally in supporting growth and prion propagation in Saccharomyces cerevisiae. Genetics. 2006;172:851–861. doi: 10.1534/genetics.105.048926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Molle W, Wielockx B, Mahieu T, Takada M, Taniguchi T, Sekikawa K, Libert C. HSP70 protects against TNF-induced lethal inflammatory shock. Immunity. 2002;16:685–695. doi: 10.1016/s1074-7613(02)00310-2. [DOI] [PubMed] [Google Scholar]
- Wacker JL, Zareie MH, Fong H, Sarikaya M, Muchowski PJ. Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat Struct Mol Biol. 2004;11:1215–1222. doi: 10.1038/nsmb860. [DOI] [PubMed] [Google Scholar]
- Warrick JM, Paulson HL, Gray-Board GL, Bui QT, Fischbeck KH, Pittman RN, Bonini NM. Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell. 1998;93:939–949. doi: 10.1016/s0092-8674(00)81200-3. [DOI] [PubMed] [Google Scholar]
- Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- Williams AJ, Knutson TM, Colomer Gould VF, Paulson HL. In vivo suppression of polyglutamine neurotoxicity by C-terminus of Hsp70-interacting protein (CHIP) supports an aggregation model of pathogenesis. Neurobiol Dis. 2009;33:342–353. doi: 10.1016/j.nbd.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willingham S, Outeiro TF, DeVit MJ, Lindquist SL, Muchowski PJ. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science. 2003;302:1769–1772. doi: 10.1126/science.1090389. [DOI] [PubMed] [Google Scholar]
- Yuan SH, Qiu Z, Ghosh A. TOX3 regulates calcium-dependent transcription in neurons. Proc Natl Acad Sci U S A. 2009;106:2909–2914. doi: 10.1073/pnas.0805555106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Li SH, Li XJ. Chaperone suppression of cellular toxicity of huntingtin is independent of polyglutamine aggregation. J Biol Chem. 2001;276:48417–48424. doi: 10.1074/jbc.M104140200. [DOI] [PubMed] [Google Scholar]