

SUMMARY
The Bcl-2 family proteins are critical apoptosis regulators that associate with mitochondria and control the activation of caspases. Recently, both mammalian and C. elegans Bcl-2 proteins have been implicated in controlling mitochondrial fusion and fission processes in both living and apoptotic cells. To better understand the potential roles of Bcl-2 family proteins in regulating mitochondrial dynamics, we carried out a detailed analysis of mitochondria in animals that either lose or have increased activity of egl-1 and ced-9, two Bcl-2 family genes that induce and inhibit apoptosis in C. elegans, respectively. Unexpectedly, we found that loss of egl-1 or ced-9, or overexpression of their gene products, had no apparent effect on mitochondrial connectivity or mitochondrial size. Moreover, loss of ced-9 did not affect the mitochondrial morphology observed in a drp-1 mutant, where mitochondrial fusion occurs but mitochondrial fission is defective, or in a fzo-1 mutant, where mitochondrial fission occurs but mitochondrial fusion is restricted, suggesting that ced-9 is not required for either the mitochondrial fission or fusion process in C. elegans. Taken together, our results argue against an evolutionarily conserved role for Bcl-2 proteins in regulating mitochondrial fission and fusion.
RESULTS
Mitochondrial morphogenesis is not affected in egl-1(lf) or ced-9(lf) mutants
Recently, the C. elegans pro-apoptotic BH3-only Bcl-2 protein EGL-1 has been implicated in promoting mitochondria fission during apoptosis [1]. In addition, the C. elegans anti-apoptotic Bcl-2 protein CED-9 was shown to mediate mitochondria fission during apoptosis in one study [1] but was found to promote mitochondria fusion in healthy cells in another [2], calling into question of the exact physiological roles of C. elegans Bcl-2 family proteins in regulating mitochondria dynamics. To address the critical issue of whether Bcl-2 proteins regulate normal mitochondrial fission or fusion process in C. elegans, we carried out a comprehensive analysis of mitochondria morphology and structure in animals that either lose or have increased activity of egl-1 or ced-9. First, we visualized mitochondria in early C. elegans embryos that were stained with the mitochondria specific dye, tetramethylrhodamine ethyl ester (TMRE); the large blastomere size in early embryos permits clear visualization of the mitochondrial network. In N2 (wild type) animals, where mitochondrial fission and fusion processes are balanced, mitochondria appeared as a large network, evenly distributed through out each cell (Figure 1A) [1, 3–7]. In drp-1(tm1108) mutant animals, which are null for the DRP-1 protein expression and defective in mitochondrial fission [7], mitochondria appeared as highly connected clusters and asymmetrically distributed in individual blastomeres (Figure 1B), which results from ongoing mitochondrial fusion in the absence of mitochondrial fission [6]. In contrast, in fzo-1(tm1133) animals, which harbor a deletion in the fzo-1 gene and where mitochondrial fusion is compromised but mitochondrial fission continues [7], the mitochondrial network was disrupted into highly fragmented, punctiform organelles (Figure 1C). Thus, a defect in either the mitochondrial fission or fusion process is clearly identifiable in this assay.
Figure 1.
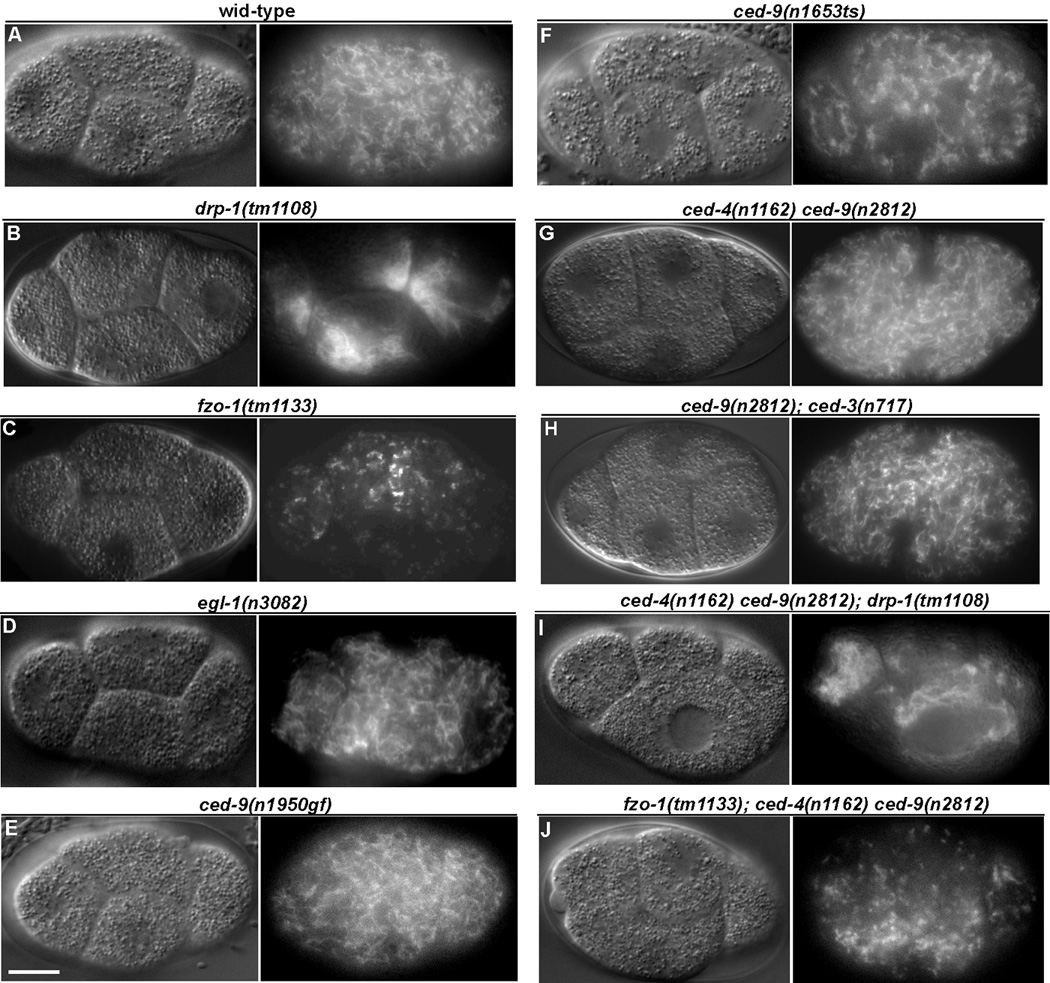
The mitochondrial network is altered in fzo-1 and drp-1 mutants but unaffected by mutations in egl-1 and ced-9. Animals were stained with tetramethylrhodamine ethyl ester (TMRE), a mitochondrial specific dye, and blastomeres at the four-cell embryonic stage were imaged. Embryos were visualized by Differential Interference Contrast (DIC, left) and rhodamine fluorescence (right) microscopy. Representative images are shown. Compared to wild type embryos (A), drp-1(tm1108) embryos (B) have a highly connected mitochondrial network, whereas mitochondria appeared highly fragmented in fzo-1(tm1133) embryos (C). Mitochondria in egl-1(n3082) (D), ced-9(n1950gf) (E), ced-9(n1653ts) at the restrictive temperature (F), ced-4(n1162) ced-9(n2812) (G), and ced-9(n2812); ced-3(n717) (H) embryos were indistinguishable from those observed in wild type embryos. Loss of ced-9 has no effect on the mitochondria morphology in drp-1(tm1108) or fzo-1(tm1133) animals. The mitochondrial network in the ced-4(n1162) ced-9(n2812); drp-1(tm1108) embryo (I) and in the fzo-1(tm1133); ced-4(n1162) ced-9(n2812) embryo (J) is similar to that seen in drp-1(tm1108) embryos (B) and fzo-1(tm1133) embryos (C), respectively. Scale bar represents 10 µ m.
Mitochondria in egl-1(n3082) animals, which carry a strong loss-of-function (lf) mutation in egl-1, appeared undistinguishable from those in wild type animals (Figure 1D), although somatic programmed cell death is abolished in these animals [8]. Similarly, the mitochondrial network appeared unaffected in ced-9(n1950gf) animals (Figure 1E), which carry a gain-of-function (gf) mutation (a G169E substitution) in the ced-9 gene that prevents EGL-1 from binding to CED-9 [9, 10] and thus blocks C. elegans programmed cell death [11]. We also analyzed mitochondria morphology in two ced-9(lf) mutants: ced-9(n1653ts) and ced-9(n2812). The n1653 mutation causes a Y149N substitution in CED-9 that reduces its association with CED-4 at the restrictive temperature (25°C) and compromises its apoptosis inhibitory activity [12], leading to ectopic apoptosis. n2812 results in an early nonsense mutation in the ced-9 gene [13] that abolishes expression of ced-9 in C. elegans [14] and is a putative null allele. ced-9(n282) animals are embryonic lethal due to excessive apoptosis but can be maintained and analyzed in the ced-3(lf) or ced-4(lf) mutant background, which blocks apoptosis [11]. As shown in Figure 1F–H, we observed no significant difference in mitochondrial morphology in ced-9(n1653ts), ced-4(n1162) ced-9(n2812), or ced-9(n2812); ced-3(n717) embryos compared to that in N2 embryos or that in ced-3(n717) or ced-4(n1162) embryos or ced-9(n1653ts) embryos at the permissive temperature (Figure S1). We quantified the connectivity of mitochondria in N2, drp-1(tm1108), fzo-1(tm1133), egl-1(n3082), and ced-9(n2812); ced-3(n717) blastomeres by generating line intensity plots and calculating the frequency of major TMRE fluorescent spikes (Figure S2; method described in Experimental Procedures). In N2 blastomeres, TMRE fluorescent signals varied in frequency, with an average of 0.49 fluorescent spikes/µm (Figure S2). TMRE fluorescent signals were very broad and of low spike frequency in drp-1(tm1108) blastomeres (average frequency of 0.16 fluorescent spikes/µm; Figure S2), consistent with large clumps of mitochondria asymmetrically distributed within cells. In contrast, fzo-1(tm1133) embryos displayed high frequency of TMRE signal spikes, averaging 2.29 spikes/µm, delineating punctiform mitochondria evenly distributed throughout the cells (Figure S2). The frequency of TMRE signal spikes in egl-1(n3082), ced-9(n1950gf), or ced-9(n2812); ced-3(n717) blastomeres was similar to that of N2 animals [an average frequency of 0.44 spikes/µm and 0.48 spikes/µm in egl-1(n3082) and ced-9(n2812); ced-3(n717) blastomeres; Figure S2]. Taken together, these results suggest that loss of egl-1 or ced-9 function does not affect mitochondria dynamics and morphology in C. elegans
Of note, a recent report showed that mitochondria appeared highly fragmented in ced-9(n1653ts) embryos at the restrictive temperature [2]. However, in that study, embryos were examined at a later stage of development and the mitochondrial fragmentation observed could have been the result of widespread ectopic apoptosis [1, 7], rather than a requirement for ced-9 to maintain the integrity of the mitochondrial network. Importantly, CED-9 protein is ubiquitously expressed in embryos as early as the two cell stage [14]. If CED-9 is required to maintain normal mitochondrial networks, its role should be uncovered in early embryos. The expression pattern of EGL-1 is not well understood, but egl-1 transcription has been shown to be upregulated in several cells destined to die [15]. Nonetheless, our results suggest that the activity of egl-1 is not required for normal mitochondrial morphogenesis.
We carried out electron microscopy (EM) analysis to confirm the TMRE staining results in Figure 1 and to investigate whether egl-1 or ced-9 might play subtle roles in regulating mitochondrial dynamics. In 2-dimensional images of EM sections from N2 embryos, mitochondria appeared in a variety of shapes and sizes, ranging from small spherical organelles to longer dumbbell shaped organelles (Figure 2A), and with a mean longitudinal length of 0.94 µm (Figure 2F). As expected, mitochondria in drp-1(tm1108) embryos were very long, with fewer individual mitochondria observed in each cell (Figure S3A) and a mean mitochondrial length of 2.28 µm (Figure 2F)[7]. fzo-1(tm1133) embryos displayed only small and spherical mitochondria, with a mean mitochondrial length of 0.38 µm (Figure S3B and Figure 2F). However, mitochondria in egl-1(n3082), ced-9(n1950gf), ced-9(n1653ts), and ced-9(n2812); ced-3(n717) embryos appeared similar to those observed in N2 embryos and in all cases had mean longitudinal mitochondrial lengths that were not significantly different from that of N2 animals (Figure 2B–E). Mitochondria in the germline, gut, and muscle cells of adult egl-1(lf), ced-9(lf); ced-3(lf), or ced-9(gf) mutants also appeared normal (data not shown). The mitochondrial morphology in N2, drp-1(tm1108), fzo-1(tm1133), egl-1(n3082), and ced-9(n2812); ced-3(n717) animals was confirmed by serial EM sectioning and 3-dimensional reconstruction from the serial images (Figure 3 and Figure S4). Again, mitochondria in N2, egl-1(n2812), and ced-9(n2812); ced-3(n717) animals varied in shape and size and were evenly distributed throughout the cell. In contrast, mitochondria in drp-1(tm1108) embryos were long, highly interconnected, and clustered around the nucleus, whereas mitochondria in fzo-1(tm1133) embryos were small, puntiform, and evenly distributed. Altogether, these results confirm that egl-1 and ced-9 do not have a detectable role in regulating mitochondrial fission or fusion in C. elegans.
Figure 2.

Electron microscopy (EM) analysis of mitochondria morphology in egl-1 and ced-9 mutants. Representative electron micrographs of embryos from the following strains are shown: N2 (A), egl-1(n3082) (B), ced-9(n1950gf) (C), ced-9(n1653ts) at the restrictive temperature (D), ced-9(n2812); ced-3(n717) (E). Scale bar represents 1 µm. Arrows indicate representative mitochondria. (F) Quantification of the mean mitochondrial length. Randomly selected mitochondria from electron micrographs were measured along their longitudinal axis. SEM, standard error of the mean. n, the number of mitochondria scored.
Figure 3.

3-dimensional mitochondria images reconstructed from serial electron micrographs. 3D models of mitochondria (red) in various mutant embryos were generated from stacks consisting of 15∼20 serial electron micrographs (10,000X) of 80 nm thick sections (see Experimental Procedures for detail). In each panel, top-down view (left) and side view (right) of the cell are shown. Mitochondria in the drp-1(tm1108) mutant are interconnected to form a large network, whereas mitochondria in the fzo-1(tm1133) mutant are highly fragmented and spherical. No obvious difference is seen in mitochondria organization and architecture among N2, egl-1(n3082), and ced-9(n2812); ced-3(n717) embryos. Nuclei (blue) and cell outlines (white) are also shown.
ced-9 does not promote drp-1-dependent mitochondrial fission or fzo-1-dependent mitochondrial fusion
If CED-9 somehow has both pro-fission and pro-fusion activities as previously reported [1, 2], it is conceivable that loss of ced-9 in C. elegans would yield no net effect on the mitochondrial network. To address this possibility, we examined the effect of a ced-9(lf) mutation on the mitochondrial morphologies in drp-1(tm1108) and fzo-1(tm1133) mutant backgrounds, where mitochondrial fusion or fission occurs in isolation, respectively [5]. Mitochondria in fzo-1(tm1133); ced-4(n1162) ced-9(n2812) triple mutant embryos appeared highly fragmented and indistinguishable from mitochondria in fzo-1(tm1133); ced-4(n1162) or fzo-1(tm1133) embryos (Figure 1C and J, and data not shown). On the other hand, mitochondria in ced-4(n1162) ced-9(n2812); drp-1(tm1108) triple mutants were highly connected and asymmetrically distributed in the cells, just as was observed in ced-4(n1162); drp-1(tm1108) and drp-1(tm1108) animals (Figure 1B and I, and data not shown). Therefore, in a physiological setting (i.e. where ced-9, drp-1, and fzo-1 are not overexpressed), loss of ced-9 has no discernable effect on either drp-1-dependent mitochondrial fission or fzo-1-dependent mitochondrial fusion. In comparison, drp-1(tm1108) completely suppresses the highly fragmented mitochondria phenotype caused by the fzo-1(tm1133) mutation [7], indicating that drp-1 is required for the mitochondrial fragmentation observed in fzo-1 mutants.
Overexpression of CED-9 or EGL-1 does not promote mitochondrial fission or fusion
Although egl-1 and ced-9 are not required for mitochondrial fission or fusion in normal cells, they might directly affect one of these processes during apoptosis [1]. Therefore, we examined whether overexpression of either EGL-1 or CED-9 could affect mitochondrial morphology in C. elegans, in comparison with their activities in inducing or inhibiting apoptosis. We generated animals carrying an integrated transgene of egl-1 or ced-9 under the control of heat-shock promoters (P hspegl-1 and P hspced-9, respectively) and examined the mitochondrial network in young embryos before and after heat-shock treatment. Since overexpression of EGL-1 potently induces ectopic apoptosis in C. elegans [8] and overexpression of CED-9 strongly inhibits normal programmed cell death [13], we quantified the effect of induced overexpression of EGL-1 and CED-9 on cell death by counting the number of cell corpses in heat-treated Phspegl-1 and Phspced-9 embryos and the number of cells that should die but inappropriately survived in the anterior pharynx of larvae that hatched from the heat-treated Phspced-9 embryos. Heat-shock treatment of Phspegl-1 embryos resulted in the appearance of over 40 cell corpses by the comma stage of embryonic development (Figure 4A) and ultimately caused growth arrest and death of all embryos prior to hatching (data not shown). In contrast, heat-shock treatment of Phspced-9 animals greatly reduced the number of cell corpses in embryos (Figure 4A) and caused on average the survival of over nine extra cells in the anterior pharynx of the resulting larvae (Figure 4B). These results demonstrate that induced global expression of EGL-1 and CED-9 potently promotes or inhibits apoptosis in Phspegl-1 and Phspced-9 animals, respectively. However, we did not observe an obvious change in mitochondrial morphology in Phspegl-1 or Phspced-9 embryos following the heat shock treatment (Figure 4C). In contrast, drp-1(tm1108) or fzo-1(tm1133) embryos at the same developmental stage showed clear signs of mitochondrial clustering and mitochondrial fragmentation, respectively (Figure S5). Analysis of EM micrographs of heat-treated Phspegl-1 embryos revealed that in most cells mitochondria appeared normal (Figure 4D) and that the mean mitochondrial length remained at 0.92 µm, similar to what was observed in N2 animals (Figure 2F). Highly fragmented and spherical mitochondria were observed in cell corpses of Phspegl-1 embryos (Figure 4E), suggesting that mitochondria eventually fragment in dying cells, probably as a secondary event of apoptosis. Consistent with this, we did not observe evidence of widespread mitochondrial fragmentation or ectopic apoptosis when EGL-1 was overexpressed in a ced-3(lf) background, even when animals were observed up to five hours post heat-shock treatment (Figure S6; data now shown). Therefore, elevated levels of EGL-1 and CED-9 proteins do not appear to directly promote either mitochondrial fission or fusion in C. elegans. However, our results do not rule out the possibility that EGL-1 may play a role in promoting mitochondria fragmentation in normally dying cells. Of note, it was reported that ectopic egl-1 expression (using a similar Phspegl-1 construct) induced widespread mitochondrial fission, independent of ced-3 [1]. The reason for the discrepancy between that study and ours is currently unclear.
Figure 4.

Overexpression of EGL-1 or CED-9 affects programmed cell death in C. elegans but does not obviously affect mitochondrial morphogenesis. (A) N2, Phspegl-1 (smIs82), or Phspced-9 (smIs157) embryos were treated with heat-shock (right) or left untreated (left) and the number of cell corpses in the head region of 1.5-fold stage embryos was scored 2 hours post heat-shock treatment. (B) Embryos of the indicated genotypes were treated with heat-shock (right) or left untreated (left) and allowed to hatch into L4 larvae, at which point the number of extra cells that inappropriately survived in the anterior pharynx was scored. 20 animals were scored in both (A) and (B). (C) N2, Phspegl-1, or Phspced-9 embryos were stained with TMRE and either treated with heat-shock (right) or left untreated (left) and visualized by Differential Interference Contrast (DIC) and rhodamine fluorescence microscopy. Mitochondria in heat-shock treated Phspegl-1 or Phspced-9 embryos were indistinguishable from those observed in untreated embryos and wild type embryos. Scale bar represents 10 µm. (D) An EM micrograph of a heat-shock treated Phspegl-1 embryo. Arrows indicate several mitochondria of various shapes and sizes. (E) Mitochondria in a cell corpse of a heat-shock treated Phspegl-1 embryo are fragmented as a result of apoptosis. SEM: standard error of the mean.
DISCUSSION
Although several recent reports have suggested a connection between Bcl-2 proteins and the mitochondrial fission or fusion process [1, 2, 16–18], we did not find an obvious requirement for egl-1 or ced-9 in regulating either process in C. elegans. Analysis of strong loss-of-function egl-1 and ced-9 mutants and a gain-of-function ced-9 mutant, which either are defective in apoptosis or contain excessive cell deaths, revealed no detectable difference in the mitochondrial network when observed in live animals stained with mitochondria-specific dye, in high magnification EM sections, or in 3-dimensional models reconstructed from serial EM sections. Moreover, ced-9 does not affect either drp-1-dependent mitochondrial fission or fzo-1-dependent mitochondrial fusion in vivo (Figure 1B, C, I, J). These findings disagree with two recent reports that proposed a role for EGL-1 and CED-9 in regulating mitochondrial dynamics in C. elegans [1, 2]. In one report, overexpression of EGL-1 was shown to induce mitochondrial fragmentation, independent of ced-3, and mitochondrial fission induced by ectopic drp-1 expression was blocked by both ced-9(n1950gf) and ced-9(n2812lf) mutations [1]. However, we did not observe widespread mitochondrial fragmentation following EGL-1 induction (Figure 4) and loss of ced-9 does not affect mitochondria fission or fusion on its own or affect drp-1-dependent mitochondrial fission when examined in a physiological context (i.e. in the fzo-1 mutant background when drp-1 was not overexpressed) (Figure 1C, J). Mitochondria are fragmented in cell corpses following EGL-1 induction, but this is likely the result of a downstream caspase-dependent apoptotic process [7] rather than a process that is regulated by EGL-1 or CED-9 to activate apoptosis. Indeed, we did not detect any obvious cell death defect in the strong loss-of-function drp-1(tm1108) mutant, where mitochondria are highly fused, or in animals deficient in fzo-1 or eat-3, where mitochondria are highly fragmented [7], indicating that the mitochondria fusion or fission process does not play a role in apoptosis activation. Similarly, we found that overexpression of CED-9 in C. elegans does not induce mitochondrial fission or fusion, although its overexpression was reported to cause excessive mitochondrial fusion in cultured mammalian cells [2]. CED-9 therefore may target proteins or signaling pathways in mammals that don’t exist in the worm. Taken together, our findings indicate that EGL-1 and CED-9 do not have a discernable role in promoting either mitochondrial fission or fusion in C. elegans and challenge the hypothesis that regulation of mitochondrial morphogenesis is an evolutionarily conserved feature of Bcl-2 proteins [2].
In mammals, overexpression of Bcl-xL can induce both mitochondrial fission and fusion depending on its level of expression [2, 17] and can increase mitochondrial biomass [17], whereas overexpression of Bcl-2 promotes mitochondrial fusion [2] and overexpression of Bax induces mitochondrial fission and apoptosis [16]. Paradoxically, Bax and Bak are also reported to be important for mitochondrial fusion in healthy cells [18], raising an interesting question of how mammalian Bcl-2 proteins achieve opposing functions in mitochondria dynamics. Bcl-2 proteins might affect mitochondria fusion or fission in mammals by interacting with the mitochondrial fusion machinery, since Bcl-xL was shown to interact with Mfn2 [2] and Bax/Bak were found to be required for Mfn2 to form discrete foci in the outer mitochondrial membrane [18]. Therefore, it appears that mammalian Bcl-2 proteins may have evolved a role to directly or indirectly regulate mitochondrial fission/fusion and such a role does not appear to exist in their worm counterparts. Further analysis of mitochondria dynamics under physiological settings will be critical to understand the roles and mechanisms of mammalian Bcl-2 proteins in regulating mitochondria fusion/fission.
Supplementary Material
Supplemental materials include Experimental Procedures and six additional
ACKNOWLEDGEMENTS
We thank Caenorhabditis Genetics Center for strains and Tom Giddings for assistance with EM operation. This research was supported in part by fellowships from the Jane Coffins Child Memorial Fund for Medical Research and the Canadian Institutes of Health Research (D.G.B), start-up funds from the University of Florida (B.H.K), the Burroughs Wellcome Fund Career Award (D.X.), and the NIH R01 GM059083 and GM079097 grants (D.X.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Jagasia R, Grote P, Westermann B, Conradt B. DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature. 2005;433:754–760. doi: 10.1038/nature03316. [DOI] [PubMed] [Google Scholar]
- 2.Delivani P, Adrain C, Taylor RC, Duriez PJ, Martin SJ. Role for CED-9 and EGL-1 as regulators of mitochondrial fission and fusion dynamics. Mol. Cell. 2006;21:761–773. doi: 10.1016/j.molcel.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 3.Ichishita R, Tanaka K, Sugiura Y, Sayano T, Mihara K, Oka T. An RNAi screen for mitochondrial proteins required to maintain the morphology of the organelle in C. elegans. J. Biochem. 2008;143:449–454. doi: 10.1093/jb/mvm245. [DOI] [PubMed] [Google Scholar]
- 4.Chan DC. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell. Dev. Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- 5.Okamoto K, Shaw JM. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu. Rev. Genet. 2005;39:503–536. doi: 10.1146/annurev.genet.38.072902.093019. [DOI] [PubMed] [Google Scholar]
- 6.Labrousse AM, Zappaterra MD, Rube DA, van der Bliek AM. C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol. Cell. 1999;4:815–826. doi: 10.1016/s1097-2765(00)80391-3. [DOI] [PubMed] [Google Scholar]
- 7.Breckenridge DG, Kang B-H, Kokel DK, Mitani S, Staehelin LA, Xue D. Caenorhabditis elegans drp-1 and fis-2 regulate distinct cell death execution pathways downstream of ced-3 and independent of ced-9. Mol. Cell. 2008;31:586–597. doi: 10.1016/j.molcel.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conradt B, Horvitz HR. The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell. 1998;93:519–529. doi: 10.1016/s0092-8674(00)81182-4. [DOI] [PubMed] [Google Scholar]
- 9.Hengartner MO, Horvitz HR. Activation of C. elegans cell death protein CED-9 by an amino-acid substitution in a domain conserved in Bcl-2. Nature. 1994;369:318–320. doi: 10.1038/369318a0. [DOI] [PubMed] [Google Scholar]
- 10.Parrish J, Metters H, Chen L, Xue D. Demonstration of the in vivo interaction of key cell death regulators by structure-based design of secondsite suppressors. Proc. Natl. Acad. Sci U.S.A. 2000;97:11916–11921. doi: 10.1073/pnas.210391597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hengartner MO, Ellis RE, Horvitz HR. Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature. 1992;356:494–499. doi: 10.1038/356494a0. [DOI] [PubMed] [Google Scholar]
- 12.Spector MS, Desnoyers S, Hoeppner DJ, Hengartner MO. Interaction between the C. elegans cell-death regulators CED-9 and CED-4. Nature. 1997;385:653–656. doi: 10.1038/385653a0. [DOI] [PubMed] [Google Scholar]
- 13.Hengartner MO, Horvitz HR. C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell. 1994;76:665–676. doi: 10.1016/0092-8674(94)90506-1. [DOI] [PubMed] [Google Scholar]
- 14.Chen F, Hersh BM, Conradt B, Zhou Z, Riemer D, Gruenbaum Y, Horvitz HR. Translocation of C. elegans CED-4 to nuclear membranes during programmed cell death. Science. 2000;287:1485–1489. doi: 10.1126/science.287.5457.1485. [DOI] [PubMed] [Google Scholar]
- 15.Peden E, Killian D, Xue D. Cell death specification in C. elegans. Cell Cycle. 2008;7:2479–2484. doi: 10.4161/cc.7.16.6479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, Youle RJ. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J. Cell Biol. 2002;159:931–938. doi: 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berman SB, Chen Y-B, Qi B, McCaffery JM, Rucker EB, Goebbels S, Nave KA, Arnold BA, Jonas EA, Pineda Fj, Hardwick MJ. Bcl-xL increases mitochondrial fission, fusion, and biomass in neurons. J. Cell. Biol. 2009 doi: 10.1083/jcb.200809060. Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–662. doi: 10.1038/nature05111. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental materials include Experimental Procedures and six additional