

Abstract
There is much speculation whether extravascular inflammation accelerates atherosclerosis. We tested this hypothesis in apoE−/− mice using three well characterized models of non-autoimmune chronic inflammation: croton oil-induced skin inflammation, Aspergillus fumigatus antigen-induced allergic lung disease, and A. fumigatus antigen-induced peritonitis. The croton oil model produced recurrent inflammatory skin ulceration, and marked increases in plasma levels of IL-6 and serum amyloid A (SAA). The allergic lung disease model showed strong local inflammation with eosinophilic infiltration and serum IgE induction. The recurrent peritonitis model was accompanied by mild elevation in plasma SAA levels. Aortic atherosclerosis was quantified by computer-assisted morphometry of en face arteries in apoE−/− mice at 33 weeks for the croton-oil model, 26 and 42 weeks for the allergic lung disease model, and 26 weeks for the peritonitis model. We found that all three forms of chronic extravascular inflammation had no effect on the rate of atherosclerosis development.
Keywords: Inflammation, Atherosclerosis, Dermatitis, Lung Disease, Peritonitis
Introduction
Atherosclerosis is the commonest underlying pathology of most cardiovascular diseases (CVD), the major cause of mortality and morbidity in developed countries [1]. Dyslipidemia is a dominant risk factor in atherosclerosis development [2]. There is increasing evidence, however, that inflammation mediates many of the ill effects of dyslipidemia [3]. Furthermore, atherosclerotic lesions are themselves loci of inflammation and a source of proinflammatory cytokines [4].
We speculated that extravascular inflammatory processes, by diverse mechanisms, including leukocyte activation, immunomodulation, and increased circulating inflammatory mediators, could modulate the inflammatory response in lesions and contribute directly to atherosclerosis development. In support of this notion, several epidemiological studies have found associations between inflammatory processes resulting from chronic infectious diseases, i.e. of the respiratory system [5; 6] or gums (periodontitis) [5; 7], or from non-infectious autoimmune disorders, such as rheumatoid arthritis (RA) [8; 9] and systemic lupus erythematosus (SLE) [10-12], and atherosclerosis and/or CVD. Furthermore, circulating levels of markers of inflammation, such as interleukin-6 (IL-6) and C-reactive protein (CRP), have been found to be predictors of atherosclerosis and cardiovascular events [13; 14]. However, the contention that extravascular inflammation is itself a causative factor for accelerated atherosclerosis remains unproven. For example, chronic infections and autoimmune diseases are often associated with increased cardiovascular risk profiles [11; 15; 16]. Furthermore, some pathogens that are thought to promote atherosclerosis, such as Chlamydia pneumoniae, cytomegalovirus and other herpesviridae, can actually infect vascular cells [17], and autoimmune diseases are associated with vasculitis, arterial stiffness and vascular dysfunction [18; 19]. It is important to determine if chronic inflammatory conditions in extravascular sites can actually cause or predispose to atherosclerotic cardiovascular disease especially in the presence of a proatherogenic dyslipidemia.
Mouse models constitute a powerful tool to study the pathogenesis of atherosclerosis, and can help to study the link between chronic inflammation and atherosclerosis [20]. In fact, recent reports have shown that different mouse strains with mutations that result in a lupus-susceptible immune system, a systemic disorder, can present with accelerated atherosclerosis [21-24]. If indeed extravascular inflammation is the underlying mechanism leading to accelerated atherosclerosis, patients with other forms of extravascular inflammation could also benefit from strict management of their cardiovascular risk factors. In this study, we assessed the effect of non-autoimmune chronic inflammation on atherosclerosis development in apoE−/− mice, a widely used genetic animal model of atherosclerosis[25], by applying three well characterized models of experimental inflammation: (1) croton oil-induced chronic skin lesions [26; 27]: (2) A. fumigatus antigen-induced allergic lung disease [28]: and (3) A. fumigatus antigen-induced peritonitis.
Materials and Methods
Animals
Female apoE−/− mice (3-5 weeks age) were obtained from The Jackson Laboratory, housed in Baylor's Transgenic Mouse Facility, and given access to water and standard mouse chow ad libitum. Mice were separated into matched control and treatment groups based on serum cholesterol levels measured just before the beginning of treatments. Protocols for handling and treatment of animals were approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine.
Croton Oil-Induced Skin Inflammation Model
The lower backs of the mice were shaved using a scalpel under intraperitoneal (i.p.) anesthesia with avertin (2 mg/ml tribromoethanol [Aldrich] dissolved in tert-amyl alcohol, resuspended in 0.9% NaCl, 1:39, vol:vol), and a solution of 10% croton oil (Sigma) in acetone:olive oil (4:1, vol:vol) was applied to the exposed skin, resulting in focal ulcers and inflammation [26; 27]. The skin ulcers healed completely with full return of hair within a month, after which time the treatment was repeated. Accordingly, croton oil treatments were performed beginning at 8 weeks of age and repeated every 4 weeks, i.e., week 12, 16, 20, 24, 28, and 32, until the mice were sacrificed by isoflurane inhalation for aortic lesion quantitation and skin histopathology at 34 weeks of age. After euthanasia the dorsal pelt was removed and fixed in 10% neutral buffered formaldehyde. The skin was trimmed to 2-4 mm wide strips cut parallel to the direction of hair growth. The trimmed skin specimens were paraffin embedded, sectioned at 4 μm and stained with hematoxylin and eosin (H/E).
A. Fumigatus Antigen-Induced Allergic Lung Disease Model
The A. fumigatus preparation was made and administered intranasally (i.n.) to the treatment group as previously described [28], while the control group was left untreated. The time course of administration of A. fumigatus antigen was as follows: treatments at 10, 11, 12, and 14 weeks of age, followed by treatments every 4 weeks, i.e., at weeks 18, 22, 26, 30, 34, 38, 42. Following completion of the treatment schedule, mice were sacrificed by overdose of pentobarbital. Bronchoalveolar lavage (BAL) fluid was collected from lungs by rinsing twice with 1 ml of PBS, then the lungs were removed and preserved in phosphate-buffered saline (PBS)/10% formalin. The heart and aorta were isolated for atherosclerosis measurements. Two hundred microliters of BAL fluid were placed in a cytospin apparatus and the cells were sedimented onto microscope slides, followed by Wright-Giemsa staining and differential cell counting [28]. Serum IgE levels were measured by ELISA as previously described [28]. Histological evaluation of formalin-fixed lung tissues was performed after H/E and Periodic Acid Schiff (PAS) staining of paraffin embedded tissue.
A. Fumigatus Antigen-Induced Peritonitis Model
Another set of mice was divided into plasma cholesterol matched controls and treatment groups in which the latter received A. fumigatus antigen by i.p. injection as described [29; 30]. The i.p. treatments were performed at 10, 11, 12, and 14 weeks of age, followed by treatments every 4 weeks, i.e., at weeks 18, 22, 26. After the last treatment, the mice were sacrificed and aortas were harvested for lesion quantitation.
Plasma Inflammation Markers and Cholesterol Levels
Periodically, EDTA-blood was obtained by retro-orbital bleeding under avertin anesthetic to obtain leukocyte counts. Plasma levels of cytokines IL-6, tumor necrosis factor-α (TNF-α), and interleukin-1β (IL-1β) and acute phase protein serum amyloid A (SAA) were measured by ELISA kits (Biosource). Plasma cholesterol levels were measured by enzymatic kit (Sigma).
Aortic Lesion Quantitation (Atherosclerosis Measurements)
Mice were sacrificed by overdose with anesthetic isoflurane or pentobarbital, as specified. The heart and abdominal aorta were removed and cleaned under a dissecting microscope. The aortas were opened longitudinally and pinned to cardboard, fixed overnight in PBS/10% formalin, and then stained with Oil Red O. En face lesion area quantitation using computer-assisted morphometry was performed as previously described [31].
Statistical Analysis
Experimental groups were compared to controls or each other using Student's t-test or Mann-Whitney Rank Sum test. Values are reported as mean ± standard deviation and a P value of equal or less than 0.05 was considered significant.
Results
Croton Oil-Induced Chronic Skin Inflammation
Croton oil treatment of the shaved backs of the mice led to erythema and then ulceration of the area of application (approximately 2 cm2 and not exceeding 2 cm in diameter) in the first few days after treatment. This was followed by a period of wound repair where the skin ulcer was red and scabbed or crusted. After about 1 week, the superficial crust and necrotic tissue peeled off, revealing intact skin by 10 to 14 days, and by 3 weeks, most of the fur had regrown. Histology revealed necrosis of epidermis and dermis, followed by ulceration with moderate to intense suppurative inflammation (Fig. 1A), then inflammation diminished as re-epithelialization (healing) progressed. In re-epithelialized or healed specimens there was epidermal and dermal thickening due to epidermal hyperplasia and dermal fibrosis, and there were scattered inflammatory cells in the dermis and subcutis. In older, multiply treated animals, there was xanthomatous change in the subcutis, sometimes with associated fat necrosis and foci of pyogranulomatous inflammation.
Figure 1.
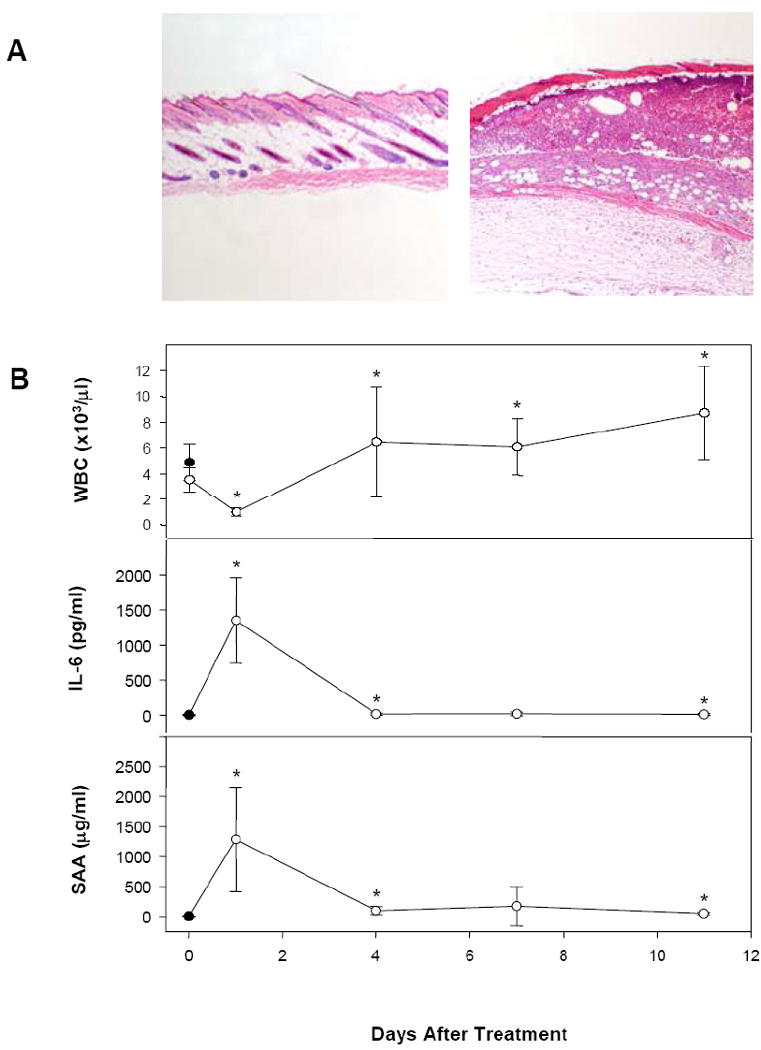
A. Histopathology of skin sections from control (left) and day 7 croton oil-treated (right) mice (40×) showing ulceration, inflammation, and necrosis in treated mice. B. Leukocyte counts (top), IL-6 levels (middle), and SAA (bottom) levels from plasma of untreated control (solid symbol) and croton oil-treated (open symbols) mice at the indicated times after treatment.
We measured various blood and plasma markers in order to further characterize the inflammatory process during croton oil treatment. One day after croton oil treatment, white blood cell (WBC) counts dropped, while by 4 days post-treatment and thereafter, the counts were slightly elevated (Fig. 1B, top). Levels of the proinflammatory cytokine IL-6 increased markedly, from undetectable to1,352 pg/ml, one day after croton oil treatment, but returned to near baseline within 4 days (Fig. 1B, middle). Coinciding with the changes in IL-6 level was a marked increase in the acute phase protein SAA, from a baseline level of 9.2 μg/ml to 1,287 μg/ml one day post-treatment, decreasing to about 5-10 times above baseline from 4 to 11 days post-treatment (Fig. 1B, bottom). Neither TNF-α or IL-1β levels were affected by croton oil treatment (data not shown). Shown measurements were taken at the last treatment interval before sacrifice of the animal for atherosclerotic lesion measurements. Measurements taken at an intermediate treatment timepoint yielded similar results.
The mice were sacrificed at 34 weeks of age, two weeks after the final croton oil treatment. As shown in Table 1, there were no differences in plasma cholesterol and body weights between control and croton oil-treated mice. Lesions in apoE−/− mice at this age consist mainly of intermediate lesions that present with macrophage infiltration, fibrosis and some necrotic cores [32]. Importantly, computer-assisted en face lesion quantitation revealed no difference in total lesional area between control and inflammation groups (control mean 10.19 mm2 vs. treated mean 10.21 mm2, P = 0.942). Thus, repeated croton oil treatments (7 cycles) induced the formation of a chronic inflammatory skin lesion interspersed with transient healing, as well as a strong acute phase response, with pro-inflammatory cytokines detectable in the plasma and stimulation of hepatic production of acute phase proteins. However, this chronic dermatitis model did not influence the extent of atherosclerosis development in the aorta in apoE−/− mice.
Table 1.
Body weight and plasma cholesterol levels
| Control Group | Inflammation Group | |
|---|---|---|
| 1. Chronic Skin Lesion Study | ||
| (6 weeks) | ||
| Weight (g) | 17.8 ± 0.8 | 18.1 ± 1.2 |
| Cholesterol (mg/dL) | 439 ± 84 | 439 ± 66 |
| (33 weeks) | ||
| Weight (g) | 23.0 ± 0.7 | 22.5 ± 0.6 |
| Cholesterol (mg/dL) | 494 ± 69 | 523 ± 76 |
| 2. Allergic lung disease Study | ||
| (8 weeks) | ||
| Weight (g) | 17.9 ± 0.9 | 16.7 ± 0.8a |
| Cholesterol (mg/dL) | 432 ± 129 | 475 ± 147 |
| (26 weeks) | ||
| Weight (g) | 22.4 ± 0.9 | 20.7 ± 1.3a |
| Cholesterol (mg/dL) | 452 ± 81 | 526 ± 86 |
| (42 weeks) | ||
| Weight (g) | N.D. | N.D. |
| Cholesterol (mg/dL) | 454 ± 55 | 447 ± 77 |
| 3. Peritonitis Study | ||
| (8 weeks) | ||
| Weight (g) | 17.9 ± 0.9 | 17.4 ± 1.1 |
| Cholesterol (mg/dl) | 432 ± 129 | 454 ± 103 |
| (26 weeks) | ||
| Weight (g) | 22.4 ± 0.9 | 21.5 ± 0.6b |
| Cholesterol (mg/dL) | 452 ± 81 | 442 ± 52 |
N.D. not determined
P < 0.01
P < 0.05
Aspergillus Fumigatus Allergic lung disease Study
Next, we assessed the rate of atherosclerosis development in a model of asthma-like disease termed allergic lung disease. We induced airway hyperreactivity by repeated i.n. delivery of A. fumigatus antigen (7 and 11 times over the course of 16 and 32 weeks, respectively). This is a well characterized model in terms of local cytokine production both in BAL fluids and in vitro by lung cells, and inflammatory cellular infiltration into the lungs. Intranasal treatment of the mice with A. fumigatus antigen did not produce detectable alterations in plasma IL-6 levels or TNF-α levels one day (data not shown), and SAA levels were mildly increased from a baseline of 0.9 ± 0.8 μg/ml to 65 ± 28 μg/ml. However, as previously described [28], plasma IgE, was markedly higher in animals receiving A. fumigatus by i.n. route (44.2 ± 7.7 μg/ml) versus untreated animals (2.9 ± 3.3 μg/ml). Histological analysis revealed marked lung inflammation characterized by increased eosinophil infiltration (detected by H/E staining) and the presence of enlarged mucous cells (seen by PAS staining) in A. fumigatus antigen treated animals (Fig. 3A). Finally, cytospin analysis of BAL fluids showed a drastic increase in the proportion of eosinophils, as well as some increase in neutrophils, in fluid from A. fumigatus antigen treated animals (Fig. 3B). Thus, this allergic lung disease model resulted in a strong inflammatory response in the lung, with only mild changes in the level of circulating pro-inflammatory cytokines or acute phase reactants.
Figure 3.
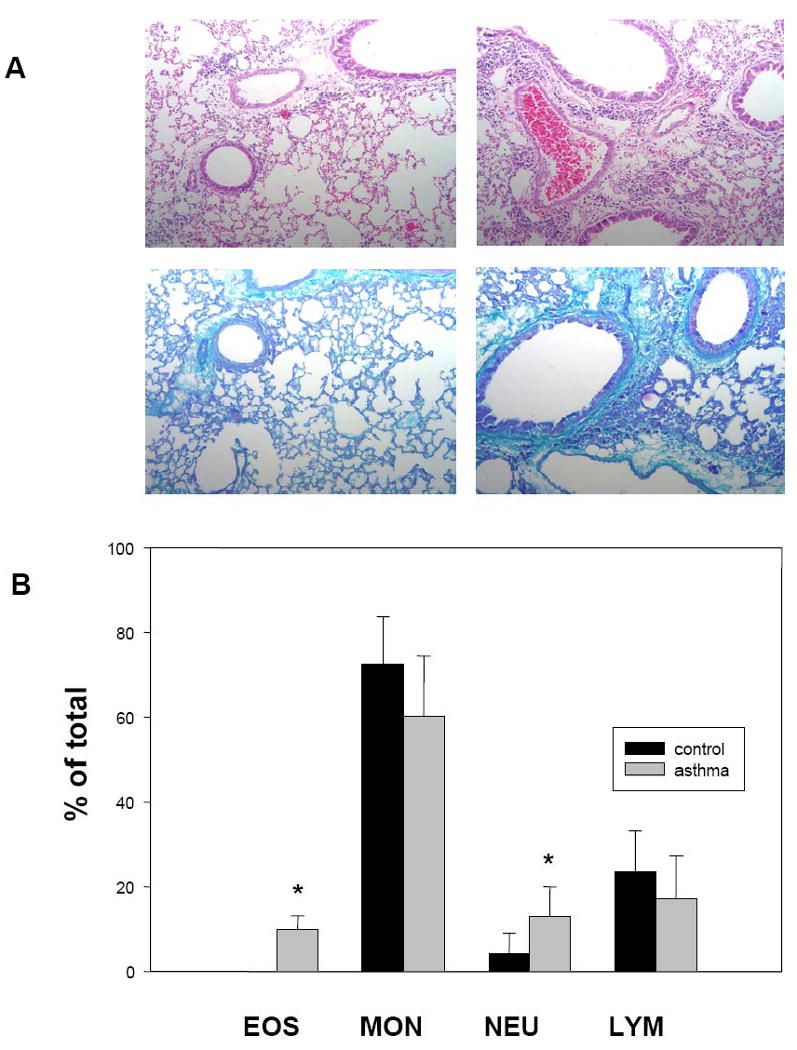
A. Histological evaluation of lung inflammation following intranasal challenge with A. fumigatus allergen. Comparison of H/E stained lung sections from control (top left) and A. fumigatus antigen-challenged (top right) animals (20×), showing increased eosinophil infiltration in animals one day after the final allergen challenge. Comparison of PAS stained control (bottom left) and treated (bottom right) sections (20×) shows enlarged mucous cells in challenged animals. B. Percentage of leukocytes in BAL fluid from control and A. fumigatus antigen-challenged groups. Proportions of eosinophils (EOS), monocytes (MON), neutrophils (NEU), and lymphocytes (LYM) were determined one day after the final challenge.
The mice were sacrificed at 26 and 42 weeks of age and aortas were harvested for atherosclerosis measurements. Plasma cholesterol levels did not differ between control and treated groups at either time-point. Body weight was decreased in the treatment group by 8% for the 26 week time-point, while no changes were observed at 42 weeks (Table 1). Again, similar to the chronic dermatitis model, despite the inflammatory changes observed in the lungs, at neither time point was there any difference in total lesional area between controls and mice with A. fumigatus antigen-induced allergic lung disease (Fig. 4A).
Figure 4.
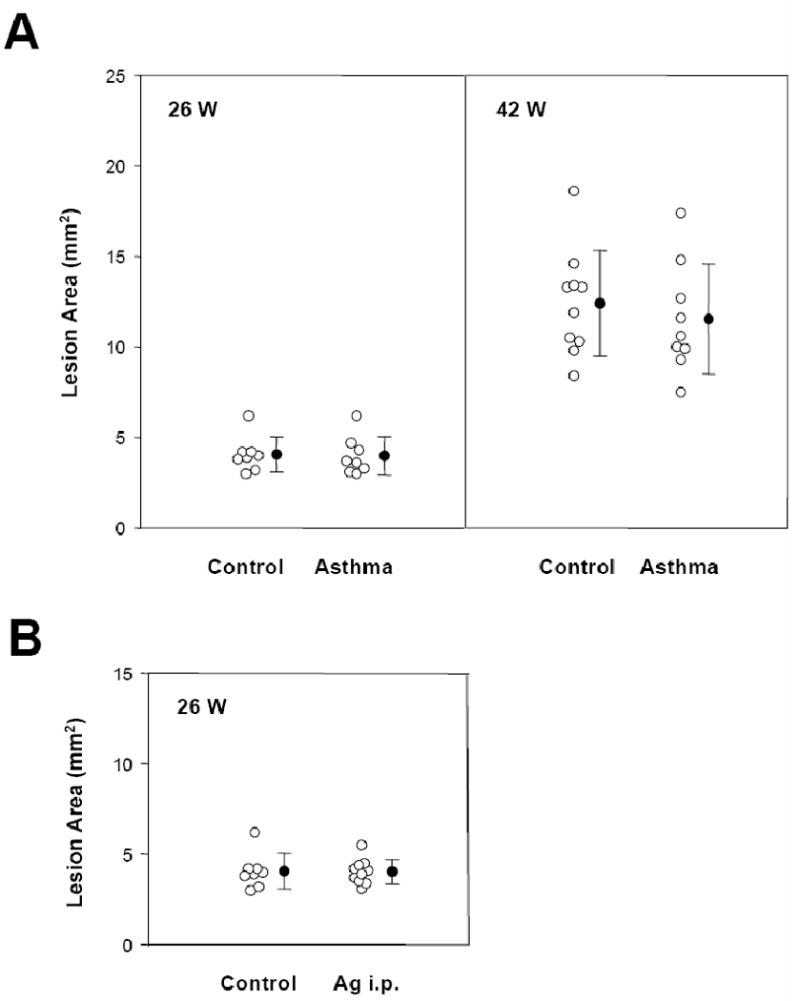
A. Effect of inflammatory allergic lung disease on atherosclerosis in apoE−/−.Lesion area was determined in control and A. fumigatus antigen-challenged groups at 26 (left) or 42 (right) weeks of age. B. Effect of inflammatory peritonitis on atherosclerosis in apoE−/− mice. Total aortic en face lesion area was determined in control and A. fumigatus antigen-i.p. challenge groups at 26 weeks of age.
Aspergillus Fumigatus Peritonitis Study
At the same time that we investigated the allergic lung disease animals, we subjected another set of mice to repeated i.p. delivery of the same antigen in order to induce an inflammatory peritonitis. This treatment consisted of 7 administrations of antigen over the course of 16 weeks, i.e. out to 26 weeks of age (see Materials and Methods).
This method of antigen delivery is sufficient to induce marked increases in serum antigen-specific antibodies [29]. Intraperitoneal treatment of the mice with A. fumigatus antigen did not alter IL-6 or TNF-α levels in the plasma after one day (data not shown), but SAA levels were significantly increased above the baseline of 0.9 ± 0.8 μg/ml to 111 ± 93 μg/ml in the peritonitis model. Similar to the croton oil model, WBC counts dropped one day after antigen treatment, with gradual increase toward pretreatment levels (data not shown). There was no significant difference in serum IgE level in animals that received the i.p. antigen (3.5 ± 4.7 μg/ml) versus that in untreated animals (2.9 ± 3.3 μg/ml). Plasma cholesterol was similar in control and treated mice, while body weight decreased 4% in the chronic inflammation group (Table 1). Once more, quantitative morphometry revealed no difference in atherosclerotic lesion area between apoE−/− mice afflicted by the induced peritonitis compared with that in untreated animals (Fig. 4B).
Discussion
In this study, we tested three different models of experimental recurrent or chronic inflammation on atherosclerosis development in apoE−/− mice. The three models presented with local inflammation at different sites (skin, lungs and intraperitoneal), and different patterns of plasma markers of inflammation, with a robust increase in plasma IL-6 and SAA in croton oil-induced chronic skin inflammation, and more modest but significant increases in plasma SAA in both the Aspergillus fumigatus-induced allergic lung disease and peritonitis. However, none of the three models of inflammation were able to influence the rate of atherosclerosis development in apoE−/− mice.
Epidemiological studies have shown a strong association between some inflammatory diseases, particularly autoimmune rheumatic diseases, and atherosclerosis [11; 16]. Although a confounding factor in the epidemiological studies is that autoimmune rheumatic diseases are commonly associated with increases in “traditional” cardiovascular risk factors [11; 16], the rates of cardiovascular events remain higher in patients with these diseases even after adjustment for baseline risk profiles [33; 34]. Furthermore, several independent studies performed in different lupus mouse models, in which many of the “traditional” atherosclerotic risk factors can be controlled, support a connection between SLE and atherosclerosis [21-24]. We note that, like the current investigation, these studies were performed in dyslipidemic mice (LDL receptor deficiency for the first study, and apoE deficiency for the other two studies). We employed apoE−/− mice as our model, but failed to detect any effect of three different non-autoimmune chronic inflammatory processes on atherosclerosis development. A possible explanation for this difference is that SLE and other systemic autoimmune inflammatory diseases affect multiple organs and tissues and are primarily associated with chronic vasculitis [16], which could be a major proatherogenic factor [35; 36]. Furthermore, there is substantial overlap in the histopathology of vasculitis and atherosclerosis, itself a form of chronic vascular inflammation. Another factor noted in both SLE patients and mouse models is an increase in circulating anti-oxLDL and anti-cardiolipin antibodies that, together with the secretion of lupus-specific autoantibodies, could also contribute to modulate atherogenesis by mechanisms that could involve lipoprotein uptake by foam cells and/or immunomodulation at the lesion site [23; 37]. Therefore, although autoimmune processes could share some common mechanisms of action with non-autoimmune chronic extravascular inflammation, such as the release of soluble mediators or the activation of leukocytes transiting through vessels perfusing the inflamed tissues, these may not be the main mechanisms responsible for the proatherogenic effects associated with the autoimmune disorders. We cannot rule out the possibility that other forms of chronic extravascular non-autoimmune inflammation may predispose to atherosclerosis in apoE−/− mice. However, the fact that none of 3 different models of chronic or recurrent inflammation is associated with accelerated atherosclerosis indicates that extravascular inflammation is unlikely to be a common risk factor for accelerated atherosclerotic cardiovascular diseases.
Figure 2.
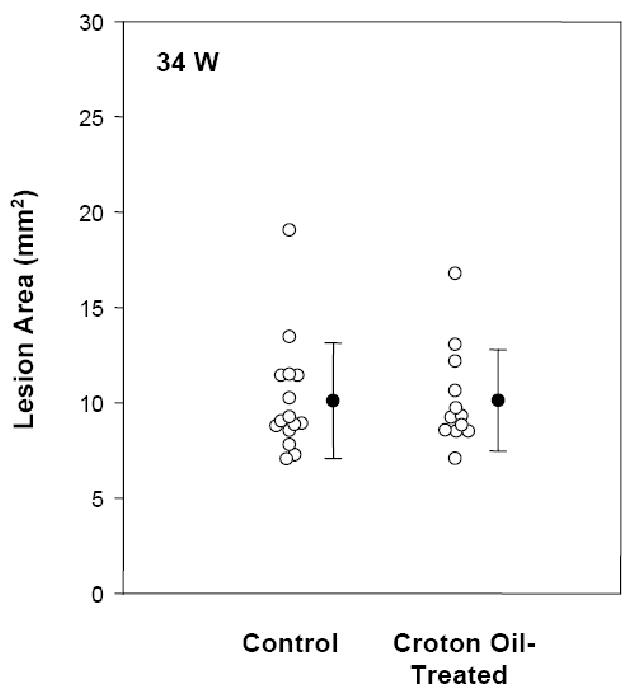
Effect of inflammation by chronic skin lesion on atherosclerosis in apoE−/− mice. Total aortic en face lesion area was determined in control and croton-oil treated groups at 34 weeks of age.
Acknowledgments
We thank Javier Martinez-Botas and Aksam Merched for assistance with some of the experimental procedures, and Leslie Wu for administrative assistance. This work was supported by a grant from the National Institutes of Health HL-51586 and by the Betty Rutherford Chair, St. Luke's Episcopal Hospital and Baylor College of Medicine, Houston, Texas (to Lawrence Chan). Antoni Paul was supported in part by a Scientist Development Grant from the American Heart association 0535118N.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rosamond W, Flegal K, Friday G, Furie K, Go A, Greenlund K, Haase N, Ho M, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M, Meigs J, Moy C, Nichol G, O'Donnell CJ, Roger V, Rumsfeld J, Sorlie P, Steinberger J, Thom T, Wasserthiel-Smoller S, Hong Y American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics--2007 Update: A Report From the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007;115:e69–171. doi: 10.1161/CIRCULATIONAHA.106.179918. [DOI] [PubMed] [Google Scholar]
- 2.Berliner JA, Navab M, Fogelman AM, Frank JS, Demer LL, Edwards PA, Watson AD, Lusis AJ. Atherosclerosis: basic mechanisms. Oxidation, inflammation, and genetics. Circulation. 1995;91:2488–2496. doi: 10.1161/01.cir.91.9.2488. [DOI] [PubMed] [Google Scholar]
- 3.Willerson JT, Ridker PM. Inflammation as a cardiovascular risk factor. Circulation. 2004;109:II2–10. doi: 10.1161/01.CIR.0000129535.04194.38. [DOI] [PubMed] [Google Scholar]
- 4.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 5.Kiechl S, Egger G, Mayr M, Wiedermann CJ, Bonora E, Oberhollenzer F, Muggeo M, Xu Q, Wick G, Poewe W, Willeit J. Chronic infections and the risk of carotid atherosclerosis: prospective results from a large population study. Circulation. 2001;103:1064–1070. doi: 10.1161/01.cir.103.8.1064. [DOI] [PubMed] [Google Scholar]
- 6.Meier CR, Jick SS, Derby LE, Vasilakis C, Jick H. Acute respiratory-tract infections and risk of first-time acute myocardial infarction. Lancet. 1998;351:1467–1471. doi: 10.1016/s0140-6736(97)11084-4. [DOI] [PubMed] [Google Scholar]
- 7.Paquette DW. The periodontal infection-systemic disease link: a review of the truth or myth. J Int Acad Periodontol. 2002;4:101–109. [PubMed] [Google Scholar]
- 8.Naz SM, Symmons DP. Mortality in established rheumatoid arthritis. Best Pract Res Clin Rheumatol. 2007;21:871–883. doi: 10.1016/j.berh.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 9.Solomon DH, Goodson NJ, Katz JN, Weinblatt ME, Avorn J, Setoguchi S, Canning C, Schneeweiss S. Patterns of cardiovascular risk in rheumatoid arthritis. Ann Rheum Dis. 2006;65:1608–1612. doi: 10.1136/ard.2005.050377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kao AH, Sabatine JM, Manzi S. Update on vascular disease in systemic lupus erythematosus. Curr Opin Rheumatol. 2003;15:519–527. doi: 10.1097/00002281-200309000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Hahn BH, Grossman J, Chen W, McMahon M. The pathogenesis of atherosclerosis in autoimmune rheumatic diseases: roles of inflammation and dyslipidemia. J Autoimmun. 2007;28:69–75. doi: 10.1016/j.jaut.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Bruce IN. ‘Not only…but also’: factors that contribute to accelerated atherosclerosis and premature coronary heart disease in systemic lupus erythematosus. Rheumatology (Oxford) 2005;44:1492–1502. doi: 10.1093/rheumatology/kei142. [DOI] [PubMed] [Google Scholar]
- 13.Ridker PM, Rifai N, Stampfer MJ, Hennekens CH. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation. 2000;101:1767–1772. doi: 10.1161/01.cir.101.15.1767. [DOI] [PubMed] [Google Scholar]
- 14.Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002;347:1557–1565. doi: 10.1056/NEJMoa021993. [DOI] [PubMed] [Google Scholar]
- 15.Steptoe A, Shamaei-Tousi A, Gylfe A, Henderson B, Bergstrom S, Marmot M. Socioeconomic status, pathogen burden and cardiovascular disease risk. Heart. 2007;93:1567–1570. doi: 10.1136/hrt.2006.113993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haque S, Mirjafari H, Bruce IN. Atherosclerosis in rheumatoid arthritis and systemic lupus erythematosus. Curr Opin Lipidol. 2008;19:338–343. doi: 10.1097/MOL.0b013e328304b65f. [DOI] [PubMed] [Google Scholar]
- 17.Muhlestein JB, Anderson JL. Infectious serology and atherosclerosis: how burdensome is the risk? Circulation. 2003;107:220–222. doi: 10.1161/01.cir.0000043909.78380.a0. [DOI] [PubMed] [Google Scholar]
- 18.Roman MJ, Devereux RB, Schwartz JE, Lockshin MD, Paget SA, Davis A, Crow MK, Sammaritano L, Levine DM, Shankar BA, Moeller E, Salmon JE. Arterial stiffness in chronic inflammatory diseases. Hypertension. 2005;46:194–199. doi: 10.1161/01.HYP.0000168055.89955.db. [DOI] [PubMed] [Google Scholar]
- 19.Datta D, Ferrell WR, Sturrock RD, Jadhav ST, Sattar N. Inflammatory suppression rapidly attenuates microvascular dysfunction in rheumatoid arthritis. Atherosclerosis. 2007;192:391–395. doi: 10.1016/j.atherosclerosis.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 20.Breslow JL. Mouse Models of Atherosclerosis. Science. 1996;272:685–688. doi: 10.1126/science.272.5262.685. [DOI] [PubMed] [Google Scholar]
- 21.Aprahamian T, Rifkin I, Bonegio R, Hugel B, Freyssinet JM, Sato K, Castellot JJ, Jr, Walsh K. Impaired clearance of apoptotic cells promotes synergy between atherogenesis and autoimmune disease. J Exp Med. 2004;199:1121–1131. doi: 10.1084/jem.20031557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stanic AK, Stein CM, Morgan AC, Fazio S, Linton MF, Wakeland EK, Olsen NJ, Major AS. Immune dysregulation accelerates atherosclerosis and modulates plaque composition in systemic lupus erythematosus. Proc Natl Acad Sci USA. 2006;103:7018–7023. doi: 10.1073/pnas.0602311103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng X, Li H, Rumbin AA, Wang X, La CA, Brechtelsbauer K, Castellani LW, Witztum JL, Lusis AJ, Tsao BP. ApoE-/-Fas-/- C57BL/6 mice: a novel murine model simultaneously exhibits lupus nephritis, atherosclerosis, and osteopenia. J Lipid Res. 2007;48:794–805. doi: 10.1194/jlr.M600512-JLR200. [DOI] [PubMed] [Google Scholar]
- 24.Ma Z, Choudhury A, Kang SA, Monestier M, Cohen PL, Eisenberg RA. Accelerated atherosclerosis in ApoE deficient lupus mouse models. Clin Immunol. 2008;127:168–175. doi: 10.1016/j.clim.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meir KS, Leitersdorf E. Atherosclerosis in the Apolipoprotein E-Deficient Mouse: A Decade of Progress. Arterioscler Thromb Vasc Biol. 2004;24:1006–1014. doi: 10.1161/01.ATV.0000128849.12617.f4. [DOI] [PubMed] [Google Scholar]
- 26.Berg DJ, Leach MW, Kuhn R, Rajewsky K, Muller W, Davidson NJ, Rennick D. Interleukin 10 but not interleukin 4 is a natural suppressant of cutaneous inflammatory responses. J Exp Med. 1995;182:99–108. doi: 10.1084/jem.182.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mizgerd JP, Bullard DC, Hicks MJ, Beaudet AL, Doerschuk CM. Chronic inflammatory disease alters adhesion molecule requirements for acute neutrophil emigration in mouse skin. J Immunol. 1999;162:5444–5448. [PubMed] [Google Scholar]
- 28.Grunig G, Corry DB, Leach MW, Seymour BW, Kurup VP, Rennick DM. Interleukin-10 is a natural suppressor of cytokine production and inflammation in a murine model of allergic bronchopulmonary aspergillosis. J Exp Med. 1997;185:1089–1099. doi: 10.1084/jem.185.6.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corry DB, Grunig G, Hadeiba H, Kurup VP, Warnock ML, Sheppard D, Rennick DM, Locksley RM. Requirements for allergen-induced airway hyperreactivity in T and B cell-deficient mice. Mol Med. 1998;4:344–355. [PMC free article] [PubMed] [Google Scholar]
- 30.Murali PS, Kumar A, Choi H, Banasal NK, Fink JN, Kurup VP. Aspergillus fumigatus antigen induced eosinophilia in mice is abrogated by anti-IL-5 antibody. J Leukoc Biol. 1993;53:264–267. doi: 10.1002/jlb.53.3.264. [DOI] [PubMed] [Google Scholar]
- 31.Guevara NV, Kim HS, Antonova EI, Chan L. The absence of p53 accelerates atherosclerosis by increasing cell proliferation in vivo. Nat Med. 1999;5:335–339. doi: 10.1038/6585. [DOI] [PubMed] [Google Scholar]
- 32.Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–140. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- 33.Esdaile JM, Abrahamowicz M, Grodzicky T, Li Y, Panaritis C, du BR, Cote R, Grover SA, Fortin PR, Clarke AE, Senecal JL. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum. 2001;44:2331–2337. doi: 10.1002/1529-0131(200110)44:10<2331::aid-art395>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 34.del RI, Williams K, Stern MP, Freeman GL, Escalante A. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum. 2001;44:2737–2745. doi: 10.1002/1529-0131(200112)44:12<2737::AID-ART460>3.0.CO;2-%23. [DOI] [PubMed] [Google Scholar]
- 35.Bacon PA, Stevens RJ, Carruthers DM, Young SP, Kitas GD. Accelerated atherogenesis in autoimmune rheumatic diseases. Autoimmun Rev. 2002;1:338–347. doi: 10.1016/s1568-9972(02)00100-3. [DOI] [PubMed] [Google Scholar]
- 36.Guillevin L, Dorner T. Vasculitis: mechanisms involved and clinical manifestations. Arthritis Res Ther. 2007;9 2:S9. doi: 10.1186/ar2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frostegard J, Svenungsson E, Wu R, Gunnarsson I, Lundberg IE, Klareskog L, Horkko S, Witztum JL. Lipid peroxidation is enhanced in patients with systemic lupus erythematosus and is associated with arterial and renal disease manifestations. Arthritis Rheum. 2005;52:192–200. doi: 10.1002/art.20780. [DOI] [PubMed] [Google Scholar]