

Abstract
Phosphatidic acid (PA) has been increasingly recognized as an important signaling lipid regulating cell growth and proliferation, membrane trafficking, and cytoskeletal reorganization. Recent studies indicate that the signaling PA generated from phospholipase D (PLD) and diacylglycerol kinase (DGK) plays critical roles in regulating the activity of some members of Ras superfamily of small guanosine triphosphatases (GTPases), such as Ras, Rac and Arf. Change of PA levels regulates the activity of small GTPases by modulating membrane localization and activity of small GTPase regulatory proteins, guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs). In addition, PA also targets some small GTPases to membranes by direct binding. This review summarizes the roles of PLD and DGK in regulating the activity of several Ras superfamily members and cellular processes they control. Some future directions and the implication of PA regulation of Ras small GTPases in pathology are also discussed.
Keywords: Phospholipids, Phosphatidic acid, Phospholipase D, Diacylglycerol kinase, Ras, Small GTPases
1. Introduction
1.1 Phosphatidic acid and signaling
Phosphatidic acid (PA) has been long proposed as a pleiotropic bioactive lipid regulating cell growth and proliferation, membrane trafficking, and cytoskeletal reorganization [1, 2]. The cellular signaling PA is produced through two major pathways: the hydrolysis of phosphatidylcholine (PC) by phospholipase D (PLD), and the phosphorylation of diacylglycerol (DAG) by diacylglycerol kinase (DGK).
The mammalian PLD family consists of two related gene products, PLD1 and PLD2 [3–6]. PLD1 is directly regulated by classic protein kinase C (PKC) and members of the ADP-ribosylation factor (Arf) and Rho family small GTPases in conjunction with phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) [7]. In contrast to PLD1, PLD2 is constitutively active in the presence of PI(4,5)P2 and localized to the plasma membrane in most cell types [8, 9]. Subsequent studies have shown that PLD2 can also be activated in intact cells by agonists and can be regulated by small GTPases and PKC isoforms [5, 10, 11].
DGK family is defined by a conserved catalytic domain (DAGKc) located at the C-terminal half of the protein, and comprises 10 distinct enzymes [12, 13]. Based on the distinct regulatory domains, DGKs can be grouped into five subtypes: type I DGK (α, β and γ), type II DGK (δ, η and κ), type III DGK (ε), type IV DGK (ζ andι), and type V DGK (θ) [12, 14]. Whereas all DGK catalyzes the same reaction, the presence of non-conserved regulatory regions appears to confer specificity to the distinct DGK isoforms by restricting their subcellular site of action and/or defining their activation mechanism [12, 14]. By regulating the level of both DAG and PA in a reciprocal manner, DGK can potentially act as terminators of DAG-mediated signals as well as activators of PA-mediated signals [12, 14].
The activity of both PLD and DGK is tightly regulated by external stimuli. However, compared to phosphatidylinositides and DAG, the signaling role of PA has not been fully appreciated until recently. PA is generally believed to be less stable, and is metabolized to DAG by phosphatidic acid phosphatase (PAP). In the studies published in the 1980’s and early 1990’s, the major role assigned to PLD and DGK was the control of intracellular DAG level, and many biological effects of modulating PLD and DGK activities were interpreted as a result of PKC overstimulation [14]. Recent studies by us and other groups have demonstrated that PA directly regulates the activities of some key signaling molecules, such as mTOR, Ras, and myosin II [9, 15–17], suggesting that PA itself is an important signaling lipid in cells.
1.2. Regulation of the activity of Ras superfamily of small guanosine triphosphatases
The Ras superfamily of small guanosine triphosphatases (GTPases) contains over 150 human members. Based on sequence and functional similarities, the Ras superfamily of small GTPases is divided into at least five distinct branches: Ras, Rho, Rab, Arf, and Ran [18, 19]. Upon extracellular stimulation, activation of Ras GTPases controls a diversity of downstream cytoplasmic signaling cascades and cellular functions. These small GTPases share a common biochemical mechanism and act as binary molecular switches. They exhibit high-affinity binding for GDP and GTP, and possess low intrinsic GTP hydrolysis and GDP/GTP exchange activities. GDP/GTP binding is controlled by two main classes of regulatory proteins: guanine nucleotide exchange factors (GEFs) promote formation of the active, GTP-bound form, whereas GTPase activating proteins (GAPs) accelerate the intrinsic GTPase activity to promote formation of the inactive GDP-bound form [18, 19]. The fine regulation of Ras superfamily of small GTPases facilitates their key involvement in a widely diverse spectrum of biochemical and biological processes.
Earlier studies on PLD mainly focused on the regulation of PLD activity by small GTPases. Recent studies have shown that PLD and DGK also act upstream to regulate the activities of some small GTPases. We have recently reported that PA directly binds to the pleckstrin homology (PH) domain of Sos, a Ras GEF, and thus recruits Sos to the plasma membrane and converts Ras-GDP to Ras-GTP [17]. This and other similar findings in the literature have suggested that regulation of GEFs and GAPs by PA metabolism might be a common mechanism for controlling the activities of small GTPases in response to signal stimulation. The source of PA and its relative contribution to the activity of individual small GTPases in a particular cellular context, however, remain to be further investigated. We have summarized the regulation of some members of Ras superfamily by PA and PA-metabolizing enzymes in Table 1.
Table 1.
Small GTPases regulated by PLD and DGK signaling
| Small GTPases | Enzymes | Signaling lipids | Lipid-binding proteins | Effect on small GTPases | Cellular stimulation | Biological functions | Refs |
|---|---|---|---|---|---|---|---|
| Ras | PLD2 | PA | Sos | Activation | EGF | Transformation of NIH3T3 cells | [17] |
| DAG | RasGRP1 | Activation | Anti-CD28 & LFA-1 | T cell activation | [16] | ||
| DAGα | DAG | RasGRP1 | Inhibition | Anti-CD3 & anti-CD28 | T cell activation | [27] | |
| RasGRP1 | Inhibition | Anti-CD3 & anti-CD28, PMA | T cell anergy | [34] | |||
| DGKζ | DAG | RasGRP1 | Inhibition | Anti-CD3 | T cell activation | [29] | |
| Not examined | Inhibition | Anti-CD3 | T cell activation, proliferation | [33] | |||
| Not examined | Inhibition | IgE/DNP-HSA | Mast cell degranulation, cytokine production | [32] | |||
| DGKα & DGKζ | DAG & PA | Not examined | Inhibition | Whole animal | T cell maturation | [30] | |
| DGKι | RasGRP3 | Activation | Thrombin, PMA | Tumor formation | [35] | ||
| Rac1 | PLD1 & PLD2 | PA | Rac1 | Membrane targeting | Integrin | Cell spreading and migration | [39] |
| DGKα | ? | Not examined | Activation | HGF | Cell scatter, migration, spreading, lamellipodia formation and membrane ruffling | [40] | |
| DGKγ | PA | β2-chimaerin | Inhibition | EGF | Lamellipodia formation and membrane ruffling | [42, 43] | |
| DGKζ* | N/A | N/A | Activation | Neurite outgrowth | [44] | ||
| Arf1 | ? | PA | AGAP1 | Inhibition | [47] | ||
| Arf1 & Arf5 | ? | PA | ASAP1 | Inhibition | [45, 46] | ||
| Arf6 | ? | PA | ACAPs | Inhibition | [48] | ||
| PA | Arf6 | Membrane targeting | [49] |
LFA-1, Lymphocyte function-associated antigen-1; Arf, ADP-ribosylation factor; PMA, phorbol 12-myristate 13-acetate (PMA).
DGKζ binds directly to Rac1 and recruits to membranes.
2. Ras activation
Ras activation is tightly regulated by guanine nucleotide exchange factors (GEFs). To date, four subfamilies of GEF molecules have been identified: Sos, RasGRF, RasGRP and CNRasGEF [18, 19]. The ubiquitously expressed Sos signals downstream of receptor tyrosine kinases (RTKs). RasGRF proteins are expressed predominantly in the central nervous system. The family of RasGRP proteins is expressed primarily in haematopoietic cell lineages and function downstream of non-receptor tyrosine kinases [20]. It is worth mentioning that these four subfamilies have different domain structures, thus the likely mechanism of their activation may be significantly different.
2.1. PA promotes the plasma membrane recruitment and activation of Sos upon EGFR activation
RTK-mediated Ras activation requires recruitment of the Ras GEF Sos to the plasma membrane where it catalyzes the conversion of Ras-GDP to Ras-GTP. In a classical model established in 1993, Sos membrane translocation is mediated by the formation of a ternary complex with the activated RTK and Grb2, through the binding of the SH3 domain of adaptor molecule Grb2 to a proline-rich sequence in the C-terminus of Sos. However, this model failed to explain some questions rising from several genetic and biomedical studies. The Grb2-binding deficient mutants still retain the normal subcellular distribution and transforming activity of Sos in both biochemical and in vivo studies [21, 22]. These and other studies have led to the proposal that Grb2 is not directly involved in Sos translocation but may function to overcome a negative regulation of Sos by its C-terminus [21], thus implicated the Sos N-terminal PH and Dbl homology (DH) domains as the critical determinant of its function [21–24].
The PH domain has been well known for its ability to bind to phosphatidylinositides and target proteins to membranes. The PH domain of Sos was first found to bind to PI(4,5)P2 [20]. However, the membrane localization of Sos-PH is not dependent on its ability to bind PI(4,5)P2 [17], indicating the presence of other mechanisms responsible for Sos membrane targeting and activation. Intrigued by homology with the PA-binding site in p47phox, a NADPH oxidase component, we have demonstrated that the PH domain of Sos binds to PA, through a PA-binding site independent of the PI(4,5)P2-binding site, at an affinity higher than the PI(4,5)P2 binding [17].
In contrast to PI(4,5)P2, the PA-binding appears to be critical for the plasma membrane recruitment of Sos-PH. A membrane-permeable form of PA is sufficient to recruit GFP-tagged Sos-PH to the plasma membrane, but fails to induce plasma membrane recruitment of the PA-binding deficient mutant, Sos-PH-HR/EE [17]. Furthermore, a carboxy-terminally truncated Sos protein (SosΔC), which is unable to bind Grb2, is recruited normally to the plasma membrane in response to EGF receptor (EGFR) activation and activates Ras. In contrast, SosΔC with the PA-binding mutations (HR/EE) is not recruited to the plasma membrane and does not stimulate Ras GTP loading [17]. These results clearly demonstrate a primary role for PA, and not Grb2–EGFR, as the critical plasma membrane anchor of Sos that mediates Ras activation.
PLD2 is activated by EGFR and mediates MAPK activation [25, 26], implying that it is the source of EGFR-generated PA. Ectopic expression of PLD2 was sufficient to recruit SosΔC to the plasma membrane and to activate Ras, whereas knockdown of endogenous PLD2 or ectopic expression of a PLD2 dominant negative mutant, had no effect on EGFR activation, but completely blocked EGF-stimulated Sos recruitment and Ras activation [17]. Thus these results show that PLD2 operates between EGFR and Sos and that PA is both necessary and sufficient for Sos plasma membrane recruitment. Finally, the PLD2-dependent and Grb2-independent mechanism for Sos-mediated Ras activation is important for the transforming activity of Ras. The combination of Ras, SosΔC and PLD2 resulted in a marked increase in foci number, and this synergistic effect was abolished when the PA binding-deficient Sos mutant was used instead [17].
The recent findings of acidic phospholipids regulation of Sos membrane targeting and activation has led to a revised model for the Sos-mediated Ras activation (Figure 1) [see further details in 20]: upon growth factor stimulation, Sos is recruited to the plasma membrane through two independent sites, the C-terminal Grb2-binding site and its PA- and PI(4,5)P2-binding PH domain. In addition to the membrane anchorage function, the interactions also induce conformational changes and overcome the negative regulation of Sos by its C-terminus and DH-PH domains, resulting in binding of Ras-GDP to an allosteric site on Sos. The binding induces a low-level activity of Sos, converting Ras-GDP to Ras-GTP, and leads to a maximal Sos activity. Moreover, interactions of Sos either with Ras-GDP or Ras-GTP at the allosteric regulatory site further stabilize the membrane localization of Sos (Figure 1). The signaling PA level and PLD activity are regulated by extracellular signaling [4, 5, 27], providing a fine control of Ras activity. It still remains unclear what the relative contributions of PA and PI(4,5)P2 are, and the enzymes metabolizing these lipids, to the Sos-mediated Ras activation. It is likely that binding of PA and PI(4,5)P2 to Sos are additive or synergistic, since Sos binds to these two phospholipids at separate sites in its PH domain [17].
Figure 1.
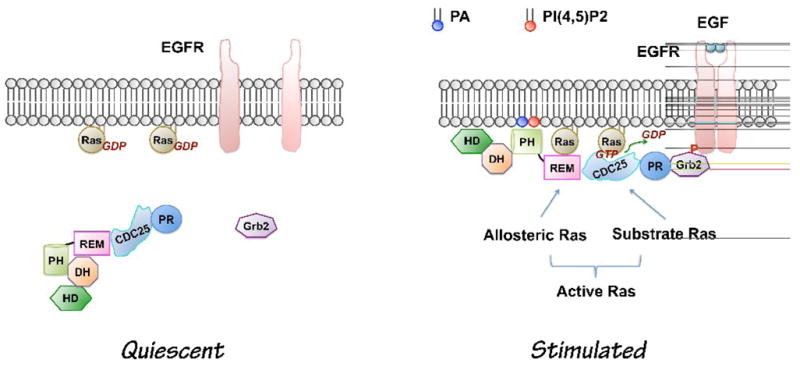
Current model of growth factor-induced Sos activation (Modified based on [20]). In quiescent cells, PLD activity is low. The DH-PH domains block the allosteric binding site for Ras. Upon growth factor stimulation, Sos is recruited to the plasma membrane through at least two independent sites: C-terminal Grb2-binding site and its lipid PA- and PI(4,5)P2 -binding PH domain. Interaction of the PH domain with phospholipids leads to conformational changes allowing binding of allosteric Ras, which lead to the maximal activation of Sos. PA level is increased by receptor activation of PLD2 [17]. The role of PA from other sources in growth factor-induced Sos activation has not been tested yet. CDC25, CDC25-homology domain, DH, Dbl homology domain; EGF, Epidermal growth factor; EGFR, Epidermal growth factor receptor; HD, Histone-like domain; PH, pleckstrin homology domain; PR, Proline-rich motif; REM, Ras exchanger motif; Phosphatidic acid, PA; Phosphatidylinositol-4,5-bisphosphate; PI(4,5)P2.
2.2. Regulation of RasGRP-mediated Ras activation by PA-DAG metabolism
Most of the studies on the RasGRP subfamily of Ras GEF were performed in immune system because of their primary expression in haematopoietic cell lineages [12, 14]. The RasGRP proteins have distinct regulatory domains compared to the Sos subfamily. They do not have a PH domain, but instead possess a C-terminal C1 domain which is regulated by calcium and DAG [12, 14]. Thus, it would be reasonable to speculate that signaling pathways leading to change of the DAG level have an impact on Ras activity. Indeed, both PLD and DGKs have been reported to regulate RasGRP activation by either increasing or decreasing DAG level.
2.2.1. PLD2-PAP-mediated RasGRP1 membrane translocation
Using a fluorescent reporter for activated Ras, YFP–RBD, Mor et al. found that TCR activation with or without co-stimulation of CD28 led to activation of Ras only on the Golgi apparatus, whereas co-stimulation with lymphocyte function-associated antigen-1 (LFA-1) induced Ras activation on both the Golgi and the plasma membrane in Jurkat and primary mouse T cells [16]. The Ras activation co-stimulated through LFA-1 requires RasGRP1, which is activated by DAG on the plasma membrane. However, the pool of DAG augmented at the plasma membrane through LFA-1 signaling is PLCγ1-independent and sufficient to recruit RasGRP1 in the absence of PLCγ1-stimulated calcium.
DAG can also be produced through PAP conversion of PLD-generated PA [4–6]. Further experiments show that, when TCR activation is accompanied by activation of the LFA-1, PLD2 is activated and generates PA at the plasma membrane. The PLD2-generated PA is then converted to DAG by PAP. The increased plasma membrane DAG activates Ras through recruiting RasGRP1 to the plasma membrane. Inhibition of PLD with low concentrations of primary butanol and small hairpin RNA blocked recruitment of RasGRP1 to the plasma membrane and Ras activation at the plasma membrane, after co-stimulation of the TCR and LFA-1. Inhibition of PAP by propranolol also blocked Ras activation at the plasma membrane. These results suggest that PA and PA-derived DAG are required for Ras activation by LFA-1 signaling [16].
2.2.2. Complex regulation of Ras activity by different DGK isoforms
The regulation of Ras activity by DGKs is mainly through RasGRPs. It appears that Ras activity can be either positively or negatively regulated by specific RasGRP isoforms in different cell types, although the mechanism for the discrepancy is not clear yet. In T cells, both DGKα and ζ isoforms negatively control DAG level and the Ras-ERK signaling pathway after TCR engagement. Sanjuan et al. found that T cell activation by anti-CD3 and anti-CD28 was rapidly followed by RasGRP1 and DGKα translocation to the plasma membrane. Transfection of the dominant-negative DGKα increased GTP-Ras levels compared with those in control cells, even in the absence of T cell activation. After TCR stimulation, Ras activation was further increased in the cells transfected with this mutant [28]. On the contrary, when cells were transfected with the constitutively active DGKα isoform, Ras activation decreased dramatically at the two time points tested. These experiments confirm that modification of DGKα activity has a profound effect on the DAG-based signals proceeding from the TCR [28]. The negative regulation of Ras pathway in T cells is supported by other studies whereby the expression of an inactive mutant of DGKα and ζ [29, 30], and knockout of DGKα and ζ, reduced activation of the Ras signaling [31–35].
In contrast to T cells, Ras activity is lower in embryonic fibroblasts lacking DGKι, suggesting that DGKι activates Ras signaling in this cell type [36]. DGKι was bound to RasGRP3 and inhibited its activation of Rap1 by metabolizing DAG. Because Rap1 can antagonize the function of Ras, these data support a model that DGKι regulates RasGRP3 with a predominant effect on Rap1 activity [36].
It was originally believed that DAG was the only signaling molecule mediating the function of DGKs. Recent findings of the signaling functions of DGK-produced PA have added to the complexity of DGK-regulated Ras pathways [13, 14, 37]. For example, DGKζ-generated PA promotes TLR-induced IL-12 production by negatively regulating the PI3K-AKT pathway, and plays a critical role in host defense against Toxoplasma gondii [32]. In another study, Guo et al. report that DGKα and ζ synergistically promote T cell maturation in the thymus. The developmental blockage in DGKα (−/−)ζ(−/−) mice can be partially overcome by treatment with PA [31]. Although it is very technically challenging, it would be critical to examine the relative contribution of DAG and PA in DGK-mediated physiological and pathological functions in the future studies.
3. PLD and DGK regulation of Rac1
Rho family GTPases are key regulators of cytoskeletal dynamics and affect many cellular processes, including cell polarity, migration, vesicle trafficking and cytokinesis [38, 39]. Most of the functional information on Rho family proteins has come from studies of RhoA, Rac1 and Cdc42. In addition to Ras, some recent publications have suggested that PA regulates the activity of Rac1 small GTPase, which is well known for its activity in promoting actin polymerization and the formation of lamellipodia [38, 39]. Interestingly, Rho family proteins including Rac1 are also well characterized activators of PLD [3–6], indicating a potential signaling feedback loop of Rhos-PLDs/DGKs-PA.
Changes in the PA level produced by PLD does not seem to directly affect the GTP loading of Rac1 during cell spreading in two independent studies published recently [9, 40]. Instead of the GTP loading, Chae et al. proposed that PLD-generated PA modulates Rac1 downstream signaling by targeting it to the plasma membranes through its C-terminal polybasic motif [40]. In our study, we found that Rac signaling is independent of PLD activity. During early cell spreading, downregulation of PLD2 activity is required for membrane protrusion activity [9]. This process is controlled by PLD2-regulated myosin II activity at the cell periphery [9]. Changes of PLD activity did not affect the GTP-bound Rac1 and its membrane targeting. Furthermore, although the constitutively activated mutant of Rac1 itself is sufficient to promote membrane protrusion, it failed to rescue the delay of early cell spreading induced by PLD2 overexpression [9].
The DGK regulation of Rac1 activity is likely different from PLD. It was reported that some DGK isoforms directly promote the activation of Rac1, whereas the others inhibit it. It is not clear whether the difference is caused by individual DGK isoforms or by different experimental systems. Chianale et al. showed that inhibition of DGKα by treatment of R59949 (a DGK inhibitor), or expression of DGKα dominant negative mutants abolished HGF-induced Rac activation and membrane targeting [41]. On the contrary, another DGK family member, DGKγ was found to act as an upstream suppressor of Rac1, and suppress lamellipodium/membrane ruffle formation in NIH3T3 fibroblasts stimulated with PDGF [42]. A following study also supports the finding of DGKγ as a negative regulator of Rac1 [43]. DGKγ specifically recruits and colocalizes at the plasma membrane with β2-chimaerin, a GAP for Rac1 in EGF-stimulated Cos-7 cells. Moreover, DGKγ markedly enhances the EGF-dependent GAP activity of β2-chimaerin through its catalytic action, thus negatively regulates Rac1 activation and membrane ruffling [43]. Interestedly, PA is a potent stimulator of β2-chimaerin activity in an in vitro GAP assay using recombinant proteins [44].
Finally, one addition to the complexity is that the regulation of Rac1 activity by some DGK isoforms may be independent of their catalytic activity (PA production). DGKζ directly interacts with Rac1 through a binding site located within its C1 domains. Together with syntrophin, these proteins form a tertiary complex in N1E-115 cells. A DGKζ mutant that mimics phosphorylation of the MARCKS domain was unable to bind an activated Rac1 mutant (Rac1V12) and phorbol 12-myristate 13-acetate (PMA)-induced PKC activation inhibited the interaction of DGKζ with Rac1V12, suggesting that PKC-mediated phosphorylation of the MARCKS domain negatively regulates DGKζ binding to active Rac1. PKC-mediated phosphorylation of the MARCKS domain would favor translocation of DGKζ, together with the rest of the complex, to the membrane. This allows nucleotide exchange on Rac (GTP for GDP) and its subsequent dissociation from DGKζ and syntrophin [45].
The discrepancy described above might not simply be caused by the reagents and protocols used in different laboratories. It is more likely that there exist isoforms-specific and/or cell-specific roles for PLD and DGK. A proof of this idea is that in the same cell line, while the DGKγ mutants affected Rac1 activation and cell morphology, none of the corresponding mutants of DGKα and DGKβ, which are closely related isoforms, had the same effect [42]. It would be interesting to investigate if manipulation of specific PLD or DGK isoforms using the same reagents still leads to activation of different downstream effectors and opposite cell morphology in the same or different experimental systems.
4. PA regulation of Arf small GTPases
The Arf proteins are best known for their role in membrane trafficking, and also contribute significantly to the regulation of actin cytoskeletal organization. The significance of PA regulation of Arf activity has not been tested in any cellular context yet. However, PA, in cooperation with phosphoinositides, is an extremely potent activator for the activity of several Arf GAP proteins in vitro. The activity of ASAP1, an Arf GAP protein, is stimulated about 10, 000 fold by PI(4,5)P2 and PA. PA has been found to activate Arf GAPs with preferences for different Arf members, i.e., ASAP1 (GAP for Arf1 and Arf5) [46, 47], AGAP1 (GAP for Arf1) [48], and ACAPs (GAP for Arf6) [49]. Thus it is very likely that the PA regulation of GAP is a common mechanism for the inactivation of Arf small GTPases. Several members of Arf family are direct activators of PLD [4–6]. It is possible that PA regulation of Arf GAPs creates a negative feedback loop to terminate PLD-PA and Arf signaling. More in vivo studies in intact cells need to be performed to test this hypothesis.
In addition to regulation of activity of Arf GAPs, Arfs may bind directly to PA. Using a PA-coupled Affi-Gel 10 as an affinity reagent, several PA-binding proteins including Arf from brain cytosol were identified. Interestedly, the Arf6 binding to PA beads requires myristoylation and depends on its GTP-loading [50].
5. Future perspectives
Recent results have shown specific and direct interactions between PA and PA-binding proteins [1, 2]. These types of interactions involve binding of PA to a positively charged site on a PA-binding protein, such as Raf-1, SHP-1 and mTOR [1, 2]. However, the primary structures of these PA-binding regions reveal no significant homology. They are diverse and include sequences that had not previously been described as lipid binding domains. Future analysis of the tertiary structures of PA-binding regions will be required to reveal what a general PA- binding pocket looks like and how the specificity of the PA-binding is achieved. Nonetheless, there are a few well characterized lipid binding domains (including p47phox PX [51], Sos PH [17] and some C2 domains [52, 53]) that do have affinity for PA. Many regulatory proteins of small GTPases, such as GEFs and GAPs, contain these lipid binding domains and sequences rich in polybasic amino acids. It would not be surprising to identify more small GTPases regulated by PA or PA-derived lipids in the future.
Another longstanding question is whether PAs generated from PLD and DGK have the same signaling functions. It was proposed long time ago that PAs derived from the PLD and DGK reactions are structurally different because the initial substrates have different fatty acid components. PA produced by PLD is largely composed of saturated and mono-unsaturated fatty acids, while DGK produced PA is enriched in poly-unsaturated fatty acids. Different molecular species can have different effects on downstream targets and can be acted upon differently by enzymes [reviewed in 1]. However, no study has been done to compare the functional differences between PLD and DGK isoforms when each of these enzymes is inhibited using specific small molecule inhibitors and RNAi. It is possible that the localization of the PA-generating enzymes on particular membrane compartments is a more critical determinant than the structure of the fatty acid chains on PA. It would be interesting to examine the relative contribution of PLD and DGK isoforms to the activity of a particular small GTPase in the same experimental system.
In the past few years, the role of PA in cellular signaling and trafficking processes has been emerging [1, 2] - among these is one of the key pathways, growth receptor-induced Ras activation [17]. However, we still lack information about the precise spatial and temporal regulation of PA signaling, especially at the single cell level. Imaging of phosphatidylinositides turnover has been greatly advanced by the use of fluorescent protein-targeted protein domains that specially recognize distinct phosphatidylinositide species. To detect PA level change in living cells, the use of intracellular PA reporters or sensors is being explored. Two PA sensors have been developed, one from the PA-binding region of Raf-1 [54] and another from the PA-binding motif of Spo20p, a yeast homolog of SNAP25 [9, 55]. Both reporters are sufficient to target green fluorescent protein to membranes upon stimulations [9, 54, 55]. To understand the detailed mechanism of PA regulation of small GTPases and other signaling events, it would be critical to use the current or newer PA reporters to simultaneously detect the dynamics of PA and PA-metabolizing enzymes, as well as PA and the activity of small GTPases.
Aberrant functions of PLD, DGK and Ras superfamily of small GTPases have been implicated in a number of human diseases [14, 20, 56]. Given the emerging roles of PA in regulating the activity of small GTPases summarized in this review, an obvious question would be whether PLD- and DGK-generated PA contributes to the onset of these pathologies through regulation of small GTPases. The answer to this question will certainly help to identify drug targets and design new therapeutic strategies.
Acknowledgments
The authors thank Dr. Aimalohi Esechie for her comments on the manuscript. This work was supported by a research grant from National Institutes of Health to GD (GM071475).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang X, Devaiah SP, Zhang W, Welti R. Signaling functions of phosphatidic acid. Prog Lipid Res. 2006;45:250–278. doi: 10.1016/j.plipres.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 2.Stace CL, Ktistakis NT. Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim Biophys Acta. 2006;1761:913–926. doi: 10.1016/j.bbalip.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Frohman MA, Sung TC, Morris AJ. Phospholipase D Structure and Regulation. Biochem Biophys Acta. 1999;1439:175–186. doi: 10.1016/s1388-1981(99)00093-1. [DOI] [PubMed] [Google Scholar]
- 4.Cockcroft S. Signalling roles of mammalian phospholipase D1 and D2. Cell Mol Life Sci. 2001;58:1674–1687. doi: 10.1007/PL00000805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McDermott M, Wakelam MJ, Morris AJ. Phospholipase D. Biochem Cell Biol. 2004;82:225–253. doi: 10.1139/o03-079. [DOI] [PubMed] [Google Scholar]
- 6.Jenkins GM, Frohman MA. Phospholipase D: a lipid centric review. Cell Mol Life Sci. 2005;62:2305–2316. doi: 10.1007/s00018-005-5195-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hammond SM, Jenco JM, Nakashima S, Cadwallader K, Gu Q, Cook S, Nozawa Y, Prestwich GD, Frohman MA, Morris AJ. Characterization of two alternately spliced forms of phospholipase D1. Activation of the purified enzymes by phosphatidylinositol 4,5-bisphosphate, ADP-ribosylation factor, and Rho family monomeric GTP-binding proteins and protein kinase C-alpha. J Biol Chem. 1997;272:3860–3868. doi: 10.1074/jbc.272.6.3860. [DOI] [PubMed] [Google Scholar]
- 8.Colley WC, Sung TC, Roll R, Jenco J, Hammond SM, Altshuller Y, Bar-Sagi D, Morris AJ, Frohman MA. Phospholipase D2, a distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Curr Biol. 1997;7:191–201. doi: 10.1016/s0960-9822(97)70090-3. [DOI] [PubMed] [Google Scholar]
- 9.Du G, Frohman MA. A lipid-signaled myosin phosphatase surge disperse cortical contractile force early in cell spreading. Mol Biol Cell. 2009;20:200–208. doi: 10.1091/mbc.E08-06-0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Du G, Altshuller YM, Kim Y, Han JM, Ryu SH, Morris AJ, Frohman MA. Dual requirement for rho and protein kinase C in direct activation of phospholipase D1 through G protein-coupled receptor signaling. Mol Biol Cell. 2000;11:4359–4368. doi: 10.1091/mbc.11.12.4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hiroyama M, Exton JH. Studies of the roles of ADP-ribosylation factors and phospholipase D in phorbol ester-induced membrane ruffling. J Cell Physiol. 2005;202:608–622. doi: 10.1002/jcp.20156. [DOI] [PubMed] [Google Scholar]
- 12.Imai S, Kai M, Yasuda S, Kanoh H, Sakane F. Identification and characterization of a novel human type II diacylglycerol kinase, DGK kappa. J Biol Chem. 2005;280:39870–39881. doi: 10.1074/jbc.M500669200. [DOI] [PubMed] [Google Scholar]
- 13.Topham MK. Signaling roles of diacylglycerol kinases. J Cell Biochem. 2006;97:474–484. doi: 10.1002/jcb.20704. [DOI] [PubMed] [Google Scholar]
- 14.Merida I, Avila-Flores A, Merino E. Diacylglycerol kinases: at the hub of cell signalling. Biochem J. 2008;409:1–18. doi: 10.1042/BJ20071040. [DOI] [PubMed] [Google Scholar]
- 15.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 16.Mor A, Campi G, Du G, Zheng Y, Foster DA, Dustin ML, Philips MR. The lymphocyte function-associated antigen-1 receptor costimulates plasma membrane Ras via phospholipase D2. Nat Cell Biol. 2007;9:713–719. doi: 10.1038/ncb1592. [DOI] [PubMed] [Google Scholar]
- 17.Zhao C, Du G, Skowronek K, Frohman MA, Bar-Sagi D. Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat Cell Biol. 2007;9:706–712. doi: 10.1038/ncb1594. [DOI] [PubMed] [Google Scholar]
- 18.Mitin N, Rossman KL, Der CJ. Signaling interplay in Ras superfamily function. Curr Biol. 2005;15:R563–574. doi: 10.1016/j.cub.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 19.Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J Cell Sci. 2005;118:843–846. doi: 10.1242/jcs.01660. [DOI] [PubMed] [Google Scholar]
- 20.Buday L, Downward J. Many faces of Ras activation. Biochim Biophys Acta. 2008;1786:178–187. doi: 10.1016/j.bbcan.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 21.Wang W, Fisher EM, Jia Q, Dunn JM, Porfiri E, Downward J, Egan SE. The Grb2 binding domain of mSos1 is not required for downstream signal transduction. Nat Genet. 1995;10:294–300. doi: 10.1038/ng0795-294. [DOI] [PubMed] [Google Scholar]
- 22.McCollam L, Bonfini L, Karlovich CA, Conway BR, Kozma LM, Banerjee U, Czech MP. Functional roles for the pleckstrin and Dbl homology regions in the Ras exchange factor Son-of-sevenless. J Biol Chem. 1995;270:15954–15957. doi: 10.1074/jbc.270.27.15954. [DOI] [PubMed] [Google Scholar]
- 23.Byrne JL, Paterson HF, Marshall CJ. p21Ras activation by the guanine nucleotide exchange factor Sos, requires the Sos/Grb2 interaction and a second ligand-dependent signal involving the Sos N-terminus. Oncogene. 1996;13:2055–2065. [PubMed] [Google Scholar]
- 24.Qian X, Vass WC, Papageorge AG, Anborgh PH, Lowy DR. N terminus of Sos1 Ras exchange factor: critical roles for the Dbl and pleckstrin homology domains. Mol Cell Biol. 1998;18:771–778. doi: 10.1128/mcb.18.2.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Di Fulvio M, Lehman N, Lin X, Lopez I, Gomez-Cambronero J. The elucidation of novel SH2 binding sites on PLD2. Oncogene. 2006;25:3032–3040. doi: 10.1038/sj.onc.1209340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen Y, Xu L, Foster DA. Role for phospholipase D in receptor-mediated endocytosis. Mol Cell Biol. 2001;21:595–602. doi: 10.1128/MCB.21.2.595-602.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liscovitch M, Czarny M, Fiucci G, Tang X. Phospholipase D: molecular and cell biology of a novel gene family. Biochem J. 2000;345(Pt 3):401–415. [PMC free article] [PubMed] [Google Scholar]
- 28.Sanjuan MA, Pradet-Balade B, Jones DR, Martinez AC, Stone JC, Garcia-Sanz JA, Merida I. T cell activation in vivo targets diacylglycerol kinase alpha to the membrane: a novel mechanism for Ras attenuation. J Immunol. 2003;170:2877–2883. doi: 10.4049/jimmunol.170.6.2877. [DOI] [PubMed] [Google Scholar]
- 29.Jones DR, Sanjuan MA, Stone JC, Merida I. Expression of a catalytically inactive form of diacylglycerol kinase alpha induces sustained signaling through RasGRP. FASEB J. 2002;16:595–597. doi: 10.1096/fj.01-0762fje. [DOI] [PubMed] [Google Scholar]
- 30.Topham MK, Prescott SM. Diacylglycerol kinase zeta regulates Ras activation by a novel mechanism. J Cell Biol. 2001;152:1135–1143. doi: 10.1083/jcb.152.6.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo R, Wan CK, Carpenter JH, Mousallem T, Boustany RM, Kuan CT, Burks AW, Zhong XP. Synergistic control of T cell development and tumor suppression by diacylglycerol kinase alpha and zeta. Proc Natl Acad Sci U S A. 2008;105:11909–11914. doi: 10.1073/pnas.0711856105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu CH, Machado FS, Guo R, Nichols KE, Burks AW, Aliberti JC, Zhong XP. Diacylglycerol kinase zeta regulates microbial recognition and host resistance to Toxoplasma gondii. J Exp Med. 2007;204:781–792. doi: 10.1084/jem.20061856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olenchock BA, Guo R, Carpenter JH, Jordan M, Topham MK, Koretzky GA, Zhong XP. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat Immunol. 2006;7:1174–1181. doi: 10.1038/ni1400. [DOI] [PubMed] [Google Scholar]
- 34.Zhong XP, Hainey EA, Olenchock BA, Jordan MS, Maltzman JS, Nichols KE, Shen H, Koretzky GA. Enhanced T cell responses due to diacylglycerol kinase zeta deficiency. Nat Immunol. 2003;4:882–890. doi: 10.1038/ni958. [DOI] [PubMed] [Google Scholar]
- 35.Zha Y, Marks R, Ho AW, Peterson AC, Janardhan S, Brown I, Praveen K, Stang S, Stone JC, Gajewski TF. T cell anergy is reversed by active Ras and is regulated by diacylglycerol kinase-alpha. Nat Immunol. 2006;7:1166–1173. doi: 10.1038/ni1394. [DOI] [PubMed] [Google Scholar]
- 36.Regier DS, Higbee J, Lund KM, Sakane F, Prescott SM, Topham MK. Diacylglycerol kinase iota regulates Ras guanyl-releasing protein 3 and inhibits Rap1 signaling. Proc Natl Acad Sci U S A. 2005;102:7595–7600. doi: 10.1073/pnas.0500663102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakane F, Imai S, Kai M, Yasuda S, Kanoh H. Diacylglycerol kinases: why so many of them? Biochim Biophys Acta. 2007;1771:793–806. doi: 10.1016/j.bbalip.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 38.Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 39.Wennerberg K, Der CJ. Rho-family GTPases: it’s not only Rac and Rho (and I like it) J Cell Sci. 2004;117:1301–1312. doi: 10.1242/jcs.01118. [DOI] [PubMed] [Google Scholar]
- 40.Chae YC, Kim JH, Kim KL, Kim HW, Lee HY, Heo WD, Meyer T, Suh PG, Ryu SH. Phospholipase D activity regulates integrin-mediated cell spreading and migration by inducing GTP-Rac translocation to the plasma membrane. Mol Biol Cell. 2008;19:3111–3123. doi: 10.1091/mbc.E07-04-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chianale F, Cutrupi S, Rainero E, Baldanzi G, Porporato PE, Traini S, Filigheddu N, Gnocchi VF, Santoro MM, Parolini O, van Blitterswijk WJ, Sinigaglia F, Graziani A. Diacylglycerol kinase-alpha mediates hepatocyte growth factor-induced epithelial cell scatter by regulating Rac activation and membrane ruffling. Mol Biol Cell. 2007;18:4859–4871. doi: 10.1091/mbc.E07-02-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsushima S, Kai M, Yamada K, Imai S, Houkin K, Kanoh H, Sakane F. Diacylglycerol kinase gamma serves as an upstream suppressor of Rac1 and lamellipodium formation. J Biol Chem. 2004;279:28603–28613. doi: 10.1074/jbc.M314031200. [DOI] [PubMed] [Google Scholar]
- 43.Yasuda S, Kai M, Imai S, Kanoh H, Sakane F. Diacylglycerol kinase gamma interacts with and activates beta2-chimaerin, a Rac-specific GAP, in response to epidermal growth factor. FEBS Lett. 2007;581:551–557. doi: 10.1016/j.febslet.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 44.Caloca MJ, Wang H, Kazanietz MG. Characterization of the Rac-GAP (Rac-GTPase-activating protein) activity of beta2-chimaerin, a ‘non-protein kinase C’ phorbol ester receptor. Biochem J. 2003;375:313–321. doi: 10.1042/BJ20030727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yakubchyk Y, Abramovici H, Maillet JC, Daher E, Obagi C, Parks RJ, Topham MK, Gee SH. Regulation of neurite outgrowth in N1E-115 cells through PDZ-mediated recruitment of diacylglycerol kinase zeta. Mol Cell Biol. 2005;25:7289–7302. doi: 10.1128/MCB.25.16.7289-7302.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown MT, Andrade J, Radhakrishna H, Donaldson JG, Cooper JA, Randazzo PA. ASAP1, a phospholipid-dependent arf GTPase-activating protein that associates with and is phosphorylated by Src. Mol Cell Biol. 1998;18:7038–7051. doi: 10.1128/mcb.18.12.7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kam JL, Miura K, Jackson TR, Gruschus J, Roller P, Stauffer S, Clark J, Aneja R, Randazzo PA. Phosphoinositide-dependent activation of the ADP-ribosylation factor GTPase-activating protein ASAP1. Evidence for the pleckstrin homology domain functioning as an allosteric site. J Biol Chem. 2000;275:9653–9663. doi: 10.1074/jbc.275.13.9653. [DOI] [PubMed] [Google Scholar]
- 48.Nie Z, Stanley KT, Stauffer S, Jacques KM, Hirsch DS, Takei J, Randazzo PA. AGAP1, an endosome-associated, phosphoinositide-dependent ADP-ribosylation factor GTPase-activating protein that affects actin cytoskeleton. J Biol Chem. 2002;277:48965–48975. doi: 10.1074/jbc.M202969200. [DOI] [PubMed] [Google Scholar]
- 49.Jackson TR, Brown FD, Nie Z, Miura K, Foroni L, Sun J, Hsu VW, Donaldson JG, Randazzo PA. ACAPs are arf6 GTPase-activating proteins that function in the cell periphery. J Cell Biol. 2000;151:627–638. doi: 10.1083/jcb.151.3.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Manifava M, Thuring JW, Lim ZY, Packman L, Holmes AB, Ktistakis NT. Differential binding of traffic-related proteins to phosphatidic acid- or phosphatidylinositol (4,5)- bisphosphate-coupled affinity reagents. J Biol Chem. 2001;276:8987–8994. doi: 10.1074/jbc.M010308200. [DOI] [PubMed] [Google Scholar]
- 51.Karathanassis D, Stahelin RV, Bravo J, Perisic O, Pacold CM, Cho W, Williams RL. Binding of the PX domain of p47(phox) to phosphatidylinositol 3,4-bisphosphate and phosphatidic acid is masked by an intramolecular interaction. EMBO J. 2002;21:5057–5068. doi: 10.1093/emboj/cdf519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jose Lopez-Andreo M, Gomez-Fernandez JC, Corbalan-Garcia S. The simultaneous production of phosphatidic acid and diacylglycerol is essential for the translocation of protein kinase Cepsilon to the plasma membrane in RBL-2H3 cells. Mol Biol Cell. 2003;14:4885–4895. doi: 10.1091/mbc.E03-05-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lindsay AJ, McCaffrey MW. The C2 domains of the class I Rab11 family of interacting proteins target recycling vesicles to the plasma membrane. J Cell Sci. 2004;117:4365–4375. doi: 10.1242/jcs.01280. [DOI] [PubMed] [Google Scholar]
- 54.Rizzo MA, Shome K, Watkins SC, Romero G. The recruitment of Raf-1 to membranes is mediated by direct interaction with phosphatidic acid and is independent of association with Ras. J Biol Chem. 2000;275:23911–23918. doi: 10.1074/jbc.M001553200. [DOI] [PubMed] [Google Scholar]
- 55.Zeniou-Meyer M, Zabari N, Ashery U, Chasserot-Golaz S, Haeberle AM, Demais V, Bailly Y, Gottfried I, Nakanishi H, Neiman AM, Du G, Frohman MA, Bader MF, Vitale N. Phospholipase D1 production of phosphatidic acid at the plasma membrane promotes exocytosis of large dense-core granules at a late stage. J Biol Chem. 2007;282:21746–21757. doi: 10.1074/jbc.M702968200. [DOI] [PubMed] [Google Scholar]
- 56.Huang P, Frohman MA. The potential for phospholipase D as a new therapeutic target. Expert Opin Ther Targets. 2007;11:707–716. doi: 10.1517/14728222.11.5.707. [DOI] [PubMed] [Google Scholar]