

Abstract
Purpose
Recent clinical trials of antivascular endothelial growth factor (VEGF) agents for glioblastoma showed promising progression-free and overall survival rates. However, available clinical imaging does not separate antitumor effects from antipermeability effects of these agents. Thus although anti-VEGF agents may decrease tumor contrast-enhancement, vascularity, and edema, the mechanisms leading to improved survival in patients remain incompletely understood. Our goal was to determine whether alleviation of edema by anti-VEGF agents alone could increase survival in mice.
Methods
We treated mice bearing three different orthotopic models of glioblastoma with a VEGF-targeted kinase inhibitor, cediranib. Using intravital microscopy, molecular techniques, and magnetic resonance imaging (MRI), we measured survival, tumor growth, edema, vascular morphology and function, cancer cell apoptosis and proliferation, and circulating angiogenic biomarkers.
Results
We show by intravital microscopy that cediranib significantly decreased tumor vessel permeability and diameter. Moreover, cediranib treatment induced normalization of perivascular cell coverage and thinning of the basement membrane, as mirrored by an increase in plasma collagen IV. These rapid changes in tumor vascular morphology and function led to edema alleviation—as measured by MRI and by dry/wet weight measurement of water content—but did not affect tumor growth. By immunohistochemistry, we found a transient decrease in macrophage infiltration and significant but minor changes in tumor cell proliferation and apoptosis. Systemically, cediranib increased plasma VEGF and placenta growth factor levels, and the number of circulating CXCR4+CD45+ cells. However, by controlling edema, cediranib significantly increased survival of mice in the face of persistent tumor growth.
Conclusion
Anti-VEGF agents may be able to improve survival of patients with glioblastoma, even without inhibiting tumor growth.
INTRODUCTION
In a recent phase II study, we showed that cediranib (AZD2171, Recentin; AstraZeneca Pharmaceuticals, Wilmington, DE), a potent panvascular endothelial growth factor (VEGF) receptor tyrosine kinase inhibitor, can transiently normalize tumor vessels in recurrent glioblastoma,(1) causing rapid changes in tumor vessel structure and function and alleviating cerebral edema evaluated by magnetic resonance imaging (MRI). In addition, cediranib reduced tumor-associated contrast enhancement to less than one half of the pretreatment value and reduced tumor bulk and mass effect in the majority of patients. Similar antiedema effects and improvement in progression-free and overall survival have also been reported in other VEGF-targeted clinical trials of recurrent glioblastoma.2–4 These encouraging findings have led to randomized phase II and III studies with anti-VEGF agents in combination with chemotherapy and as monotherapy. However, the underlying mechanisms for their success remain poorly understood. Although the antiedema effects of VEGF-targeted therapy are widely accepted, its contribution to the survival benefits remains unknown. Thus these clinical data raise an important question: Is controlling edema by anti-VEGF agents sufficient to increase survival? In patients, this determination is difficult because MRI-based determinations of tumor progression are confounded by changes in vascular permeability after anti-VEGF therapy.5,6 To address this question, we used intravital microscopy, histology, molecular and cellular marker analyses, and functional MRI to investigate the effects of cediranib in three orthotopic models of glioblastoma in mice (two human gliomas—U87 and U118—and a highly invasive rat glioma, CNS1).
METHODS
Animal Models and Cell Lines
We implanted cranial windows into nude mice as previously described.7 After 1 week, we implanted small fragments (0.2 to 0.3 mm diameter) of U87 or U118 or CNS1 tumors superficially into the left cerebral cortex under the cranial window at a depth of 0.4 to 1 mm. To gain the ability to measure the tumor size in real-time, green fluorescence protein (GFP) was stably transfected into U87, U118, and CNS1 cells using a retroviral construct. All cell lines were maintained in DMEM medium with 10% fetal bovine serum. All experiments were approved by the Massachusetts General Hospital Subcommittee on Research Animal Care.
Tumor Size Monitoring and Treatment Protocols
U118-GFP, U87-GFP, and CNS1-GFP tumors were measured daily by intravital microscopy. Tumor size was measured by fitting an ellipse to the GFP signal (Appendix Fig A1, online only). Tumor volume was calculated based on the equation:
After reaching a diameter of 2.5 mm (or to a volume 6 to 8 μl) for U87 and U118 and 2 mm for CNS1—defined as day 0—treatment was started with either cediranib, dexamethasone (Baxter Healthcare Corp, Deerfield, IL), or saline. Cediranib dissolved in 1% Tween was administered via oral gavage at the dose of 6 mg/kg each day.8 Dexamethasone was administered intraperitoneally at a dose of 10 mg/kg each day. For all cediranib studies, mice bearing gliomas were treated until reaching the end point with cediranib or Tween. For all dexamethasone studies, mice bearing gliomas were treated every day until reaching the end point with dexamethasone or saline. For survival studies, lethargic mice or mice with severe neurologic symptoms were humanely euthanized.
Histology and Immunostaining
Tumor-bearing mice were perfusion-fixed by infusion of 4% paraformaldehyde through the left ventricle. For immunofluorescence analysis, mouse brains were postfixed for 1 hour in 4% formaldehyde in phosphate-buffered saline (PBS) followed by incubation in 30% sucrose in PBS overnight at 4°C and subsequent mounting in freezing media (OCT, Tissue-Tek, Torrance, CA). Brains were sectioned every 20 μm and incubated for 4 hours at room temperature in a mixture of anti-CD31 antibody (2.5 μg/mL; clone 2H8, Millipore Chemicon International, Temecula, CA) and either anti-NG2 antibody (2.5 μg/mL; Millipore), anti-CD13 antibody (1.7 μg/mL, clone R3-63, Serotec R3-63; AbD Serotec, Morphosys UK Ltd, Oxford, UK), antilaminin antibody (0.95 μg/mL; Dako, Carpinteria, CA), or anti-collagen IV antibody (0.5 μg/mL; Millipore) in 0.2% Triton-X100% and 5% normal horse serum (NHS) in PBS. After several washes in PBS, tissue sections were incubated for 1 hour at room temperature with 1:400 dilutions of Cy5-conjugated antiarmenian hamster antibody and Cy3 conjugated antirat or antirabbit antibody in 0.2% Triton-X100% and 5% NHS in PBS. After several washes in PBS, tissues were postfixed in formaldehyde and mounted with 4'-6-diamidino-2-phenylindole–containing mounting media (Vectashield, VectorLabs, Burlingame, CA) for confocal microscopy. Brain sections were stained also for CD11b and F4/80 incubating allophycocyanin (CD11b, clone M1/70) or phycoerythrin (F4/80, clone BM8) –conjugated primary antibodies (BD Biosciences Pharmingen, Franklin Lakes, NJ) for 1 hour at room temperature in 5% NHS in PBS. Apoptotic cells were detected using ApopTag Red In Situ Apoptosis Detection Kit following manufacturer's (Millipore) protocol. For the detection of cell proliferation, mice were injected intraperitoneally with 1 mg of 5-bromo-2-deoxyuridine (BrdU; Sigma-Aldrich, St Louis, MO) 24 hours before sacrifice. For the detection of the incorporated BrdU from the U87-bearing brain sections, antigen retrieval was performed by incubating tissue sections in pH6 citrate antigen retrieval solution (Dako) at 95°C for 10 minutes before staining sections with Alexa Fluor 546 conjugated anti-BrdU antibody (2 μg/mL; clone PRB-1, Invitrogen Molecular Probes, Carlsbad, CA) for 1 hour at room temperature in 0.2% Triton-X100% and 5% NHS in PBS.
Quantification of the stained area was performed using an in-house segmentation algorithm (MATLAB, The Mathworks, Natick, MA). Analysis of vascular proximity was performed by fitting the intensity profile around the vessels (determined by CD31 staining) to an exponential function (I = Ae-x/L + C), where I = pixel intensity, x = distance from vessels (1 to 10 μm), and L = characteristic length. This method was used for assessment of the perivascular cell proximity to the vessel wall, as well as basement membrane thickness.
MRI-Based Measurement of Permeability, Edema, and Tumor Growth
All magnetic resonance images were acquired on a 9.4 Tesla MRI scanner (Bruker Biospin, Billerica, MA). Animals were anesthetized with a 50:50 mixture of O2 and medical air plus 1.5% isoflurane and placed prone in a cradle. Either a custom-built 1-cm transmit/receive surface coil, positioned on the head of the animals, or a transmit/receive birdcage mouse-head coil were used to acquire the images.
T2-weighted rapid acquisition with relaxation enhancement (RARE) images were acquired to assess the tumor volume. The acquisition parameters were as follows: TE = 10, RARE factor = 16, TR = 3,000 msec, NA = 4, 11 image slices, 0.5-mm slice thickness, 150-μm in-plane resolution. Tumor volume was determined from the T2 hyperintense regions of the brain, measured using an in-house segmentation algorithm (MATLAB).
T2 relaxation maps were generated from multiecho spin-echo images and used to assess tumor edema. Acquisition parameters were as follows: TE = 10 msec, 10 echoes, TR = 2,500 msec, 11 image slices, 0.5 mm slice thickness, 150 mm in-plane resolution, NA = 2. Voxelwise exponential fitting of the image signal intensity as a function of echo-time was performed (MATLAB) to determine T2 relaxation time maps.
Tumor blood vessel permeability (Ktrans) was assessed from Dynamic Contrast Enhanced (DCE) magnetic resonance images. The DCE sequence consisted of a T1-weighted gradient-echo sequence with TE = 2.5 msec, TR = 50 msec, flip angle = 35°, FOV = 1.92 cm, matrix = 96 × 96 (in-plane resolution = 200 μm), 0.5-mm slice thickness, one image slice, 70 to 100 repetitions, temporal resolution = 4.8 seconds. A total of 50 to 100 μL of 100 mmol/L Gd-DTPA (0.2 to 0.4 mmoles/kg) was injected approximately 30 seconds after commencement of the DCE imaging sequence. The signal intensity in the tumor region of interest (ROI) was analyzed using an in-house written MATLAB program, which models the tumor signal enhancement using the two-compartment model of Tofts et al,9 to extract the volume transfer constant (Ktrans), the volume of the extravascular extracellular space (ne), and the flux rate constant between the extravascular extracellular space and the blood plasma (kep). Briefly, the time dependence of tumor ROI signal intensity was fit to the equation:
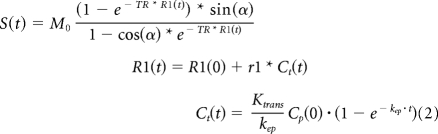 |
where R1 is the longitudinal relaxation rate, α is the flip angle, TR is the repetition time, and Ct is the tracer (contrast agent) concentration.9
Intravital Multiphoton Laser Scanning Microscopic Analysis of Vessel Diameter, Density, and Relative Hematocrit Analysis
In vivo multiphoton laser scanning microscopic analysis of glioblastoma vessels was performed as described previously.10 The tumor area was identified by analysis of GFP constitutively expressed by U87, U118, and CNS1. Vessel angiography was performed after intravenous injection of 0.1 mL of 10 mg/mL fluorescein isothiocyanate-dextran (500,000 MW; Sigma). Hematocrit analysis was performed after injection of 150 μL of 50% hematocrit fluorescent RBCs (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate, DiD; Invitrogen). Two adjacent areas were imaged by acquiring three-dimensional stacks (resolution, 2.4 × 2.4 × 2.5 μm/pixel). Tumor volume was segmented using an in-house algorithm (MATLAB). Vessels were traced as described.11 Hematocrit analysis was performed by scanning through a line perpendicular to the vessel direction and extracting RBC velocity and flux. Hematocrit was calculated based on the equation:
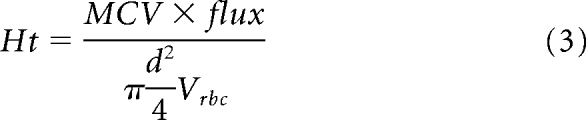 |
For vessels with velocity lower then 10 μm/s, relative hematocrit was measured by counting the number of RBCs in the vessel in a snapshot.
Water Content Analysis by Dry/Wet Weight Measurements
Anesthetized mice were euthanized by cervical dislocation and the brains were collected. Brains were dissected into several compartments: tumor, ipsilateral hemisphere, and contralateral hemisphere. Tissues were weighed immediately and dried in a vacuum for up to 2 weeks. Weights were collected throughout the drying period until the final dry weight was established. Water content was calculated as follows:
Statistical Analysis
Data are expressed as mean ± standard error of the mean. The principal statistical test was the t test (two-tailed with unequal variance). We analyzed the experiments involving multiple comparisons using repeated measures multivariate analysis of variance followed by posthoc within and between groups hypothesis testing (SYSTAT 12; SYSTAT Soft Inc, Chicago, IL). We used the nonparametric log-rank test for survival studies. We considered a P value of less than .05 to be statistically significant.
RESULTS
Monotherapy With Cediranib Significantly Increases Survival of Mice With Glioma
GFP-expressing U87, U118, and CNS1 tumors were orthotopically grown into the cerebral cortex of nude mice bearing cranial windows. We started treatment when gliomas reached a diameter of 2 to 2.5 mm, measured by intravital microscopy. Cediranib increased survival in all the tumor models (Figs 1A through 1C), even though tumor growth was not affected at the dose used (Figs 1D through 1I). Because cediranib-treated mice had longer survival without a delay in tumor growth, their tumors were twice as large as those in the control group at the experiment end point (Fig 1E). Untreated mice tended to die suddenly, despite their healthy appearance and relatively small tumor size. In contrast, mice treated with cediranib survived longer and slowly became cachectic and had to be euthanized.
Fig 1.

Cediranib treatment leads to increased survival without effects on tumor growth. Cediranib (6 mg/kg of body weight per day) treatment leads to statistically significant survival benefit in U87 (A; **P < .001, n = 6), U118 (B; **P < .01, n = 4), and CNS1 (C; **P < .01, n = 8). (D) Single animal tumor growth curves acquired by fluorescence microscopic imaging show the lack of growth delay after cediranib treatment. (E) Maximal tumor volume (measured at end point). Cediranib-treated animals survived with statistically significantly larger U87 (**P < .001, n = 6), U118 (**P < .001, n = 4), and CNS1 (**P < .001, n = 8) tumors. (F) Representative intravital fluorescence microscopic image of U87, U118, and CNS1 at first and last time points. (G) U87 tumor volume measured ex vivo at day 2 and day 8 to 11 in control and cediranib-treated animals shows the lack of tumor growth delay with cediranib treatment at day 2 (P > .05, n = 5) (H) Cediranib treatment does not affect U87 tumor growth assessed by magnetic resonance imaging (volumes measured on T2 weighted stacks 0.15 mm XY resolution × 0.5 mm Z resolution; n = 3) (I) Representative T2-weighted single brain slices of cediranib- and control-treated animals bearing U87 tumors.
Mechanism of Action of Cediranib Monotherapy Is Alleviation of Edema
Although multifactorial, one of the major causes of death of patients with glioblastoma is cerebral herniation (seen in more than 60% of patients), which is primarily caused by cerebral edema and intracranial hypertension.12 The clinical effects of cerebral edema have been extensively studied in preclinical models of vasogenic and cytotoxic edemas. Cerebral physiology was found to be hypersensitive to increase in water content, which leads to intracranial hypertension, clinical deterioration, and morbidity.13–15 A major contributor to this edema is increased vascular permeability and accumulation of inflammatory cells (macrophages). Cediranib inhibits both VEGF receptor 2 (VEGFR2) signaling in endothelial cells (a major determinant of vascular permeability16) and VEGF receptor 1 (VEGFR1) signaling (a key pathway in monocyte/macrophage recruitment to tumors17). Hence we hypothesized that the survival benefits seen in our models are primarily due to the alleviation of cerebral edema by cediranib. To investigate the mechanisms of cediranib survival benefit, we focused on the U87 model, in which the cancer cells do not express functional VEGF receptors (data not shown). We assessed edema directly by the dry/wet weight ratio of the brain ex vivo (Fig 2A) and indirectly using MRI-T2 maps in vivo (Fig 2B). Cediranib significantly decreased tumor water content at day 2, but this effect was transient, reverting to control levels at day 8 through 11 (Fig 2A). This suggests that cediranib could no longer control cerebral edema caused by the tumor enlargement at later time points.
Fig 2.

Cediranib decreases glioblastoma-induced edema, and corticosteroid-mediated edema control increases survival without affecting tumor growth. (A) Water content measured by dry/wet weight ratio in the tumor and ipsilateral and contralateral cerebrum. Cediranib significantly decreased tumor and ipsilateral water content (*P < .05, n = 5). (B) Representative T2 maps of single brain slices used to evaluate edema (T2 correlates with water content). (C) Quantification of T2 in the tumor and the cerebral cortex. Cediranib significantly decreased T2 signal in the tumor (***P < .001) and the cerebral cortex (**P < .01). (D) Dexamethasone (Dex) (10 mg/kg each day) treatment leads to a statistically significant survival benefit (*P < .05, n = 14). (E) Individual tumor growth curves acquired by fluorescence microscopic imaging showing the lack of growth delay after Dex treatment. (F) Maximal tumor volume (measured at end point). Dex-treated animals survived with significantly larger tumors (*P < .05, n = 14).
Brain Edema Alleviation Can Increase Survival Despite Tumor Enlargement
To determine whether a transient decrease in edema alone is sufficient to increase survival, we treated a separate cohort of tumor-bearing mice with dexamethasone, a corticosteroid commonly used to alleviate cerebral edema in patients with glioma. Dexamethasone treatment induced a modest enhancement of survival but also did not inhibit tumor growth (Figs 2D and 2F). Interestingly, the survival benefit provided by dexamethasone was not as pronounced as that of cediranib: the dexamethasone group had smaller tumors at end point than those in the cediranib-treated group. This is similar to an earlier report in which vascular normalization in an animal glioma model after dexamethasone treatment was also noted.18 Next, we investigated the mechanisms by which cediranib treatment led to a more pronounced survival benefit.
Edema Alleviation Is Caused by Decreased Glioblastoma Vascular Permeability Associated With Vascular Normalization
To determine whether cediranib normalizes tumor vasculature in our models, we measured changes in the tumor vascular morphology and function after cediranib treatment. Similar to patients with glioblastoma, postcontrast MRI showed that cediranib significantly decreased Ktrans—a parameter dependent on vascular permeability—in the glioblastoma xenografts (Fig 3A). Furthermore, intravital microscopy measurements demonstrated that cediranib significantly decreased tumor vessel permeability and diameter, as well as vascular hemoconcentration (elevated hematocrit, Figs 3B through 3E and 4), all hallmarks of tumor vascular normalization.19 Given the decrease in vessel diameter seen with intravital microscopic measurements after cediranib, we sought further evidence of normalization of the structure of glioblastoma vessels. Fluorescence immunohistochemistry showed that cediranib increased the proximity of perivascular to endothelial cells without significant changes in the extent of perivascular cell coverage (Figs 5A and 6). In addition, cediranib treatment led to thinning of the vascular basement membrane (Fig 5B), similar to that seen after anti-VEGFR2 antibody therapy,20 and an increase in plasma collagen IV at day 2 (Appendix Fig A3, online only). Additionally, cediranib-mediated vascular normalization was limited to a time window after which most of the functional and morphologic vascular parameters reverted to the abnormal phenotype. Specifically, at later time points, all normalization parameters reversed—including vascular permeability, vessel diameter, basement membrane thickness, and hemoconcentration—except for pericyte proximity.
Fig 3.

Cediranib treatment normalizes glioma vessel morphology and function. (A) Cediranib treatment (6 mg/kg each day) leads to a statistically significant decrease in Ktrans (arbitrary units, **P < .05, n = 4). (B) Cediranib significantly decreases vascular permeability measured by intravital microscopy (*P < .05, n = 12). (C) Representative multiphoton laser scanning microscopy (MPLSM) three-dimensional reconstruction of the tumor (red) and peritumor vessels (gray) superimposed with RBC positions acquired through analysis of injected fluorescently labeled RBC. Control animals have predominantly hemoconcentrated vessels (closed arrows). Cediranib-treated animals have predominantly low hematocrit vessels (open arrows). (D) Mean and SE of tumor vessel relative hematocrit measured by MPLSM. Cediranib significantly decreases relative hematocrit (*P < .05, n = 4). (E) Cediranib transiently but significantly decreases vessel diameter measured by MPLSM (*P < .05 n = 4).
Fig 4.

Cediranib decreases vessel diameter at early time points and decreases vessel density at later time points. (A, B) Representative multiphoton laser scanning microscopic (MPLSM) three-dimensional reconstruction of the tumor (white) and peritumor vessels (gray) showing the effects of (A) control versus (B) cediranib treatment on vessel diameter. (C) Cediranib transiently but significantly decreases vessel diameter measured by MPLSM (*P < .05, n = 4). (D) Cediranib significantly decreases vessel density (length) measured by MPLSM (*P < .05, n = 4). (E) Cediranib decreases vessel diameter at the tumor margin at day 2 (measured by immunostaining for CD31 endothelial staining; *P < .05, n = 9). (F) Cediranib decreases microvascular density at the tumor center at day 8 (measured by immunostaining for CD31; *P < .05, n = 9).
Fig 5.

Cediranib treatment decreases macrophage infiltration and normal vessel wall structure; at day 8, microvascular density is decreased, tumor cell proliferation is decreased, and apoptosis is increased. (A) Cediranib decreases the characteristic length describing the distance between perivascular cells and the vessel wall (measured by immunostaining for CD13, *P < .05, n = 5; and NG2, *P < .05, n = 9). (B) Cediranib transiently but significantly decreases basement membrane thickness at day 2 (measured by immunostaining for laminin and collagen IV; *P < .05, n = 5). (C) Cediranib decreases microvascular density at the tumor center at day 8 (measured by immunostaining for CD31 endothelial staining; *P < .05, n = 9). (D) A total of 1 mg/mouse of 5-bromo-2-deoxyuridine (BrdU) was injected intraperitoneally 24 hours before the end point. The fraction of nuclei with incorporated BrdU is quantified and plotted for each animal. Cediranib significantly decreased proliferation only at day 8 (*P < .05, n = 4). (E) Terminal deoxynucleotidyl transferase–mediated (TUNEL) staining was used to assess apoptosis of tumor cells. The fraction of TUNEL-positive nuclei was plotted for each animal. Cediranib significantly increased apoptosis at day 8 (*P < .05, n = 9). (F) Cendiranib transiently but significantly decreases macrophages infiltration at day 2 (measured by immunostaining for F4/80; *P < .05, n = 5). Myeloid cells infiltration is significantly increased at day 8 (measured by immunostaining for CD11b; **P < .0001, n = 5).
Fig 6.

Quantification of pericyte coverage and proximity. (A) Representative immunohistochemistry fields of CD31- (endothelial marker) and NG2- (pericyte marker) stained tumor vessels. The fraction of NG2-positive pixels was analyzed at various distances from the vessel wall (1 μm to 10 μm), and the data were fit to an exponential function, yielding the quantity L, the characteristic extension of stain from the wall. (B) The fraction of NG2- or CD13- (pericyte marker) positive pixels was analyzed at various distances from the vessel wall. The average signal for each animal (based on five areas) is plotted, showing a difference between cediranib-treated and control animals. (C) Representative field of CD31- (endothelial marker) and NG2- (top panels) or CD13- (bottom panels) stained tumor vessels with control or cediranib treatment at day 2. (D) Cediranib significantly decreased pericyte distance from endothelial cells without changing the extent of perivascular cell coverage (NG2, P < .05, n = 9; CD13, P < .05, n = 5).
Transient Nature of the Vascular Normalization and Edema Control May Be Caused by Activation of Inflammatory Mechanisms Independent of VEGF and Angiogenesis
In contrast to early time points, when cediranib normalized vessel function without detectable pruning of vessels, extended cediranib treatment decreased microvascular density in the center of the tumors (Fig 5C). This was associated with a modest but significant increase in tumor cell apoptosis and decrease in tumor cell proliferation (Figs 5D and 5E). However, the ratio of proliferating to apoptotic tumor cells remained high (more than 20-fold), explaining the sustained tumor growth rate. This result is consistent with other reports, in which a reduction of microvascular density was achieved without significant reduction of tumor growth.21
In addition, immunohistochemical analyses showed that cediranib significantly reduced macrophage infiltration (Fig 5F). This effect might be due to direct VEGFR1 blockade by cediranib and/or could be secondary to normalization of the tumor vessels and environment. However, infiltration of macrophages as well as other myeloid cells significantly increased at later time points during cediranib treatment (Fig 5F). The accumulation of these inflammatory cells may be involved in the escape from vascular normalization and edema control at the later time points.22
Finally, preclinical studies have also suggested that antiangiogenic therapy (eg, sunitinib) exerts systemic effects, causing elevation in multiple circulating angiogenic markers, which can promote tumor growth.17,23,24 In clinical studies, we found that cediranib increased circulating angiogenic markers in patients with recurrent glioblastoma.(1) In mice with U87 gliomas, we found that cediranib significantly increased the plasma levels of both mouse and human (tumor-derived) placenta growth factor and human VEGF but not mouse VEGF. Cediranib also rapidly increased the number of circulating CXCR4+CD45+ cells (Appendix Fig A2, online only). Future studies should establish whether activation of systemic inflammatory and/or angiogenic markers are causally related to the persistent growth of gliomas through antiangiogenic treatment.
DISCUSSION
In summary, our results show that cediranib decreases edema by normalizing tumor vasculature, increasing survival in brain tumor models—even in the face of persistent glioblastoma growth. We hypothesize that this may be a “class effect” of agents that target theVEGF signaling pathway, including tyrosine kinase inhibitors and antibodies. Moreover, our preclinical observations regarding reduction in vascular permeability, vessel diameter, and vasogenic cerebral edema mirror the clinical findings with cediranib monotherapy inpatients with recurrent glioblastoma obtained by vascular MRI. In the latter study, we also observed extensions of progression-free and overall survival, relative to historical controls, in patients with rapid decreases in vascular permeability after cediranib treatment.25 However, our observations in preclinical model systems do not exclude thepossibility that anti-VEGF therapy in human tumors may improve survival by mechanisms beyond alleviation of edema. In fact, thevascular normalization window induced by anti-VEGF therapeutics may enhance the sensitivity of tumors to the cytotoxic effects of ionizing radiation and chemotherapy by a number of potential mechanisms.8,19,20,26 Collectively, these results highlight the need for novel antiangiogenic and antitumor strategies to combat tumor resistance to anti-VEGF therapies and the need for methods to distinguish antitumor, antiedema, and antivascular effects in patients.
Acknowledgment
We thank G. Gorospe, C. Kopel, and S. Roberge for their outstanding technical assistance. We also thank J. Jurgensmeier and A. Ryan from AstraZeneca for their helpful input.
Appendix
Supplemental Materials and Methods
Measurement of plasma soluble molecules.
Peripheral blood was obtained from mice bearing orthotopic U87 tumors at baseline (pretreatment) and 2 and 8 days after cediranib treatment. Blood was collected in an EDTA-containing Vacutainer and spun down, and plasma was aliquoted and frozen immediately. Plasma analysis was carried out for circulating human vascular endothelial growth factor (VEGF) and PlGF using multiplex array plates from Meso-Scale Discovery (Gaithersburg, MD) and for mouse collagen IV, VEGF, and PlGF using enzyme-linked immunosorbent assay kits from Exocell Inc (Philadelphia, PA) and R&D System (Minneapolis, MN). Every sample was run in duplicate.
CD45+CXCR4+ hematopoietic precursor enumeration by flow cytometry.
Flow cytometric analysis was performed after immunostaining of whole blood samples with rat anti-mouse CD45-peridinin-chlorophyll-protein complex and CXCR4-phycoerythrin antibodies (BD Biosciences Pharmingen, Franklin Lakes, NJ). CXCR4+CD45+ cells were enumerated as a fraction of blood circulating CD45+ cells using an LSR-II flow cytometer (BD Biosciences).
Fig A1.

Validation of intravital measurements (IVM) of tumor volume. To validate the accuracy of the tumor volume estimation, we compared tumor volume measurements based on fluorescence microscopy imaging to actual tumor weights measured ex vivo. This comparison was performed for U87 tumors and showed that the green fluorescence protein–based estimations are highly accurate for small tumor volumes (approximately 20 μL reached at day 2, yellow circles, ie, when the comparisons with untreated tumors were performed), but showed a tendency to underestimate the tumor volume for larger tumor sizes (reached at day 8 after cediranib treatment, blue circles).
Fig A2.

Systemic effects of cediranib treatment. Levels of human- and mouse-derived vascular endothelial growth factor (VEGF) and placenta growth factor (PlGF) in plasma of mice with orthotopic U87 gliomas, plotted for control treatment (day 2) and cediranib treatment (at days 2 and 8). Cediranib significantly increased the plasma levels of both mouse and human (tumor-derived) PlGF (*P < .05) and human VEGF (*P < .05), as well as the number of circulating CXCR4+CD45+ cells (**P < .01).
Fig A3.

Changes in plasma collagen IV levels mirror thinning of the basement membrane. We hypothesized that the thinning of the vascular basement membrane owing to vascular normalization would increase the level of circulating collagen IV. Indeed, cediranib significantly increased the plasma levels of collagen IV at day 2 (***P < .001 cediranib day 2 v control). Collagen IV levels were decreased at day 8 (**P < .01 cediranib day 2 v cediranib day 8).
Footnotes
Supported in part by National Institutes of Health Grants No. P01-CA80124 and R01-CA115767 (to R.K.J.) and K24-CA125440 and R01-CA129371 (to T.T.B.), by postdoctoral fellowships from the Susan G. Komen Foundation (to W.S.K. and D.L.) and Damon Runyon Foundation (to J.L.), by a predoctoral fellowship from the United States Department of Defense (A.M.D.), and by gifts from AstraZeneca Pharmaceuticals and the Montesi Family Research Fund.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: Tracy T. Batchelor, AstraZeneca (C), Genentech (C), Millennium (C); Dan G. Duda, Takeda Pharmaceutical (C); A. Gregory Sorensen, Bayer-Schering (C), Breakaway Imaging (C), AstraZeneca (C), Eli Lilly (C), EPIX Pharmaceuticals (C), Genentech (C), General Electric Healthcare (C), Mitsubishi Pharma (C), Novartis (C), Takeda-Millennium (C), Thermal Technologies (C); Rakesh K. Jain, SynDevRx (U), AstraZeneca (C), Dyax (C), Millenium (C) Stock Ownership: None Honoraria: Tracy T. Batchelor, Vertex, Schering-Plough, Enzon; A. Gregory Sorensen, Siemens Medical Solutions, Millenium Pharmaceuticals Research Funding: A. Gregory Sorensen, Siemens Medical Solutions, General Electric Healthcare, GlaxoSmithKline, Novartis Pharmaceuticals, Merck (USA), Stem Cells Inc., Ono Pharmaceuticals, Amgen, AstraZeneca; Rakesh K. Jain, AstraZeneca Expert Testimony: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: Walid S. Kamoun, Carsten D. Ley, Christian T. Farrar, Tracy T. Batchelor, Emmanuelle di Tomaso, Dan G. Duda, Lance L. Munn, Dai Fukumura, A. Gregory Sorensen, Rakesh K. Jain
Financial support: Tracy T. Batchelor, A. Gregory Sorensen, Rakesh K. Jain
Administrative support: Tracy T. Batchelor, A. Gregory Sorensen, Rakesh K. Jain
Provision of study materials or patients: A. Gregory Sorensen, Rakesh K. Jain
Collection and assembly of data: Walid S. Kamoun, Carsten D. Ley, Christian T. Farrar, Annique M. Duyverman, Johanna Lahdenranta, Delphine A. Lacorre, Emmanuelle di Tomaso, Dan G. Duda
Data analysis and interpretation: Walid S. Kamoun, Carsten D. Ley, Christian T. Farrar, Annique M. Duyverman, Johanna Lahdenranta, Delphine A. Lacorre, Tracy T. Batchelor, Emmanuelle di Tomaso, Dan G. Duda, Lance L. Munn, Dai Fukumura, A. Gregory Sorensen, Rakesh K. Jain
Manuscript writing: Walid S. Kamoun, Dan G. Duda, Lance L. Munn, A. Gregory Sorensen, Rakesh K. Jain
Final approval of manuscript: Walid S. Kamoun, Carsten D. Ley, Christian T. Farrar, Annique M. Duyverman, Johanna Lahdenranta, Delphine A. Lacorre, Tracy T. Batchelor, Emmanuelle di Tomaso, Dan G. Duda, Lance L. Munn, Dai Fukumura, A. Gregory Sorensen, Rakesh K. Jain
REFERENCES
- 1.Batchelor TT, Sorensen AG, di Tomaso E, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cloughesy TF. A phase II, randomized, non-comparative clinical trial of the effect of bevacizumab (BV) alone or in combination with irinotecan (CPT) on 6-month progression free survival (PFS6) in recurrent, treatment-refractory glioblastoma (GBM) J Clin Oncol. 2008;26(suppl):91s. abstr 2010b. [Google Scholar]
- 3.Pope WB, Lai A, Nghiemphu P, et al. MRI in patients with high-grade gliomas treated with bevacizumab and chemotherapy. Neurology. 2006;66:1258–1260. doi: 10.1212/01.wnl.0000208958.29600.87. [DOI] [PubMed] [Google Scholar]
- 4.Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13:1253–1259. doi: 10.1158/1078-0432.CCR-06-2309. [DOI] [PubMed] [Google Scholar]
- 5.Norden AD, Young GS, Setayesh K, et al. Bevacizumab for recurrent malignant gliomas: Efficacy, toxicity, and patterns of recurrence. Neurology. 2008;70:779–787. doi: 10.1212/01.wnl.0000304121.57857.38. [DOI] [PubMed] [Google Scholar]
- 6.Sorensen AG, Batchelor TT, Wen PY, et al. Response criteria for glioma. Nat Clin Pract Oncol. 2008;5:634–644. doi: 10.1038/ncponc1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan F, Salehi HA, Boucher Y, et al. Vascular permeability and microcirculation of gliomas and mammary carcinomas transplanted in rat and mouse cranial windows. Cancer Res. 1994;54:4564–4568. [PubMed] [Google Scholar]
- 8.Wedge SR, Kendrew J, Hennequin LF, et al. AZD2171: A highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005;65:4389–4400. doi: 10.1158/0008-5472.CAN-04-4409. [DOI] [PubMed] [Google Scholar]
- 9.Tofts PS, Brix G, Buckley DL, et al. Estimating kinetic parameters from dynamic contrast-enhanced T(1)-weighted MRI of a diffusable tracer: Standardized quantities and symbols. J Magn Reson Imaging. 1999;10:223–232. doi: 10.1002/(sici)1522-2586(199909)10:3<223::aid-jmri2>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 10.Brown EB, Campbell RB, Tsuzuki Y, et al. In vivo measurement of gene expression, angiogenesis and physiological function in tumors using multiphoton laser scanning microscopy. Nat Med. 2001;7:864–868. doi: 10.1038/89997. [DOI] [PubMed] [Google Scholar]
- 11.Tyrrell JA, di Tomaso E, Fuja D, et al. Robust 3-D modeling of vasculature imagery using superellipsoids. IEEE Trans Med Imaging. 2007;26:223–237. doi: 10.1109/TMI.2006.889722. [DOI] [PubMed] [Google Scholar]
- 12.Silbergeld D, Rostomily R, Alvord EJ. The cause of death in patients with glioblastoma is multifactorial: Clinical factors and autopsy findings in 117 cases of supratentorial glioblastoma in adults. J Neurooncol. 1991;10:179–185. doi: 10.1007/BF00146880. [DOI] [PubMed] [Google Scholar]
- 13.Manley GT, Fujimura M, Ma T, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- 14.Thiagarajah JR, Papadopoulos MC, Verkman AS. Noninvasive early detection of brain edema in mice by near-infrared light scattering. J Neurosci Res. 2005;80:293–299. doi: 10.1002/jnr.20439. [DOI] [PubMed] [Google Scholar]
- 15.Papadopoulos MC, Manley GT, Krishna S, et al. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004;18:1291–1293. doi: 10.1096/fj.04-1723fje. [DOI] [PubMed] [Google Scholar]
- 16.Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: A critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002;20:4368–4380. doi: 10.1200/JCO.2002.10.088. [DOI] [PubMed] [Google Scholar]
- 17.Fischer C, Jonckx B, Mazzone M, et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007;131:463–475. doi: 10.1016/j.cell.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 18.Badruddoja MA, Krouwer HG, Rand SD, et al. Antiangiogenic effects of dexamethasone in 9L gliosarcoma assessed by MRI cerebral blood volume maps. Neuro Oncol. 2003;5:235–243. doi: 10.1215/S1152851703000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jain RK. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 20.Winkler F, Kozin SV, Tong RT, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: Role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004;6:553–563. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 21.Svensson A, Backman U, Fuchs D, et al. Angiogenesis can be reduced without significant reduction of tumor growth. Anticancer Res. 2007;27:3883–3889. [PubMed] [Google Scholar]
- 22.Shojaei F, Ferrara N. Refractoriness to antivascular endothelial growth factor treatment: Role of myeloid cells. Cancer Res. 2008;68:5501–5504. doi: 10.1158/0008-5472.CAN-08-0925. [DOI] [PubMed] [Google Scholar]
- 23.Ebos JM, Lee CR, Christensen JG, et al. Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with antitumor efficacy. Proc Natl Acad Sci U S A. 2007;104:17069–17074. doi: 10.1073/pnas.0708148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaked Y, Henke E, Roodhart JM, et al. Rapid chemotherapy-induced acute endothelial progenitor cell mobilization: Implications for antiangiogenic drugs as chemosensitizing agents. Cancer Cell. 2008;14:263–273. doi: 10.1016/j.ccr.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Batchelor TT, di Tomaso E, Duda DG, et al. A multidisciplinary phase II study of AZD2171 (cediranib), an oral pan-VEGF receptor tyrosine kinase inhibitor. patients with recurrent glioblastoma; American Association for Cancer Research Annual Meeting; April 12-16, 2008; San Diego, CA. abstr LB-247. [Google Scholar]
- 26.Padera TP, Kuo AH, Hoshida T, et al. Differential response of primary tumor versus lymphatic metastasis to VEGFR-2 and VEGFR-3 kinase inhibitors cediranib and vandetanib. Mol Cancer Ther. 2008;7:2272–2279. doi: 10.1158/1535-7163.MCT-08-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]