

Abstract
Fibromyalgia syndrome (FMS) is a chronic widespread pain syndrome that is estimated to affect 4 to 8 million U.S. adults. The exact molecular mechanisms underlying this illness remain unclear, rendering most clinical treatment and management techniques relatively ineffective. It is now known that abnormalities in both nociceptive and central pain processing systems are necessary (but perhaps not sufficient) to condition the onset and maintenance of FMS. These same systemic abnormalities are thought to be responsible for the loss of cephalic gray matter density observed in all FMS patients groups studied to date. The current scope of FMS treatment focuses largely on analgesia and does not clearly address potential neuroprotective strategies. This article proposes a combined treatment of pregabalin and memantine to decrease the pain and rate of gray matter atrophy associated with FMS. This dual-drug therapy targets the voltage-gated calcium ion channel (VGCC) and the N-methyl D-aspartate receptor (NMDAR) (respectively), two primary components of the human nociceptive and pain processing systems.
INTRODUCTION
Fibromyalgia syndrome (FMS) is a complex chronic pain syndrome that is defined by widespread pain for more than 3 months and the presence of at least 11 of 18 tender points [1]. It is characterized by central nervous system (CNS) malfunction and often overlaps with other related functional somatic syndromes, inducing additional symptoms such as pronounced fatigue, sleep abnormalities, sensitivity to stress, and cognitive difficulties [2]. FMS is estimated to affect 4 to 8 million adults in the United States, 90% of whom are women [3].
The etiology and pathophysiology of FMS are not well defined, rendering most current treatment methods largely ineffective. It is now known that systemic nociceptive and central pain processing abnormalities are necessary (although perhaps not sufficient) to condition the onset and maintenance of FMS. Recent studies suggest that prolonged exposure to a high stress environment combined with polymorphisms in genes involved in stress, anxiety, and pain response systems [4] may play a significant role in the development of chronic FMS pain through physical and functional modifications of the CNS [5], although much of the underlying biomolecular pathology remains to be elucidated. Overstimulation of the N-methyl D-aspartate receptor (NMDAR) and the voltage-gated calcium ion channel (VGCC) are of particular interest, as these biomolecules play central roles in the development and maintenance of chronic pain [5, 6]. This overstimulation is also capable of inducing excitotoxic neurodegeneration [7], which may be responsible for the gray matter atrophy frequently observed comorbidly with chronic pain [8–13].
Although FMS patients exhibit a chronic pain state and a loss of cephalic gray matter density consistent with NMDAR- and VGCC-overstimulation, current treatment strategies fail to target both of these molecular pathways in unison. This article reviews these molecular mechanisms underlying chronic pain and excitotoxic neurodegeneration, and discusses their relevance to FMS symptomology. A combined treatment of pregabalin (a VGCC blocker) and memantine (an NMDAR antagonist) is then proposed as a novel FMS treatment strategy. This dual-drug therapy promises to safely and simultaneously decrease the activity of VGCCs and NMDARs, likely reducing the pain and rate of gray matter density loss associated with FMS.
DEVELOPMENT OF CHRONIC PAIN
The perception of pain depends on input from the nociceptive system, a physiological component of homeostasis maintenance mediated by a dual afferent sensory network. Nociceptive stimulation activates two primary types of receptors – low-threshold nociceptors connected to fast conducting A-delta (first pain) afferent nerve fibers, and high-threshold nociceptors that conduct impulses in slow, unmyelinated, C-fiber (second pain) afferent nerve fibers. Synaptic transmission synapses these A-delta and C fibers within the dorsal horn of the spinal cord, and neurotransmitters and excitatory amino acids (e.g., substance P, aspartate, glutamate, etc.) modulate further transmission of nociceptive signals to supraspinal sites (thalamus, anterior cingulated cortex, insular cortex, and somatosensory cortex) via the ascending pathways [14 and assoc. refs.].
Because the nervous system is plastic, strong or repeated noxious stimulation of dorsal horn neurons can condition a state of increased neuronal responsiveness or hyperexcitability, also known as central sensitization. Sensitization of the CNS causes the chronic pain sensations of hyperalgesia (exaggerated perception of painful stimuli) and allodynia (perception of innocuous stimuli as painful) by altering the function of endogenous chemical, electrophysiological, and pharmacological systems. Central sensitization is thought to mediate chronic pain via temporal summation of second pain, or “wind-up,” “a central spinal mechanism in which repetitive noxious stimulation results in a slow temporal summation that is experienced in humans as increased pain” [14]. During wind-up, transmission of second pain signals to dorsal horn nociceptive neurons by unmyelinated C-fibers activates NMDARs. This induces a subsequent neurochemical cascade of pain modulators, including nitric oxide, prostaglandins, activated protein kinases (PKC), and other cytosolic signaling molecules, eventually leading to altered gene expression patterns [15]. Nociceptive biomolecules, including substance P, nitric oxide, pronociceptive cytokines, and prostaglandins, have been found at pathologically high levels in FMS patients, suggesting a fundamental role for central sensitization via wind-up in the maintenance, and possibly development, of FMS [5, 16].
While biochemical data support the involvement of hyperactive pronociceptive pathways in FMS, studies also suggest that hypoactivity of the serotonergic-noradrenergic and opioidergic antinociceptive pathways may contribute to FMS pain [17]. Harris and colleagues [18] report decreased central mu-opioid receptor availability in FMS patients, a finding that may help to explain why opioid pharmaceuticals and elevated levels of endogenous opioids have little or no affect on FMS pain [18, 19]. The mu-opioid receptor has been shown to heterodimerize with the substance P receptor [20], suggesting a mechanism by which elevated levels of substance P may lead to the observed decrease in mu-opioid receptor availability in FMS patients. Pharmaceuticals that reduce endogenous levels of pronociceptive biomolecules, particularly substance P, may thus reduce FMS pain through downregulation of pronociceptive pathways and upregulation of antinociceptive pathways (Fig. 1).
Figure 1.
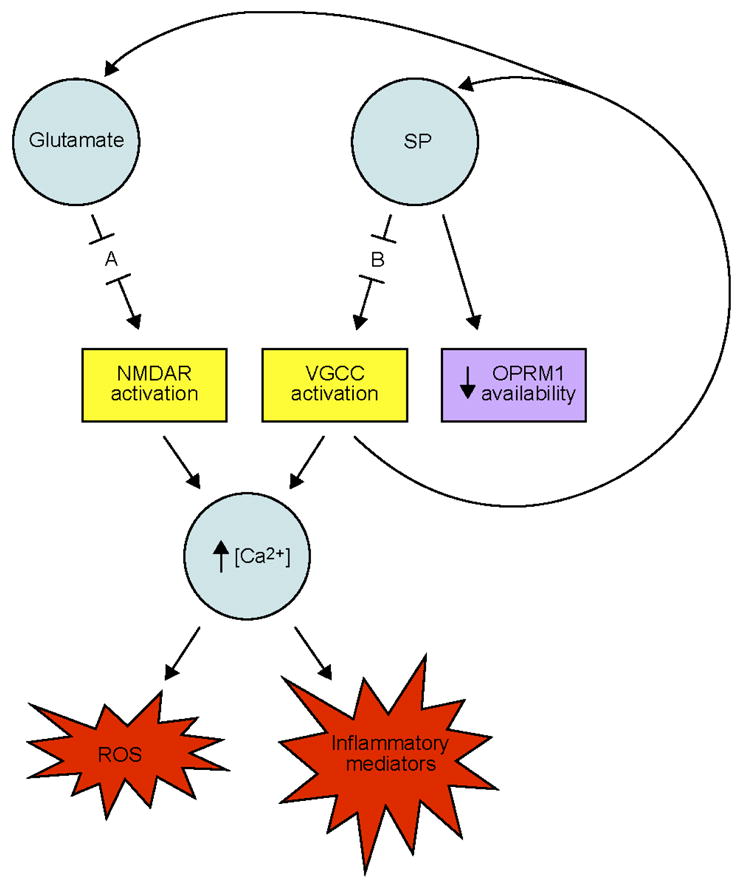
Proposed calcium ion-dependent molecular mechanisms of FMS. Abbreviations: A, site of action of memantine; B, site of action of pregabalin; NMDAR, N-methyl D-aspartate receptor; OPRM1, mu-opioid receptor; ROS, reactive oxygen species; SP, substance P; VGCC, voltage-gated calcium ion channel.
NEUORPHYSIOLOGICAL CONSEQUENCES OF CHRONIC PAIN
Once established, chronic pain itself seems to induce significant physiological changes, particularly in the CNS. Apkarian and colleagues [21] observed a 5–11% decrease in neocortical gray matter volume in chronic back pain patients compared to controls. This decrease in volume is equivalent to the gray matter volume lost in 10–20 years of normal aging and is related to pain duration; their results indicate 1.3 cm3 loss of gray matter for every year of chronic pain. The localization of gray matter loss to the bilateral dorsolateral prefrontal cortical and right thalamic regions suggests a role of thalamocortical pain processes in this localized neurodegeneration. A separate group later confirmed this hypothesis by identifying a negative correlation between pain unpleasantness/intensity and loss of gray matter in chronic back pain patients [22].
A similar, more striking pattern of neurodegeneration has been observed in female FMS patients. Kuchinad et al. [9] used voxel-based morphometry (VBM) to analyze cephalic MRI images of female FMS patients and healthy controls. Their findings indicate a significant reduction in gray matter and total brain volume in FMS patients. The patient group exhibited significantly less gray matter density in several brain regions, including the left parahippocampal gyrus, bilateral mid/posterior cingulate gyrus, left insula, and medial frontal cortex. The FMS patients displayed an age associated gray matter loss 3.3 times greater than that of controls (~3.7 cm3/year compared to 1.1 cm3/year). The investigators also observed a negative correlation between gray matter volume and FMS duration, similar to that observed for chronic back pain [21]. Patients exhibited a 10.5 cm3 decrease in gray matter with each year of diagnosed FMS, a loss equivalent to 9.5 times that observed in normal aging. A separate VBM study by Schmidt-Wilcke et al. [12] reported additional gray matter reductions in the right superior temporal gyral and left posterior thalamic regions of FMS patient brains compared to controls.
Similar reductions in gray matter density have been observed in several other chronic pain and stress disorders, including tension-type headache [11], chronic fatigue syndrome [8, 10], and posttraumatic stress disorder [13]. The patterns of gray matter loss differ among these illnesses, however, despite their extensive comorbidity. It has been proposed by Kuchinad et al. [9] that similar underlying mechanisms condition the atrophy observed in each illness, and that the variation in affected regions explains the differences (and similarities) among the illnesses’ associated symptoms. Although an accelerated loss of gray matter density is not always indicative of neuronal apoptosis, cognitive and biochemical profiles of FMS patients studied to date support the role of neurodegenerative mechanisms in the illness [5, 16, 23, 24].
CHRONIC PAIN-RELEVANT MECHANISMS OF EXCITOTOXIC NEURODEGENERATION
Release of neuropeptides (e.g., substance P) and excitatory amino acids (e.g., aspartate, glutamate) in the dorsal horn of the spinal cord initiates and facilitates the maintenance of central sensitization through complex biochemical cascades. Both types of ligands are also capable of inducing excitotoxic neurodegeneration via two separate calcium-dependent pathways.
Neurodegeneration can be initiated and maintained by a pathological process known as glutamate-induced excitotoxicity. Glutamate is a potent excitatory neurotransmitter that causes over-stimulation of ionotropic glutamate receptors (particularly NMDARs) when present at abnormally high levels in the CNS. NMDAR activation causes a subsequent increase in intracellular Ca2+ concentration, inducing a neurochemical cascade that increases levels of reactive oxygen species (ROS) and inflammatory mediators throughout the brain and spinal cord [25]. Increased levels of these destructive biomolecules are known to cause neuronal apoptosis, particularly in gray matter regions of the brain [26] and in inhibitory interneurons in the dorsal horns of the spinal cord [27]. It has been suggested that excessive stimulation of glutamate receptors, particularly NMDARs, is responsible for the neurodegeneration associated with Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, human immunodeficiency virus (HIV)-associated dementia, multiple sclerosis, amyotrophic lateral sclerosis (ALS), and glaucoma, as well as chronic neuropathic pain [28].
The second neurodegenerative pathway leads to cell death by producing the same harmful biomolecules as the glutamate/NMDAR pathway. Pathologically high levels of neuropeptides (particularly substance P) continuously activate VGCCs in the CNS via neurokinin-G protein-coupled receptor binding. This chronic opening of Ca2+ channels further amplifies the concentration of intracellular calcium, leading to increased generation of inflammatory mediators and harmful ROS [29]. Activation of VGCCs also induces the release of glutamate and substance P [30], conditioning a cycle of neuronal excitation in the CNS through chronic NMDAR and VGCC stimulation (Fig. 1).
Biochemical profiles of FMS patients are consistent with these mechanisms of neurodegeneration. Abnormally high cerebrospinal fluid levels of glutamate, substance P, inflammatory cytokines, and ROS are hallmarks of FMS [31–35]. Interestingly, anti-inflammatory medications have little or no effect on FMS pain [17], implying that the elevated cytokine levels observed in FMS patients are not produced by typical cyclooxygenase- or prostaglandin-mediated pathways (Table 1). This evidence supports the involvement of a CNS-based (rather than a tissue-based) inflammatory process. Clinical observations also suggest a limited effect of dietary-based antioxidant therapies on oxidative stress (OS) levels in FMS, implicating the activity of potent, intrinsic ROS-generating mechanisms such as NMDAR- and VGCC-overstimulation in the illness.
Table 1.
Examples and characteristics of common pharmacologic agents currently used in chronic pain management [77].
| Type | Class | Example(s) | Molecular Mechanism(s) |
|---|---|---|---|
| Nonopioid analgesic | Nonselective NSAIDa | Asprin Naproxen Ibuprofen |
COXb-l,2 inhibition |
| Nonopioid analgesic | Selective NSAIDa | Celecoxib | COXb-2 inhibition |
| Nonopioid analgesic | N/A | Acetaminophen | Prostaglandin synthesis inhibition (mechanism unknown) |
| Scheduled opioid analgesic | Agonist | Morphine1 Oxymorphone2 Codeine3 Fentanyl4 |
mu-opioid receptor binding (strong)1,2,3,4 kappa-opioid receptor binding (weak)1,3,4 delta-opioid receptor binding (weak)1,2,3 |
| Scheduled opioid analgesic | Partial agonist | Buprenorphine | mu-opioid receptor binding (strong) kappa-opioid receptor binding (strong) delta-opioid receptor binding (strong) |
| Scheduled opioid analgesic | Agonist-antagonist | Pentazocine Butorphanol |
kappa-opioid receptor binding (strong agonist) mu-opioid receptor binding (weak antagonist) |
| Nonscheduled opioid analgesic | Agonist | Tramadol | mu-opioid receptor binding; norepinephrine/serotonin reuptake inhibition |
| Nonscheduled opioid analgesic | Agonist-antagonist | Nalbuphine | kappa-opioid receptor binding (strong agonist) mu-opioid receptor binding (partial antagonist) delta-opioid receptor binding (weak) |
| Nonscheduled opioid analgesic | Antagonist | Naloxone | mu-opioid receptor binding (strong) kappa-opioid receptor binding (strong) delta-opioid receptor binding (strong) |
| Adjuvant analgesic (Antidepressant) | Tricyclic antidepressant | Amitriptyline Nortriptyline |
Na+ ion channel inhibition; NMDAc receptor blockade (non-neuroprotective) |
| Adjuvant analgesic (Antidepressant) | sSNRId | Venlafaxine Duloxetine |
Serotonin/norepinephrine reuptake inhibition |
| Adjuvant analgesic (Anticonvulsant) | N/A | Carbamazepine1 Pregabalin2 Neurontin2 |
Na+ ion channel inactive state stabilization1 VGCCe alpha-2-delta subunit binding2 |
| Skeletal muscle relaxant | N/A | Cyclobenzaprine | 5-HT2 receptor antagonist |
| Local anesthetic | N/A | Lidocane 5% patch | Na+ ion channel blockade |
nonsteroidal anti-inflammatory drugs
cyclooxygenase
N-methyl-D-aspartate
selective Serotonin/Norepinephrine Reuptake Inhibitor
voltage-gated calcium ion channel
PROPOSED PHARMACEUTICALS
Pregabalin
The non-steroidal anti-inflammatory and opioid medications listed in Table 1 are often successful in managing chronic peripheral pain. In contrast, the recommended pharmaceutical strategy for the treatment of chronic central pain, such as that involved in FMS, indicates neuroactive compounds that downregulate sensory processing [17]. Two such medications are pregabalin (Lyrica) and gabapentin (Neurontin). These anticonvulsants are used widely in the treatment of various chronic pain conditions, including painful diabetic neuropathy, postherpetic neuralgia, chronic post-operative pain, and FMS [17, 36–44]. These adjuvant analgesics exert their therapeutic effects by binding to, and decreasing the activity of, the alpha-2-delta subunit of the VGCC [45]. Over-stimulation of this subunit is thought to play an important role in the hypersensitization process, suggesting a mechanism of action for the drugs’ analgesic properties [30].
Pregabalin’s binding affinity for the alpha-2-delta subunit is six times greater than that of gabapentin, rendering pregabalin more clinically effective at lower doses [30]. Stochastic simulation modeling by Vera-Llonch, et al. [46] also suggests that pregabalin (375 mg/day) may provide better analgesic outcomes than gabapentin (1200 mg/day and 1800 mg/day) over a 12-week period. This increased efficacy, as well as its favorable pharmacokinetic profile, renders pregabalin the preferred anticonvulsant adjuvant analgesic for chronic pain treatment [30]. As of this writing, pregabalin is one of two prescription medications currently approved by the FDA (Food and Drug Administration) for the treatment of FMS. The use of pregabalin in FMS treatment is further supported by the drug’s ability to decrease excitotoxic neurodegeneration induced by the mechanisms described above [47], perhaps in part through its role as an opener of ATP-sensitive potassium ion (KATP) channels [48] that mediate neuroprotection [49].
Memantine
NMDAR antagonists possess significant pain-reducing and neuroprotective properties and are used widely in clinical practice [see 50 for a review]. Dextromethorphan and ketamine have shown particular pain-reducing efficacy in FMS [51, 52], although their use as longitudinal treatments is limited for two reasons. First, relatively little data exist regarding the safety of these drugs with chronic use, as would likely be required for FMS treatment. Secondly, high-dose administration of these and other high-affinity NMDAR antagonists (such as MK-801) can cause over-antagonization of the glutamatergic system, which itself leads to neurodegeneration [53].
The NMDAR antagonist memantine (Namenda) is an amantadine derivative that has been used to treat Parkinson’s disease, spasticity, convulsions, vascular dementia, and Alzheimer’s disease with an excellent clinical safety record for over 20 years. It is an uncompetitive open-channel blocker that dissociates relatively rapidly from the channel, enabling it to limit pathological activity of the NMDAR while sparing normal synaptic activity [54]. Memantine exhibited an extremely low incidence of side effects in human clinical trials [55, 56], and a recent trial extension demonstrated the drug’s clinical tolerability even with prolonged use [57]. The clinically approved human dosage of memantine begins at 5 mg per day and increases gradually over several weeks, which may contribute to the drug’s lack of clinical side effects [54].
While decreased NMDAR affinity contributes to memantine’s safety and efficacy as a neuroprotective agent, it also renders it less effective than high-affinity antagonists (e.g., ketamine) in chronic pain management [58, 59]. However, new research has highlighted memantine’s effectiveness in the treatment of complex regional pain syndrome [60] and phantom limb pain [61], suggesting that its quality of pain reduction is dependent on the type of pain being treated. Memantine may also show increased efficacy in the treatment of FMS-associated chronic pain. Kim and colleagues [62] reported an increased expression of NMDAR subunit 2D in the skin of FMS patients with fibromyalgia, which could be indicative of a more generalized increase of the receptor in other peripheral nerves. The increased availability of this molecular target would, in effect, improve memantine’s effectiveness in FMS patients [63 and assoc. refs.].
Memantine may also suppress neuronal excitability and confer neuroprotection in a manner similar to pregabalin. In neuropathic pain, opening of neuroprotective KATP channels is suppressed, at least in part, by reduced channel regulation conferred by decreased calcium/calmodulin-dependent protein kinase II (CaMKII) activity [64]. Memantine has been shown to activate CaMKII [65] and, thus, may further enhance neuroprotection and suppress neuronal excitability by restoring regulation and opening of KATP channels.
As discussed above, there is good reason to believe that pharmaceuticals capable of downregulating pronociceptive pathways, such as pregabalin and memantine, may provide a significant reduction in FMS pain. As shown in Figure 1, downregulation of pronociceptive pathways is expected to increase the availability of mu-opioid receptors, thereby increasing the activity of the opioidergic antinociceptive pathway and enhancing the efficacy of endogenous and exogenous opioids. Combining pregabalin and memantine with an opioid medication is thus expected to enhance the analgesic effects of all three drugs. Indeed, memantine has been shown to enhance opioid analgesia while simultaneously preventing opioid tolerance, reducing a primary concern of chronic opioid use [66]. The benefits of memantine in FMS treatment are thus expected to be threefold: 1) neuroprotection via antagonism of NMDARs, 2) analgesia through normalization of disregulated pro- and antinociceptive pathways, and 3) enhanced analgesia and prevention of opioid tolerance in a combinatorial analgesic approach.
DISCUSSION
Voxel-based morphometry studies have identified distinct regions of accelerated gray matter loss in the brains of FMS patients. These findings may be of significant clinical relevance, as neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and ALS are characterized by similar losses of gray matter [67–70]. The neurobiological basis of the FMS gray matter reductions remains unclear, however, as the observed atrophy is not well defined on a microscopic level. A decrease in cephalic gray matter volume does not necessarily indicate neuronal apoptosis. Alternative morphometric explanations include decreases in neuron size, glial cell shrinkage or apoptosis, loss of neuronal synapses, and decreases in blood flow or interstitial fluid [71]. Decreased cerebral blood flow in the thalamic and cortical regions has been reported in some FMS patients [72–74], lending support to these alternative explanations.
It is also still unclear whether gray matter atrophy is a cause or a consequence of FMS symptom chronification, although circumstantial evidence suggests the latter. Gray matter loss in FMS is observed primarily in brain regions related to stress (parahippocampal gyrus) and pain processing (cingulate, insular, and prefrontal cortices; right superior temporal gyrus; left posterior thalamus) [9, 12]. The negative correlation between FMS duration and extent of gray matter loss suggests that these structural changes may reflect a chronic exposure to pain and stress. Gray matter decreases in regions such as the frontal and parahippocampal cortices also appear consistent with cognitive difficulties common in FMS, lending further support to the idea that chronic pain and stress condition the longitudinal gray matter loss observed in FMS patients [9].
Additional longitudinal studies are indicated to determine 1) whether the observed accelerated regional loss of gray matter is the cause or consequence of FMS, 2) whether these observed structural changes are the result of neuronal apoptosis or other morphometric explanations, and 3) whether suppression of neuronal apoptosis via inhibition of nociception-induced neurotoxic mechanisms may consequently prevent loss of gray matter and clinical manifestations of FMS. However, taken together, the current imaging, biochemical, and behavioral data suggest that these structural changes are conditioned, at least in part, by harmful levels of inflammation and ROS in the CNS produced by a pathological increase in intracellular calcium. Successful treatments will thus likely include pharmaceuticals that target two primary components of calcium-dependent excitotoxic neurodegeneration, namely, neuropeptide-induced opening of VGCCs and glutamate-induced NMDAR activation.
As can be seen in Table 1, common analgesic pharmaceuticals fail to target both calcium-dependent molecular pathways thought to contribute to the neurological symptoms of FMS. Consequently, the independent prescription of these drugs does not adequately treat both mechanisms of excitotoxicity thought to underlie FMS pain and presumed neuronal apoptosis. An intuitive pharmaceutical combination to decrease this excitotoxicity is an NMDAR antagonist, such as memantine, and a VGCC blocker, such as pregabalin (Fig. 1).
Pregabalin and memantine have previously been proposed as independent FMS treatments [41, 75]. Clinical trials have demonstrated that both pharmaceuticals are safe and well tolerated, even in healthy volunteers who exhibit no signs of pathological excitotoxic neurodegeneration [42, 76]. An adverse co-interaction between pregabalin and memantine is unlikely; pregabalin has not been associated with any pharmacokinetic interactions and, based on its pharmacokinetic profile, none are expected [30, 42]. However, pregabalin and memantine are expected to produce additive, albeit moderate, CNS depressive effects, and their coincident usage has not been well documented to date. Clinical trials of their adjunctive use are therefore still indicated to ensure patient safety with the combined use of these pharmaceuticals in FMS treatment.
CONCLUSION AND FUTURE DIRECTIONS
Fibromyalgia syndrome is a heterogeneous illness of complex etiology. Although each incidence is thought to be initiated by a unique combination of genetic and environmental interactions, the disease state itself appears to be maintained by similar biomolecular networks, some of which are capable of inducing excitotoxic neurodegeneration. Current treatment methods do not target both calcium-dependent components of these underlying networks and thus fail to adequately reduce FMS pain and associated gray matter atrophy. The combinatorial treatment of pregabalin and memantine addresses this issue by targeting the VGCC and the NMDAR, respectively.
Chronic pain may now be viewed in part as a neurodegenerative disease, and clinical treatment strategies should be adjusted to reflect this new knowledge. The centrality of the NMDAR and VGCC to chronic pain and excitotoxic neurodegeneration make them potentially ideal targets for pharmacotherapy in FMS. Thus, it is clear that the dual-drug treatment of pregabalin and memantine proposed in this article warrants clinical investigation. Pending confirmation of the safety of the adjunctive use of these drugs, longitudinal studies examining the effect of this combinatorial treatment on the chronic pain and gray matter loss in FMS patients would be of particular interest.
This dual-network approach promises to produce additive analgesic, anti-inflammatory, and antioxidant effects in the CNS, decreasing the pain and rate of gray matter loss associated with FMS. However, the heterogeneity of the FMS patient population makes it unlikely that this treatment method will relieve all of the symptoms of FMS. Concurrent treatment with pregabalin and memantine should therefore be viewed as a potential part of an individualized, polymodal treatment program. Because of the negative correlation between FMS duration and extent of gray matter loss, timely clinical consideration of this treatment is indicated to potentially reduce further risk of gray matter atrophy in FMS patients. Further research efforts are warranted to clearly characterize the safety and efficacy of the combined use of pregabalin and memantine in the treatment of FMS.
Acknowledgments
This work was supported by the NSF IGERT training grant # 0221625. The author would like to thank Drs. Susan Ackerman, Carol Bult, Yichang Jia, and Julie Wells for helpful discussion and review of the manuscript. This article is dedicated to Lisa Hosie, without whom this work would not have been possible.
References
- 1.Wolfe F, Smythe HA, Yunus MB, et al. The American College of Rheumatology 1990 criteria for the classification of fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis Rheum. 1990;33:160–172. doi: 10.1002/art.1780330203. [DOI] [PubMed] [Google Scholar]
- 2.Barsky AJ, Borus JF. Functional Somatic Syndromes. Ann Intern Med. 1999;130:910–921. doi: 10.7326/0003-4819-130-11-199906010-00016. [DOI] [PubMed] [Google Scholar]
- 3. [Accessed Sept 28, 2008];Fibromyalgia. 2004 < http://www.niams.nih.gov/Health_Info/Fibromyalgia/default.asp>.
- 4.Buskila D, Sarzi-Puttini P. Review: Biology and therapy of fibromyalgia. Genetic aspects of fibromyalgia syndrome. Arthritis Research & Therapy. 2006;8:218. doi: 10.1186/ar2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Staud R. Biology and therapy of fibromyalgia: pain in fibromyalgia syndrome. Arthritis Research & Therapy. 2006;8:208. doi: 10.1186/ar1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cao YQ. Voltage-gated calcium channels and pain. Pain. 2006;126:5–9. doi: 10.1016/j.pain.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 7.Sattler R, Xiong Z, Lu WY, MacDonald JF, Tymianski M. Distinct Roles of Synaptic and Extrasynaptic NMDA Receptors in Excitotoxicity. J Neurosci. 2000;20:22–23. doi: 10.1523/JNEUROSCI.20-01-00022.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Lange FP, Kalkman JS, Bleijenberg G, et al. Gray matter volume reduction in the chronic fatigue syndrome. Neuroimage. 2005;26:777–781. doi: 10.1016/j.neuroimage.2005.02.037. [DOI] [PubMed] [Google Scholar]
- 9.Kuchinad A, Schweinhardt P, Seminowicz DA, et al. Accelerated Brain Gray Matter Loss in Fibromyalgia Patients: Premature Aging of the Brain? J Neurosci. 2007;27:4004–4007. doi: 10.1523/JNEUROSCI.0098-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okada T, Tanaka M, Kuratsune H, Watanabe Y, Sadato N. Mechanisms underlying fatigue: a voxel-based morphometric study of chronic fatigue syndrome. BMC Neurology. 2004;4:14. doi: 10.1186/1471-2377-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidt-Wilcke T, Leinisch E, Straube A, et al. Gray matter decrease in patients with chronic tension type headache. Neurology. 2005;65:1483–1486. doi: 10.1212/01.wnl.0000183067.94400.80. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt-Wilcke T, Luerding R, Weigand T, et al. Striatal grey matter increase in patients suffering from fibromyalgia - A voxel-based morphometry study. Pain. 2007;132:S109–S116. doi: 10.1016/j.pain.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 13.Villarreal G, Hamilton DA, Petropoulos H, et al. Reduced hippocampal volume and total white matter volume in posttraumatic stress disorder. Biol Psychiatry. 2002;52:119 –125. doi: 10.1016/s0006-3223(02)01359-8. [DOI] [PubMed] [Google Scholar]
- 14.Meeus M, Nijs J. Central sensitization: a biopsychosocial explanation for chronic widespread pain in patients with fibromyalgia and chronic fatigue syndrome. Clin Rheumatol. 2007;26:465–473. doi: 10.1007/s10067-006-0433-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ji RR, Strichartz G. Cell signaling and the genesis of neuropathic pain. Sci STKE. 2004;2004:reE14. doi: 10.1126/stke.2522004re14. [DOI] [PubMed] [Google Scholar]
- 16.Staud R. Evidence of involvement of central neural mechanisms in generating fibromyalgia pain. Current Rheumatology Reports. 2002;4:299–305. doi: 10.1007/s11926-002-0038-5. [DOI] [PubMed] [Google Scholar]
- 17.Dadabhoy D, Clauw DJ. Therapy Insight: fibromyalgia--a different type of pain needing a different type of treatment. Nature Clinical Practice Rheumatology. 2006;2:364–372. doi: 10.1038/ncprheum0221. [DOI] [PubMed] [Google Scholar]
- 18.Harris RE, Clauw DJ, Scott DJ, et al. Decreased Central mu-Opioid Receptor Availability in Fibromyalgia. J Neurosci. 2007;27:10000–10006. doi: 10.1523/JNEUROSCI.2849-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baraniuk JN, Whalen G, Cunningham J, Clauw DJ. Cerebrospinal fluid levels of opioid peptides in fibromyalgia and chronic low back pain. BMC Musculoskelet Disord. 2004;5:48. doi: 10.1186/1471-2474-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfeiffer M, Kirscht S, Stumm R, et al. Heterodimerization of substance P and mu-opioid receptors regulates receptor trafficking and resensitization. J Biol Chem. 2003;278:51630–51637. doi: 10.1074/jbc.M307095200. [DOI] [PubMed] [Google Scholar]
- 21.Apkarian AV, Sosa Y, Sonty S, et al. Chronic Back Pain Is Associated with Decreased Prefrontal and Thalamic Gray Matter Density. J Neurosci. 2004;24:10410–10415. doi: 10.1523/JNEUROSCI.2541-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmidt-Wilcke T, Leinisch E, Ganßbauer S, et al. Affective components and intensity of pain correlate with structural differences in gray matter in chronic back pain patients. Pain. 2006;125:89 –97. doi: 10.1016/j.pain.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 23.Glass JM, Park DC. Cognitive dysfunction in fibromyalgia. Current Rheumatology Reports. 2001;3:123–127. doi: 10.1007/s11926-001-0007-4. [DOI] [PubMed] [Google Scholar]
- 24.Park DC, Glass JM, Minear M, Crofford LJ. Cognitive function in fibromyalgia patients. Arthritis Rheum. 2001;44:2125–2133. doi: 10.1002/1529-0131(200109)44:9<2125::AID-ART365>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 25.Strong J, Unruh AM, Wright A, Baxter GD, Wall PD. Pain: A Textbook for Therapists. Elsevier Health Sciences; 2002. [Google Scholar]
- 26.Klein JA, Ackerman SL. Oxidative stress, cell cycle, and neurodegeneration. J Clin Invest. 2003;111:785–793. doi: 10.1172/JCI18182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Apkarian AV, Scholz J. Shared mechanisms between chronic pain and neurodegenerative disease. Drug Discovery Today: Disease Mechanisms. 2006;3:319–326. [Google Scholar]
- 28.Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nature Reviews Drug Discovery. 2006;5:160–170. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- 29.Warfield CA, Bajwa ZH. Principles and Practice of Pain Medicine. McGraw-Hill Professional; 2003. [Google Scholar]
- 30.Gajraj NM. Pregabalin: its pharmacology and use in pain management. Anesth Analg. 2007;105:1805–1815. doi: 10.1213/01.ane.0000287643.13410.5e. [DOI] [PubMed] [Google Scholar]
- 31.Larson AA, Giovengo SL, Russell IJ, Michalek JE. Changes in the concentrations of amino acids in the cerebrospinal fluid that correlate with pain in patients with fibromyalgia: implications for nitric oxide pathways. Pain. 2000;87:201–211. doi: 10.1016/S0304-3959(00)00284-0. [DOI] [PubMed] [Google Scholar]
- 32.Sarchielli P, Mancini ML, Floridi A, et al. Increased levels of neurotrophins are not specific for chronic migraine: evidence from primary fibromyalgia syndrome. Journal of Pain. 2007;8:737–745. doi: 10.1016/j.jpain.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 33.Frazer KA, Wade CM, Hinds DA, et al. Segmental phylogenetic relationships of inbred mouse strains revealed by fine-scale analysis of sequence variation across 4.6 mb of mouse genome. Genome Res. 2004;14:1493–1500. doi: 10.1101/gr.2627804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ozgocmen S, Ozyurt H, Sogut S, Akyol O. Current concepts in the pathophysiology of fibromyalgia: the potential role of oxidative stress and nitric oxide. Rheumatol Int. 206;26:585–597. doi: 10.1007/s00296-005-0078-z. [DOI] [PubMed] [Google Scholar]
- 35.Bagis S, Tamer L, Sahin G, et al. Free radicals and antioxidants in primary fibromyalgia: an oxidative stress disorder? Rheumatol Int. 2005;25:188–190. doi: 10.1007/s00296-003-0427-8. [DOI] [PubMed] [Google Scholar]
- 36.Wiffen PJ, McQuay HJ, Rees J, Moore RA. Gabapentin for acute and chronic pain. Cochrane Database of Systematic Reviews. 2005:CD005452. doi: 10.1002/14651858.CD005452. [DOI] [PubMed] [Google Scholar]
- 37.Rosenstock J, Tuchman M, Lamoreaux L, Sharma U. Pregabalin for the treatment of painful diabetic peripheral neuropathy: a double-blind, placebo-controlled trial. Pain. 2004;110:628–638. doi: 10.1016/j.pain.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 38.Freynhagen R, Strojek K, Griesing T, Whalen E, Balkenohl M. Efficacy of pregabalin in neuropathic pain evaluated in a 12-week, randomised, double-blind, multicentre, placebo-controlled trial of flexible- and fixed-dose regimens. Pain. 2005;115:254–263. doi: 10.1016/j.pain.2005.02.032. [DOI] [PubMed] [Google Scholar]
- 39.Sabatowski R, Galvez R, Cherry DA, et al. Pregabalin reduces pain and improves sleep and mood disturbances in patients with post-herpetic neuralgia: results of a randomised, placebo-controlled clinical trial. Pain. 2004;109:26–35. doi: 10.1016/j.pain.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 40.Arnold LM, Goldenberg DL, Stanford SB, et al. Gabapentin in the treatment of fibromyalgia; a randomized, double-blind, placebo-controlled multicenter trial. Arthritis Rheum. 2007;56:1336–1344. doi: 10.1002/art.22457. [DOI] [PubMed] [Google Scholar]
- 41.Crofford LJ, Rowbotham MC, Mease PJ, et al. Pregabalin for the Treatment of Fibromyalgia Syndrome. Arthritis Rheum. 2005;52:1264–1273. doi: 10.1002/art.20983. [DOI] [PubMed] [Google Scholar]
- 42.Lyseng-Williamson KA, Siddiqui MA. Pregabalin: a review of its use in fibromyalgia. Drugs. 2008;68:2205–2223. doi: 10.2165/00003495-200868150-00009. [DOI] [PubMed] [Google Scholar]
- 43.Fassoulaki A, Patris K, Sarantopoulos C, Hogan Q. The analgesic effect of gabapentin and mexiletine after breast surgery for cancer. Anesth Analg. 2003;95:985–991. doi: 10.1097/00000539-200210000-00036. [DOI] [PubMed] [Google Scholar]
- 44.Fassoulaki A, Triga A, Melemeni A, Sarantopoulos C. Multimodal analgesia with gabapentin and local anesthetics prevents acute and chronic pain after breast surgery for cancer. Anesth Analg. 2005;101:1427–1432. doi: 10.1213/01.ANE.0000180200.11626.8E. [DOI] [PubMed] [Google Scholar]
- 45.Sarantopoulos C, McCallum B, Kwok WM, Hogan Q. Gabapentin decreases membrane calcium currents in injured as well as in control mammalian primary afferent neurons. Reg Anesth Pain Med. 2002;27:47–57. doi: 10.1053/rapm.2002.29124. [DOI] [PubMed] [Google Scholar]
- 46.Vera-Llonch M, Dukes E, Argoff C, Oster G. Analgesic outcomes in patients with painful diabetic neuropathy or post-herpetic neuralgia receiving pregabalin versus gabapentin. Journal of Pain. 2005;6:S33. [Google Scholar]
- 47.Ha KY, Kim YH, Rhyu KW, Kwon SE. Pregabalin as a neuroprotector after spinal cord injury in rats. Eur Spine J. 2008;17:864–872. doi: 10.1007/s00586-008-0653-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang CW, Huang CC, Wu SN. The opening effect of pregabalin on ATP-sensitive potassium channels in differentiated hippocampal neuron-derived H19-7 cells. Epilepsia. 2006;47:720–726. doi: 10.1111/j.1528-1167.2006.00498.x. [DOI] [PubMed] [Google Scholar]
- 49.Sun HS, Feng ZP, Miki T, Seino S, French RJ. Enhanced neuronal damage after ischemic insults in mice lacking Kir6.2-containing ATP-sensitive K+ channels. J Neurophysiol. 2006;95:2590–2601. doi: 10.1152/jn.00970.2005. [DOI] [PubMed] [Google Scholar]
- 50.Bountra C, Munglani R, Schmidt WK. Informa Health Care. 2003. Pain: Current Understanding, Emerging Therapies, and Novel Approaches to Drug Discovery. [Google Scholar]
- 51.Staud R, Vierck CJ, Robinson ME, Price DD. Effects of the N-Methyl-D-Aspartate Receptor Antagonist Dextromethorphan on Temporal Summation of Pain Are Similar in Fibromyalgia Patients and Normal Control Subjects. Journal of Pain. 2005;6:323–332. doi: 10.1016/j.jpain.2005.01.357. [DOI] [PubMed] [Google Scholar]
- 52.Graven-Nielsen T, Kendall SA, Henriksson KG, et al. Ketamine reduces muscle pain, temporal summation, and referred pain in fibromyalgia patients. Pain. 2000;85:483–491. doi: 10.1016/S0304-3959(99)00308-5. [DOI] [PubMed] [Google Scholar]
- 53.Low SJ, Roland CL. Review of NMDA antagonist-induced neurotoxicity and implications for clinical development. Int J Clin Pharmacol Ther. 2004;42:1–14. doi: 10.5414/cpp42001. [DOI] [PubMed] [Google Scholar]
- 54.Chen HSV, Lipton SA. The chemical biology of clinically tolerated NMDA receptor antagonists. J Neurochem. 2006;97:1611–1626. doi: 10.1111/j.1471-4159.2006.03991.x. [DOI] [PubMed] [Google Scholar]
- 55.Reisberg B, Doody R, Stöffler A, et al. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med. 2003;348:1333–1341. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- 56.Tariot PN, Farlow MR, Grossberg GT, et al. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291:317–324. doi: 10.1001/jama.291.3.317. [DOI] [PubMed] [Google Scholar]
- 57.Reisberg B, Doody R, Stoffler A, et al. A 24-week open-label extension of memantine in moderate to severe Alzheimer’s disease. Arch Neurol. 2006;63:49–54. doi: 10.1001/archneur.63.1.49. [DOI] [PubMed] [Google Scholar]
- 58.Nikolajsen L, Gottrup H, Kristensen AGD, Jensen TS. Memantine (a N-Methyl-D-Aspartate Receptor Antagonist) in the Treatment of Neuropathic Pain After Amputation or Surgery: A Randomized, Double-Blinded, Cross-Over Study. Anesth Analg. 2000;91:960–966. doi: 10.1097/00000539-200010000-00036. [DOI] [PubMed] [Google Scholar]
- 59.Weinbroum AA, Rudick V, Paret G, Ben-Abraham R. The role of dextromethorphan in pain control. Can J Anaesth. 2000;47:585–596. doi: 10.1007/BF03018952. [DOI] [PubMed] [Google Scholar]
- 60.Sinis N, Birbaumer N, Gustin S, et al. Memantine treatment of complex regional pain syndrome: a preliminary report of six cases. Clin J Pain. 2007;23:237–243. doi: 10.1097/AJP.0b013e31802f67a7. [DOI] [PubMed] [Google Scholar]
- 61.Hackworth RJ, Tokarz KA, Fowler IM, Wallace SC, Stedje-Larsen ET. Profound Pain Reduction After Induction of Memantine Treatment in Two Patients with Severe Phantom Limb Pain. Anesth Analg. 2008;107:1377–1379. doi: 10.1213/ane.0b013e31817f90f1. [DOI] [PubMed] [Google Scholar]
- 62.Kim SH, Jang TJ, Moon IS. Increased expression of N-Methyl-D-aspartate receptor subunit 2D in the skin of patients with fibromyalgia. J Rheumatol. 2006;33:785–788. [PubMed] [Google Scholar]
- 63.Chen X, Moore-Nichols D, Nguyen H, Michaelis EK. Calcium Influx Through NMDA Receptors, Chronic Receptor Inhibition by Ethanol and 2-Amino-5-Phosphonopentanoic Acid, and Receptor Protein Expression. J Neurochem. 1999;72:1969–1980. doi: 10.1046/j.1471-4159.1999.0721969.x. [DOI] [PubMed] [Google Scholar]
- 64.Sarantopoulos C, Kawano T, Zoga V, et al. Suppressed regulation of peripheral sensory neuronal KATP channels by the Ca2+-Calmodulin-CaMKII pathway mediates hyperalgesia after painful nerve injury. Abstracts, Anaesthetic Research Society 2008 Meeting; November 20–21, 2008; London, UK. 2008. p. 18. [Google Scholar]
- 65.Almeida RC, Souza DG, Soletti RC, et al. Involvement of PKA, MAPK/ERK and CaMKII, but not PKC in the acute antidepressant-like effect of memantine in mice. Neurosci Lett. 2006;395:93–97. doi: 10.1016/j.neulet.2005.10.057. [DOI] [PubMed] [Google Scholar]
- 66.Harris AC, Rothwell PE, Gewirtz JC. Effects of the NMDA receptor antagonist memantine on the expression and development of acute opiate dependence as assessed by withdrawal-potentiated startle and hyperalgesia. Psychopharmacology (Berl) 2008;196:649–660. doi: 10.1007/s00213-007-0998-2. [DOI] [PubMed] [Google Scholar]
- 67.Thompson PM, Hayashi KM, de Zubicaray G, et al. Dynamics of gray matter loss in Alzheimer’s disease. J Neurosci. 2003;23:994–1005. doi: 10.1523/JNEUROSCI.23-03-00994.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nagano-Saito A, Washimi Y, Arahata Y, et al. Cerebral atrophy and its relation to cognitive impairment in Parkinson disease. Neurology. 2005;64:224–229. doi: 10.1212/01.WNL.0000149510.41793.50. [DOI] [PubMed] [Google Scholar]
- 69.Summerfield C, Junque C, Tolosa E, et al. Structural brain changes in Parkinson disease with dementia: a voxel-based morphometry study. Arch Neurol. 2005;62:281–285. doi: 10.1001/archneur.62.2.281. [DOI] [PubMed] [Google Scholar]
- 70.Ellis CM, Suckling J, Amaro E, Jr, et al. Volumetric analysis reveals corticospinal tract degeneration and extramotor involvement in ALS. Neurology. 2001;57:1571–1578. doi: 10.1212/wnl.57.9.1571. [DOI] [PubMed] [Google Scholar]
- 71.May A. Chronic pain may change the structure of the brain. Pain. 2008;137:7–15. doi: 10.1016/j.pain.2008.02.034. [DOI] [PubMed] [Google Scholar]
- 72.Bradley LA, Sotolongo A, Alberts KR, et al. Abnormal Regional Cerebral Blood Flow in the Caudate Nucleus Among Fibromyalgia Patients and Non-Patients Is Associated with Insidious Symptom Onset. Journal of Musculoskeletal Pain. 1999;7:285–292. [Google Scholar]
- 73.Kwiatek R, Barnden L, Tedman R, et al. Regional Cerebral Blood Flow in Fibromyalgia. Arthritis Rheum. 2000;43:2823–2833. doi: 10.1002/1529-0131(200012)43:12<2823::AID-ANR24>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 74.Wik G, Fischer H, Bragee B, Kristianson M, Fredrikson M. Retrosplenial cortical activation in the fibromyalgia syndrome. NeuroReport. 2003;14:619–621. doi: 10.1097/00001756-200303240-00019. [DOI] [PubMed] [Google Scholar]
- 75.DeMaria S, Jr, Hassett AL, Sigal LH. N-Methyl-D-Aspartate Receptor-Mediated Chronic Pain: New Approaches to Fibromyalgia Syndrome Etiology and Therapy. Journal of Musculoskeletal Pain. 2006;15:33–44. [Google Scholar]
- 76.Mobius HJ, Stoffler A, Graham SM. Memantine hydrochloride: pharmacological and clinical profile. Drugs Today (Barc) 2004;40:685–695. [PubMed] [Google Scholar]
- 77.Katz WA, Barkin RL. Dilemmas in chronic/persistent pain management. Am J Ther. 2008;15:256–264. doi: 10.1097/MJT.0b013e3181671c5a. [DOI] [PubMed] [Google Scholar]