

Abstract
Calcium (Ca2+) influx is required for the sustained secretion of insulin and is accompanied by a large rate of energy usage. We hypothesize that the energy usage reflects a process [Ca2+/metabolic coupling process (CMCP)] that couples Ca2+ to insulin secretion by pancreatic islets. The aim of the study was to test this hypothesis by testing the effect of inhibiting candidate Ca2+-sensitive proteins proposed to play a critical role in the CMCP. The effects of the inhibitors on oxygen consumption rate (OCR), a reflection of ATP usage, and insulin secretion rate (ISR) were compared with those seen when L-type Ca2+ channels were blocked with nimodipine. We reasoned that if a downstream Ca2+-regulated site was responsible for the OCR associated with the CMCP, then its inhibition should mimic the effect of nimodipine. Consistent with previous findings, nimodipine decreased glucose-stimulated OCR by 36% and cytosolic Ca2+ by 46% and completely suppressed ISR in rat pancreatic islets. Inhibitors of three calmodulin-sensitive proteins (myosin light-chain kinase, calcineurin, and Ca2+/calmodulin-dependent protein kinase II) did not meet the criteria. In contrast, KN-62 severed the connection between Ca2+ influx, OCR, and ISR without interfering with Ca2+ influx. In the presence of nimodipine or KN-62, potentiators of ISR, acetylcholine, GLP-1, and arginine had little effect on insulin secretion, suggesting that the CMCP is also essential for the amplification of ISR. In conclusion, a KN-62-sensitive process directly mediates the effects of Ca2+ influx via L-type Ca2+ channels on OCR and ISR, supporting the essential role of the CMCP in mediating ISR.
Keywords: oxygen consumption, calmodulin, islet, KN-62
calcium (Ca2+) is the major intracellular determinant of insulin secretion, and glucose-stimulated secretion does not occur in the absence of extracellular Ca2+ (12, 17). Although many Ca2+-sensitive proteins have been identified in the β-cell, the exact mechanism mediating Ca2+'s effect on insulin secretion is unclear, and this void in knowledge represents a major hurdle in understanding the etiology of islet dysfunction in type 2 diabetes. An important clue as to how Ca2+ stimulates insulin secretion may come from the intimate relationship between Ca2+, oxygen consumption rate (OCR), and insulin secretion rate (ISR). Blocking Ca2+ influx by L-type Ca2+ channels leads to a 35–40% reduction in glucose-stimulated OCR, a reflection of ATP turnover, and complete inhibition of glucose-stimulated insulin secretion (42, 45). To explain this fundamental and intrinsic property of islets, we have hypothesized that the potent effects of Ca2+ influx on OCR reflect an essential downstream and highly energetic process that couples Ca2+ influx through L-type Ca2+ channel with ISR. We have termed this process the Ca2+/metabolic coupling process (CMCP) and have sought to develop and test a conceptual model of its operation (Fig. 1).
Fig. 1.

Conceptual design of study to test hypothesized model of the role of calcium (Ca2+)/metabolic coupling process (CMCP) in glucose-stimulated insulin secretion. A model by Henquin (18) proposed that influx of Ca2+ triggers the release of insulin. Into this framework, we define a CMCP that reflects the large energy (ATP) usage that occurs upon activation of both Ca2+ influx through L-type Ca2+ channels and metabolic rate (as depicted by the circular symbol denoting summation of the 2 input signals). The model reflects the observation that energy usage associated with amplification and exocytosis of secretory granules is small, and, like triggering of Ca2+ influx, activation of the CMCP is essential for insulin secretion to occur. The hypothesis was tested in this study by screening inhibitors of Ca2+-sensitive kinases for their ability to mimic the effects of nimodipine on insulin secretion rate (ISR) and oxygen consumption rate (OCR) without affecting Ca2+ influx. In addition, the absolute dependency of ISR on CMCP activation was tested by measuring the potentiation of ISR by glucagon-like peptide-1 (GLP-1), acetylcholine (Ach), and arginine (Arg) in the presence of nimodipine or 1-[N,O-bis-(5-isoquinolinesulfonyl)-N-methyl-l-tyrosyl]-4-phenylpiperazine (KN-62) (see text for more detailed description of model and study design).
Our previous studies defining the relationship between cytosolic Ca2+, OCR, and ISR have identified three important features that contributed to the formulation of this model. First, Ca2+-sensitive OCR appears to depend more strongly on the usage of ATP rather than changes in the TCA cycle that contribute to ATP production. This is based on the finding that, when glucose-stimulated Ca2+ influx was blocked by nimodipine or diazoxide, NADH and cytochrome c reduction (markers of metabolic rate in mitochondria) did not change significantly despite large changes in cytosolic Ca2+ and OCR (42, 45). This indicates that although Ca2+ may effect TCA cycle activity (25), this cannot be the major driving force mediating the sustained changes in Ca2+-sensitive OCR in islets. Likewise, it is also unlikely that Ca2+-mediated changes in mitochondrial volume thought to occur in response to increased K+ permeability make a large contribution to Ca2+-sensitive changes in OCR (15). This is based on observations that islets respond to 30 mM KCl with an increase in both OCR and cytochrome c reduction (Gilbert M and Sweet IR, unpublished observations). Since blocking Ca2+ influx does not affect cytochrome c reduction, K+-induced changes in cytochrome c reduction must not be involved. Consistent with the scenario that energy usage mediates the effect, ATP/ADP ratio, a known inhibitor of OCR in mitochondria (5), was reciprocally related to alterations in Ca2+ influx brought about by blockers and activators of L-type Ca2+ channels (8). It is not yet known what Ca2+-sensitive processes are utilizing the bulk of the ATP, but some expected candidates do not appear to make a significant contribution. Ca2+ influx by the channels is a non-energy-dependent process, indicating that the ATP usage corresponding to the OCR must be caused by a process triggered by Ca2+. The amount of ATP used in secretion of insulin makes only a minor contribution to overall ATP turnover in the islets (45) as an agent that activates protein kinase C [12-O-tetradecanoylphorbol 13-acetate (TPA)] or inhibits cAMP phosphodiesterase activity [3-isobutyl-1-methylxanthine (IBMX)] (thereby stimulating cAMP-dependent pathways such as protein kinase A), increasing ISR but having no effect on OCR (14, 45). Pumping of Ca2+ out of the cell remains a possibility, although blocking sarcoendoplasmic reticulum Ca2+ ATPase (SERCA), an ATPase that pumps Ca2+ at rates similar to Ca2+ efflux, did not have an effect on OCR (45).
Second, Ca2+-sensitive OCR, but not total glucose-stimulated OCR or ATP/ADP in the β-cell (9), is strongly correlated with glucose-stimulated ISR across the entire range of glucose concentrations (45). This does not prove causality, but no instances where sustained insulin secretion occurred in the absence of activation of Ca2+-sensitive OCR have been observed (14, 45). The obligatory increase in ATP/ADP that is well established to be essential for insulin secretion is nearly saturated above 12 mM glucose (9), whereas insulin secretion is most sensitive to changes between 12 and 20 mM glucose (45).
Third, activation of Ca2+-sensitive OCR (i.e., the CMCP) is under dual control by a metabolic factor and Ca2+ influx exclusively via L-type Ca2+ channels (14). Ca2+ release from the endoplasmic reticulum has only a relatively minor contribution to OCR and ISR (14, 45). These findings are consistent with the notion that microdomains around the L-type Ca2+ channel provide preferential access to exocytotic sites (3, 28, 38) or regulatory proteins (47), bestowing greater efficacy on ISR than Ca2+ derived from other sources.
Taken together, these data support the scenario that a Ca2+-sensitive process, which utilizes large amounts of energy, is an essential regulatory factor in determining ISR (14). We have endeavored in this study to provide further support that 1) the CMCP is operational in the islet and 2) the operation of the CMCP is essential for insulin secretion. The strategy taken was to measure OCR and ISR in the presence of blockers of proposed Ca2+-dependent proteins. If the CMCP is indeed downstream of Ca2+ influx via L-type Ca2+ channels, then data obtained in the presence of the inhibitor should satisfy two criteria: the inhibitor will mimic the effects of nimodipine on OCR and ISR, independent of effects on Ca2+ turnover, and in the presence of nimodipine, no effect of the blocker will be seen. One inhibitor, 1-[N,O-bis-(5-isoquinolinesulfonyl)-N-methyl-l-tyrosyl]-4-phenylpiperazine (KN-62), with actions on Ca2+/calmodulin-dependent protein kinase (CaMK) II and a number of calmodulin-dependent and -independent proteins (1, 23, 29) was found to meet these criteria in a dose-dependent fashion. Thus, it appears that the model is further supported by demonstration of the existence of a regulatory process downstream of Ca2+. Moreover, the sensitivity of Ca2+'s interrelation with ISR and OCR to KN-62 could serve as the basis for strategic screening of candidates that play a role as the Ca2+ sensor in the β-cell.
MATERIALS AND METHODS
Chemicals.
Krebs-Ringer bicarbonate (KRB) solution supplemented with 0.1% fraction V bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO) and 1% penicillin-streptomycin-fungizone (Invitrogen, Carlsbad, CA) was used for the perifusion experiments and was made up as described previously (45). Antimycin A, carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), KN-62, nimodipine, tacrolimus (FK-506), S(−)-Bay K 8644, glucagon-like peptide-1 (GLP-1), arginine, acetylcholine, potassium cyanide (KCN), and diazoxide were purchased from Sigma-Aldrich. ML-7, cell-permeable autocamtide-2-related inhibitory peptide II (AIP2), and myristoylated AIP were from EMD Biosciences (Gibbstown, NJ), and fura 2-AM and 20% pluronic F-127 were from Invitrogen. 45Ca2+ was purchased from PerkinElmer (Boston, MA).
Rat islet isolation and culture.
Islets were harvested from Sprague-Dawley rats (≈250 g; Charles River) and anesthetized by intraperitoneal injection of pentobarbital sodium (150 mg/kg rat). All procedures were approved by the University of Washington Internal Animal Care and Use Committee. Islets were prepared and purified as described (26, 42) and then cultured for 18 h in a 37°C/5% CO2 incubator prior to the experiments in RPMI medium 1640 supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen).
Measurement of OCR and ISR.
A flow culture system containing 600–750 islets in each of two chambers that concomitantly measures OCR while collecting outflow fractions for subsequent measurement of ISR was used (described previously in Refs 42, 44, and 46). OCR was calculated as the flow rate (80–90 μl/min) times the difference between inflow and outflow oxygen tension measured by detecting the phosphorescence lifetime (Tau Theta) of an oxygen-sensitive porphyrin dye that was painted on the inside of the perifusion chamber (45). Insulin was measured by ELISA per the manufacturer's instructions (Mercodia).
Imaging and quantification of cytosolic Ca2+.
Cytosolic Ca2+ was measured as reflected by fluorescence detection of fura 2-AM (Invitrogen) after islets were incubated in 2 μM fura 2-AM and 0.02% pluronic acid for 40–50 min in a 37°C/5% CO2 incubator. Subsequently, the islets were pipetted into a temperature-controlled, 250-μl perifusion dish (Bioptechs, Butler, PA) that was mounted onto the stage of a Nikon Eclipse TE-200 inverted microscope, and KRB (containing 5 mM NaHCO3) was pumped through the dish at a flow rate of 150 μl/min. Fluorescent emission was detected at 510 nm by a Photometrics Cool Snap EZ camera (Photometrics, Tucson, AZ) during alternating excitation at either 340 or 380 nm. Results are displayed as the ratio of the fluorescent intensities during excitation at these two wavelengths (F340/F380).
Imaging and quantification of NAD(P)H.
NAD(P)H autofluorescence was measured similarly to Ca2+, except there was no need for loading with dye, and the excitation and emission wavelengths were 360 and 460 nm, respectively. To calibrate the relative fluorescence units (RFU), at the end of the experiments the steady-state RFU in the presence of 3 mM KCN/20 mM glucose and subsequently 10 μM FCCP/3 mM glucose was measured. The normalized fluorescence of NAD(P)H was then calculated as
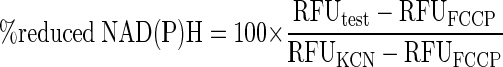 |
where RFUFCCP and RFUKCN equaled the average of the final 10 time points where each agent was present.
Measurement of Ca2+ influx.
Ca2+ influx was measured by first preincubating islets in KRB containing 3 mM glucose for 60 min in a 37°C/5% CO2 incubator. Subsequently, islets were picked into 12 × 75-mm test tubes containing 90 μl of KRB (with 0.5 mM Ca2+) and indicated additions (except 20 mM glucose) and incubated for 30 min, and then 10 μl of 45Ca2+ (1 μCi) was added by use of a repeater pipette. For islets that were to be exposed to 20 mM glucose, the supplemental glucose was added immediately (within 1 min) after the 45Ca2+ so that the Ca2+ influx measurement would include the initial stimulation by high glucose. At ∼14 min after addition of 45Ca2+, the islet suspension was transferred to 0.4-ml centrifuge tubes containing 100 μl of an oil mixture, and then the free and the cell-associated radioactivity were separated at exactly 15 min by spinning the islets through the oil layer and counting, as has been described previously (43).
Static measurement of ISR.
ISR was determined statically with multiple conditions, as described previously (30). Briefly, islets were handpicked into a petri dish containing KRB, 0.1% BSA, and 3 mM glucose and incubated at 37°C/5% CO2 for 60 min. Subsequently, islets were picked into wells of 96-well plates containing desired amounts of glucose and agents as indicated and incubated for an additional 60 min. At the end of this period, supernatant was assayed for insulin. For solutions containing 10 mM arginine, the pH was adjusted back to 7.4 by the addition of HCl.
Selection of inhibitor concentrations.
Inhibitor concentrations were selected to be ≥20 times greater than the IC50. In previous studies, the IC50 for AIP2, ML-7, and FK-506 have been determined to be 4 (20), 400 (22), and 1 nM (39), respectively.
Perifusion protocols and data analysis.
The basic protocol for the OCR, ISR, Ca2+, and NAD(P)H perifusion experiments entailed a 90-min baseline period at 3 mM glucose, followed by stimulation with 20 mM glucose (45 min). The subsequent steps in the protocols involved the addition of effectors at intervals of 45 min or as indicated. All perifusion data were then displayed as the change relative to baseline, calculated as the average of values obtained between −15 and 0 min (Figs. 2, 3, 5, 6, 7, and 9), and represent the average ± SE. In addition, steady-state changes induced by the first and second effectors were calculated from these kinetic data by taking the difference between average of the final 15 min of each subsequent period. Statistical significance (P values) for steady-state results were generated from Student's paired t-tests (using Excel; Microsoft, Redmond, WA), and for static insulin release and Ca2+ influx results (Figs. 4 and 8), ANOVA with a post hoc Bonferonni test was carried out (using Kaleidagraph; Synergy Software, Reading, PA).
Fig. 2.

Effect of diazoxide, Bay K 8644, and nimodipine on ISR, OCR, NAD(P)H, and Ca2+. Islets were perifused in the presence of 3 mM glucose for 90 min. Glucose was increased to 20 mM at time 0, and diazoxide (50 μM) and Bay K 8644 (10 μM) were applied at 45 and 90 min, respectively. OCR and ISR data were obtained concomitantly from the same perifusions. Samples for ISR were collected every 5 min and measured for indicated time points. The cytosolic Ca2+ [represented as ratio of fluorescence (F340/F380)] and normalized NAD(P)H levels were imaged using an inverted fluorescence microscope and digital camera (see materials and methods).
Fig. 3.

Effect of various doses of KN-62 on OCR and ISR. Islets were perifused in the presence of 3 mM glucose for 90 min, and subsequently glucose was increased to 20 mM at time 0. At 45-min intervals, KN-62 was added sequentially at 0.1, 1, and 10 μM, and then to test whether its effects were irreversible, KN-62 was washed out of the perifusion chamber.
Fig. 5.
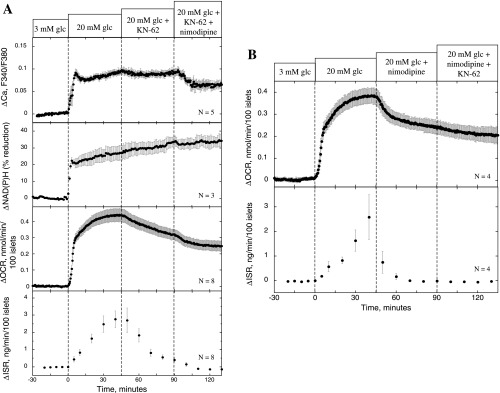
Effect of KN-62 and nimodipine on ISR, OCR, NAD(P)H, and Ca2+. Conditions were as described in the legend of Fig. 2, except that the protocol used involved applying KN-62 (10 μM) and nimodipine (5 μM) at 45 and 90 min, respectively (A), or nimodipine (5 μM) and KN-62 (10 μM) at 45 and 90 min, respectively (B).
Fig. 6.

Effect of KN-62 and nimodipine on Ca2+ (A) and ISR and OCR (B). A: islets were perifused in the presence of 3 mM glucose for 90 min. Glucose was increased to 20 mM at time 0, and at 45 min, nimodipine (10 nM), KN-62 (10 μM), or control solution was applied. Subsequently, at 90 min, 5 μM nimodipine was applied to the islets that had received the test agents. Cytosolic Ca2+ is plotted as the fractional change relative to stimulation by 20 mM glucose. Each curve represents the average of multiple perifusions (n = 3, control; n = 4, nimodipine; n = 5, KN-62). B: for measurements of OCR and ISR, conditions were as described in the legend of Fig. 2, except that the protocol used involved applying 10 nM and 5 μM nimodipine at 45 and 90 min, respectively.
Fig. 7.

Effect of KN-62 and nimodipine on ISR, OCR, and Ca2+ on unstimulated islets. A: conditions were as described in the legend of Fig. 2, except that the protocol used involved adding KN-62 (10 μM) to the inflow media 45 min prior to glucose being increased to 20 mM and nimodipine (5 μM) being added 45 min after glucose was increased. B: nimodipine (5 μM) was added in the presence of 3 mM glucose.
Fig. 9.

Effect of KN-62 and nimodipine on stimulation of ISR, OCR, and Ca2+ by Bay K 8644. Conditions were as described in the legend of Fig. 2, except that the protocol used involved applying KN-62 (10 μM) to glucose-stimulated islets, and subsequently, Bay K 8644 (10 μM) and nimodipine (5 μM) were applied at 45-min intervals.
Fig. 4.

Effect of nimodipine and KN-62 on glucose-stimulated Ca2+ influx. Rates of Ca2+ uptake were measured as described in materials and methods in the presence of indicated agents. Results are the average of 3 separate experiments, where results from each individual experiment were normalized to the islet-associated radioactivity obtained at 20 mM glucose. Statistical analysis was performed using ANOVA with a post hoc Bonferonni test. Ca2+ influx in the presence 20 mM glucose was significantly different from Ca2+ influx in the presence of 3 mM glucose. The presence of nimodipine, but not KN-62, significantly decreased Ca2+ influx.
RESULTS
Activation of Ca2+ influx via L-type Ca2+ channels stimulates oxidative metabolism in islets.
We (42) and others (32) have demonstrated previously that closure of ATP-sensitive K+ (KATP) channels by glibenclamide or tolbutamide results in an increase in OCR. To demonstrate that the L-type Ca2+ channel activity, independent of the KATP channel-induced depolarization, is tightly coupled to changes in ATP usage and ISR (42, 45), we measured the effects of directly activating L-type Ca2+ channels using the combination of diazoxide and Bay K 8644. The protocol involved first establishing a baseline at 3 mM glucose, followed by an increase to 20 mM, and then L-type Ca2+ channels were deactivated by hyperpolarizing the β-cell membrane potential with 50 μM diazoxide, an agent that opens KATP channels. All four measured parameters, cytosolic Ca2+, NAD(P)H, OCR, and ISR, increased in response to the change in glucose concentration (Fig. 2). It should be noted that for the flow systems used for the OCR and ISR measurements, as well as the imaging system for Ca2+ and NAD(P)H, the temporal resolution is limited to ∼3–4 min due to mixing in the tubing and chamber. Therefore, differences in the initial rapid responses to glucose between these four parameters cannot be resolved, including first-phase insulin secretion release. Therefore, in this study, conclusions are based primarily on the steady-state responses. For all measurements made at 3 and 20 mM glucose (n = 46), steady-state values of OCR were 0.39 ± 0.027 and 0.74 ± 0.032 nmol·min−1·100 islets−1 (means ± SE), and steady-state values of ISR were 0.17 ± 0.030 and 2.48 ± 0.20 ng·min−1·100 islets−1, respectively. In response to diazoxide, Ca2+ transiently decreased by 38% and, in response to 10 μM Bay K 8644, increased and remained above glucose-stimulated levels (Fig. 2). In parallel experiments, OCR decreased by 0.061 ± 0.006 nmol·min−1·100 islets−1 (P < 0.005) and ISR by 92 ± 2% (P < 0.005) due to the prevention of voltage-dependent Ca2+ influx by diazoxide (Fig. 2). Subsequent activation of Ca2+ influx by Bay K 8644 reversed diazoxide's inhibition of both OCR and ISR [OCR increased by 0.069 ± 0.01 nmol·min−1·100 islets−1 (P < 0.005), and ISR returned to 63% of its stimulated level (P < 0.05)]. Thus, Ca2+ influx through L-type Ca2+ channels and OCR is tightly coupled, and we have termed this relation CMCP. To test whether the CMCP was driven by changes in TCA cycle activity, NAD(P)H was measured as a reflection of energy production. Glucose elicited a 35% increase in NAD(P)H (Fig. 2), whereas NAD(P)H was unaffected by either compound, precluding the possibility that Ca2+ is driving the changes in OCR by increasing TCA cycle activity.
Three blockers of calmodulin-sensitive proteins did not interact with nimodipine's mechanism of action.
To demonstrate that the CMCP is a downstream target of Ca2+ entry, the effects of several inhibitors of Ca2+-sensitive regulatory proteins were tested. On the basis of previous studies indicating that CaMKII is an important mediator of insulin secretion (11), we first endeavored to test whether the suppression of OCR and ISR by nimodipine was mediated by this protein. Islets were exposed to 1 μM AIP2, an agent that inhibits autophosphorylation (and therefore the activation) of CaMKII (20). This inhibitor did decrease OCR slightly [steady-state ΔOCR was −0.053 ± 0.008 nmol·min−1·100 islets−1 (n = 4, P < 0.05)] but did not decrease ISR [steady-state ΔISR was 1.36 ± 0.72 ng·min−1·100 islets−1 (n = 4, not significant)] (kinetic data not shown). In addition, the decrement in OCR and ISR induced by subsequent exposure to nimodipine was not diminished in the presence of AIP2, indicating a mechanism of action of AIP2 that was independent from that mediating the effects of Ca2+ influx. Similar results were obtained with a myristoylated form of AIP (data not shown). Using identical protocols, blockers of two other Ca2+-sensitive proteins that had previously been suggested as playing a role in insulin secretion (33, 35), myosin light-chain kinase (10 μM ML-7) and calcineurin (10 μM FK-506), were assessed, but neither had any effect on ISR, nor did they prevent nimodipine's inhibition of OCR (data not shown). Thus, blockers of CaMKII, myosin light-chain kinase, and calcineurin did not affect pathways mediating the effects of nimodipine.
KN-62 mimicked the effects of blocking L-type Ca2+ channels on glucose-stimulated OCR and ISR.
We next tested the effects of KN-62, a blocker of calmodulin-dependent kinases that in previous studies potently inhibited insulin secretion (1, 23, 29). As expected, KN-62 inhibited ISR in a dose-dependent fashion, but notably, OCR decreased in parallel with similar sensitivity (Fig. 3). To test the possibility that KN-62's mechanism of action is simply via inhibition of Ca2+ influx, measurements of glucose-stimulated Ca2+ uptake in the presence of nimodipine (5 μM) or KN-62 (10 μM) were made (Fig. 4). Blocking L-type Ca2+ channels with nimodipine reduced glucose-stimulated influx by 81% (P < 0.001), whereas KN-62 had no effect. Thus, it appeared that KN-62 had similar effects on OCR and ISR as nimodipine but without acting on Ca2+ channels in the plasma membrane.
To rigorously compare the effects of nimodipine and KN-62, experiments were carried out with matching protocols. At a concentration of KN-62 that completely inhibited ISR (10 μM), glucose-stimulated OCR was decreased by 0.11 ± 0.011 nmol·min−1·100 islets−1 (n = 8, P < 0.005; Fig. 5A), an amount comparable with the decrease observed with nimodipine (0.13 ± 0.011 nmol·min−1·100 islets−1, n = 4, P < 0.005; Fig. 5B). Both agents potently inhibited ISR, 91 vs. 93% for KN-62 and nimodipine, respectively, whereas neither agent had an effect on NAD(P)H, ruling out inhibition of metabolism by the TCA as the driving force for the decrease in OCR. To fully test whether KN-62 was acting through a mechanism common to the effect of blocking L-type Ca2+ channels, nimodipine was applied subsequent (Fig. 5A) and prior to KN-62 treatment (Fig. 5B). Importantly, in the presence of nimodipine, the effect of KN-62 on OCR was reduced by 85%, and in the presence of KN-62 the effect of nimodipine on OCR was reduced by 65%, suggesting that KN-62 blocked a downstream target of Ca2+.
Despite the fact that no effect of KN-62 on Ca2+ influx was observed, KN-62 did have a minor effect on cytosolic Ca2+, albeit only a small percentage relative to the actions of nimodipine and diazoxide. To test whether a change in Ca2+ of this magnitude could contribute to the effect of KN-62 on OCR and ISR, we assessed the effect of nimodipine at a concentration that elicited a slightly bigger change in Ca2+ than that produced by KN-62. After islets with 20 mM glucose were stimulated, the decrement in cytosolic Ca2+ in response to 10 nM nimodipine was 35% greater than that caused by KN-62 (Fig. 6A). At this concentration of nimodipine, the agent had no detectable affects on OCR and ISR (Fig. 6B). Subsequent administration of 5 μM nimodipine demonstrated the necessity of more substantial decreases in cytosolic Ca2+ for the inhibition of the Ca2+-dependent OCR and ISR to occur.
At basal glucose levels (3 mM), which are associated with only minimal L-type Ca2+ channel activity, KN-62 and nimodipine had only small effects on Ca2+, OCR, and ISR (Fig. 7, A and B). In the presence of KN-62 for 45 min, 20 mM glucose increased OCR by 0.31 ± 0.02 nmol·min−1·100 islets−1 (n = 3), which was 79% of that elicited by glucose in the absence of KN-62. The kinetics of the Ca2+ response to glucose in the presence of KN-62 appeared normal. Note that the magnitude of the increase is not directly interpretable due to intrinsic interexperimental variability of fluorescence. KN-62 blocked ISR in response to 20 mM glucose, and, as in Fig. 5A, the subsequent addition of nimodipine had only a small effect on OCR with KN-62 present. In summary, KN-62 was able to mimic the action of L-type Ca2+ channel blockade on OCR and ISR without significantly affecting Ca2+ influx, and these actions are not explained by the small changes in cytosolic Ca2+ that were observed.
KN-62 mimicked the effects of blocking L-type Ca2+ channels on potentiation of insulin secretion.
In addition to suppressing glucose-stimulated ISR, blocking L-type Ca2+ channels also prevents the potentiation of insulin secretion by acetylcholine (14), GLP-1 (24), arginine (27), and KCl (31). To further test our hypothesized role of the CMCP in the control of ISR, the ability of nimodipine and KN-62 to interfere with potentiation of ISR by these agents was assessed by static assays of ISR (Fig. 8, A and B). As can be seen, all four agents amplified glucose-stimulated ISR as expected, but similar to nimodipine, these effects were abolished by pretreatment of the islets with KN-62. Thus, the potentiation of ISR induced by acetylcholine, GLP-1, and arginine is dependent on a step inhibited by KN-62, demonstrating the essentiality of activating CMCP for sustained ISR to occur. However, in contrast to the ability of KN-62 to prevent the KCl potentiation of ISR, pretreatment with KN-62 did not significantly block stimulation of ISR by an agonist of L-type Ca2+ channel Bay K 8644 (Fig. 8B).
Fig. 8.

Effect of potentiators of glucose-stimulated insulin secretion in the presence and absence of nimodipine (A) or KN-62 (B) on ISR. ISR was measured on islets incubated in wells of 96-well plates containing the indicated concentrations of glucose, GLP-1 (100 nM), Arg (10 mM), Ach (10 μM), KCl (30 mM), and Bay K 8644 (10 μM) without (filled bars) or with 5 μM nimodipine (A, open bars) or 10 μM KN-62 (B, open bars). Statistical analysis was performed using ANOVA with a post hoc Bonferonni test, and differences were considered significant if P < 0.005. ISR for 3 mM glucose was significantly different from all ISRs in the presence of 20 mM glucose in the absence of inhibitors. ISRs in the presence of 20 mM glucose with inhibitors were statistically different from all ISRs in the absence of inhibitors except for Bay K 8644 + or − KN-62. In the absence of inhibitors, ISRs in the presence of GLP-1, Arg, Ach, KCl, and Bay K 8644 were statistically different from 20 mM glucose alone.
Stimulatory effect of Bay K 8644 on OCR and ISR observed in the presence of KN-62.
To further investigate the unexpected ability of Bay K 8644 to override the inhibitory effects of KN-62 on ISR, the kinetics of OCR, Ca2+, and ISR were measured in response to Bay K 8644 and subsequently nimodipine in the presence of KN-62 (Fig. 9). After recapitulating the inhibitory effects of KN-62 on OCR and ISR, Bay K 8644 stimulated all three measured parameters in the presence of KN-62. However, despite the ability of Bay K 8644 to override the effects of KN-62, nimodipine subsequently reversed Bay K 8644's effects. In summary, KN-62 can block downstream effects of Ca2+ influx, including KCl's effects on increased Ca2+ influx, but its effects can be overridden by Bay K 8644. This suggests that Bay K 8644's actions are mediated by a mechanism that is more involved than simply increasing Ca2+ influx, but the concomitant increase in OCR and ISR by Bay K 8644 provides further support that activation of the CMCP is imperative for the stimulation of sustained insulin secretion.
DISCUSSION
A novel model relating Ca2+, energy usage, and insulin secretion.
Ca2+ influx through L-type Ca2+ channels is essential for glucose-stimulated insulin secretion (18). To understand how Ca2+'s actions on insulin secretion are mediated, we have focused previously on the effect of Ca2+ influx on OCR (45). Remarkably, about 35–40% of glucose-stimulated OCR is directly linked to the influx of Ca2+ through L-type Ca2+ channels, a process that by itself does not utilize energy, whereas about 25% is linked to protein synthesis (45). Contrary to expectations, the exocytosis of insulin granules does not contribute significantly to the Ca2+-dependent OCR (10, 45). Since protein synthesis is not sensitive to Ca2+, and ion pumping by Na+-K+ and SERCA pumps was not found to contribute significantly to the effect of Ca2+ influx, the mechanism mediating the usage of energy by this Ca2+-sensitive process remains unidentified. On the basis of these considerations, we postulated previously that Ca2+-sensitive OCR reflects a highly energetic biochemical cascade that couples Ca2+ influx to insulin secretion (14) and have termed this process CMCP until its underlying mechanism can be identified (Fig. 1). The major findings of the present study provide further support for this cascade in islets by demonstrating that an inhibitor of calmodulin-dependent proteins, KN-62, mimicked the effects of L-type Ca2+ channel blockade on both ISR and OCR by a mechanism independent of Ca2+ turnover. These findings provide further support for the conceptual model proposed based on the results of our previous studies (Fig. 1) (14), which depict the KN-62-sensitive CMCP activated exclusively by Ca2+ entering through L-type Ca2+ channels, which in turn increases OCR and ISR. Importantly, our data support the absolute requirement for activation of CMCP for significant ISR to occur since KN-62 blocked ISR even in the presence of potentiators of glucose-stimulated ISR such as GLP-1, arginine, and acetylcholine.
KN-62 inhibited an energy-requiring step downstream of L-type Ca2+ channels.
The design to test the CMCP hypothesis was based on the stipulation that blocking a downstream target of Ca2+ influx via L-type Ca2+ channels would mimic the effect of blocking L-type Ca2+ channels on OCR and ISR. Of the four inhibitors tested, KN-62, a compound whose major action is best recognized as a blocker of CaMKII (19), uniquely satisfied these criteria; inhibition of OCR and ISR by KN-62 and nimodipine was similar, and after pretreatment with the other agent, the effects of nimodipine or KN-62 on OCR and ISR were minimal. The effect of diazoxide on OCR was slightly less than that of nimodipine. However, a lower concentration of diazoxide than what is typical to avoid its inhibitory effect on F0F1 ATPase synthesis was used (6). Central to the validity of the study, KN-62 had little effect on Ca2+ influx in the intact rat islets used in our study. The influx of Ca2+ as measured by the uptake of 45Ca2+ does underestimate the rate of influx since some of the 45Ca2+ leaks out during the accumulation of tracer, and therefore, it is not a true unidirectional flux. However, the method was chosen so that the experiments could be done with intact islets under conditions closely matching those that were used for the OCR measurements, and the clear demonstration of a nimodipine effect validated the approach.
It should be noted that three previous studies have reported that KN-62 leads to a decrease in Ca2+ influx (1, 2, 23). Using patch clamping on single mouse islet cells, studies by Ammälä et al. (1) found that Ca2+ influx was reduced by 50% during the 10-s duration of the experiment. A previous study in HIT cells showed an effect of KN-62 on Ca2+ influx through L-type Ca2+ channels (23). However, 1 μM KN-62 completely inhibited nutrient- and KCl-stimulated Ca2+ rise and insulin secretion in the study, a concentration that had little effect on ISR by the rat islets in our experiments. So it appears that HIT cells are particularly sensitive to KN-62. In studies conducted by Bhatt et al. (2), 10 μM KN-62 suppressed KCl-induced increases in cytosolic Ca2+ by INS-1 cells. Despite the fact that these studies suggested that there might be an effect of KN-62 on Ca2+ turnover in rat islets, we observed no effect of KN-62 on Ca2+ influx, and we observed only an effect on cytosolic Ca2+ that was small compared with that induced by maximally effective concentrations of nimodipine. Importantly, this small effect on cytosolic Ca2+ was unlikely to account for the similar effects of the two agents on OCR and ISR since these parameters remained unchanged in the face of slightly bigger decreases in cytosolic Ca2+ induced with low concentrations of nimodipine. This being the case, our results demonstrate the existence of a molecular target of Ca2+ that couples the effect of Ca2+ through L-type Ca2+ channels on OCR and ISR independently of Ca2+ turnover and support the existence of the CMCP.
Potentiation of insulin secretion stimulated by acetylcholine, GLP-1, and arginine downstream of CMCP.
Similar to Ca2+ influx through L-type Ca2+ channels, the KN-62-sensitive step was also essential for insulin secretion to occur in the presence of strong potentiators of insulin secretion. Thus, KN-62 abolished ISR in the presence of acetylcholine, GLP-1, or arginine. Acetylcholine and GLP-1 are thought to act by altering the efficacy with which cytosolic Ca2+ stimulates insulin secretion (18). Despite the lack of understanding of how amplification occurs, our data suggest that the CMCP is linked to the triggering of insulin secretion rather than amplification. Amplification of insulin secretion by activation of protein kinase C (by TPA) or cAMP-dependent signals such as protein kinase A (by IBMX) occurs in the absence of changes in OCR (10, 14, 45). However, KN-62 mimics both the OCR and ISR effects of blocking L-type Ca2+ channels, and amplification is ineffectual in the absence of triggering. Therefore, KN-62 is blocking a step downstream of the triggering step by L-type Ca2+ channels but upstream of amplification.
Bay K 8644 has multiple mechanisms of action on islet CMCP.
In contrast to stimulation of ISR by KCl, which was fully inhibited by KN-62, Bay K 8644 stimulation of OCR and ISR overcame the inhibitory effects of KN-62. This indicates that Bay K 8644 has two effects, one to increase Ca2+ influx by its known interaction with the dihydropyridine binding site on the L-type Ca2+ channel (50) and one that prevents KN-62 action. This could occur if Bay K 8644 affected separate proteins, each of which would carry out these roles, or, alternatively, Bay K 8644 binding to the L-type Ca2+ channel has two or more actions. To clarify, although taken singly, the inability of KN-62 to block Bay K 8644 could be interpreted as inconsistent with the concept of the CMCP. However, in the context of KN-62's strong effect to block the effects of KCl-induced Ca2+ influx on ISR, the data indicate that Bay K 8644 has an additional effect above and beyond a direct effect on increasing Ca2+ influx. Taken together, in response to nimodipine, KN-62, and Bay K 8644, changes in Ca2+ influx, OCR, and ISR correlated well, a central tenet of the CMCP postulate where activation of the CMCP is essential for insulin secretion to occur.
Target of Ca2+ traversing L-type Ca2+ channels.
Certainly, a long-term goal of this research is to identify the Ca2+-sensitive target(s) that controls the Ca2+-mediated increase in OCR and ISR induced by glucose. The search for a “Ca2+ sensor” has been ongoing for many years, and candidates include myosin light-chain kinase (33), Ca2+-dependent protein kinases (11), calcineurin (35), and, more recently, synaptotagmins (13). Our view is that characterization of functional correlates like Ca2+-sensitive OCR and sensitivity to KN-62 and Bay K 8644 provides critical evidence that will allow for positive identification of the protein or proteins that comprise the Ca2+ sensor and the CMCP. However, despite intensive study (1, 2, 23, 29, 37, 48), the mechanism mediating KN-62's inhibitory effect on insulin secretion remains unresolved. Some studies have suggested a significant role for CaMKII, consistent with the known action of KN-62 on this enzyme (11, 48). Accordingly, we tested another blocker of CaMKII, AIP2, but in contrast to KN-62, it increased ISR. It is important to note that, despite the common referral of KN-62 in the literature as a CaMKII inhibitor (19), the blocker appears to target a number of calmodulin-dependent and calmodulin-independent proteins such as CaMKIV and -V (19), glycogen synthase kinase 3b, p38-regulated/activated kinase, and mitogen-activated protein kinase-activated protein kinase 2 (7). We also cannot rule out the possibility that the actions of KN-62 are being mediated by other calmodulin-binding proteins such as adenylyl cyclases (16) or actin polymerization (49), whose sensitivity to KN-62 is not known, or an effect unrelated to calmodulin binding. A known calmodulin-independent effect of KN-62 is inhibition of P2 purinergic receptors (40), an effect that has been shown to decrease cytosolic Ca2+ (34). However, interaction with this receptor is unlikely to be mediating KN-62's effects on ISR and OCR, since the decrease in Ca2+ concentration caused by KN-62 was not high enough to elicit changes in these two parameters. Another potential mediator of Ca2+'s effect on OCR and ISR is pumping Ca2+ out of the cell by plasma membrane Ca2+ ATPase (PMCA), the rate of which may also be activated by calmodulin (41). To determine the contribution of PMCA to Ca2+-sensitive OCR, it would be ideal to acutely block PMCA activity. However, no specific inhibitors exist. Thus, the data in this study demonstrate that the CMCP is sensitive to KN-62, and this sensitivity can be overcome by Bay K 8644, but the identity of the protein(s) that mediates the CMCP remains unknown.
High-energy usage an intrinsic control feature of insulin secretion.
A basic model of triggering and amplification of insulin secretion by Henquin (18) has postulated that an increase in cytosolic Ca2+ is an essential factor enabling the stimulation of insulin secretion to occur and the efficacy of the cytosolic Ca2+ to be amplified. Our working model (Fig. 1) expands on this model by incorporating the energy usage associated with the activation of the secretory process. Indeed, high-energy usage is an intrinsic characteristic of the β-cell but one whose contribution to secretory function remains unknown. Islets receive 10% of the pancreatic blood flow (21) despite comprising only 1–2% of the mass (4). The high usage of energy may make the islets susceptible to overstimulation, which has been imputed as a major factor in loss of islet function in type 2 diabetes (36). Thus it will be critical to understand the role of energy usage in addition to ATP generation to elucidate how β-cells operate and the factors that contribute to their failure in type 2 diabetes. The data generated in this study of KN-62 actions provide further support that the processes of Ca2+ influx, a metabolic factor, and insulin secretion are coupled by a highly energetic process that is essential for amplification of insulin secretion to occur.
GRANTS
This research was funded by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (DK-17047 and DK-063986) and a grant from the National Science Foundation (SBIR IIP-0750508).
Acknowledgments
Special thanks to Dr. Les Satin for sage editorial input.
REFERENCES
- 1.Ammälä C, Eliasson L, Bokvist K, Larsson O, Ashcroft FM, Rorsman P. Exocytosis elicited by action potentials and voltage-clamp calcium currents in individual mouse pancreatic B-cells. J Physiol 472: 665–688, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhatt HS, Conner BP, Prasanna G, Yorio T, Easom RA. Dependence of insulin secretion from permeabilized pancreatic beta-cells on the activation of Ca(2+)/calmodulin-dependent protein kinase II. A re-evaluation of inhibitor studies. Biochem Pharmacol 60: 1655–1663, 2000. [DOI] [PubMed] [Google Scholar]
- 3.Bokvist K, Eliasson L, Ammälä C, Renström E, Rorsman P. Co-localization of L-type Ca2+ channels and insulin-containing secretory granules and its significance for the initiation of exocytosis in mouse pancreatic B-cells. EMBO J 14: 50–57, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52: 102–110, 2003. [DOI] [PubMed] [Google Scholar]
- 5.Chance B Electron transfer: pathways, mechanisms, and controls. Annu Rev Biochem 46: 967–980, 1977. [DOI] [PubMed] [Google Scholar]
- 6.Comelli M, Metelli G, Mavelli I. Downmodulation of mitochondrial F0F1 ATP synthase by diazoxide in cardiac myoblasts: a dual effect of the drug. Am J Physiol Heart Circ Physiol 292: H820–H829, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351: 95–105, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Detimary P, Gilon P, Henquin JC. Interplay between cytoplasmic Ca2+ and the ATP/ADP ratio: a feedback control mechanism in mouse pancreatic islets. Biochem J 333: 269–274, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Detimary P, Jonas JC, Henquin JC. Possible links between glucose-induced changes in the energy state of pancreatic B cells and insulin release. Unmasking by decreasing a stable pool of adenine nucleotides in mouse islets. J Clin Invest 96: 1738–1745, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doliba NM, Qin W, Vatamaniuk MZ, Buettger CW, Collins HW, Magnuson MA, Kaestner KH, Wilson DF, Carr RD, Matschinsky FM. Cholinergic regulation of fuel-induced hormone secretion and respiration of SUR1−/− mouse islets. Am J Physiol Endocrinol Metab 291: E525–E535, 2006. [DOI] [PubMed] [Google Scholar]
- 11.Easom RA CaM kinase II: a protein kinase with extraordinary talents germane to insulin exocytosis. Diabetes 48: 675–684, 1999. [DOI] [PubMed] [Google Scholar]
- 12.Frankel BJ, Atwater I, Grodsky GM. Calcium affects insulin release and membrane potential in islet β-cells. Am J Physiol Cell Physiol 240: C64–C72, 1981. [DOI] [PubMed] [Google Scholar]
- 13.Gauthier BR, Wollheim CB. Synaptotagmins bind calcium to release insulin. Am J Physiol Endocrinol Metab 295: E1279–E1286, 2008. [DOI] [PubMed] [Google Scholar]
- 14.Gilbert M, Jung SR, Reed BJ, Sweet IR. Islet oxygen consumption and insulin secretion tightly coupled to calcium derived from L-type calcium channels but not from the endoplasmic reticulum. J Biol Chem 283: 24334–24342, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Halestrap AP The regulation of the matrix volume of mammalian mitochondria in vivo and in vitro and its role in the control of mitochondrial metabolism. Biochim Biophys Acta 973: 355–382, 1989. [DOI] [PubMed] [Google Scholar]
- 16.Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol 41: 145–174, 2001. [DOI] [PubMed] [Google Scholar]
- 17.Henquin JC Relative importance of extracellular and intracellular calcium for the two phases of glucose-stimulated insulin release: studies with theophylline. Endocrinology 102: 723–730, 1978. [DOI] [PubMed] [Google Scholar]
- 18.Henquin JC Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49: 1751–1760, 2000. [DOI] [PubMed] [Google Scholar]
- 19.Hidaka H, Yokokura H. Molecular and cellular pharmacology of a calcium/calmodulin-dependent protein kinase II (CaM kinase II) inhibitor, KN-62, and proposal of CaM kinase phosphorylation cascades. Adv Pharmacol 36: 193–219, 1996. [DOI] [PubMed] [Google Scholar]
- 20.Ishida A, Shigeri Y, Tatsu Y, Uegaki K, Kameshita I, Okuno S, Kitani T, Yumoto N, Fujisawa H. Critical amino acid residues of AIP, a highly specific inhibitory peptide of calmodulin-dependent protein kinase II. FEBS Lett 427: 115–118, 1998. [DOI] [PubMed] [Google Scholar]
- 21.Jansson L, Hellerström C. Stimulation by glucose of the blood flow to the pancreatic islets of the rat. Diabetologia 25: 45–50, 1983. [DOI] [PubMed] [Google Scholar]
- 22.Krarup T, Jakobsen LD, Jensen BS, Hoffmann EK. Na+-K+-2Cl− cotransport in Ehrlich cells: regulation by protein phosphatases and kinases. Am J Physiol Cell Physiol 275: C239–C250, 1998. [DOI] [PubMed] [Google Scholar]
- 23.Li G, Hidaka H, Wollheim CB. Inhibition of voltage-gated Ca2+ channels and insulin secretion in HIT cells by the Ca2+/calmodulin-dependent protein kinase II inhibitor KN-62: comparison with antagonists of calmodulin and L-type Ca2+ channels. Mol Pharmacol 42: 489–498, 1992. [PubMed] [Google Scholar]
- 24.Lu M, Wheeler MB, Leng XH, Boyd AE 3rd. The role of the free cytosolic calcium level in beta-cell signal transduction by gastric inhibitory polypeptide and glucagon-like peptide I(7–37). Endocrinology 132: 94–100, 1993. [DOI] [PubMed] [Google Scholar]
- 25.Luciani DS, Misler S, Polonsky KS. Ca2+ controls slow NAD(P)H oscillations in glucose-stimulated mouse pancreatic islets. J Physiol 572: 379–392, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsumoto S, Shibata S, Kirchhof N, Hiraoka K, Sageshima J, Zhang XW, Gilmore T, Ansite J, Zhang HJ, Sutherland DE, Hering BJ. Immediate reversal of diabetes in primates following intraportal transplantation of porcine islets purified on a new histidine-lactoioniate-iodixanol gradient (Abstract). Transplantation 67: S220, 1999. [Google Scholar]
- 27.McClenaghan NH, Barnett CR, O'Harte FP, Flatt PR. Mechanisms of amino acid-induced insulin secretion from the glucose-responsive BRIN-BD11 pancreatic B-cell line. J Endocrinol 151: 349–357, 1996. [DOI] [PubMed] [Google Scholar]
- 28.Mears D Regulation of insulin secretion in islets of Langerhans by Ca(2+)channels. J Membr Biol 200: 57–66, 2004. [DOI] [PubMed] [Google Scholar]
- 29.Niki I, Okazaki K, Saitoh M, Niki A, Niki H, Tamagawa T, Iguchi A, Hidaka H. Presence and possible involvement of Ca/calmodulin-dependent protein kinases in insulin release from the rat pancreatic beta cell. Biochem Biophys Res Commun 191: 255–261, 1993. [DOI] [PubMed] [Google Scholar]
- 30.Niswender CM, Willis BS, Wallen A, Sweet IR, Jetton TL, Thompson BR, Wu C, Lange AJ, McKnight GS. Cre recombinase-dependent expression of a constitutively active mutant allele of the catalytic subunit of protein kinase A. Genesis 43: 109–119, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Ohta M, Nelson J, Nelson D, Meglasson MD, Erecinska M. Effect of Ca++ channel blockers on energy level and stimulated insulin secretion in isolated rat islets of Langerhans. J Pharmacol Exp Ther 264: 35–40, 1993. [PubMed] [Google Scholar]
- 32.Panten U, Zunkler BJ, Scheit S, Kirchhoff K, Lenzen S. Regulation of energy metabolism in pancreatic islets by glucose and tolbutamide. Diabetologia 29: 648–654, 1986. [DOI] [PubMed] [Google Scholar]
- 33.Penn EJ, Brocklehurst KW, Sopwith AM, Hales CN, Hutton JC. Ca2+-Calmodulin dependent myosin light-chain phosphorylating activity in insulin-secreting tissues. FEBS Lett 139: 4–8, 1982. [DOI] [PubMed] [Google Scholar]
- 34.Poulsen CR, Bokvist K, Olsen HL, Hoy M, Capito K, Gilon P, Gromada J. Multiple sites of purinergic control of insulin secretion in mouse pancreatic beta-cells. Diabetes 48: 2171–2181, 1999. [DOI] [PubMed] [Google Scholar]
- 35.Renström E, Ding WG, Bokvist K, Rorsman P. Neurotransmitter-induced inhibition of exocytosis in insulin-secreting beta cells by activation of calcineurin. Neuron 17: 513–522, 1996. [DOI] [PubMed] [Google Scholar]
- 36.Ritzel RA, Hansen JB, Veldhuis JD, Butler PC. Induction of beta-cell rest by a Kir6.2/SUR1-selective K(ATP)-channel opener preserves beta-cell insulin stores and insulin secretion in human islets cultured at high (11 mM) glucose. J Clin Endocrinol Metab 89: 795–805, 2004. [DOI] [PubMed] [Google Scholar]
- 37.Rucha A, Verspohl EJ. Heterologous desensitization of insulin secretion by GIP (glucose-dependent insulinotropic peptide) in INS-1 cells: the significance of Galphai2 and investigations on the mechanism involved. Cell Biochem Funct 23: 205–212, 2005. [DOI] [PubMed] [Google Scholar]
- 38.Satin LS, Tavalin SJ, Kinard TA, Teague J. Contribution of L- and non-L-type calcium channels to voltage-gated calcium current and glucose-dependent insulin secretion in HIT-T15 cells. Endocrinology 136: 4589–4601, 1995. [DOI] [PubMed] [Google Scholar]
- 39.Schwaninger M, Blume R, Krüger M, Lux G, Oetjen E, Knepel W. Involvement of the Ca(2+)-dependent phosphatase calcineurin in gene transcription that is stimulated by cAMP through cAMP response elements. J Biol Chem 270: 8860–8866, 1995. [DOI] [PubMed] [Google Scholar]
- 40.Sluyter R, Shemon AN, Barden JA, Wiley JS. Extracellular ATP increases cation fluxes in human erythrocytes by activation of the P2X7 receptor. J Biol Chem 279: 44749–44755, 2004. [DOI] [PubMed] [Google Scholar]
- 41.Strehler EE, Zacharias DA. Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol Rev 81: 21–50, 2001. [DOI] [PubMed] [Google Scholar]
- 42.Sweet IR, Cook DL, DeJulio E, Wallen AR, Khalil G, Callis J, Reems J. Regulation of ATP/ADP in pancreatic islets. Diabetes 53: 401–409, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Sweet IR, Cook DL, Lernmark A, Greenbaum CJ, Wallen AR, Marcum ES, Stekhova SA, Krohn KA. Systematic screening of potential beta-cell imaging agents. Biochem Biophys Res Commun 314: 976–983, 2004. [DOI] [PubMed] [Google Scholar]
- 44.Sweet IR, Cook DL, Wiseman RW, Greenbaum CJ, Lernmark A, Matsumoto S, Teague JC, Krohn KA. Dynamic perifusion to maintain and assess isolated pancreatic islets. Diabetes Technol Ther 4: 67–76, 2002. [DOI] [PubMed] [Google Scholar]
- 45.Sweet IR, Gilbert M. Contribution of calcium influx in mediating glucose-stimulated oxygen consumption in pancreatic islets. Diabetes 55: 3509–3519, 2006. [DOI] [PubMed] [Google Scholar]
- 46.Sweet IR, Khalil G, Wallen AR, Steedman M, Schenkman KA, Reems JA, Kahn SE, Callis JB. Continuous measurement of oxygen consumption by pancreatic islets. Diabetes Technol Ther 4: 661–672, 2002. [DOI] [PubMed] [Google Scholar]
- 47.Trus M, Corkey RF, Nesher R, Richard AM, Deeney JT, Corkey BE, Atlas D. The L-type voltage-gated Ca2+ channel is the Ca2+ sensor protein of stimulus-secretion coupling in pancreatic beta cells. Biochemistry 46: 14461–14467, 2007. [DOI] [PubMed] [Google Scholar]
- 48.Wenham RM, Landt M, Walters SM, Hidaka H, Easom RA. Inhibition of insulin secretion by KN-62, a specific inhibitor of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem Biophys Res Commun 189: 128–133, 1992. [DOI] [PubMed] [Google Scholar]
- 49.Yuan J, Shi GX, Shao Y, Dai G, Wei JN, Chang DC, Li CJ. Calmodulin bound to stress fibers but not microtubules involves regulation of cell morphology and motility. Int J Biochem Cell Biol 40: 284–293, 2008. [DOI] [PubMed] [Google Scholar]
- 50.Zahradníková A, Minarovic I, Zahradník I. Competitive and cooperative effects of Bay K8644 on the L-type calcium channel current inhibition by calcium channel antagonists. J Pharmacol Exp Ther 322: 638–645, 2007. [DOI] [PubMed] [Google Scholar]