

Abstract
The corpus luteum (CL) plays a central role in the maintenance of pregnancy in rodents, mainly by secreting progesterone. Female mice lacking prolactin (PRL) receptor (R) are sterile due to a failure of embryo implantation, which is a consequence of decreased luteinizing hormone (LH) receptor expression in the CL and inadequate levels of progesterone. We attempted to treat PRLR−/− females with human chorionic gonadotropin (hCG) and showed a de novo expression of LHR mRNA in the corpora lutea. Binding analysis confirmed that the LHR in hCG-treated PRLR−/− animals was functional. This was accompanied with increased expression of steroidogenic enzymes involved in progesterone synthesis. Despite these effects, no embryo implantation was observed because of high expression of 20α-hydroxysteroid dehydrogenase. To better appreciate the molecular mechanisms underlying maintenance of the CL, a series of mRNA expression-profiling experiments was performed on isolated corpora lutea of PRLR−/− and hCG-treated PRLR−/− mice. This approach revealed several novel candidate genes with potentially pivotal roles in ovarian function, among them, p27, VE-cadherin, Pten, and sFRP-4, a member of the Wnt/frizzled family. This study showed the differential role of PRL and LH in CL function and identified new targets of these hormones in luteal cells.
Keywords: prolactin receptor, luteinizing hormone receptor, corpus luteum
the corpus luteum (CL) plays a central role in the maintenance of pregnancy. This function is carried out largely by progesterone, which is the main steroid synthesized by this transient endocrine gland (27). Activation of the luteinizing hormone (LH) receptor (R) in follicular cells by the preovulatory LH surge causes ovulation and rapidly initiates a program of terminal differentiation of the ovulated follicle into a CL through a process termed luteinization. There are structural and genomics changes that lead to the terminal differentiation of follicular cells into nondividing progesterone-producing luteal cells. Cells undergoing luteinization must stop dividing and begin expressing a new set of molecules that will allow luteal cells to survive in a different hormonal environment. Thus the final luteal cell phenotype depends on a specific combination of genes encoding for regulatory proteins such as receptors, transcription factors, and signaling proteins, which ensures the expression of only those genes necessary for luteal cell function. One of the most important changes during luteinization is the alteration in the cellular responsiveness to external signals allowing luteal cells to respond to a new set of hormones, the most important being prolactin (PRL) and LH. The expression of the LHR increases after luteinization and becomes highly abundant in the CL. This upregulation of LHR during CL formation has been shown to be due to PRL both in vivo (7, 24) and in vitro (21). In rodents, semicircadian surges of PRL secretion are induced by cervical stimuli, which are believed to be responsible for the conversion of the CL to the corpus luteum of pregnancy, leading to an increase in the life span of this gland and its capacity to secrete a sufficient amount of progesterone to maintain pregnancy (10, 11). This conversion is thought to involve distinct luteotropic effects of PRL: upregulation of LH receptor expression as well as progesterone secretion (8) and repression of 20α-hydroxysteroid dehydrogenase (20α-HSD) (1), the enzyme involved in progesterone catabolism. On the other side, other factors, like prostaglandin F2α (PGF2α), are involved in the regulation of luteolysis. PGF2α and PRL seem to act in an opposite manner to regulate the expression of key luteal genes, particularly 20α-HSD (25, 26). The development of the PRLR knockout mouse model has contributed to the understanding of PRL functions (20) and has mainly highlighted its essential role in reproduction. We previously demonstrated that PRLR−/− females are infertile due to a failure of implantation (20). Luteinization and CL formation do take place in the absence of PRLR. However, 2 days after mating, PRLR knockout mice exhibited corpora lutea undergoing regression and displaying strong DNA cleavage associated with low indications of vascularization and absence of sufficient progesterone to support implantation and subsequent placental development and maintenance. Because the CL is the main source of progesterone production in rodents, progesterone administration was able to rescue preimplantation egg development and embryo implantation in PRLR−/− females (3). The PRLR is thus a key component regulating ovarian function and governing the regulation of progesterone secretion, and, in fact, PRL triggers an early signal that induces the survival and steroidogenic capacity of the CL.
We studied the luteal expression of LHR and steroidogenesis in PRLR−/− mice treated by human chorionic gonadotropin (hCG) to determine whether the stimulation of LHR and progesterone is independently mediated by PRL, or whether progesterone production depends solely on high expression of LHR and LH stimulation. We found that LH/hCG can stimulate the expression of its cognate receptor in the CL and stimulates the expression of enzymes involved in steroidogenesis in the absence of PRL signaling. It, however, does not prevent the expression of 20α-HSD, maintain progesterone levels, nor rescue the fertility of the PRLR−/− mice. Next, to better appreciate the molecular mechanisms underlying maintenance of the CL, we performed a series of mRNA expression profiling experiments using macroarrays. This approach revealed several novel candidate genes with pivotal roles in ovarian function.
MATERIALS AND METHODS
Experimental animals.
Wild-type and PRLR−/− female mice were generated by intercrosses of heterozygous mice (PRLR+/−), as described previously (20). The PRLR+/− mice used for the interbreeding study were inbred 129/Sv. PCR analysis of tail DNA determined the genotypes of the offspring as described previously (3). Females were mated with fertile males to induce pregnancy; the day that the vaginal plug was first observed was considered day 0.5 of pregnancy. Animals were housed in a 12:12-h day-night cycle at 22°C and 50% relative humidity with food and water provided ad libitum. Experimental designs and procedures are in agreement with the guidelines of the animal ethics committee of the Ministère de l'Agriculture, France. This study was reviewed and approved by the Comite Regional D'Ethique sur L'Experimentation Animale d'ile de France René Descartes.
Animal handling and hormonal treatment.
Wild-type or PRLR−/− female mice (6–8 wk of age) were mated with fertile males of the same strain to induce pregnancy. The morning a vaginal plug was seen was designated day 0.5 of pregnancy. PRLR−/− female mice were treated intrperitoneally two times daily with 0.125 IU hCG chorulon (Intervet). Ovaries were collected on either day 2.5 or day 5.5 of pregnancy and fixed on either Bouin's fixative for hematoxylin-eosin-safran staining or with 4% paraformaldehyde for immunohistochemistry. Corpora lutea were isolated by microdissection on day 2.5 of pregnancy and were immediately conserved at −80°C. To examine for number of implantation sites, mice were treated by hCG on day 0.5 of gestation until day 5.5, when the number of implantation sites was recorded by monitoring the localized uterine vascular permeability after intravenous injection of saline Chicago blue dye solution (Sigma, St. Louis, MO).
In situ hybridization.
Ovaries were flash-frozen and serially sectioned (5 μm) using a Zeiss cryostat. Sections were mounted on positively charged glass slides, fixed in 4% paraformaldehyde (pH 7.4) for 10 min at room temperature, washed in 2× PBS for 5 min, incubated in 0.2% acetic anhydride in 0.1 M triethanolamine (pH 8.0) for 10 min, and rinsed in 2× saline-sodium citrate (SSC) 2 × 2 min as previously described (9). [35S]UTP-labeled sense and anti-sense probes were synthesized using SP6 or T7 RNA polymerase (Promega, Madison, WI). Sections were coated with the labeled probe and then incubated overnight at 45°C. After hybridization, slides were washed at high stringency and treated with ribonuclease A (20 μg/ml) at 37°C for 20 min, washed for 30 min at 60°C in 1× SSC, and dehydrated through an ethanol series. After drying, the slides were dipped in photographic NTB-2 emulsion (Eastman Kodak) and exposed at 4°C for 5–15 days. Slides were developed with Kodak D-19 developer and fixer (Eastman Kodak) and then stained with hematoxylin.
Binding assays on histological sections.
Ovaries were collected and were coated with cryoprotectant embedding medium (Tissue-Tek, Miles, Elkhart, IN), frozen in cold isopentane, and then stored at −20°C. The binding of 125I-labeled hCG to ovarian frozen sections was studied by an autoradiographic method, as described previously (6). Briefly, ovaries were serially sectioned at a thickness of 5 μm with a cryostat. After fixation for 10 min at 4°C in picric acid-formaldehyde, sections were stored at −20°C and then circled with Depex (Gurr, BDH, Poole, UK). hCG was iodinated by the Iodogen method (Sigma, St. Quentin, France) and purified by Sephadex G-50 chromatography. Sections were incubated in a drop of PBS (0.1% BSA, pH 7.4) containing 125I-labeled hCG (4 × 105 cpm/50 μl). To determine nonspecific binding, for the tested ligand, an adjacent serial section of ovary was incubated with an excess of unlabeled ligand (500 ng/50 μl hCG). At the end of the incubation period, the sections were washed two times in PBS, postfixed in 3% glutaraldehyde-PBS, washed in PBS, air-dried, and stained with Feulgen. For autoradiography, they were then dipped in Kodak NTB-2 emulsion diluted 1:1 with distilled water, air-dried, exposed for 2 wk at 4°C, then developed and fixed by classical procedures.
Progesterone measurements.
Serum progesterone (P4) was measured by RIA (Immunotech Beckman Coulter France). The analytical sensitivity was 0.05 ng/ml, and the antibody used in the immunoassay was highly specific for progesterone. Intra-assay coefficient varied from 3 to 6%, whereas interassay coefficient varied from 5 to 7% (3, 15).
Semiquantitative RT-PCR analysis.
Total RNA was extracted from CL at day 2.5 by the Nucleospin RNA kit II (BD biosciences). One microgram was reverse transcribed using 1 μg oligo(dT) (Amersham Pharmacia Biotech), 200 U of Moloney murine leukemia virus reverse transcriptase (GIBCO-BRL), 1 mM of dNTP, and 40 units of RNasin (GIBCO-BRL) for 30 min at 37°C. The amplification of LHR, 20α-HSD, P450scc, Frizzled 3 (Fz-3), Frizzled 4 (Fz-4), Wnt-4, secreted frizzled-related protein-4 (sfrp-4), VE-cadherin, p27, Phosphatase and Tensin Homolog (Pten), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transcripts was performed with oligonucleotides primers (Eurogentec) listed below (Table 1) using Taq polymerase (GIBCO-BRL) on 1 μl of the RT reaction in a mix containing 0.5 mM of dNTP and 2.5 mM of MgCl2 in 20 μl final. PCR was carried out for 15, 22, 25, or 28 cycles to ensure the linear amplification.
Table 1.
Primer sequences of genes analyzed in PCR and RT-PCR
| Primer | Primer Sequence | Product Size, bp | Tannealing, °C |
|---|---|---|---|
| LHR sense | CTTATACATAACCACCATAC CAG | 516 | 61 |
| LHR anti | ATCCCAGCCACTGAGTTCA TTC | ||
| 20α-HSD sense | GCAGGATTGGCCAAGTCC ATC | 440 | 61 |
| 20α-HSD anti | GTACCGAAGATTTCTGTC CAG | ||
| P450scc sense | GCCAACATTACCGAGATGC | 426 | 61 |
| P450scc anti | ACACCCCAGCCAAAGCC | ||
| Pten sense | AAGGCACAAGAGGCCCTAGAT | 230 | 61 |
| Pten anti | CCTGATCACAAAGACTTCCAT | ||
| GAPDH sense | GAGGGATCTCGCTCCTGGAAGA | 458 | 61 |
| GAPDH anti | GGTGAAGGTCGGAGTCAACGGA | ||
| Fz-3 sense | CCGTACCAGGTTACTCAGATGAGT | 604 | 57 |
| Fz-3 anti | GCACTGGTTCCATCCTCCTCAATA | ||
| Fz-4 sense | CTGGGCACTCTTTCGATATTCTGC | 482 | 55 |
| Fz-4 anti | TCCAAGCTGTGACCTGCTAAAGTG | ||
| Wnt-4 sense | TTCTCACAGTCCTTTTGGACG | 310 | 65 |
| Wnt-4 anti | TCTGTATGTGGCTTGAACTGTG | ||
| Sfrp-4 sense | CATCAAGCCCTGCAAGTCTG | 766 | 60 |
| Sfrp-4 anti | TAAGGGTGGCTCCATCACAG | ||
| VE-cadherin sense | TCTCTTCATCGATGTGCATT | 154 | 60 |
| VE-cadherin anti | ACGGACAAGATCAGCTCCTC | ||
| p27 sense | CAGAATCATAAGCCCCTGGA | 224 | 57 |
| p27 antisense | TCTGACGAGTCAGGCATTTG |
Tannealing, annealing temperature; LHR, leutenizing hormone receptor; 20α-HSD, 20α-hydroxysteroid dehydrogenase; Pten, phosphatase and tensin homolog; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Fz, frizzled; Sfrp, secreted frizzled-related protein.
Products of LHR, 20α-HSD, and P450scc were resolved by electrophoresis on 1% agarose gel, which was stained with ethidium bromide. Data were analyzed with the EDAS-120 system (Kodak digital imaging system) and software. Primers for mouse Fz-4, Wnt-4, sfrp-4, p27, and Pten were added to one aliquot, and primers for GAPDH were added to the other aliquot as an internal control. [32P]deoxy-CTP (ICN, Los Angeles, CA), Taq polymerase, and Thermocycle buffer (Promega) were also included in the PCRs. Mouse Fz-4, Wnt-4, sfrp-4, p27, and Pten were amplified at 22 cycles, 25 cycles for VE-cadherin, and 28 cycles for Fz-3 (94 C for 1 min, 60 C for 2 min, 72 C for 3 min). The amplified cDNA products were resolved on a 5% polyacrylamide gel, which was dried and exposed to film. The radioactive PCR product bands were quantified by using a Storm 860 PhosphorImager (Molecular Dynamics, Sunnyvale, CA). The intensity of signals was standardized by the use of GAPDH, which was included in each experiment as an internal control.
Immunohistochemistry.
Tissues were fixed overnight in 4% paraformaldehyde and embedded in paraffin, and 5-μm-thick sections were prepared. Following deparaffinization, antigen retrieval was performed by maintaining slides in 10 mM citrate buffer, pH 6.0 at 750 W, and microwaved two times for 5 min, followed by cooling on ice for 30 min. Slides were rinsed with Tris-buffered saline-0.1% Tween (TBST), saturated by 0.6% hydrogen peroxide for 10 min, and blocked with 5% goat serum in TBST for 30 min at room temperature. Thereafter, sections were incubated overnight with mouse polyclonal anti-Pten antibodies [1:400 (catalog no. 9661; Cell Signaling, Ozyme) in 5% goat serum-TBS (RB-072-P1; NeoMarkers, Microm) or with rabbit monoclonal phospho (p)-protein kinase B (Akt) (Thr308) antibodies (1:80) in 5% goat serum-TBS (244F9; Cell Signaling, Ozyme). Rabbit polyclonal cleaved-caspase 3 (Asp175) (9661; Cell Signaling, Ozyme) and rabbit polyclonal VE-cadherin (BMS158; Tebu Bio) were used in a 1:400 dilution overnight at 4°C. For negative control slides, no primary antibodies were added to 5% goat serum. The sections were washed in TBST, incubated for 30 min with biotinylated goat anti-rabbit IgG (BA-1000; Vector Laboratories), and then incubated with streptavidin-peroxidase complex using a Vectastain ABC Elite kit (Vector Laboratories). Subsequently, the chromogenic reaction was carried out with the 3,3′-diaminobenzidine tetrahydrochloride-plus kit (00-2020; Zymed Laboratories), and the reaction was terminated by tap water. Slides were counterstained with hematoxylin before permanent mounting with Aquatex (Merck) and then evaluated under a light microscope.
RNA isolation, probe preparation, and hybridization.
Mouse corpora lutea were microdissected under a microscope, and the total RNA was isolated by using a nucleospin RNA kit II (BD Bioscience) following the manufacturer's instructions. cDNA probes were generated from 5 μg of total RNA reverse transcriptase reaction using a mix of dCTP, dGTP, and dTTPs plus [α-32P]dATP (Amersham) and a mixture of primers specific to each of the genes present in the arrays. All probes used were 107 cpm, and the difference between control and experimental probes in each assay was <10%. cDNA were hybridized to an Atlas Mouse 1.2 array (no. 7853–1) nylon membrane (Clontech). Hybridization and posthybridization washes were performed according to the manufacturer's protocol. The signal was scanned with a PhosphorImager (Molecular Dynamics) after 5 days exposure. Control and experimental RNA were always processed in parallel. Spot intensities from scanned membranes were analyzed using the AtlasImage 1.5 software (Clontech). Grids were oriented manually and adjusted to ensure optimal spot recognition using AtlasImage's fine tuning tools, discarding spots with dust or locally high background. In all experiments, only variations of RNA levels larger than twofold with respect to controls were accepted.
Statistical analysis.
Each RT-PCR experiment was independently repeated at least three times with three different animals each day of gestation. All values are expressed as means ± SE of combined data from the replicate experiments. Analyses were processed with Statview version 5 (Abacus Concepts, Berkeley, CA). Descriptive statistics were performed for each variable; quantitative results are presented as median (range). Hormonal parameters were compared using the Mann-Whitney test. Proportions for the two groups were compared using the χ2-test. Values of P < 0.05 were considered statistically significant.
RESULTS
Analysis of histological morphology of ovaries from hCG-treated mice.
We attempted to treat PRLR−/− females with low doses of hCG to rescue LHR expression. The gross appearance of the whole ovary at day 2.5 of pregnancy revealed that hCG treatment was able to preserve the structure of the CL with well-organized histological morphology (Fig. 1, A–C). On day 2.5 of pregnancy, cleaved caspase 3 could be detected by immunohistochemistry only on sections from ovaries of PRLR−/− in contrast to hCG-treated females and wild-type females, indicating that hCG administration prevented the severe apoptosis seen in corpora lutea of PRLR−/− mice (Fig. 1, D–F).
Fig. 1.
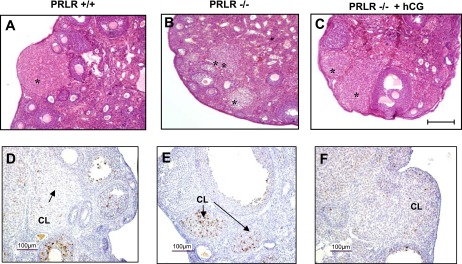
Histological analysis of ovaries from prolactin receptor (PRLR)+/+, PRLR−/−, and human chorionic gonadotropin (hCG)-treated PRLR−/− mice. Representative view of sections of ovaries stained with hematoxylin-eosin from PRLR+/+ (A), PRLR−/− (B), or hCG-treated PRLR−/− (C) of 8-wk-old female mice. Ovaries contain all developmental stages of follicle development. *Corpus luteum (CL). Bar scale: 200 μm. Immunodetection of cleaved caspase-3 in CL of PRLR+/+ (D), PRLR−/− (E), or hCG-treated PRLR−/− (F) mice at 2.5 days postcoitum (dpc). Bar scale 100 μm.
Ovarian LH-r expression in hCG-treated mice.
PRL is known to be essential to the LHR expression, and not surprisingly the PRLR−/− mice showed a decreased expression of the LHR mRNA expression in the CL at day 2.5 of pregnancy. Treatment with hCG of PRLR−/− mice during 2 days revealed a clear de novo expression of LHR mRNA in the corpora lutea compared with those of wild-type ovaries (Fig. 2, A–D). Moreover, LHR mRNA expression levels were confirmed by RT-PCR analysis from isolated corpora lutea RNA (Fig. 2E). To examine whether the LH receptor protein was synthesized and able to bind the hormone, we performed binding analysis on ovarian sections. The results showed similar levels of LH binding activity in corpora lutea of both wild-type and hCG-treated PRLR−/− mice (Fig. 2, F–H).
Fig. 2.
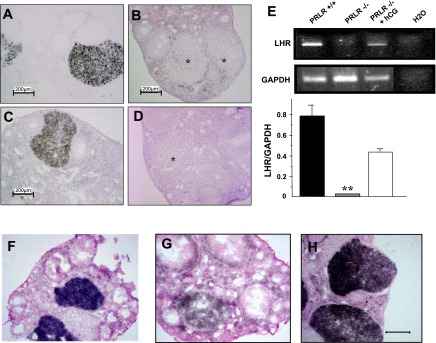
Expression of luteinizing hormone (LH) receptor mRNA. In situ hybridization of LHR in ovarian sections at 2.5 dpc of PRLR+/+ and PRLR−/− or hCG-treated PRLR−/− mice. A: representative section is shown to demonstrate the histology and highlights the hybridization signal in wild-type ovaries. B: in the absence of the PRLR, the expression of LHR is not seen. C: however, LHR expression is present in hCG-treated PRLR−/− mice. D: negative control sections probed with sense riboprobe showed no defined pattern of probe hybridization. Bar scale = 200 μm. E: representative results for transcripts by RT-PCR analysis of LHR mRNA in PRLR+/+, PRLR−/−, and hCG-treated PRLR−/− microdissected CL at 2.5 dpc, using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal control. The bar graph (bottom) shows the average relative amplicon band intensity, which was determined by densitometry (n = 3; **P < 0.01, t-test). Values are expressed as means ± SE. The binding of 125I-labeled hCG to ovarian frozen sections was studied by an autoradiographic method and performed on sections from wild-type (F), PRLR−/− (G), and hCG-treated PRLR−/− (H) ovaries at 2.5 dpc.
Progesterone levels and fertility of PRLR−/− mice treated with hCG.
We examined whether hCG stimulation of LHR expression in the CL of PRLR−/− mice is accompanied by an increase in progesterone and implantation. Because CL function is required for efficient implantation, five PRLR−/− female mice were treated with hCG for 5 days, and blastocyst implantation was analyzed. No site of implantation was observed in the treated mice. Indeed, similar progesterone levels were observed in PRLR−/− (5.48 ± 3.15 ng/ml, n = 8) and hCG-treated PRLR−/− (5.07 ± 2.65 ng/ml, n = 8) mice, whereas these levels were higher in wild-type animals (15.44 ± 4.79 ng/ml, n = 7, P < 0.05).
Effect of hCG on steroidogenic enzymes in PRLR−/− mice.
Because LH is known to acutely upregulate progesterone production via the stimulation of cholesterol transport to P450 side chain cleavage (P450scc), expression of steroidogenic enzymes was assessed in CL at day 2.5 of gestation by semiquantitative RT-PCR.
As shown in Fig. 3, deletion of PRLR causes, together with the drop of LHR, a severe inhibition in the expression of P450scc, one steroidogenic enzyme known to be upregulated by LH. However, no effect of hCG was observed on 20α-HSD, an enzyme involved in progesterone catabolism and negatively controlled by PRL. This enzyme, powerfully repressed by PRL, is highly expressed in corpora lutea of PRLR−/− and PRLR−/− treated with hCG.
Fig. 3.

Expression of P450scc and 20α-HSD mRNA. Representative results of the analysis for transcript RT-PCR analysis of P450scc and 20α-hydroxysteroid dehydrogenase (20α-HSD) mRNA in PRLR+/+, PRLR−/−, and hCG-treated PRLR−/− microdissected CL at 2.5 dpc, using GAPDH as an internal control. The bar graph (bottom) shows the average relative amplicon band intensity, which was determined by densitometry (n = 3; **P < 0.01, t-test). Values are expressed as a percentage of wild-type (WT) values and as means ± SE.
Macroarray analysis.
To further examine the genes regulated by LH/hCG independently of PRL, mRNA isolated from corpora lutea of wild-type, PRLR−/−, and hCG-treated PRLR−/− mice at day 2.5 of pregnancy were subjected to hybridization to an Atlas Mouse 1.2 array (no. 7853-1) nylon membrane (Clontech), which represents ∼2,000 genes. Signal intensities from membranes were analyzed using the AtlasImage 1.5 software (Clontech). The overall hybridization revealed an expression of a specific set of 102, 39, and 82 genes for wild-type, PRLR−/−, and hCG-treated PRLR−/− mice, respectively. On membrane hybridized with cDNAs from WT ovaries, PRLR expression was considered as a positive control. Inhibin A expression and the absence of the inhibin B on all three membranes confirmed the good quality of the microdissected isolated corpora lutea. The comparative analysis of gene expression between the three experimental conditions was assessed. To be as stringent as possible, in all experiments, we focused on those genes displaying a mean fold induction greater then or equal to two. Macroarray analysis performed with corpora lutea showed that PRL increases the expression of 26 genes and decreases the expression of only one detected in WT but not in PRLR−/− mice. Fourteen genes appeared to be upregulated by hCG, whose expression was detected in PRLR−/− hCG-treated mice but not in PRLR−/− mice (Table 2). It is of course impossible to discern if the effects of PRL in the control group compared with the PRLR−/− group are direct or indirect.
Table 2.
Putative prolactin and LH target genes
| Ratio | ||
|---|---|---|
| Putative upregulated prolactin target gene | ||
| M58566 | Butyrate response factor 1 | >10 |
| X53476 | High-mobility group protein 14 | >10 |
| X91753 | Transcription factor 4 | >10 |
| Y12880 | Ring finger protein 2 | >10 |
| U29762 | D site albumin promoter-binding protein | >10 |
| X83930 | Cadherin 5 | >10 |
| L060039 | Platelet/endothelial cell adhesion molecule | >10 |
| Y00769 | Integrin-β1 (fibronectin receptor-β) | >10 |
| X68193 | expressed in nonmetastatic cells 2, protein (NM23B) | >10 |
| X53584 | Heat shock protein, 60-kDa 1 | >10 |
| D30697 | Glutathione S-transferase,pi 2 | >10 |
| M14757 | ATP-binding cassette, subfamily B (MDR/TAP) member 1 | >10 |
| U17162 | BclII-binding athanogene 1 (BAG1) | >10 |
| AA041883 | Cytoplasmid dynein light chain 1; protein inhibitor of neuronal nitric oxide synthase (mPIN) | >10 |
| U88327 | Cytokine-inducible SH2-containing protein 2 | >10 |
| U43205 | Frizzled homolog 3 FZD-3 FZ-3 | >10 |
| Z71173 | Inositol 1,4,5-triphosphate receptor 5 | >10 |
| X61432 | Calmodulin | >10 |
| U28423 | Protein kinase, interferon-inducible double-stranded RNA-dependent inhibitor | >10 |
| U03184 | Cortactin | >10 |
| U57311 | Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, η-polypeptide | >10 |
| X53337 | Cathepsin D | >10 |
| D00611 | Basigin | >10 |
| S69407 | Endoglin | >10 |
| X96618 | Recombination-activating gene 1 gene activation | >10 |
| D49429 | RAD 21 homolog | >10 |
| Putative downregulated prolactin target gene | ||
| J05196 | Calcium-binding protein intestinal | 0.1 |
| Putative upregulated LH target gene | ||
| U10440 | Cyclin-dependent kinase inhibitor 1B (P27) | 6 |
| U94479 | Integrin-linked kinase | 3 |
| U43512 | Dystroglycan 1 | 10 |
| M36829 | Heat shock protein, 84 kDa 1 | 9 |
| M36830 | Heat shock protein, 86 kDa 1 | 7 |
| J03752 | Microsomal glutathione S-transferase (MGST1; GST12) | 2 |
| J04696 | Glutathione S-transferase, μ2 | 9 |
| U83628 | Defender against cell death 1 | 6 |
| X95403 | Ras-related protein RAB2 | 2 |
| X70296 | Serine protease inhibitor 4 | 7 |
| X51438 | Vimentin | 3 |
| X92411 | RAD23b homolog (Saccharomyces cerevisiae) | 3 |
| M64292 | B-cell translocation gene 2 antiproliferative | 2 |
| U92437 | Phosphatase and tensin homolog | 6 |
Among the genes upregulated by PRL, we focused our attention on VE-cadherin, an adhesion molecule involved in CL formation and angiogenesis (17). To assess the role of this gene in the luteinization process, RT-PCR was performed on this transcript and confirmed the downregulation of VE-cadherin in the absence of PRLR even in mRNA expression and protein expression. hCG treatment did not affect the expression of this, gene indicating that PRL stimulation of VE-cadherin expression does not involve LH/hCG (Fig. 4A).
Fig. 4.

Expression of VE-cadherin, frizzled (Fz)-4, secreted frizzled-related protein (sFRP)-4, and Wnt-4 mRNA. A: analysis for VE-cadherin mRNA and immunolocalization of the protein. B: analysis for Fz-4, sFRP-4, and Wnt-4 mRNA transcripts by RT-PCR analysis in PRLR+/+, PRLR −/−, and hCG-treated PRLR−/− microdissected CL at 2.5 dpc. The bar graph shows the average relative amplicon band intensity, which was determined by densitometry in PRLR+/+ (black bars), PRLR−/− (gray bars), and hCG-treated PRLR−/− (open bars) (n = 4; **P < 0.01 and ***P < 0.001, t-test) ovaries. Values are expressed as a percentage of WT values and as means ± SE.
Interestingly, we focused our attention on a member of the Wnt/Frizzled family, Fzd-3, potentially regulated by PRL. Because of the potential implication of these factors in CL development and maintenance in rodents, some other members of this family were tested by RT-PCR (Fig. 4B). The expression of sFRP-4 was then shown to be elevated in CL of wild-type mice compared with CL from PRLR−/− female mice. However, no significant change of Fzd-3 (data not shown), Fzd-4, and Wnt-4 mRNA expression was observed in WT CL compared with PRLR−/− mice, with or without hCG treatment.
Finally, we identified that two other major genes, p27 and Pten, whose expression was markedly decreased in CL of PRLR−/− female mice and rescued by hCG treatment in PRLR−/− mice. Both genes are involved in the cell cycle and differentiation. These potential candidate target genes were validated by RT-PCR (Fig. 5) where the Pten and p27 expression was about of the same order in dissected CL from PRLR+/+ and hCG-treated PRLR−/− mice. Moreover, PTEN protein was observed by immunohistochemistry in corpora lutea of PRLR+/+ mice (Fig. 6A), whereas PTEN staining was very faint in PRLR−/− corpora lutea (Fig. 6B). PTEN protein product is a lipid phosphatase that antagonizes phosphatidylinositol 3-kinase (PI 3-kinase) function and consequently inhibits downstream signaling through Akt. Next, we examined the state of Akt activation. We found PTEN absence to be correlated with strong Akt phosphorylation immunostaining (p-Akt), suggesting that activation of the PI 3-kinase pathway might be involved in this process (Fig. 6, D and E). Overall, the present data show that hCG treatment enhanced PTEN expression and prevented p-Akt expression (Fig. 6, C and F).
Fig. 5.

Expression of phosphatase and tensin homolog (Pten) and p27 mRNA. Representative results of the analysis of RT-PCR of Pten and p27 mRNA in microdissected CL at 2.5 dpc. The bar graph (bottom) shows the average relative amplicon band intensity that was determined by densitometry in PRLR+/+ (black bars), PRLR −/− (gray bars), and hCG-treated PRLR−/− (open bars) (n = 3; *P < 0.05, t-test) ovaries. Values are expressed as a percentage of WT values and as means ± SE.
Fig. 6.
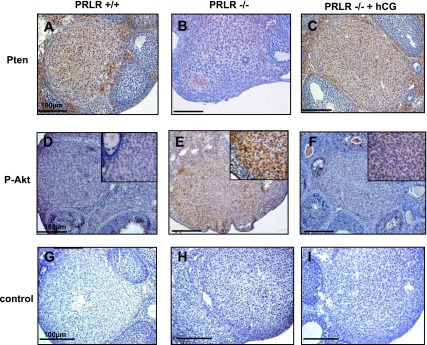
Immunolocalization of PTEN and phospho (Thr308)-protein kinase B (Akt) proteins in mouse ovaries at 1.5 dpc. Representative sections of PRLR+/+ (A, D, and G), PRLR−/− (B, E, and H), and hCG-treated PRLR−/− (C, F, and I) ovaries are shown. Staining is mainly seen in cells of the CL. The strongest immunoreactivity for PTEN is observed in wild-type CL (A) and hCG-treated (C) ovaries, whereas phosphorylated Akt is positive only in PRLR−/− (E). Insets represent higher magnification expressing or not expressing phosphorylated (p)-Akt. G–I: negative control (section incubated with nonimmune serum). Scale bar: 100 μm.
DISCUSSION
This study shows that, in PRLR−/− females, the CL function as determined by progesterone secretion was not enhanced by hCG despite the increase in LHR and the decrease of apoptosis. In these conditions, implantation fails, probably because of the activity of 20α-HSD and consequent catabolism of progesterone. We have also identified new PRL and LH target genes in CL, especially genes involved in the proliferation/differentiation pathway and in vascularization, all processes known to be crucial in CL development and maintenance.
The formation, function, and regression of the CL involve a wide variety of highly complex and multifaceted biological processes. In rodents, the CL of pregnancy is highly dependent on the action of PRL and PRL-like hormones, which play key roles in causing luteal cell hypertrophy and in maintaining progesterone production (23). PRL is known to affect a number of genes in which expression is important in the general functioning of the CL and that may secondarily lead to increased progesterone secretion and the proper maintenance of gestation. It has been shown early on that PRL is essential for LHR expression in rodent CL (7, 22, 24), and the generation of the PRLR knockout mouse model further substantiated these findings (9). However, whereas extensive investigations have defined the mechanism of LHR downregulation at the promoter and receptor levels (2), no information defining the molecular mechanism of PRL-mediated stimulation of LHR is available to date. LH is an important modulator of CL function in a number of species, including rodents. In the rat CL, LH is capable of acute stimulation of progesterone synthesis, while the long-term effects of LH are mediated through its maintenance of luteal estradiol production (23). In this report, we provide novel evidence indicating that LH/hCG can induce, if administered early during CL formation, the expression and the binding activity of its cognate receptor in the CL in the complete absence of PRL signaling. Using this mouse model, we show that LH/hCG can also stimulate the expression of steroidogenic enzymes in the absence of PRL signaling. Yet, despite the positive effect of low doses of hCG administered a day after mating to PRLR−/− mice, and although the CL was perfectly organized showing no apoptotic signs, levels of progesterone secreted were too low to allow blastocyst implantation. These results suggest that, although the activation of steroidogenesis by the hCG treatment occurs, the rapid catabolism of progesterone by 20α-HSD persists, which might explain implantation failure. Indeed, this was not the result of the use of a low dose of hCG, since higher doses of hCG were unable to rescue progesterone levels (data not shown). This enzyme catabolizes progesterone in luteal cells to a weak metabolite unable to support pregnancy (1) and is clearly repressed by PRL. The specific expression of this enzyme in the CL may explain why LH alone cannot sustain progesterone production in rodents as it does in many species with CL devoid of 20α-HSD. However, we cannot exclude, on one hand based on our data, that hCG induces a partial reduction in 20α-HSD and on the other hand that a defect of vascularization could also participate to the reduced level of progesterone.
Luteinization is a differentiation process associating granulosa and theca cells, without any cellular proliferation. To investigate the biological action of PRL and to define the major targets of PRL in CL, we performed a comparative analysis of gene expression under the three conditions and identified a set of genes potentially regulated by PRL and/or LH.
First, we identified VE-cadherin, also known as cadherin 5, as a potential target of PRL. This is of high interest, since the development of the CL is a process that is critically dependent on angiogenesis (18). New blood vessels provide substrate for steroidogenesis and allow for delivery of endocrine and paracrine factors to and from the follicle and CL. It has been shown that inhibition of VE-cadherin with an immunoneutralizing antibody during gonadotropin-induced ovarian stimulation results in inhibition of follicular and CL angiogenesis (17). The vascular area of the CL was reduced by 90% in the treated group compared with controls. The number and size of CL as well as progesterone levels in treated mice were also significantly reduced in the treated group. The downregulation of VE-cadherin, in the absence of PRL signaling, could then explain the low vascularization of corpora lutea observed in PRLR−/− mice.
We next identified sFRP-4 as a potential target of PRL in corpora lutea. This factor is one member of the Wnt/fzd family, with the WNT proteins acting locally to control diverse developmental processes such as proliferation and differentiation by transducing their signal after binding to G protein-coupled receptors of the Fzd family (19). These factors are known to play a role in luteal function. Indeed, it has been shown that Wnt-4 expression increased after hCG treatment and remained elevated in the CL during pregnancy (13). Fzd-4 has been implicated in CL formation since Fzd-4 null mice failed to form functional CL despite normal follicular development and ovulation (12). We confirmed previous findings that sFRP-4 gene expression is induced by hCG and to be further upregulated by PRL (14). This factor may be important after ovulation for remodeling and differentiation of the CL. However, this study shows only the transcriptional level of this gene.
Two genes, p27 and Pten, are markedly stimulated in the CL of PRLR−/− mice by in vivo treatment with hCG. p27 was previously shown to be stimulated by LH during luteinization. Indeed, we have previously described that, 2 days after ovulation, the CL of PRLR−/− mice shows an elevated degree of apoptosis together with decreased levels of p27 and steroidogenic enzymes (9). These data support the importance of PRL and LH as antiapoptotic hormones needed for luteal survival.
The Pten gene encodes a protein that is a member of the tyrosine phosphatase family, a phosphoinositide-3-phosphatase, and plays an important role in cell proliferation, cell survival, and apoptosis. In particular, it inhibits cell cycle progression by downregulating cyclin D1 and activates proapoptotic molecules through Akt-dependent and -independent pathways (28). Pten acts by inactivating PI 3-kinase-dependent signaling, including Akt (16). The expression of Pten was shown to increase during terminal follicular growth in the sheep and was able to upregulate the activity of the p27 promoter (5). This increase of expression of Pten in large preovulatory ovine follicles suggests that it is induced by LH, corroborating our data showing the increase in Pten expression by low doses of hCG in the CL of PRLR−/− mice. However, in our animal model, Pten expression was decreased in CL of PRLR−/− female mice, but not abolished, and rescued by hCG treatment. This seems not to be in accordance with a recent study that has shown that loss of PTEN in granulosa cells results in facilitated ovulation and also in the persistence of nonsteroidogenic luteal structures in the adult mouse ovary (4). One explanation must be that PRL and LH are the major regulators of CL function, independently of Pten expression. Whether p-Akt is coexpressed with caspases or is localized in steroidogenic cells is not known yet and remains to be investigated.
In conclusion, we have demonstrated that PRL signaling through its receptor is the most potent regulator of CL formation, mainly by regulating progesterone catabolism by 20α-HSD, and that low levels of hCG can, in a PRLR−/− model, stimulate LHR expression and prolong the life span of the CL by silencing caspase-3 activity. LH/hCG can also stimulate the luteal expression of steroidogenic enzymes independently of PRL. However, hCG cannot rescue CL function even though decreased apoptosis and partial replacement of LHR was affected probably because of the persistent high 20α-HSD activity. Reduced vascular supply could also contribute to the loss of CL function. Furthermore, we have highlighted several new genes implicated in CL development and maintenance, under the direct or indirect regulation of LH and PRL. How these genes are regulated, the signaling pathways of the multiple hormones and cytokines involved, and their actions and interactions need further investigation.
GRANTS
This study was supported by grants from INSERM. J. Beaufaron and A. Bachelot received a fellowship from IPSEN Biotech and Fondation pour la Recherche Médicale, respectively. G. Gibori was supported by National Institute of Child Health and Human Development Grant HD-11119 and a Fullbright Grant.
Acknowledgments
We are indebted to Sophie Berissi of Institut National de la Santé et de la Recherche Médicale (INSERM) Unit 845 for providing histological sections.
REFERENCES
- 1.Albarracin CT, Parmer TG, Duan WR, Nelson SE, Gibori G. Identification of a major prolactin-regulated protein as 20 alpha- hydroxysteroid dehydrogenase: coordinate regulation of its activity, protein content, and messenger ribonucleic acid expression. Endocrinology 134: 2453–2460, 1994. [DOI] [PubMed] [Google Scholar]
- 2.Ascoli M, Fanelli F, Segaloff DL. The lutropin/choriogonadotropin receptor, a 2002 perspective. Endocr Rev 23: 141–174, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Binart N, Helloco C, Ormandy CJ, Barra J, Clement-Lacroix P, Baran N, Kelly PA. Rescue of preimplantatory egg development and embryo implantation in prolactin receptor-deficient mice after progesterone administration. Endocrinology 141: 2691–2697, 2000. [DOI] [PubMed] [Google Scholar]
- 4.Fan HY, Liu Z, Cahill N, Richards JS. Targeted disruption of Pten in ovarian granulosa cells enhances ovulation and extends the life span of luteal cells. Mol Endocrinol 22: 2128–2140, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Froment P, Bontoux M, Pisselet C, Monget P, Dupont J. PTEN expression in ovine granulosa cells increases during terminal follicular growth. FEBS Lett 579: 2376–2382, 2005. [DOI] [PubMed] [Google Scholar]
- 6.Froment P, Seurin D, Hembert S, Levine JE, Pisselet C, Monniaux D, Binoux M, Monget P. Reproductive abnormalities in human IGF binding protein-1 transgenic female mice. Endocrinology 143: 1801–1808, 2002. [DOI] [PubMed] [Google Scholar]
- 7.Gafvels M, Bjurulf E, Selstam G. Prolactin stimulates the expression of luteinizing hormone/chorionic gonadotropin receptor messenger ribonucleic acid in the rat corpus luteum and rescues early pregnancy from bromocriptine-induced abortion. Biol Reprod 47: 534–540, 1992. [DOI] [PubMed] [Google Scholar]
- 8.Gibori G, Richards JS. Dissociation of two distinct luteotropic effects of prolactin: regulation of luteinizing hormone-receptor content and progesterone secretion during pregnancy. Endocrinology 102: 767–774, 1978. [DOI] [PubMed] [Google Scholar]
- 9.Grosdemouge I, Bachelot A, Lucas A, Baran N, Kelly PA, Binart N. Effects of deletion of the prolactin receptor on ovarian gene expression. Reprod Biol Endocrinol 1: 1–12, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gunnet JW, Freeman ME. The mating-induced release of prolactin: a unique neuroendocrine response. Endocrinol Rev 4: 44–61, 1983. [DOI] [PubMed] [Google Scholar]
- 11.Gunnet JW, Freeman ME. Hypothalamic regulation of mating-induced prolactin release. Effect of electrical stimulation of the medial preoptic area in conscious female rats. Neuroendocrinology 38: 12–16, 1984. [DOI] [PubMed] [Google Scholar]
- 12.Hsieh M, Boerboom D, Shimada M, Lo Y, Parlow AF, Luhmann UF, Berger W, Richards JS. Mice null for Frizzled4 (Fzd4−/−) are infertile and exhibit impaired corpora lutea formation and function. Biol Reprod 73: 1135–1146, 2005. [DOI] [PubMed] [Google Scholar]
- 13.Hsieh M, Johnson MA, Greenberg NM, Richards JS. Regulated expression of Wnts and Frizzleds at specific stages of follicular development in the rodent ovary. Endocrinology 143: 898–908, 2002. [DOI] [PubMed] [Google Scholar]
- 14.Hsieh M, Mulders SM, Friis RR, Dharmarajan A, Richards JS. Expression and localization of secreted frizzled-related protein-4 in the rodent ovary: evidence for selective up-regulation in luteinized granulosa cells. Endocrinology 144: 4597–4606, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Kuttenn F, Couillin P, Girard F, Billaud L, Vincens M, Boucekkine C, Thalabard JC, Maudelonde T, Spritzer P, Mowszowicz I. Late-onset adrenal hyperplasia in hirsutism. N Engl J Med 313: 224–231, 1985. [DOI] [PubMed] [Google Scholar]
- 16.Leslie NR, Downes CP. PTEN function: how normal cells control it and tumour cells lose it. Biochem J 382: 1–11, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakhuda GS, Zimmermann RC, Bohlen P, Liao F, Sauer MV, Kitajewski J. Inhibition of the vascular endothelial cell (VE)-specific adhesion molecule VE-cadherin blocks gonadotropin-dependent folliculogenesis and corpus luteum formation and angiogenesis. Endocrinology 146: 1053–1059, 2005. [DOI] [PubMed] [Google Scholar]
- 18.Neeman M, Abramovitch R, Schiffenbauer YS, Tempel C. Regulation of angiogenesis by hypoxic stress: from solid tumours to the ovarian follicle. Int J Exp Pathol 78: 57–70, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science 303: 1483–1487, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ormandy CJ, Camus A, Barra J, Damotte D, Lucas BK, Buteau H, Edery M, Brousse N, Babinet C, Binart N, Kelly PA. Null mutation of the prolactin receptor gene produces multiple reproductive defects in the mouse. Genes Dev 11: 167–178, 1997. [DOI] [PubMed] [Google Scholar]
- 21.Piquette GN, LaPolt PS, Oikawa M, Hsueh AJ. Regulation of luteinizing hormone receptor messenger ribonucleic acid levels by gonadotropins, growth factors, and gonadotropin-releasing hormone in cultured rat granulosa cells. Endocrinology 128: 2449–2456, 1991. [DOI] [PubMed] [Google Scholar]
- 22.Polan ML, Laufer N, Dlugi AM, Tarlatzis BC, Haseltine FP, DeCherney AH, Behrman HR. Human chorionic gonadotropin and prolactin modulation of early luteal function and luteinizing hormone receptor-binding activity in cultured human granulosa-luteal cells. J Clin Endocrinol Metab 59: 773–779, 1984. [DOI] [PubMed] [Google Scholar]
- 23.Risk M, Gibori G. Mechanisms of luteal cell regulation by prolactin. In: Prolactin, edited by Horseman ND. Boston, MA: Kluwer, 2001, p. 265–295.
- 24.Segaloff DL, Wang HY, Richards JS. Hormonal regulation of luteinizing hormone/chorionic gonadotropin receptor mRNA in rat ovarian cells during follicular development and luteinization. Mol Endocrinol 4: 1856–1865, 1990. [DOI] [PubMed] [Google Scholar]
- 25.Stocco C, Callegari E, Gibori G. Opposite effect of prolactin and prostaglandin F(2 alpha) on the expression of luteal genes as revealed by rat cDNA expression array. Endocrinology 142: 4158–4161, 2001. [DOI] [PubMed] [Google Scholar]
- 26.Stocco C, Djiane J, Gibori G. Prostaglandin F(2alpha) (PGF(2alpha)) and prolactin signaling: PGF(2alpha)-mediated inhibition of prolactin receptor expression in the Corpus luteum. Endocrinology 144: 3301–3305, 2003. [DOI] [PubMed] [Google Scholar]
- 27.Stocco C, Telleria C, Gibori G. The molecular control of corpus luteum formation, function and regression. Endocr Rev 28: 117–149, 2007. [DOI] [PubMed] [Google Scholar]
- 28.Weng L, Brown J, Eng C. PTEN induces apoptosis and cell cycle arrest through phosphoinositol-3-kinase/Akt-dependent and -independent pathways. Hum Mol Genet 10: 237–242, 2001. [DOI] [PubMed] [Google Scholar]