

Abstract
1α,25(OH)2 vitamin D3 [1,25(OH)2D3] increases serum Ca2+ concentration in vivo, an action counteracted by activation of the Ca2+-sensing receptor (CaSR), which decreases parathyroid hormone (PTH) secretion and increases renal Ca2+ excretion. Relatively little is known of the role the CaSR plays in this response through its potentially direct actions in kidney, gut, and bone independently of PTH. We report PTH-independent roles of the CaSR in modulating the response to exogenous 1,25(OH)2D3 in mice with targeted disruption of both the CaSR and PTH genes (C−P−) compared with that in mice with disruption of the PTH gene alone (C+P−) or wild-type mice (C+P+). After intraperitoneal injection of 0.5 ng/g body wt 1,25(OH)2D3, peak calcemic responses were observed at 24 h in all three genotypes in association with 1) a greater increase in serum Ca2+ in C−P− mice than in the other genotypes on a Ca2+-replete diet that was attenuated by a Ca2+-deficient diet and pamidronate, 2) increased urinary Ca2+-to-creatinine ratios (UCa/Cr) in the C+P− and C+P+ mice but a lowered ratio in the C−P− mice on a Ca2+-replete diet, and 3) no increase in calcitonin (CT) secretion in the C+P+ and C+P− mice and a small increase in the C−P− mice. PTH deficiency had the anticipated effects on the expression of key genes involved in Ca2+ transport at baseline in the duodenum and kidney, and injection of 1,25(OH)2D3 increased gene expression 8 h later. However, the changes in the genes evaluated did not fully explain the differences in serum Ca2+ seen among the genotypes. In conclusion, mice lacking the full-length CaSR have increased sensitivity to the calcemic action of 1,25(OH)2D3 in the setting of PTH deficiency. This is principally from enhanced 1,25(OH)2D3-mediated gut Ca2+ absorption and decreased renal Ca2+ excretion, without any differences in bone-related release of Ca2+ or CT secretion among the three genotypes that could explain the differences in their calcemic responses.
Keywords: vitamin D, kidney
the calcium-sensing receptor (CaSR) controls the secretion of parathyroid hormone (PTH), which, in turn, via direct and/or indirect actions on kidney, bone, and intestine, maintains normal extracellular ionized calcium (Ca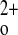 ) levels (3). There is limited understanding of direct functions of the CaSR expressed in tissues other than the parathyroid gland that are involved in overall Ca
) levels (3). There is limited understanding of direct functions of the CaSR expressed in tissues other than the parathyroid gland that are involved in overall Ca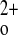 homeostasis (27). Several studies have supported a role of the CaSR in regulating the function of calcitonin (CT)-secreting C cells (10), and the CaSR plays several putative roles in regulating renal function (2, 14), but the evidence implicating the receptor in regulating the functions of bone (6–8, 20) and/or the gastrointestinal (GI) tract (4) is limited and/or controversial. Recent studies, however, in mice with conditional knockout of the receptor in chondrocytes or osteoblasts have implicated the CaSR as a key regulator of these two cell types (5).
homeostasis (27). Several studies have supported a role of the CaSR in regulating the function of calcitonin (CT)-secreting C cells (10), and the CaSR plays several putative roles in regulating renal function (2, 14), but the evidence implicating the receptor in regulating the functions of bone (6–8, 20) and/or the gastrointestinal (GI) tract (4) is limited and/or controversial. Recent studies, however, in mice with conditional knockout of the receptor in chondrocytes or osteoblasts have implicated the CaSR as a key regulator of these two cell types (5).
1,25(OH)2 vitamin D3 [1,25(OH)2D3] has a central role in Ca2+ homeostasis by virtue of its capacity to increase bone turnover, GI Ca2+ absorption, and renal tubular Ca2+ reabsorption as well as to inhibit parathyroid function (1). PTH, by promoting 1α-hydroxylation of 25-hydroxyvitamin D3, increases the circulating level of 1,25(OH)2D3, which then acts on its target tissues. The genomic effects of 1,25(OH)2D3 on Ca2+ metabolism are mediated by interaction of the ligand-vitamin D receptor (VDR) complex with vitamin D-responsive elements upstream of genes involved in Ca2+ homeostasis (1). Interestingly, some studies suggest a direct regulatory effect of Ca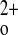 on renal vitamin D metabolism that might be exerted through the CaSR (11).
on renal vitamin D metabolism that might be exerted through the CaSR (11).
In preliminary studies characterizing mice with homozygous knockout of exon 5 of the CaSR and/or the PTH genes, we observed an altered homeostatic response of mice with knockout of both genes to exogenous 1,25(OH)2D3 administration that was related to the absence of the CaSR gene. On the basis of these studies, we sought to investigate in more detail the in vivo functional and molecular aspects of CaSR-mediated, PTH-independent modulation of responses to exogenous 1,25(OH)2D3. We used three genotypes of mice [(CaSR+/+ PTH−/−, called C+P−), (CaSR−/−PTH−/−, C−P−) or wild-type mice (CaSR+/+PTH+/+, C+P+)] to evaluate responses to 1,25(OH)2D3 in vivo and to measure the expression of key genes expressed in the kidney and duodenum that are involved in Ca2+ transport and 1,25(OH)2D3 catabolism. We compared the C+P− and C−P− mice to assess roles of the CaSR that are independent of the effects of PTH and compared the C+P− and C+P+ mice to determine actions of PTH per se.
MATERIALS AND METHODS
Materials.
1,25(OH)2D3 and primers for genotyping experimental mice were obtained from Sigma-Aldrich (St. Louis, MO). Pamidronate was obtained from Toronto Research Chemicals (Toronto, ON, Canada). Serum calcitonin (CT) levels were determined using an immunoradiometric rat/mouse CT assay (ALPCO Diagnostics, Salem, NH). Commercial kits were used for measuring serum and urine Ca2+ levels (Eagle Diagnostics, De Soto, TX). Urine and serum creatinine levels were measured using the Stanbio creatinine kit (Stanbio Laboratory, Boerne, TX). Serum 1,25(OH)2D3 levels were determined using a competitive enzyme immunoassay (IDS, Fountain Hills, AZ). Total RNA and cDNA were prepared from harvested tissues using kits obtained from Qiagen (Valencia, CA). Proprietary TaqMan exon-spanning primers for real-time PCR with a final concentration of 900 nM and a 6-FAM dye-labeled TaqMan MGB probe with a final concentration of 250 nM were obtained from Applied Biosystems (Foster City, CA).
Rationale behind use of the exon 5 CaSR/PTH and PTH knockout mouse models.
The neonatal lethality of the homozygous CaSR knockout (KO) mice has limited their utility for detailed studies of the CaSR's physiological roles in vivo. However, several other mouse models are available for such investigations (14, 28). One such model “rescues” the homozygous CaSR KO mice by crossing heterozygous CaSR KO mice with mice in which the PTH gene has been knocked out, and this model has established that the CaSR contributes importantly to the fine control of the serum Ca2+ concentration (14). None of these models have evaluated in any detail the role of the CaSR in C-cell, kidney, and other tissues in maintaining Ca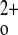 homeostasis in not only the absence but also the presence of PTH (e.g., given via infusion).
homeostasis in not only the absence but also the presence of PTH (e.g., given via infusion).
This rescued mouse model was developed using the original mouse KO model in which exon 5 of the CaSR was disrupted by insertion of the neo gene (14). It has subsequently become clear, however, that an alternatively spliced CaSR, lacking exon 5, can be generated in some tissues in homozygous KO mice of this genotype (23, 24). It has not been possible to express this truncated receptor at high levels on the cell surface in heterologous systems, precluding direct documentation that it actually has activity, but chondrocytes from exon 5 CaSR KO mice still respond to Ca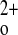 , indirectly suggesting that the CaSR lacking exon 5 may have biological activity in certain tissues.
, indirectly suggesting that the CaSR lacking exon 5 may have biological activity in certain tissues.
Recent investigations have addressed this latter point by utilizing mice with conditional KO of the CaSR in chondrocytes and osteoblasts, which were generated by crossing mice with floxed exon 7 of the CaSR with appropriate Cre-expressing mice (5). These studies have shown much more severe skeletal phenotypes than the global exon 5 KO mice, suggesting that the global CaSR−/− mice lacking exon 5 are hypomorphic with respect to functions of the CaSR in cartilage and bone, i.e., the CaSR lacking exon 5 can partially or fully rescue the function of these two cell types. Curiously, the mice utilized in the studies of Chang et al. (5) appear to have PTH-dependent hypercalcemia (∼11.5–13 mg/dl), which is of uncertain significance.
Nevertheless, the original “unrescued” exon 5 KO mice clearly have severely deranged Ca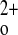 -regulated PTH release and parathyroid cellular proliferation and relative hypocalciuria that leads to death shortly after birth, as is seen in human neonatal severe hyperparathyroidism due to homozygous inactivating mutations of the CaSR. Moreover, the C−P− mice show a nearly complete loss of high Ca
-regulated PTH release and parathyroid cellular proliferation and relative hypocalciuria that leads to death shortly after birth, as is seen in human neonatal severe hyperparathyroidism due to homozygous inactivating mutations of the CaSR. Moreover, the C−P− mice show a nearly complete loss of high Ca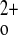 -stimulated CT secretion and a near total failure to increase renal Ca2+ excretion during marked hypercalcemia induced by a PTH infusion (13). An important advantage of the “rescued” global exon 5 KO mice is that they permit studies of how various CaSR-regulated tissues participating in Ca
-stimulated CT secretion and a near total failure to increase renal Ca2+ excretion during marked hypercalcemia induced by a PTH infusion (13). An important advantage of the “rescued” global exon 5 KO mice is that they permit studies of how various CaSR-regulated tissues participating in Ca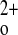 homeostasis interact and contribute to the derangements in Ca2+ metabolism. In effect, normal homeostatic loops maintaining the stability of Ca
homeostasis interact and contribute to the derangements in Ca2+ metabolism. In effect, normal homeostatic loops maintaining the stability of Ca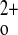 , including CaSR regulation of PTH secretion, CT secretion, and 1,25(OH)2D3 production, have been interrupted, providing a “stripped down” model in which these hormones can be added back in a controlled fashion, if desired. Although tissue-specific KO models clearly have their merits, it is likely that conditionally knocking out the CaSR in one Ca2+ homeostatic tissue (e.g., kidney) will initiate homeostatic/compensatory responses, such as changes in PTH secretion, in others that may modify or mask the primary phenotype resulting from tissue-specific KO of the CaSR.
, including CaSR regulation of PTH secretion, CT secretion, and 1,25(OH)2D3 production, have been interrupted, providing a “stripped down” model in which these hormones can be added back in a controlled fashion, if desired. Although tissue-specific KO models clearly have their merits, it is likely that conditionally knocking out the CaSR in one Ca2+ homeostatic tissue (e.g., kidney) will initiate homeostatic/compensatory responses, such as changes in PTH secretion, in others that may modify or mask the primary phenotype resulting from tissue-specific KO of the CaSR.
Animal breeding, care, and handling.
Mice with targeted disruption of the CaSR or PTH genes were utilized for the generation of C+P− and C−P− mice as has been reported previously. Animals used in the present studies were obtained following extensive backcrossing on a mixed genetic background with contributions from C57B/L6, 129/SvJ, and 129/SvEv strains. Animal protocols were approved by the Institutional Animal Care and Use Committee at Harvard Medical School and were in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Mice were housed in isolator cages or small metabolic cages in a pathogen-free facility according to the regulations of the Harvard Medical School Center for Animals Resources and Comparative Medicine, including during the collection of blood and urine samples. All animals were fed a normal Ca2+ diet [0.8% Ca2+ (wt/wt)] or a Ca2+-deficient diet [0.01% Ca2+ (wt/wt), 0.4% phosphate, Harlan Teklad-TD99224; Harlan-Teklad, Madison, WI] and plain water ad libitum. As a result of PTH gene KO and consequent hypoparathyroidism, the C+P− and C−P− mice were hypocalcemic to the same extent at baseline before the start of all experiments.
Genotyping.
For the purpose of genotyping during generation of the three genotypes of experimental mice from the double heterozygotes CaSR+/−PTH+/−, genomic DNA was isolated using a Qiagen DNeasy kit. To determine the genotype at both the Pth and CaSR loci, we carried out four PCR amplifications for each animal. To document the presence of the wild-type Casr allele, we amplified DNA samples with Casr forward primer (5′-TCATTGATGAACAGTCTTTCTCCCT-3′) and Casr reverse primer (5′-TCTGTTCTCTTTAGGTCCTGAAACA-3′). To document the presence of the KO CaSR allele, we used Neo forward primer (5′-TCTTGATTCCCACTTTGTGGTTCTA-3′) with Casr reverse primer (5′-TCATTGATGAACAGTCTTTCTCCCT-3′). The wild-type Pth allele was detected using PTH forward primer (5′-GATGTCTGCAAACACCGTGGCTAA-3′) and PTH reverse primer (5′-TCCAAAGTTTCATTACAGTAGAAG-3′). The null Pth allele was documented using a Neo forward primer (5′-TCTTGATTCCCACTTTGTGGTTCTA-3′) and PTH reverse primer (5′-TCCAAAGTTTCATTACAGTAGAAG-3′). All PCR reactions were performed using ABI GeneAmp PCR System 9700.
In vivo testing.
Several sets (n = 8–12/set) of 3- to 4-mo-old, nonpregnant female mice homozygous for KO of the CaSR and/or PTH genes or neither (group 1, wild type C+P+; group 2, C+P−; and group 3, C−P−) were administered 0.5 ng/g body wt 1,25(OH)2D3 by intraperitoneal (ip) injection. All experiments were started in the early morning, and the calcemic responses over 72 h were analyzed by obtaining serial 5-μl serum samples by cheek bleeding. Serum 1,25(OH)2D3 levels were measured at baseline and at 1 and 24 h by obtaining 100-μl serum samples on a separate cohort of mice in keeping with good animal experimentation practices. Serum CT levels were analyzed at baseline and at 24 h by obtaining 100-μl samples on yet another cohort of mice. Spot UCa/Cr ratios were analyzed at baseline and at 24 h in several cohorts of mice used for experimentation. Ten- to twenty-microliter samples of acidified clean-catch urine at the appropriate time points were obtained during spontaneous voiding or bladder compression by gentle lower abdominal compression. Injections were given to mice with free access to a Ca2+-replete diet [0.8% Ca2+ (wt/wt) chow and plain water (e.g., without any added Ca2+)]. This was repeated in separate groups of mice with or without pretreatment with pamidronate (2.5 μg/g body wt ip daily for 2 wk) and subsequent conditioning for 48–72 h on a Ca2+-deficient diet (0.01% Ca2+ chow and plain water) before 1,25(OH)2D3 administration. This dose was based on earlier studies, which had shown that pamidronate administered at somewhat lower doses than we used significantly inhibited bone resorption in rats and mice (12, 16).
Real-time PCR.
Total RNA was isolated from kidney and duodenum from several sets of animals separate from those used for physiological measurements under conditions of normal chow and plain water as well as 8 h after ip administration of 0.5 ng/g body wt 1,25(OH)2D3 using Qiagen RNAeasy kits according to the manufacturer's protocol and subsequently treated with RNase-free DNase to remove genomic DNA. These time points were chosen on the basis of prior studies, which have shown that renal vitamin D3-24-hydroxylase, a rate-limiting enzyme in the catabolism of vitamin D metabolites, has a peak mRNA expression at 8 h following 1,25(OH)2D3 ip injection in rats, with return of expression to baseline by 24 h (15, 17). Similar time courses for maximal expression have been reported in other studies for the transient receptor potential vanilloid (TRPV) channels 5 and 6 in rat intestine (15, 26). Reverse transcription using 2 μg of DNase-treated RNA was performed using Qiagen Omniscript kits on an ABI GeneAmp PCR System 9700.
To determine the relative number of cDNA molecules in the reverse-transcribed samples, we performed quantitative real-time PCR analyses using an ABI 7000 sequence detection system (Applied Biosystems) with 1 μl of cDNA, 10 μl of ABI TaqMan master mix, 1 μl of proprietary TaqMan gene assay, and 8 μl of H2O to a final volume of 20 μl. Data were normalized with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels as an endogenous control in test samples using a proprietary ABI TaqMan 6-FAM/MGB, non-primer-limited mouse GAPDH endogenous control probe. All amplifications were carried out in duplicate, and the threshold cycle (Ct) scores were averaged for calculations of relative expression values (18). The Ct scores for genes of interest in each sample were normalized against the Ct scores for the corresponding GAPDH gene in that sample, and relative expression was determined using the following calculation where the amount of target gene of interest is normalized to GAPDH in control C+P+ mice tissue (18): relative expression of target gene in the tissue of interest = 2−ΔΔCt, where ΔΔCt = (ΔCt of experimental group) − (ΔCt of C+P+ group) and ΔCt = mean Ct of target gene in the tissue of interest − mean Ct of GAPDH in the same tissue. Summary information profiles for each proprietary ABI TaqMan gene assay used for real-time PCR are displayed in Table 1.
Table 1.
Summary information profiles for each ABI proprietary TaqMan gene primer used for real-time PCR
| Gene (Symbol) | NCBI Gene Reference | Abbreviation in Text | Assay ID | Target Exon Boundry |
|---|---|---|---|---|
| Nuclear vitamin D receptor (VDR) | NM_009504.3 | VDR | Mm00437297_m1 | 3–4 |
| Calbindin D9k (S100G) | NM_009789.2, | CalD9K | Mm00486654_m1 | 2–3 |
| 1,25-Dihydroxy vitamin D3-24-hydroxylase (CYP24A1) | NM_009996.3 | CYP24A1 | Mm00487244_m1 | 7–8 |
| Sodium-calcium exchanger-1 (SLC8A1) | NM_001112798.1 | NCX1 | Mm01232254_m1 | 7–8 |
| Claudin-16 (CLDN-16) | NM_053241.4 | CLDN-16 | Mm00475025_m1 | 2–3 |
| Epithelial calcium channel-2 (TRPV6) | NM_022413.3 | TRPV6 | Mm00499069_m1 | 1–2 |
| Epithelial calcium channel-1(TRPV5) | NM_001007572.2 | TRPV5 | Mm01166036_m1 | 7–8 |
| Sodium-potassium-chloride cotransporter (SLC12A2) | NM_009194.2 | NKCC | Mm00436554_m1 | 17–18 |
ABI, Applied Biosystems International; TRPV, transient receptor potential vanilloid channel.
Statistics.
We evaluated the differences among the three genotypes at baseline and at experimental end points using one-way ANOVA or t-tests as appropriate. All values are means ± SE. Graphical illustrations were generated using Microsoft Excel 2007 software, and statistical analyses were performed using SPSS software (SPSS, Chicago, IL).
RESULTS
Baseline serum Ca2+ levels in wild-type and KO mice.
On normal chow [0.8% Ca2+ (wt/wt)] and plain water, wild-type mice remained normocalcemic (9.01 ± 0.44 mg/dl), and the C+P− and C−P− mice were hypocalcemic (6.44 ± 0.38 and 5.98 ± 0.53 mg/dl, respectively). Therefore, as noted previously, loss of the CaSR does not protect against the development of hypocalcemia in the absence of PTH.
Pharmacokinetic profile after 1,25(OH)2D3 administration.
We evaluated possible differences in pharmacokinetics of 1,25(OH)2D3 among the various genotypes that could explain differences in their responses to 1,25(OH)2D3 by measuring serum 1,25(OH)2D3 levels at baseline and 1 and 24 h after administration of exogenous 1,25(OH)2D3 by ip injection. The pharmacokinetic responses to a single dose of 1,25(OH)2D3 are displayed in Fig. 1. At baseline, presumably due to PTH deficiency, the KO genotypes had significantly lower 1,25(OH)2D3 levels compared with wild-type mice (C+P+, 110.4 ± 18 pM; C+P−, 75.4 ± 24 pM; C−P−, 61.1 ± 18.5 pM; ANOVA, P = 0.02). After injection of 0.5 ng/g body wt 1,25(OH)2D3, there was a >10-fold increase in serum levels of 1,25(OH)2D3 at 1 h postinjection in all genotypes with no significant differences among genotypes (C+P+, 1,248.1 ± 156.4 pM; C+P−, 1,103 ± 94.8 pM, C−P−, 1,239.2 ± 167.9 pM; ANOVA, P = 0.1). Levels returned to near baseline at 24 h with no difference across the genotypes (C+P+, 104.4 ± 12 pM; C+P−, 91.5 ± 15.8 pM; C−P−, 81.9 ± 13.5 pM; ANOVA, P = 0.07). There were little or no differences between baseline and 24-h postinjection 1,25(OH)2D3 levels within genotypes (C+P+, P = 0.7; C+P−, P = 0.2; C−P−, P = 0.045) that could explain the hypercalcemic response in the C−P− mice. Therefore, there were minimal, if any, differences in pharmacokinetics among the genotypes at the time points examined.
Fig. 1.

Pharmacokinetic profile of serum 1α,25(OH)2 vitamin D3 [1,25(OH)2D3] levels at baseline as well as 1 and 24 h after a single intraperitoneal (ip) dose of 1,25(OH)2D3 in 3 genotypes of mice: Ca2+-sensing receptor (CaSR)+/+parathyroid hormone (PTH)−/− (C+P− mice), CaSR−/−PTH−/− (C−P− mice), or wild-type CaSR+/+PTH+/+ (C+P+ mice). Data are means ± SE (n = 8–12/genotype). ○P < 0.02, ANOVA, baseline comparisons across genotypes. •P > 0.05, baseline comparison, C+P− vs. C−P−. ⧫P = 0.07 across-group comparisons at 1 h postinjection. ##P > 0.05 within-genotype comparison of baseline and 24-h 1,25(OH)2D3 levels. *P = 0.045, within-group comparison of baseline and 24-h 1,25(OH)2D3 levels.
Calcemic response after administration of a single dose of 1,25(OH)2D3 on a normal chow diet and plain water.
To assess the relative sensitivities of wild-type, C+P−, and C−P− mice to a (weight based) single dose of 1,25(OH)2D3 under Ca2+-replete feeding conditions, i.e., normal chow [0.8% Ca2+ (wt/wt)] and plain water, we administered 0.5 ng/g body wt 1,25(OH)2D3 by ip injection and measured serum Ca2+ levels every 12 h for 72 h (Fig. 2). There was an ∼7 mg/dl increase in serum Ca2+ in the C−P− mice at 24 h. The increase in serum Ca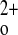 was much less marked in the C+P− mice (∼3 mg/dl) and even less so in the C+P+ mice (∼2 mg/dl; see Fig. 2) (independent t-test comparison between C−P− and C+P− at 24 h postinjection, P = 0.001). There also was a faster return of serum Ca2+ to preinjection levels in the C−P− mice compared with that in wild-type or C+P− mice. There was no change in the serum creatinine levels of the three genotypes 24 h postinjection, confirming that there were no significant changes in glomerular filtration rate as a result of 1,25(OH)2D3 administration that would explain the subsequent increase in serum Ca2+ (data not shown).
was much less marked in the C+P− mice (∼3 mg/dl) and even less so in the C+P+ mice (∼2 mg/dl; see Fig. 2) (independent t-test comparison between C−P− and C+P− at 24 h postinjection, P = 0.001). There also was a faster return of serum Ca2+ to preinjection levels in the C−P− mice compared with that in wild-type or C+P− mice. There was no change in the serum creatinine levels of the three genotypes 24 h postinjection, confirming that there were no significant changes in glomerular filtration rate as a result of 1,25(OH)2D3 administration that would explain the subsequent increase in serum Ca2+ (data not shown).
Fig. 2.

Effect of a single ip dose (0.5 ng/g body wt) of 1,25(OH)2D3 in mice fed a Ca2+-replete diet and plain water on serum Ca2+ concentrations (conc) over 72 h. Data are means ± SE (n = 8–12/genotype). *P = 0.001, C−P− vs. C+P− at 24 h.
Relative sensitivity to repeated administration of 1,25(OH)2D3 on a normal chow and plain water diet.
To assess the relative sensitivities of the three genotypes to repeated administration of 1,25(OH)2D3, we determined the dose necessary using an alternate-day dosing schedule to achieve normocalcemia from baseline hypocalcemia in the C+P− and C−P− mice (Fig. 3). C−P− mice required 0.0625 ng/g body wt 1,25(OH)2D3 to achieve and maintain normocalcemia compared with a dose of 0.4 ng/g body wt 1,25(OH)2D3 in C+P− mice. Wild-type mice were able to maintain normocalcemia on the same dose used to achieve normocalcemia in the C+P− mice. The difference in the sensitivity to 1,25(OH)2D3 of the C−P− mice relative to the other two genotypes was even more obvious with attempts to induce moderate hypercalcemia (serum Ca2+ concentrations between 12 and 13 mg/dl), where the C−P− mice required 0.125 ng/g body wt whereas the C+P+ and C+P− mice required up to 4 ng/g body wt 1,25(OH)2D3 (data not shown).
Fig. 3.

Relative sensitivities to repeated ip administration of 1,25(OH)2D3 in wild-type and knockout (KO) mice at doses designed to maintain normocalcemia using an alternate-day dosing schedule. C+P− mice needed 0.4 ng/g body wt on alternate days to achieve normocalcemia, whereas C−P− mice needed only 0.0625 ng/g body wt to achieve normocalcemia. Wild-type mice were able to maintain normocalcemia despite receiving 0.4 ng/g body wt. Arrows represent times of administration of 1,25(OH)2D3. Data are means ± SE (n = 6/genotype).
Calcemic response after administration of a single dose of 1,25(OH)2D3 on a Ca2+-deficient diet.
To determine the contribution of GI Ca2+ absorption to the serum Ca2+ responses seen in mice on the Ca2+-replete diet, we repeated the same experiments after conditioning the animals for 48–72 h on a Ca2+-deficient diet [0.01% Ca2+ (wt/wt), i.e., an 80-fold reduction in Ca2+ content] and plain water (Fig. 4). On the Ca2+-deficient diet, the calcemic response at 24 h in the C−P− mice was less robust (∼5 mg/dl) than on the Ca2+-replete diet and was similar to the change seen in the C+P− mice (4.9 mg/dl) but, again, was greater than that in the C+P+ mice (∼2.5 mg/dl). Therefore, the difference in the responses of the C−P− and C+P− mice to 1,25(OH)2D3 on the Ca2+-replete diet can apparently be accounted for by a difference in GI absorption of Ca2+.
Fig. 4.

Effect of a single dose (0.5 ng/g body wt) of 1,25(OH)2D3 in mice fed a Ca2+-deplete diet and plain water on serum Ca2+ levels over 72 h. Data are means ± SE (n = 8–12/genotype). #P > 0.05, C+P− vs. C−P− at 24 h.
Calcemic response after administration of a single dose of 1,25(OH)2D3 on a Ca2+-deficient diet with pamidronate pretreatment.
Because of the known stimulatory effect of 1,25(OH)2D3 on bone resorption, we evaluated the contribution of bone release of Ca2+ to the observed calcemic responses in mice on a Ca2+-deficient diet (Fig. 5). For this, animals were given 2.5 μg/g body wt pamidronate daily by ip injection for 2 wk, a dose higher than those previously shown to inhibit bone resorption in rats and mice (12, 16). During the last 48–72 h of this 2-wk period, the animals were placed on a Ca2+-deficient diet before injection of 0.5 ng/g body wt 1,25(OH)2D3. The mean maximal change in serum Ca2+ response to 1,25(OH)2D3 administration in the C−P− mice was 0.5 mg/dl, compared with 1.4 and 0.4 mg/dl, respectively, in the C+P− and C+P+ mice. There was a significant, albeit small, difference between C+P− and C−P− mice in serum Ca2+ level at 24 (0.9 mg/dl, P = 0.03) and 36 h (0.5 mg/dl, P = 0.04).
Fig. 5.

Effect of a single ip dose (0.5 ng/g body wt) of 1,25(OH)2D3 in mice fed a Ca2+-deplete diet and plain water after 2 wk of pretreatment with pamidronate on serum Ca2+ levels over 72 h. Data are means ± SE (n = 8–12/genotype). *P < 0.05, C+P− vs. C−P−.
CT responses after administration of a single dose of 1,25(OH)2D3 on normal chow and plain water.
To evaluate the CT responses of the three genotypes to acute 1,25(OH)2D3-mediated hypercalcemia, we evaluated circulating levels of CT in wild-type, C+P−, and C−P− mice 24 h after a single dose of 1,25(OH)2D3 (Fig. 6). There were no statistically significant changes in CT levels in wild-type and C+P− mice at 24 h compared with baseline and only a small absolute, although statistically significant, increase in serum CT in the C−P− mice, presumably reflecting, at least in part, the known inhibitory effect of 1,25(OH)2D3 on CT gene expression and secretion (22, 25).
Fig. 6.

Serum calcitonin (CT) response to a single ip dose (0.5 ng/g body wt) of 1,25(OH)2D3 at baseline and 24 h afterwards. Data are means ± SE (n = 8–12/genotype). *P < 0.05, C−P− at baseline vs. C−P− at 24 h after 1,25(OH)2D3 administration.
Urinary Ca2+ response after administration of a single dose of 1,25(OH)2D3 on normal chow and plain water.
Because of the critical role of the kidney in Ca2+ homeostasis and to define a PTH-independent role of the CaSR in the kidney in vivo, we evaluated the possible contribution of altered renal Ca2+ handling to the exaggerated calcemic response of the C−P− mice after administration of 0.5 ng/g body wt 1,25(OH)2D3 (Fig. 7). As expected, even in the absence of severe hypercalcemia in the wild-type and C+P− mice after 1,25(OH)2D3 injection (serum Ca2+ level ranging between 9.1 and 11.5 mg/dl at 24 h postinjection), we saw significant increases in the mean UCa/Cr values at 24 h compared with baseline in the wild-type (0.44 ± 0.18 to 1.54 ± 0.34, P = 0.02) and C+P− mice (0.36 ± 0.11 to 0.61 ± 0.15, P = 0.03). However, there was an actual decrease in UCa/Cr value (0.33 ± 0.13 to 0.11 ± 0.04, P = 0.03) despite frank hypercalcemia in the C−P− mice (mean serum Ca2+ level 12.4 ± 0.6 mg/dl).
Fig. 7.

Urinary Ca2+ excretion at baseline and 24 h after ip administration of 1,25(OH)2D3. Trend lines for urinary Ca2+-to-creatine ratio (UCa/Cr) vs. serum Ca2+ level are shown for C+P+ (line A), C+P− (line B), and C−P− (line C). Note the rightward and downward shift in urinary Ca2+ excretion corrected for urinary creatinine as serum Ca2+ level increases after 1,25(OH)2D3 administration. Data are presented as a scatter plot (n = 8–12/genotype).
Changes in renal gene expression 8 h after administration of a single dose of 1,25(OH)2D3 on normal chow and plain water.
The effect of 1,25(OH)2D3 on in vivo expression of genes involved in active [Na2+/Ca2+ exchanger 1 (NCX1), calbindin D9k (CalD9k), TRPV5] and passive [Na+-K+-2Cl− cotransporter (NKCC), claudin-16 (CLDN-16)] transport of Ca2+ in the distal convoluted and thick ascending limb of the kidney, respectively, as well as the action (VDR) and metabolism of vitamin D3 [1,25-dihydroxy vitamin D3-24-hydroxylase (CYP24A1)] were investigated (Fig. 8). 1,25(OH)2D3 administration resulted in statistically significant upregulation of NCX1, CalD9k, NKCC, and TRPV5 mRNA expression in all genotypes compared with baseline. There were no differences in baseline or 8-h gene expression levels of CalD9k, NKCC, and TRPV5 between the C+P− and C−P− mice. However, there was a significantly higher expression of NCX1 in the C−P− mice compared with the wild-type or C+P− mice after 1,25(OH)2D3 administration. There was no change in CLDN-16 gene expression in wild-type and C+P− mice after 1,25(OH)2D3 administration, but there was a fall in CLDN-16 gene expression in C−P− mice. There also were no differences in CYP24A1 expression levels between the genotypes, which rose substantially in all three genotypes after 1,25(OH)2D3 administration.
Fig. 8.

Expression profiles of transient receptor potential vanilloid channel 5 (TRPV5), vitamin D receptor (VDR), Na+/Ca2+ exchanger 1 (NCX1), Na+-K+-2Cl− cotransporter (NKCC), claudin-16, and 1,25-dihydroxy vitamin D3-24-hydroxylase (CYP24A1) in mouse kidney at baseline (solid bars) on a Ca2+-replete diet and plain water and 8 h after ip 1,25(OH)2D3 administration (shaded bars). The mRNA quantified by real-time PCR was calculated as a fold-change ratio relative to the mRNA for GAPDH in wild-type (C+P+) mice. Data are means ± SE (n = 6–8/genotype per condition). *P < 0.05, baseline vs. 8 h within genotypes. Y P < 0.05, baseline vs. 8 h within C−P−. #P < 0.05, baseline comparison, C+P+ vs. C+P−. ⧫ P < 0.05, baseline comparison, C−P− vs. C+P−. ¶P < 0.05, baseline comparison, C+P+ vs. C+P−. TP < 0.05; W P < 0.05; G P < 0.05, 8-h comparisons. There were no significant differences for other comparisons for which P values are not given.
Changes in duodenal gene expression after administration of a single dose of 1,25(OH)2D3 on normal chow and plain water.
Two key genes involved in the transport of Ca2+ in the duodenum, CalD9k and TRPV6, were evaluated (Fig. 9). As expected, there were significant increases in TRPV6 mRNA expression in wild-type (2-fold, P = 0.03), C+P− (8-fold, P = 0.01), and C−P− mice (10-fold, P = 0.01). There were similar increases in CalD9k mRNA expression after 1,25(OH)2D3 administration across all genotypes but no differences in expression between genotypes.
Fig. 9.

Expression profile of TRPV6 and calbindin D9k in mouse duodenum at baseline (solid bars) on a Ca2+-replete diet and plain water and 8 h after ip 1,25(OH)2D3 administration (shaded bars). Expression assays were performed as described in the legend for Fig. 8. Data are means ± SE (n = 6–8/genotype per condition). *P < 0.05, baseline vs. 8 h within genotypes. ¶ P < 0.05, baseline comparison, C+P+ vs. C+P−. T P < 0.05; W P < 0.05, 8-h comparisons. There were no significant differences for other comparisons for which P values are not given.
DISCUSSION
We examined changes in known hormonal feedback loops and the relative roles of various organs participating in the response to exogenous 1,25(OH)2D3 administration in vivo at the functional and molecular levels in mice with or without knockout of the CaSR and/or PTH genes.
We had previously defined the amount of supplemental Ca2+ in the drinking water needed to raise the serum Ca2+ level of the C+P− and C−P− mice to normocalcemic levels [1.5 and 0.75% (wt/vol), respectively] (13). In our preliminary work for this report, however, we noted that the exaggerated response of the C−P− mice to ip injection of 1,25(OH)2D3 was readily seen when they were hypocalcemic on normal chow and plain water, which avoided differences in the GI load of Ca2+ to which the various genotypes were exposed. These initial observations prompted evaluation of the relative contributions of the organs and hormonal systems involved in Ca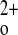 homeostasis in wild-type and KO animals to the exaggerated hypercalcemic response of the C−P− mice to 1,25(OH)2D3 to determine functions of the CaSR independent of CaSR-regulated PTH secretion.
homeostasis in wild-type and KO animals to the exaggerated hypercalcemic response of the C−P− mice to 1,25(OH)2D3 to determine functions of the CaSR independent of CaSR-regulated PTH secretion.
The pharmacokinetic profile observed after administration of a single dose of 1,25(OH)2D3 is consistent with results in other studies (21), where ip administration of 1,25(OH)2D3 yielded rapid (peak concentration within 1 h) bioavailability of >90% of the injected dose with no significant differences in plasma clearance. There were no significant differences between baseline and 24-h postinjection 1,25 (OH)2D3 levels for the C+P+ and C+P− mice and a small but significant difference for the C−P− mice of uncertain physiological relevance. There were no significant differences across the genotypes in serum 1,25(OH)2D3 levels at 1 or 24 h, suggesting that differences, if any, in clearance of 1,25(OH)2D3 were not critical in defining the differences in calcemic responses seen among the different genotypes. An explanation of the role of the CaSR independent of PTH in dampening the calcemic response to exogenous vitamin D is not provided by comparing the 24-h 1,25 dihydroxyvitamin D levels between the C+P− and C−P− mice, particularly since the C−P− mice had the highest serum Ca2+ level and lowest serum 1,25(OH)2D3 level at 24 h among the three genotypes Moreover, the more rapid fall in serum Ca2+ concentration in the C−P− compared with the C+P− mice also speaks against a reduced rate of clearance of 1,25(OH)2D3 and/or prolongation of its biological actions as factors contributing to the greater calcemic response of the C−P− mice. Therefore, it was more likely that variable sensitivities of the genotypes to genomic and/or nongenomic effects of 1,25(OH)2D3 were responsible for the observed differences in calcemic responses.
We then analyzed the calcemic responses to a single ip dose of 1,25(OH)2D3 on a Ca2+-replete/plain water diet and observed a marked calcemic response in the C−P− mice to frankly hypercalcemic levels, in contrast to the responses of the C+P+ and the C+P− mice, which remained within the normocalcemic range. The exaggerated response in C−P− mice also persisted after three consecutive doses given 48 h apart as shown in Fig. 3, supporting our observations of 1) a distinctly different potency of 1,25(OH)2D3 in inducing a calcemic response in the various genotypes, with the C−P− mice markedly more sensitive than the other two genotypes and the C+P− and C+P+ mice showing similar sensitivities, and 2) an effective defense against hypercalcemia conferred by the presence of the CaSR in the absence (in the C+P− mice) or in the presence of PTH (in the C+P+ mice) after 1,25(OH)2D3 administration. We also confirmed that an increase in CT was not a major factor in the defense against 1,25(OH)2D3-induced hypercalcemia in the C+P− and C+P+ mice (see Fig. 6), which supports in vitro studies showing that 1,25(OH)2D3 downregulates CT gene expression within hours of its administration (22, 25). The competing effects of 1,25(OH)2D3 to decrease CT mRNA expression and of increases in serum Ca2+ to increase CT secretion in vivo likely explain the lack of either a clear rise or fall in CT levels. This is in contrast to our observations of intact and robust increases (>10-fold) in serum CT in response to an increased dietary Ca2+ load in the drinking water in the C+P+ and C+P− mice and a markedly blunted response in C−P− mice (13).
The GI contribution to the observed responses in the C−P− mice to 1,25(OH)2D3 administration was significant as revealed by performing the same experiment on a mice fed a markedly Ca2+-deficient diet, although it is possible that there is some residual Ca2+ absorption of the small amount Ca2+ in the Ca2+-deficient diet (80-fold lower than Ca2+-replete diet). Under conditions of a Ca2+-deplete diet, the exaggerated calcemic response of C−P− mice to exogenous 1,25(OH)2D3 was lost compared with C+P+ and C+P− mice, although the two KO genotypes still showed a substantial calcemic response (Fig. 4). Therefore, by an uncertain mechanism, the CaSR apparently dampens GI Ca2+ absorption in response to 1,25(OH)2D3 administration. Given the known bone-resorptive effect of 1,25(OH)2D3, the possible contribution of bone to the calcemic responses observed in mice on a Ca2+-depleted diet was then evaluated by assessing the calcemic response to 1,25(OH)2D3 after inhibition of osteoclastic bone resorption using pamidronate, a nitrogen-containing bisphosphonate, in mice on a Ca2+-deficient diet (Fig. 5). Although the quantitative relationship between bone resorption and release of Ca2+ is unknown, the marked reduction in the calcemic response of the C−P− and C+P− mice to 1,25(OH)2D3 with pamidronate treatment confirmed that bone resorption was an important contributor to the rise in serum Ca2+ on the Ca2+-deplete diet and indicates that residual Ca2+ absorption on a Ca2+-deficient diet was likely not a significant contributor to the calcemic response observed in the three genotypes when receiving this diet without pamidronate. The modestly greater residual calcemic response to 1,25(OH)2D3 in the C+P− than in the C−P− mice is consistent with our earlier data showing that there is impaired osteoclastogenesis in vitro using marrow from C−P− mice compared with wild-type mice (18). Thus the absence of the full-length CaSR in the C−P− mice may have blunted their resorptive response to 1,25(OH)2D3 administration. In osteoblasts (and chondrocytes), in contrast, the CaSR lacking exon 5 appears to be able to compensate for loss of the full-length CaSR (5).
An assessment of spot UCa/Cr values at baseline and 24 h after 1,25(OH)2D3 injection on normal chow and plain water confirmed the role of the CaSR in increasing urinary Ca2+ excretion in response to an increased filtered load in the C+P+ and C+P− mice. After 1,25(OH)2D3 injection, the UCa/Cr value increased approximately threefold in the wild-type mice and twofold in the C+P− mice compared with a threefold decrease in UCa/Cr in the C−P− mice despite frank hypercalcemia in the latter (Fig. 7). The increases in urinary Ca2+ excretion in the C+P− mice in response to exogenous 1,25(OH)2D3 demonstrates that suppression of PTH is not required to see the increase in urinary Ca2+ accompanying increases in serum Ca2+. The absence of full-length CaSR, as seen in the C−P− mice, greatly impaired renal excretion of Ca2+ in response to hypercalcemia induced by 1,25(OH)2D3 administration. 1,25(OH)2D3 has a role, at least in mice, in enhancing renal Ca2+ reabsorption analogous to that of PTH, and the CaSR may blunt not only the GI but also the renal contribution [e.g., 1,25(OH)2D3-induced increase in tubular reabsorption of Ca2+] to the calcemic response to 1,25(OH)2D3 administration.
It is important to note in discussing the gene responses to exogenous 1,25(OH)2D3 administration (on normal chow and plain water) that wild-type mice were normocalcemic at baseline, whereas the C+P− and C−P− mice were hypocalcemic at baseline due to lack of PTH. It is, therefore, possible that the expression of genes such as VDR and CLDN-16 are increased in the C+P− and C−P− mice compared with wild-type mice at baseline in the kidney, presumably in an attempt to correct hypocalcemia. In the case of the VDR, however, the reductions in both serum Ca2+ (and presumably in CaSR activity, at least in the C+P− mice) and 1,25(OH)2D3 would have been expected to decrease the expression of the VDR (19). Therefore, the mechanism(s) producing the increased levels of the VDR in the hypocalcemic C+P− and C−P− mice is uncertain. However, for unclear reasons, there were no differences across all genotypes in baseline expression of TRPV5, NCX1, and NKCC genes in the kidney. The differences in baseline serum Ca2+ levels also may explain differences in the duodenum of baseline expression of PTH/vitamin D-inducible genes TRPV6 and CalD9k (Fig. 9), which were higher in the wild-type mice compared with the C+P− and C−P− mice.
Bearing in mind the variable relationship between gene transcription and translation, as well as protein trafficking/turnover/activity of transporters and channels that were not evaluated in this study, an evaluation of acute (dynamic) changes in gene expression 8 h after a single dose of 1,25(OH)2D3 was performed. This revealed predictable upregulation of TRPV5, NCX1, VDR, and CYP24A1 in the kidney and upregulation of TRPV6 and CalD9k gene expression in the duodenum in all genotypes. We also observed interesting increases in gene expression of NKCC that may suggest that 1,25(OH)2D3 stimulates not only active, transcellular reabsorption of Ca2+ but possibly also paracellular absorption of Ca2+ in the cortical thick ascending limb of the loop of Henle. There were no significant changes in the expression of CLDN-16 in wild-type and C+P− mice, but there was a significant decrease in expression of CLDN-16 in C−P− mice that is consistent with a report of 1,25(OH)2D3 decreasing CLDN-16 expression in HEK-293 cells in vitro, which do not express the CaSR endogenously (9).
There were differences in basal CYP24A1 (24-hydroxylase) expression among the genotypes. The physiological importance of this observation at baseline under hypocalcemic conditions on the subsequent calcemic responses seen after vitamin D administration is unknown, because there were no significant differences across genotypes in the relative levels of gene expression of the CYP24A1 gene 8 h after 1,25(OH)2D3 administration that could be attributable to the CaSR or PTH. The latter observation at 8 h postinjection, however, is consistent with the similar circulating 1,25(OH)2D3 levels in the three genotypes at 24 h after administration of 1,25(OH)2D3.
The known upregulation of renal CaSR expression in vivo in response to 1,25(OH)2D3, promoting urinary Ca2+ excretion, and perhaps in the intestine, thereby inhibiting intestinal Ca2+ absorption, could explain the ability of the wild-type and C+P− mice to dampen the hypercalcemic effects of 1,25(OH)2D3 administration.
The CaSR lacking exon 5 also may be upregulated by 1,25(OH)2D3 due to the presence of an intact promoter site, and it might be functional in mediating responses to Ca2+ in some or all tissues studied presently. However, if this is true, the CaSR lacking exon 5 in our C−P− mice is insufficient to dampen the hypercalcemic effect of 1,25(OH)2D3, and as such, our results may even underestimate the role of the CaSR in this response. Interestingly, evaluation of the expression of several key genes known to be involved in renal Ca2+ transport before and 8 h after 1,25(OH)2D3 administration showed upregulation of only the NCX1 gene in C−P− mice as a possible explanation of the decreased urinary Ca2+ excretion in the C−P− mice 24 h after 1,25(OH)2D3 administration. It is possible that variable kinetics of gene turnover for the genes analyzed in this study have made the search for culprit genes difficult to ascertain in our studies. Furthermore, other transporters or channels not evaluated, such as PCMA1b or CLDN-19, may be culprits. Finally, the CaSR may be involved in the regulation of trafficking of key Ca2+ transporters and channels to the cell surface as has been described for aquaporin-2 in vivo and in vitro.
In conclusion, the full-length CaSR, independent of PTH, is required to dampen the calcemic response to exogenous 1,25(OH)2D3 in vivo. In this exon 5 CaSR KO mouse model, in the absence of full-length CaSR, hypercalcemia from exogenous 1,25(OH)2D3 administration is likely due to exaggerated renal Ca2+ reabsorption and intestinal Ca2+ absorption and not from altered bone or CT responses. Renal NCX1 expression in mice lacking the full-length CaSR is significantly increased and may contribute to the PTH-independent, relative hypocalciuria observed 24 h after a single dose of 1,25(OH)2D3. These observations have potentially important implications for therapy involving the use of exogenous vitamin D3 in humans and provide more insight into PTH-independent roles of the CaSR in overall Ca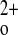 homeostasis.
homeostasis.
GRANTS
These studies were supported by National Institutes of Health (NIH) Career Development Award DK076733 (to O. Egbuna) and the Clinical Investigator Training Program at Beth Israel Deaconess Medical Center and Harvard/MIT School of Health Sciences and Technology in collaboration with Pfizer, Inc. and Merck & Co. E. Brown is supported by NIH Grant DK078331.
REFERENCES
- 1.Dusso A, Brown A, Slatoposky E. Vitamin D. Am J Physiol Renal Physiol 289: F8–F28, 2005. [DOI] [PubMed] [Google Scholar]
- 2.Ba J, Friedman PA. Calcium-sensing receptor regulation of renal mineral ion transport. Cell Calcium 35: 229–237, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Brown EM Calcium sensing by endocrine cells. Endocr Pathol 15: 187–220, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Buchan AM, Squires PE, Ring M, Meloche RM. Mechanism of action of the calcium-sensing receptor in human antral gastrin cells. Gastroenterology 120: 1128–1139, 2001. [DOI] [PubMed] [Google Scholar]
- 5.Chang W, Tu C, Chen TH, Bikle D, Shoback D. The extracellular calcium-sensing receptor (CaSR) is a critical modulator of skeletal development. Sci Signal 1: ra1, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chattopadhyay N, Brown EM. Role of calcium-sensing receptor in mineral ion metabolism and inherited disorders of calcium-sensing. Mol Genet Metab 89: 189–202, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Chertow GM, Pupim LB, Block GA, Correa-Rotter R, Drueke TB, Floege J, Goodman WG, London GM, Mahaffey KW, Moe SM, Wheeler DC, Albizem M, Olson K, Klassen P, Parfrey P. Evaluation of Cinacalcet Therapy to Lower Cardiovascular Events (EVOLVE): rationale and design overview. Clin J Am Soc Nephrol 2: 898–905, 2007. [DOI] [PubMed] [Google Scholar]
- 8.Dvorak MM, Chen TH, Orwoll B, Garvey C, Chang W, Bikle DD, Shoback DM. Constitutive activity of the osteoblast Ca2+-sensing receptor promotes loss of cancellous bone. Endocrinology 148: 3156–3163, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Efrati E, Arsentiev-Rozenfeld J, Zelikovic I. The human paracellin-1 gene (hPCLN-1): renal epithelial cell-specific expression and regulation. Am J Physiol Renal Physiol 288: F272–F283, 2005. [DOI] [PubMed] [Google Scholar]
- 10.Fudge NJ, Kovacs CS. Physiological studies in heterozygous calcium sensing receptor (CaSR) gene-ablated mice confirm that the CaSR regulates calcitonin release in vivo. BMC Physiol 4: 5, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoenderop JG, Chon H, Gkika D, Bluyssen HA, Holstege FC, St-Arnaud R, Braam B, Bindels RJ. Regulation of gene expression by dietary Ca2+ in kidneys of 25-hydroxyvitamin D3-1 alpha-hydroxylase knockout mice. Kidney Int 65: 531–539, 2004. [DOI] [PubMed] [Google Scholar]
- 12.Jara A, Lee E, Stauber D, Moatamed F, Felsenfeld AJ, Kleeman CR. Phosphate depletion in the rat: effect of bisphosphonates and the calcemic response to PTH. Kidney Int 55: 1434–1443, 1999. [DOI] [PubMed] [Google Scholar]
- 13.Kantham L, Quinn S, Egbuna O, Pang J, Butters R, Pollak M, Brown EM. The calcium sensing receptor defends against hypercalcemia independent of its regulation of PTH. Abstracts of the 29th Annual Meeting of the American Society for Bone and Mineral Research, Honolulu, Hawaii. Washington, DC: Am Soc Bone Min Res, 2007.
- 14.Kos CH, Karaplis AC, Peng JB, Hediger MA, Goltzman D, Mohammad KS, Guise TA, Pollak MR. The calcium-sensing receptor is required for normal calcium homeostasis independent of parathyroid hormone. J Clin Invest 111: 1021–1028, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kutuzova GD, Deluca HF. Gene expression profiles in rat intestine identify pathways for 1,25-dihydroxyvitamin D3 stimulated calcium absorption and clarify its immunomodulatory properties. Arch Biochem Biophys 432: 152–166, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee CT, Huynh VM, Lai LW, Lien YH. Cyclosporine A-induced hypercalciuria in calbindin-D28k knockout and wild-type mice. Kidney Int 62: 2055–2061, 2002. [DOI] [PubMed] [Google Scholar]
- 17.Lemay J, Demers C, Hendy GN, Delvin EE, Gascon-Barre M. Expression of the 1,25-dihydroxyvitamin D3-24-hydroxylase gene in rat intestine: response to calcium, vitamin D3 and calcitriol administration in vivo. J Bone Miner Res 10: 1148–1157, 1995. [DOI] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods 25: 402–408, 2001. [DOI] [PubMed] [Google Scholar]
- 19.Maiti A, Hait NC, Beckman MJ. Extracellular calcium-sensing receptor activation induces vitamin D receptor levels in proximal kidney HK-2G cells by a mechanism that requires phosphorylation of p38alpha MAPK. J Biol Chem 283: 175–183, 2008. [DOI] [PubMed] [Google Scholar]
- 20.Mentaverri R, Yano S, Chattopadhyay N, Petit L, Kifor O, Kamel S, Terwilliger EF, Brazier M, Brown EM. The calcium sensing receptor is directly involved in both osteoclast differentiation and apoptosis. FASEB J 20: 2562–2564, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Muindi JR, Modzelewski RA, Peng Y, Trump DL, Johnson CS. Pharmacokinetics of 1alpha,25-dihydroxyvitamin D3 in normal mice after systemic exposure to effective and safe antitumor doses. Oncology 66: 62–66, 2004. [DOI] [PubMed] [Google Scholar]
- 22.Naveh-Many T, Silver J. Regulation of calcitonin gene transcription by vitamin D metabolites in vivo in the rat. J Clin Invest 81: 270–273, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oda Y, Tu CL, Chang W, Crumrine D, Komuves L, Mauro T, Elias PM, Bikle DD. The calcium sensing receptor and its alternatively spliced form in murine epidermal differentiation. J Biol Chem 275: 1183–1190, 2000. [DOI] [PubMed] [Google Scholar]
- 24.Oda Y, Tu CL, Pillai S, Bikle DD. The calcium sensing receptor and its alternatively spliced form in keratinocyte differentiation. J Biol Chem 273: 23344–23352, 1998. [DOI] [PubMed] [Google Scholar]
- 25.Peleg S, Abruzzese RV, Cooper CW, Gagel RF. Down-regulation of calcitonin gene transcription by vitamin D requires two widely separated enhancer sequences. Mol Endocrinol 7: 999–1008, 1993. [DOI] [PubMed] [Google Scholar]
- 26.Schoeber JP, Hoenderop JG, Bindels RJ. Concerted action of associated proteins in the regulation of TRPV5 and TRPV6. Biochem Soc Trans 35: 115–119, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Tfelt-Hansen J, Brown EM. The calcium-sensing receptor in normal physiology and pathophysiology: a review. Crit Rev Clin Lab Sci 42: 35–70, 2005. [DOI] [PubMed] [Google Scholar]
- 28.Tu Q, Pi M, Karsenty G, Simpson L, Liu S, Quarles LD. Rescue of the skeletal phenotype in CasR-deficient mice by transfer onto the Gcm2 null background. J Clin Invest 111: 1029–1037, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]