

Abstract
Studies in animal models form the basis for much of our current understanding of the pathophysiology of asthma, and are central to the preclinical development of drug therapies. No animal model completely recapitulates all features of the human disease, however. Research has focused primarily on ways to generate allergic inflammation by sensitizing and challenging animals with a variety of foreign proteins, leading to an increased understanding of the immunological factors that mediate the inflammatory response and its physiological expression in the form of airways hyperresponsiveness. Animal models of exaggerated airway narrowing are also lending support to the notion that asthma may represent an abnormality of the airway smooth muscle. The mouse is now the species of choice for asthma research involving animals. This presents practical challenges for physiological study because the mouse is so small, but modern imaging methodologies, coupled with the forced oscillation technique for measuring lung mechanics, have allowed the asthma phenotype in mice to be precisely characterized.
Keywords: allergic inflammation, mouse, airway smooth muscle, lung impedance
asthma is a prime example of a “complex disease,” a term that has become popular for characterizing those pathologies that appear to have multifaceted etiologies and an entanglement of underlying mechanisms. The moniker of complex disease, however, often tends to belie a general lack of understanding about what is going on, and asthma is no exception. Indeed, asthma is more properly regarded as a syndrome than a disease because it is defined on the basis of clinical characteristics rather than underlying mechanisms, and there remains a great deal of controversy about which of the possible mechanisms are the most important (20). The principal characteristics of asthma are reversible airflow obstruction, hyperresponsiveness of the lungs to challenge with smooth muscle agonists, and airway inflammation. Not surprisingly, allergy heads the list of causative suspects, but it is by no means alone. Exercise, cold air, and emotional stress are also known triggers of asthma (1), leading to the notion that asthma itself represents merely the common clinical endpoint of a number of distinct disease processes (42, 53, 91).
As with most human diseases, studies in laboratory animals have produced much of what we currently think we know about the mechanisms responsible for asthma. Obviously, the relevance and validity of these studies are tied to how well we can produce accurate animal equivalents of human asthma. The development of such “animal models” is still very much a work in progress; although many of the various features of asthma have been convincingly recapitulated in animals, invariably every animal model misses some important aspect of the human syndrome (97). Also, very few animals spontaneously develop a condition with any similarity to asthma, the most reminiscent being an allergic syndrome in cats (72) and the condition known as heaves in horses (24). If asthma is a condition peculiar to humans for some reason, then having it manifest in its entirety in a laboratory animal may simply be impossible. On the other hand, given that we still do not fully understand what asthma in humans actually is, it remains difficult to know whether an animal really has it or not. Accordingly, much of the challenge in studying animal models of asthma lies in phenotyping them properly, particularly as asthma is defined in humans in terms of phenotype rather than underlying pathology. In this review, we therefore focus on the issue of how to assess the relevant function and structure in the lungs of laboratory animals, and what clues this has given us about the possible mechanistic underpinnings of asthma in humans.
Development of Animal Models
Early animal models of asthma were developed in a variety of species (90) and focused on the phenomenon of airways hyperresponsiveness (AHR), defined as excessive bronchoconstriction in response to a standardized challenge. The challenging agent was usually a smooth muscle agonist such as methacholine or histamine. These agonists were intended to mimic the actions of mediators released as part of an overexuberant immune response thought to be behind an attack of allergic asthma. In a similar vein, some animal models employed hyperpnea to mimic the asthma brought on by exercise (35). In any case, the motivation behind these models arose from the idea that asthma is primarily a matter of excessive shortening of the airway smooth muscle (98). This notion was also reflected in the predominant use of β-agonists to relax smooth muscle as frontline therapy for most asthma cases.
In the early 1980s, a greater awareness of the role of inflammation in asthma developed, driven by an increased understanding of allergic immunology together with observations that human asthmatics frequently exhibited marked symptoms when challenged with antigens of various kinds. The first inflammatory cell to be linked firmly to asthma pathogenesis was the eosinophil (27, 63), followed soon after by the T cell (66). This led to increasing use of corticosteroids in the routine treatment of asthma. Current asthma guidelines (1) call for combining corticosteroids with long-acting β-agonists in treating persistent asthma, which seems appropriate given its current definition comprising both obstructive and inflammatory components. In fact, it now seems that treating asthma with a β-agonist alone may be inadvisable. Indeed, monotherapy with some long-acting forms has recently been designated as unacceptable by the Food and Drug Administration.
As a result of studies with animal models, we now know that allergic asthma involves a complex interplay between the innate and adaptive immune systems. The involvement of adaptive immunity begins with naïve CD4 T cells that differentiate into T helper (Th) cells with the potential to regulate the fate, function, and location of a variety of other immune cell components. Different subsets of Th cells have been defined on the basis of the cytokines they secrete, with Th1 and Th2 cells being the best known examples. The production of Th1 cells is promoted primarily by IL-12, whereas differentiation into Th2 cells occurs in the presence of IL-4. In turn, Th1 cells produce IFNγ, whereas Th2 cells produce IL-4, IL-5, and IL-13. Allergic asthma is associated with a Th2 type of immune response, since the Th2 cytokines are known to cause many of the features of the disease (36, 52). Th2 cytokines, particularly IL-5 and to certain extent IL-4, are also responsible for linking adaptive to innate immunity by promoting eosinophil proliferation in bone marrow and subsequent migration to the lung. Eosinophilia in the airways has long been linked to allergic asthma, although exactly why has been controversial. Nevertheless, their pivotal role in altering lung function in allergic mice has been demonstrated in experiments with eosinophil knockout animals (58).
The Th2 cytokine IL-4 is also implicated in the dominance of antigen-specific IgE over other antibody isotypes in allergic asthma. Although a number of cytokines such as IL-21 are involved in promoting B cells to produce antibody and generation of plasma cells, only IL-4 and IL-13 are known to promote isotype switching to IgE (71). Binding of antigen-specific IgE to its Fcɛ receptor (FcɛR) on mast cells then promotes degranulation, leading to secretion of a number of bronchoactive mediators including histamine. Increased IgE levels would seem to contribute to the pathogenesis of asthma, although clinical trials with anti-IgE therapy (Xolair, omalizumab) show only modest therapeutic efficacy (19).
Th2 cells are not the only T cell variants associated with allergic airway inflammation, however. Recently, a role has been demonstrated for natural killer T (NKT) cells, which are characterized (55) by the presence of an invariant T cell receptor (Vα14 Jα18 associated with Vβ8, Vβ7, and Vβ2) and are restricted by the CD1d MHC I-like molecule and recognize glycolipids instead of protein antigen (16). Despite their relatively low numbers, NKT cells have the capacity to rapidly produce high levels of various cytokines upon antigen recognition (16) and are found in the lung. Transfer of NKT cells from mice with allergic airway inflammation to naïve mice is sufficient to induce airway hyperreactivity (3).
Another CD4 subset that has emerged within the last two years is the Th17 cell. Both TGFβ and IL-6 are necessary, although not sufficient, for the differentiation of Th17 cells (17, 62, 86). These cells produce mainly IL-17, some IL-22 and IL-21, small amounts of IFNγ, and no IL-4 (46, 69). Although IL-17 is clearly detectable in allergically inflamed lungs, its role in the pathogenesis of the allergic response remains unclear. It seems that IL-17 may be needed for the initial development of airway inflammation in mice, but administration of IL-17 decreases eosinophilia. Also, adoptive transfer of Th17 cells induces neutrophilia and imparts resistance to steroid therapy, suggesting that IL-17 may function to promote neutrophil recruitment (55) and could therefore be particularly important in severe asthma (64). IL-17 has also been shown to interfere with epithelial cell production of eotaxin (77), which plays a major role in the recruitment and activation of eosinophils (70). IL-17 may thus regulate the eosinophil-neutrophil balance in the lung.
Despite the wealth of knowledge we now have about the role of immunity in allergic asthma, the complex pathophysiology of the disease caused researchers to have a difficult time agreeing over whether asthma is due primarily to altered immune status or to an abnormality of airway smooth muscle, or even both simultaneously (5). Work has thus proceeded along both fronts, driven by the respective convictions of the scientists involved. The development of animal models of asthma has been correspondingly schizophrenic. Nevertheless, the asthma world has come together on one thing regarding animal models: the species. As with most animal-based biomedical research, mice now dominate the scene because of the immunological and molecular tools available to study them, together with their obvious practical advantages related to cost and gestation period (23, 50). This is not to say, of course, that the mouse is the only species used to model asthma. Rat models have also been widely employed (21, 84), notably the Brown-Norway strain because of its propensity to exhibit a later allergic response following antigen challenge (84). Monkeys have also attracted attention as a useful model that bridges the gap to human relevance more than rodent species (28, 39, 61). Nevertheless, most asthma-related research continues to be pursued in the mouse. The development of transgenic mice that exhibit various lung pathologies is now a huge research enterprise, and mouse models of lung disease have been the subject of a number of recent reviews that cover their various pathophysiological features in detail (11, 23, 49, 50, 54, 67).
Mouse Models of Allergic Asthma
Allergic mouse models of asthma are generated by first sensitizing an animal to a foreign protein, most commonly ovalbumin. This is typically done by injecting the protein intraperitoneally along with an adjuvant, typically aluminum hydroxide, that serves to enhance the protein's immunogenicity for reasons that are complex and not entirely understood (56). After the immune system has had a chance to mount a reaction against the antigenic protein, which takes several days, the animal receives a further antigen exposure either directly to the lungs in the form of an aerosol or via postnasal drip following nasal instillation. This elicits an inflammatory reaction in the lungs characterized by an influx of eosinophils, epithelial thickening, and AHR. There is wide variation, however, in the precise recipe used by different investigators to perform these various steps. A typical scenario is to sensitize an animal with two intraperitoneal injections spaced a week or two apart, wait another week, and then challenge the animal by exposure to a 1% ovalbumin aerosol for 30 min each day for 3 days (87). The features of allergic inflammation typically reach their peak a day or two after the final exposure, although the time course of this process has not been fully characterized.
Many variations on all aspects of the above theme have been tried. For example, inhalational exposure to NO2 has been used as a means of bolstering the immune response in place of an intraperitoneal adjuvant (18), invoking the notion that airborne pollutants may play a role in the development of asthma in industrialized societies. Also, questions about the relevance of ovalbumin to human asthma have led to the use of antigens such as extracts of house dust mite (30) and aspergillus (40), which are more representative of those that occur naturally. Of particular recent interest has been the issue of chronicity of antigen exposure. The sensitization and challenge procedure described above is very acute, yet asthma in humans is typically a chronic disease that exhibits clear features of airway remodeling in its later stages (51). Researchers have thus been motivated to explore the effects of longer-term antigen exposures in mice. Somewhat disappointingly, extended exposure to ovalbumin aerosol in at least some mouse strains such as the BALB/c leads to tolerization, characterized by the waning of the inflammatory and hyperresponsive phenotypes (9, 94). There have been some reports that other antigens such as house dust mites produce a more extended asthma-like picture (30). Furthermore, simultaneous exposure to multiple antigens produces inflammation in the absence of adjuvant, and may even break through the tolerization barrier to produce a chronic phenotype (38). Nevertheless, the chronic model has yet to be optimized, particularly in view of the rather modest degree of AHR that has so far been demonstrated. Indeed, the generation of allergic mouse models of asthma remains something of a black art, no doubt due to the immensely complex and often poorly understood biological processes involved.
Fully elucidating the biological complexities of allergic mouse models of asthma is going to take some time, but it has moved ahead in recent years thanks to significant technical advances in phenotyping using both lung function assessment and imaging. The measurement of lung mechanical function in mice poses a particular challenge, of course, due to the small size of the animal (50). Measurement techniques developed in larger animals or humans frequently do not do well when scaled down to the mouse. The frustration this causes has led to a temptation to cut corners in the assessment of the mouse phenotype, the most problematic result of which has been the use of the parameter known as enhanced pause (Penh) to assess AHR (43). Due to its attractions of noninvasiveness and simplicity, as well as its availability in a commercial device (31, 32), Penh has become widely used in the study of AHR. Unfortunately, however, this use has invariably been inappropriate. Penh is merely a nonspecific feature of the breathing pattern and cannot be taken as a measure of lung mechanics, as has been discussed extensively elsewhere (7, 65). Of course, because the mechanical properties of the lung have an important bearing on the pattern of breathing (43), it is conceivable that a change in the breathing pattern, such as might be reflected in Penh, could signal the presence of some event worthy of more detailed investigation, such as activation of irritant receptors in the lung (2). On the other hand, one can just as easily screen for a change in breathing pattern using the obvious parameters of breathing frequency and/or tidal volume that seem to be just as efficacious as Penh (2) but do not suffer from the latter's obscurity of meaning. Indeed, we would go so far as to suggest that the use of Penh be avoided completely (7). In any case, the pattern of breathing can never serve as a substitute for a measure of mechanical function.
Recently, two approaches have been used to extend unrestrained plethysmography by combining it with an independent noninvasive measure of changes in lung volume to estimate specific airway resistance (14, 76). These approaches are theoretically sound and may eventually prove useful for rapid screening, but the information they provide is noisy and subject to the vagaries of the animal's spontaneous breathing pattern. The problem underlying these approaches has been ascribed to the “phenotyping uncertainty principle” (11), which states that there is a fundamental trade-off to be made between the maintenance of natural measurement conditions and precision. Penh sits at one extreme of this trade-off; a conscious animal that is free to choose its own pattern of activity and breathing exists in its most natural state, but the measurements of lung mechanics one can obtain under these conditions are noisy and very nonspecific.
At the other extreme of the phenotyping uncertainty principle lies the measurement of lung mechanics using the forced oscillation technique in anesthetized, tracheostomized, paralyzed mice (11). Here the animal's breathing pattern is entirely under the control of the experimenter, and the upper airways are bypassed so that their mechanical properties do not interfere with those of interest (i.e., the lung). On the other hand, one is left with the conundrum that the extensive interventions employed to achieve this level of experimental control may have caused the mechanical properties of the lung to depart significantly from their natural state. Nevertheless, our understanding of AHR in mice has advanced considerably in recent years using this approach. Intermediate between the above two extremes of the phenotyping uncertainty principle lie methods, such as the measurement of transfer impedance in restrained animals (47), that are based soundly in physical theory, but which are subject to greater variation as a result of fewer constraints on the behavior of the animal.
Assessing Lung Mechanics in Mice
The assessment of lung mechanics in mice, or any experimental animal for that matter, boils down, in essence, to determining how difficult (or easy) it is to drive a given volume of gas into the lungs over a given period of time. At its most basic level, this process can amount simply to examining the peak airway pressures achieved during regular mechanical ventilation (11, 29, 50). Clearly, an increased peak pressure indicates an increased impediment to lung inflation, although in an entirely nonspecific manner. Much greater specificity is achieved with the forced oscillation technique, in which an appropriate oscillatory flow signal is applied to the airway opening while the airway pressure is measured. In fact, flow signals containing a single dominant frequency determined by the number of breaths per minute have been used for decades to obtain estimates of lung resistance and elastance (6, 74). Numerical techniques based on the Fourier transform (13) have been used more recently to determine resistance and elastance at a number of different frequencies simultaneously when the oscillatory flow signal contains components at those frequencies. This multifrequency information about the respiratory system is encapsulated in a complex function of frequency known as the input impedance (Zin) (74). Being a complex function, Zin actually consists of two independent components known as the real part and the imaginary part, themselves both functions of frequency. The real part is known as the resistance because, at any particular frequency, it is equal in value to that of the conventional resistance that would be measured by oscillating the system at only that frequency. The imaginary part of Zin is known as the reactance, and is equal to conventional elastance scaled by the inverse of frequency. Obtaining Zin in a mouse is technically challenging because of the animal's small size, but is now routinely achieved using either a commercially available device known as the Flexivent that is based around a computer-controlled piston oscillator (78) or a wavetube coupled to a loudspeaker (44).
Interpreting Zin is performed on the basis of a mathematical model of the lung with a structure that is interpretable in physiological terms. The appropriate model depends on the frequency range over which Zin is measured. When frequency is kept below ∼20 Hz in mice, the model that has come to dominate the advanced investigation of lung mechanics is the so-called constant phase model introduced by Hantos and colleagues (45). This model has a particularly simple structure consisting of a single viscoelastic lung compartment served by a single airway. The airway has a resistance Raw that reflects its caliber and length. The mass of the air in the airway also provides the model with an inertance, although this is so small in mice that it can be neglected below the 20-Hz maximum frequency typically used to determine Zin (44, 75, 87). The viscoelastic compartment is characterized by two parameters, G and H, that account for the resistance and elastic properties, respectively, of the lung tissues. It has been found empirically that the impedance of the viscoelastic compartment has a special form in which the ratio of its real part to its imaginary part does not change with frequency. It is this feature that gives rise to the name “constant phase.”
The equation for the impedance of the constant phase model of the lung is
 |
(1) |
where f is frequency in Hz and
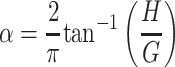 |
(2) |
RN in Eq. 1 is referred to as a Newtonian resistance because it has been found experimentally to behave like a classic resistor, but experiments with alveolar capsules in open-chest animals have confirmed that it provides an accurate measure of the overall resistance of the airway tree (83). G and H together encapsulate the mechanical properties of the lung periphery. When the lung behaves in a mechanically homogeneous fashion, G and H reflect the intrinsic dissipative and elastic properties, respectively, of the tissues. Experimental studies have shown, however, that G invariably increases proportionately more than H when the lung becomes mechanically heterogeneous in such as way that ventilation is not apportioned to different regions of the lung according to the volume of each region, but rather becomes differentially affected due to inequities in regional airway resistance and tissue stiffness. This finding is supported by theoretical analysis (8) and computational modeling (60, 82). An increase in the ratio G/H, known as hysteresivity (34), thus serves as a convenient marker for the presence of regional heterogeneities, which typically accompany most lung diseases and are also brought on transiently by induced bronchoconstriction (87). By contrast, if a fraction of the lung becomes isolated from the airway opening either by closure of the subtending airways or by atelectasis, collectively referred to as derecruitment, estimates of G and H will increase in the same proportion, leaving hysteresivity unchanged (4). The parameters RN, G, and H of the constant phase model together thus provide a means for distinguishing changes taking place in the conducting airways vs. changes in the lung periphery, and of inferring the presence of ventilation heterogeneity vs. derecruitment.
Figure 1 shows an example of the use of RN, G, and H to infer details about the nature of AHR in mice. Allergic inflammation was induced in BALB/c mice using the standard 3-day challenge protocol described above. Groups of inflamed and control animals were then exposed for 40 s to an aerosol of methacholine and then switched immediately to a 5-min protocol during which Zin was measured approximately every 15 s. All three impedance parameters increased following cessation of the challenge, with the responses from the inflamed animals being substantially greater than those in the controls. The biggest difference between inflamed and control animals, however, occurred in H, the parameter that measures lung elastance, indicating that the major effect of inflammation was manifest in the lung periphery. What is more, the ratio G/H remained largely unchanged throughout, implying that the peripheral effect that occurred was due to derecruitment of lung units. Closure of small airways was subsequently confirmed directly using microcomputed tomography (59) on the basis of images such as those shown in Fig. 2, which also serve to illustrate that the airway tree of the mouse is structurally rather different to that of the human.
Fig. 1.

The parameters of respiratory input impedance (RN, G, and H) expressed as fractional changes above baseline (ΔRN, ΔG, and ΔH, respectively) in control and allergically inflamed mice following a 40-s challenge with an aerosol of methacholine. MCh, the time of completion of delivery of methacholine. DI, the time of delivery of 2 deep lung inflations to 25 cmH2O. [From Wagers et al. (87).]
Fig. 2.

Minimum intensity projection images of a normal mouse lung (left) and an allergically inflamed mouse lung (right) following ventilation with pure oxygen obtained using microcomputed tomography. The missing basal air spaces in the inflamed lung (arrows) represent atelectatic regions that became consolidated after absorption of the oxygen by the capillary blood. Note the large bulbous appearance of the central airways and the asymmetrical branching pattern of the airway tree, which is typical of the mouse but very different in the human. [Adapted with permission from Lundblad et al. (59).]
Linking Structure to Function
Despite the technical advance represented by the forced oscillation technique over earlier methods that yielded only single values for lung resistance and elastance, the parameters of the constant phase model still constitute an extremely simplistic view of a very complicated organ. Using only measurements of pressure and flow at the airway opening, it is not currently possible to reconstruct a more detailed picture of the lung than that contained in RN, G, and H. However, one can often use computer simulation to address questions about what might be causing experimentally observed changes in these parameters. Here one tries to construct as detailed a model of the lung as anatomical and physiological knowledge will allow. The model is then used to simulate measurements of airway pressure and flow under conditions that match those used experimentally. The model thus serves as a virtual laboratory in which the feasibility of specific hypotheses can be addressed, providing in silico experimentation to complement conventional in vivo and in vitro experiments.
An example of this approach has been employed to investigate what might be responsible for the AHR observed in acutely inflamed BALB/c mice (87). A computational model of the mouse lung was constructed in which every airway in the entire airway tree was individually accounted for, with tissue units appended to each terminal bronchiole. The model was calibrated by adjusting its parameters until it exhibited changes in RN, G, and H similar to those seen in normal mice exposed to an aerosol of methacholine. To achieve this, however, it was not enough simply to simulate the effects of smooth muscle shortening by narrowing the model airways. Derecruitment of some fraction of the model lung was also required to mimic the increases in lung elastance observed experimentally. This was achieved by completely closing any airway in the model that narrowed to a specified critical radius, thereby mimicking the effects of formation of a liquid bridge across the airway lumen. In silico experimentation was then performed with the model in an attempt to make it hyperresponsive to methacholine in a way that matched experimental observations in allergically inflamed mice. Simply increasing the extent of smooth muscle shortening reproduced the observed changes in RN but failed to match the changes in G and H. However, if the degree of shortening was kept the same as in control mice, but the epithelial layer in the model was thickened to match histological observations in the inflamed animals and the critical airway closure radius was also increased somewhat, the model was able to accurately mimic the behavior of all three impedance parameters (15). This suggests that the mechanism giving rise to AHR in the acutely inflamed BALB/c mouse does not involve an abnormality of the airway smooth muscle, but instead is linked to physical changes taking place in the periphery of the lung itself. Of course, results such as these are dependent on the validity of the model used to make the simulations, and no model is a perfect representation of reality. However, the model used in this case was based reasonably closely on the anatomy of the mouse lung, so the results of such simulations can lend strong support to one hypothesis over another.
The use of computational modeling can also raise important questions for future investigation. For example, geometric amplification of airway luminal narrowing can potentially occur either by epithelial thickening or by increased mucus secretion. Certainly, epithelial hypertrophy is a characteristic feature of allergic inflammation (87), but so is mucus hypersecretion. Mucus is produced by goblet cells in the airway epithelium, and the appearance of excessive mucus in the airways can lead to significantly impaired lung function. Furthermore, IL-13, one of the Th2 cytokines, is the major inducer of epithelial cell mucus (93), signaling through the IL-13/4 receptor-α complex (92) and epidermal growth factor receptor (96). We do not know which of these two factors, epithelial thickening or excessive mucus, is dominant in allergically inflamed mice. Experimenting with mice in which these two effects are individually manipulated would thus be an important area for future research.
Mouse Models of Excessive Airway Smooth Muscle Shortening
Although allergy and inflammation are widely recognized as central to the pathogenesis of asthma, there is a school of thought that places the principal blame for asthma squarely at the feet of the airway smooth muscle. Although the jury is still out on this question, considerable effort has been devoted to identifying animal models in which AHR results from excessive smooth muscle shortening. Related to this, some degree of airway smooth muscle hypertrophy has been identified in several chronic models of allergic inflammation (57, 79). The epithelium probably also plays an important role in ASM hyperplasia and subsequent AHR; selectively activating the nuclear transcription factor NF-κB in the airway epithelium results in airway smooth muscle hyperplasia and associated AHR (73). There is also evidence that the alterations in smooth muscle associated with inflammatory remodeling result in an increase in the speed of muscle contraction (80). It is not entirely clear why this should be associated with AHR and asthma, although there is speculation that it may allow the smooth muscle to more easily move into a latch state from which escape is difficult (33).
Enhanced shortening of airway smooth muscle has been induced by treating animals with cationic proteins (Fig. 3). Mice receiving an intratracheal instillation of poly-l-lysine were found to be hypersensitive to methacholine when it was delivered as an aerosol but not as an intravenous injection (48), implicating degradation of the normal barrier function of the epithelium as the causative factor. In other words, by making the underlying smooth muscle more accessible to agonists introduced into the airways, bronchoconstriction was enhanced both in terms of magnitude and speed of onset. Poly-l-lysine is an analog of the highly charged cationic proteins found in all inflammatory cells, particularly the eosinophil, suggesting a way in which the events accompanying allergic inflammation might lead to hypersensitivity and AHR (85).
Fig. 3.

The time course of bronchoconstriction in BALB/c mice following aerosolization of 3.125, 12.5, and 50 mg/ml methacholine. The open circles are saline-treated animals; the closed circles are animals treated with poly-l-lysine. The vertical dotted lines bracket parameter values obtained following deep lung inflations given to reestablish baseline conditions. Note the exaggerated response in RN compared with G and H in the animals treated with poly-l-lysine compared with controls. This contrasts with the effects of allergic inflammation shown in Fig. 1. *Significant difference in magnitude of response; +significant difference in timing of peak. P < 0.05. [From Bates et al. (15).]
Differences in the intrinsic contractility of airway smooth muscle also arise spontaneously between different strains of animal. The A/J mouse, for example, has been shown to be hyperresponsive, and, in particular, to exhibit bronchoconstriction that develops particularly rapidly following challenge (26, 88). There are also clear differences in natural airways responsiveness between different strains of rat, the Fischer and Brown-Norway strains being key examples (37, 89). Accordingly, we now recognize that genetic factors play a major role in determining AHR.
The Role of Lung Volume
The degree of airway smooth muscle shortening elicited by challenge with a standard dose of agonist is highly dependent on transpulmonary pressure. Most of the pulmonary airways are embedded in the lung parenchyma, which transmits transpulmonary pressure from the airway lumen to the pleural surface. The parenchymal attachments to the airway wall exert an outward tethering force that opposes shortening of the airway smooth muscle. The mitigating effect of these parenchymal forces is dramatically demonstrated by the way in which airways responsiveness is decreased by an increase in transpulmonary pressure of only a few cmH2O (10, 22). Thus, a normoresponsive animal can be rendered very asthma-like simply by reducing its lung volume, just as has been demonstrated to be the case in human subjects (25). There are also some animal studies showing that chronic alterations in lung volume can lead to persistent changes in airways responsiveness, suggesting the possibility that airway smooth muscle somehow eventually adapts to a new lung volume (81). Of course, the forces of parenchymal tethering are not the only loads opposing airway smooth muscle shortening; the stiffness of the airway wall itself has also been shown to play a significant role in this regard (12, 68). In addition, the airway remodeling known to accompany some inflammatory conditions presumably has the potential to alter wall stiffness, but whether this tends to work for or against AHR remains controversial. It has recently been shown, for example, that there is no evidence of an effect on AHR due to mechanical changes in the airway wall of the acutely inflamed mouse (22). On the other hand, more chronic allergen exposures have shown evidence of an airway remodeling effect on AHR (79).
It must be mentioned that a great deal of animal work related to the hyperresponsiveness or otherwise of airway smooth muscle takes place in vitro, usually with tracheal smooth muscle, which is easily isolated and subjected to controlled force-length studies. The precision of the measurements that can be made with this preparation has led to the discovery of many intriguing phenomena such as the ability of smooth muscle to adapt its force generating capacity to baseline length (95). The biochemistry associated with the different phosphorylation states of the smooth muscle crossbridge is also rather complicated (41). It remains to be seen how significant some of these phenomena really are for asthma at the level of the entire animal or human subject. Nevertheless, computational modeling of the dynamic behavior of activated airway smooth muscle and its mechanical integration into the airway wall has shown promise in accounting for bronchoconstriction in vivo (10, 12, 22).
Mouse Models of Severe Asthma
In the animal laboratory, AHR is defined in statistical terms; a hyperresponsive animal exhibits significantly worse lung function than a control animal when subjected to a standardized challenge. Whether or not AHR is biologically significant, however, is often not considered. In fact, the degree of AHR is generally rather modest in most animal models considered to exhibit the phenomenon; a response in airway resistance of only two- to threefold above normal is not unusual (87, 88). In one sense, this may be apropos, given that much of human asthma also rumbles along chronically in a fashion that is perhaps more irritating than outright dangerous. On the other hand, in the subset of individuals who are afflicted with the severe form of the syndrome, the decrement in lung function accompanying an asthma attack can be extreme and even fatal.
What distinguishes severe asthma from its more common milder form remains an area of active research that has so far yielded few answers. It is therefore perhaps not surprising that animal models of correspondingly severe AHR are also few in number. One reason for this may be the tendency for researchers to focus on single causes, in pursuit of the most important mechanism responsible for the AHR of asthma. It has recently been suggested, however, that the key to severe asthma may be the simultaneous presence of multiple mechanisms; allergically inflamed mice treated with poly-l-lysine were shown to exhibit an extreme level of AHR that was easily fatal and that could be attributed to the synergistic interaction between an enhanced ability of smooth muscle to narrow the conducting airways and an increased propensity for liquid bridges to occlude peripheral airways (9). The mechanism behind this synergy is illustrated in Fig. 4. A severe asthma-like phenotype can also be achieved by coexpressing IL-5 and eotaxin 2 in the same animal; IL-5 leads to systemic eosinophilia, whereas selective eotaxin 2 upregulation in the airway epithelium directs chemotaxis and markedly activates the diapedesing eosinophils (70). The physiological manifestations are nearly identical to the antigen/cationic protein model (9) supporting the multiple mechanism hypothesis for the genesis of extreme AHR.
Fig. 4.

Mechanical and geometric mechanisms for airways hyperresponsiveness. A: a modest degree of shortening of the airway smooth muscle (black ring) impinging on an airway wall that thickened is due to epithelial hypertrophy and/or mucus hypersecretion (gray annulus) and leads to a substantial degree in luminal area (white). B: the same degree of luminal narrowing can be caused by accentuated smooth muscle shortening in the presence of a normal airway lining. C: both mechanisms together lead to a dramatic reduction in luminal area and may even lead to complete airway closure.
Summary and Conclusions
Asthma still appears to be a uniquely human disease despite decades of attempts to recapitulate its features in animals. Nevertheless, animal models of AHR have provided many of the key insights we currently have about the possible causes of asthma and have served as invaluable test beds for pharmacotherapy. Mouse models of asthma now represent the bulk of the scientific industry in this field because they can be explored with the most complete range of biological reagents and genomic knowledge. Recent developments in physiological phenotyping and three-dimensional imaging have added greatly to the investigative armamentarium. Combined with computational models of lung function, we are now able to make some rather precise links between structure and function in the mouse lung that enable AHR to be tied to underlying mechanisms. Of course, at the end of the day we are still going to face the issue that a mouse is not a human, so there will always be a gap in our understanding that biological relevance and experimental ethics prevent us from bridging completely. Nevertheless, we expect the development and investigation of animal models of asthma to continue for some time into the future, where a great deal of new knowledge awaits.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-67273, HL-75593, and HL-87788 and the Centers of Biomedical Research Excellence Grant P20-RR-15557 from the National Center for Research Resources.
REFERENCES
- 1.Expert Panel Report 3 (EPR-3). Guidelines for the Diagnosis and Management of Asthma–Summary Report 2007. J Allergy Clin Immunol 120: S94–S138, 2007. [DOI] [PubMed] [Google Scholar]
- 2.Adler A, Cieslewicz G, Irvin CG. Unrestrained plethysmography is an unreliable measure of airway responsiveness in BALB/c and C57BL/6 mice. J Appl Physiol 97: 286–292, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Akbari O, Stock P, Meyer E, Kronenberg M, Sidobre S, Nakayama T, Taniguchi M, Grusby MJ, DeKruyff RH, Umetsu DT. Essential role of NKT cells producing IL-4 and IL-13 in the development of allergen-induced airway hyperreactivity. Nature Med 9: 582–588, 2003. [DOI] [PubMed] [Google Scholar]
- 4.Allen G, Bates JH. Dynamic mechanical consequences of deep inflation in mice depend on type and degree of lung injury. J Appl Physiol 96: 293–300, 2004. [DOI] [PubMed] [Google Scholar]
- 5.Allen JE, Bischof RJ, Sucie Chang HY, Hirota JA, Hirst SJ, Inman MD, Mitzner W, Sutherland TE. Animal models of airway inflammation and airway smooth muscle remodelling in asthma. Pulm Pharmacol Ther. In press. [DOI] [PubMed]
- 6.Amdur MO, Mead J. Mechanics of respiration in unanesthetized guinea pigs. Am J Physiol 192: 364–368, 1958. [DOI] [PubMed] [Google Scholar]
- 7.Bates J, Irvin C, Brusasco V, Drazen J, Fredberg J, Loring S, Eidelman D, Ludwig M, Macklem P, Martin J, Milic-Emili J, Hantos Z, Hyatt R, Lai-Fook S, Leff A, Solway J, Lutchen K, Suki B, Mitzner W, Pare P, Pride N, Sly P. The use and misuse of Penh in animal models of lung disease. Am J Respir Cell Mol Biol 31: 373–374, 2004. [DOI] [PubMed] [Google Scholar]
- 8.Bates JH, Allen GB. The estimation of lung mechanics parameters in the presence of pathology: a theoretical analysis. Ann Biomed Eng 34: 384–392, 2006. [DOI] [PubMed] [Google Scholar]
- 9.Bates JH, Cojocaru A, Haverkamp HC, Rinaldi LM, Irvin CG. The synergistic interactions of allergic lung inflammation and intratracheal cationic protein. Am J Respir Crit Care Med 177: 261–268, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bates JH, Cojocaru A, Lundblad LK. Bronchodilatory effect of deep inspiration on the dynamics of bronchoconstriction in mice. J Appl Physiol 103: 1696–1705, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Bates JH, Irvin CG. Measuring lung function in mice: the phenotyping uncertainty principle. J Appl Physiol 94: 1297–1306, 2003. [DOI] [PubMed] [Google Scholar]
- 12.Bates JH, Lauzon AM. Parenchymal tethering, airway wall stiffness, and the dynamics of bronchoconstriction. J Appl Physiol 102: 1912–1920, 2007. [DOI] [PubMed] [Google Scholar]
- 13.Bates JH, Sobel BE. The conceptual basis of mathematics in cardiology III: linear systems theory and integral transforms. Coron Artery Dis 14: 185–196, 2003. [DOI] [PubMed] [Google Scholar]
- 14.Bates JH, Thompson-Figueroa J, Lundblad LK, Irvin CG. Unrestrained video-assisted plethysmography: a noninvasive method for assessment of lung mechanical function in small animals. J Appl Physiol 104: 253–261, 2008. [DOI] [PubMed] [Google Scholar]
- 15.Bates JH, Wagers SS, Norton RJ, Rinaldi LM, Irvin CG. Exaggerated airway narrowing in mice treated with intratracheal cationic protein. J Appl Physiol 100: 500–506, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Ann Rev Immunol 25: 297–336, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441: 235–238, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Bevelander M, Mayette J, Whittaker LA, Paveglio SA, Jones CC, Robbins J, Hemenway D, Akira S, Uematsu S, Poynter ME. Nitrogen dioxide promotes allergic sensitization to inhaled antigen. J Immunol 179: 3680–3688, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bousquet J, Wenzel S, Holgate S, Lumry W, Freeman P, Fox H. Predicting response to omalizumab, an anti-IgE antibody, in patients with allergic asthma. Chest 125: 1378–1386, 2004. [DOI] [PubMed] [Google Scholar]
- 20.Brusasco V, Pellegrino R. Complexity of factors modulating airway narrowing in vivo: relevance to assessment of airway hyperresponsiveness. J Appl Physiol 95: 1305–1313, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Camateros P, Tamaoka M, Hassan M, Marino R, Moisan J, Marion D, Guiot MC, Martin JG, Radzioch D. Chronic asthma-induced airway remodeling is prevented by toll-like receptor-7/8 ligand S28463. Am J Respir Crit Care Med 175: 1241–1249, 2007. [DOI] [PubMed] [Google Scholar]
- 22.Cojocaru A, Irvin CG, Haverkamp HC, Bates JH. Computational assessment of airway wall stiffness in vivo in allergically inflamed mouse models of asthma. J Appl Physiol 104: 1601–1610, 2008. [DOI] [PubMed] [Google Scholar]
- 23.Corry DB, Irvin CG. Promise and pitfalls in animal-based asthma research: building a better mousetrap. Immunol Res 35: 279–294, 2006. [DOI] [PubMed] [Google Scholar]
- 24.Couetil LL, Hoffman AM, Hodgson J, Buechner-Maxwell V, Viel L, Wood JL, Lavoie JP. Inflammatory airway disease of horses. J Vet Int Med/Am Coll Vet Int Med 21: 356–361, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Ding DJ, Martin JG, Macklem PT. Effects of lung volume on maximal methacholine-induced bronchoconstriction in normal humans. J Appl Physiol 62: 1324–1330, 1987. [DOI] [PubMed] [Google Scholar]
- 26.Duguet A, Biyah K, Minshall E, Gomes R, Wang CG, Taoudi-Benchekroun M, Bates JH, Eidelman DH. Bronchial responsiveness among inbred mouse strains. Role of airway smooth-muscle shortening velocity. Am J Respir Crit Care Med 161: 839–848, 2000. [DOI] [PubMed] [Google Scholar]
- 27.Dunn CJ, Elliott GA, Oostveen JA, Richards IM. Development of a prolonged eosinophil-rich inflammatory leukocyte infiltration in the guinea-pig asthmatic response to ovalbumin inhalation. Am Rev Respir Dis 137: 541–547, 1988. [DOI] [PubMed] [Google Scholar]
- 28.Dybas JM, Andresen CJ, Schelegle ES, McCue RW, Callender NN, Jackson AC. Deep-breath frequency in bronchoconstricted monkeys (Macaca fascicularis). J Appl Physiol 100: 786–791, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Ewart S, Levitt R, Mitzner W. Respiratory system mechanics in mice measured by end-inflation occlusion. J Appl Physiol 79: 560–566, 1995. [DOI] [PubMed] [Google Scholar]
- 30.Fattouh R, Pouladi MA, Alvarez D, Johnson JR, Walker TD, Goncharova S, Inman MD, Jordana M. House dust mite facilitates ovalbumin-specific allergic sensitization and airway inflammation. Am J Respir Crit Care Med 172: 314–321, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Finotto S, De Sanctis GT, Lehr HA, Herz U, Buerke M, Schipp M, Bartsch B, Atreya R, Schmitt E, Galle PR, Renz H, Neurath MF. Treatment of allergic airway inflammation and hyperresponsiveness by antisense-induced local blockade of GATA-3 expression. J Exp Med 193: 1247–1260, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finotto S, Neurath MF, Glickman JN, Qin S, Lehr HA, Green FH, Ackerman K, Haley K, Galle PR, Szabo SJ, Drazen JM, De Sanctis GT, Glimcher LH. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science 295: 336–338, 2002. [DOI] [PubMed] [Google Scholar]
- 33.Fredberg JJ, Inouye D, Miller B, Nathan M, Jafari S, Raboudi SH, Butler JP, Shore SA. Airway smooth muscle, tidal stretches, and dynamically determined contractile states. Am J Respir Crit Care Med 156: 1752–1759, 1997. [DOI] [PubMed] [Google Scholar]
- 34.Fredberg JJ, Stamenovic D. On the imperfect elasticity of lung tissue. J Appl Physiol 67: 2408–2419, 1989. [DOI] [PubMed] [Google Scholar]
- 35.Garland A, Jordan JE, Ray DW, Spaethe SM, Alger L, Solway J. Role of eicosanoids in hyperpnea-induced airway responses in guinea pigs. J Appl Physiol 75: 2797–2804, 1993. [DOI] [PubMed] [Google Scholar]
- 36.Georas SN, Guo J, De Fanis U, Casolaro V. T-helper cell type-2 regulation in allergic disease. Eur Respir J 26: 1119–1137, 2005. [DOI] [PubMed] [Google Scholar]
- 37.Gil FR, Zitouni NB, Azoulay E, Maghni K, Lauzon AM. Smooth muscle myosin isoform expression and LC20 phosphorylation in innate rat airway hyperresponsiveness. Am J Physiol Lung Cell Mol Physiol 291: L932–L940, 2006. [DOI] [PubMed] [Google Scholar]
- 38.Goplen N, Karim MZ, Liang Q, Gorska MM, Rozario S, Guo L, Alam R. Combined sensitization of mice to extracts of dust mite, ragweed, and Aspergillus species breaks through tolerance and establishes chronic features of asthma. J Allergy Clin Immunol 123: 925–932, e911, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gupta S, Schulz-Maronde S, Kutzleb C, Richter R, Forssmann WG, Kapp A, Forssmann U, Elsner J. Cloning, expression, and functional characterization of cynomolgus monkey (Macaca fascicularis) CC chemokine receptor 1. J Leukoc Biol 78: 1175–1184, 2005. [DOI] [PubMed] [Google Scholar]
- 40.Haczku A, Atochina EN, Tomer Y, Cao Y, Campbell C, Scanlon ST, Russo SJ, Enhorning G, Beers MF. The late asthmatic response is linked with increased surface tension and reduced surfactant protein B in mice. Am J Physiol Lung Cell Mol Physiol 283: L755–L765, 2002. [DOI] [PubMed] [Google Scholar]
- 41.Hai CM, Murphy RA. Cross-bridge phosphorylation and regulation of latch state in smooth muscle. Am J Physiol Cell Physiol 254: C99–C106, 1988. [DOI] [PubMed] [Google Scholar]
- 42.Haldar P, Pavord ID, Shaw DE, Berry MA, Thomas M, Brightling CE, Wardlaw AJ, Green RH. Cluster analysis and clinical asthma phenotypes. Am J Respir Crit Care Med 178: 218–224, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamelmann E, Schwarze J, Takeda K, Oshiba A, Larsen GL, Irvin CG, Gelfand EW. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am J Respir Crit Care Med 156: 766–775, 1997. [DOI] [PubMed] [Google Scholar]
- 44.Hantos Z, Collins RA, Turner DJ, Janosi TZ, Sly PD. Tracking of airway and tissue mechanics during TLC maneuvers in mice. J Appl Physiol 95: 1695–1705, 2003. [DOI] [PubMed] [Google Scholar]
- 45.Hantos Z, Daroczy B, Suki B, Nagy S, Fredberg JJ. Input impedance and peripheral inhomogeneity of dog lungs. J Appl Physiol 72: 168–178, 1992. [DOI] [PubMed] [Google Scholar]
- 46.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nature Immunol 6: 1123–1132, 2005. [DOI] [PubMed] [Google Scholar]
- 47.Hessel EM, Zwart A, Oostveen E, Van Oosterhout AJ, Blyth DI, Nijkamp FP. Repeated measurement of respiratory function and bronchoconstriction in unanesthetized mice. J Appl Physiol 79: 1711–1716, 1995. [DOI] [PubMed] [Google Scholar]
- 48.Homma T, Bates JH, Irvin CG. Airway hyperresponsiveness induced by cationic proteins in vivo: site of action. Am J Physiol Lung Cell Mol Physiol 289: L413–L418, 2005. [DOI] [PubMed] [Google Scholar]
- 49.Irvin CG Using the mouse to model asthma: the cup is half full and then some. Clin Exp Allergy 38: 701–703, 2008. [DOI] [PubMed] [Google Scholar]
- 50.Irvin CG, Bates JH. Measuring the lung function in the mouse: the challenge of size. Respir Res 4: 4, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.James AL, Wenzel S. Clinical relevance of airway remodelling in airway diseases. Eur Respir J 30: 134–155, 2007. [DOI] [PubMed] [Google Scholar]
- 52.Kay AB Natural killer T cells and asthma. N Engl J Med 354: 1186–1188, 2006. [DOI] [PubMed] [Google Scholar]
- 53.Kiley J, Smith R, Noel P. Asthma phenotypes. Curr Opin Pulm Med 13: 19–23, 2007. [DOI] [PubMed] [Google Scholar]
- 54.Kips JC, Anderson GP, Fredberg JJ, Herz U, Inman MD, Jordana M, Kemeny DM, Lotvall J, Pauwels RA, Plopper CG, Schmidt D, Sterk PJ, Van Oosterhout AJ, Vargaftig BB, Chung KF. Murine models of asthma. Eur Respir J 22: 374–382, 2003. [DOI] [PubMed] [Google Scholar]
- 55.Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity 21: 467–476, 2004. [DOI] [PubMed] [Google Scholar]
- 56.Kool M, Soullie T, van Nimwegen M, Willart MA, Muskens F, Jung S, Hoogsteden HC, Hammad H, Lambrecht BN. Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J Exp Med 205: 869–882, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koya T, Kodama T, Takeda K, Miyahara N, Yang ES, Taube C, Joetham A, Park JW, Dakhama A, Gelfand EW. Importance of myeloid dendritic cells in persistent airway disease after repeated allergen exposure. Am J Respir Crit Care Med 173: 42–55, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee JJ, Dimina D, Macias MP, Ochkur SI, McGarry MP, O'Neill KR, Protheroe C, Pero R, Nguyen T, Cormier SA, Lenkiewicz E, Colbert D, Rinaldi L, Ackerman SJ, Irvin CG, Lee NA. Defining a link with asthma in mice congenitally deficient in eosinophils. Science 305: 1773–1776, 2004. [DOI] [PubMed] [Google Scholar]
- 59.Lundblad LK, Thompson-Figueroa J, Allen GB, Rinaldi L, Norton RJ, Irvin CG, Bates JH. Airway hyperresponsiveness in allergically inflamed mice: the role of airway closure. Am J Respir Crit Care Med 175: 768–774, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lutchen KR, Greenstein JL, Suki B. How inhomogeneities and airway walls affect frequency dependence and separation of airway and tissue properties. J Appl Physiol 80: 1696–1707, 1996. [DOI] [PubMed] [Google Scholar]
- 61.Madwed JB, Jackson AC. Determination of airway and tissue resistances after antigen and methacholine in nonhuman primates. J Appl Physiol 83: 1690–1696, 1997. [DOI] [PubMed] [Google Scholar]
- 62.Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441: 231–234, 2006. [DOI] [PubMed] [Google Scholar]
- 63.Marsh WR, Irvin CG, Murphy KR, Behrens BL, Larsen GL. Increases in airway reactivity to histamine and inflammatory cells in bronchoalveolar lavage after the late asthmatic response in an animal model. Am Rev Respir Dis 131: 875–879, 1985. [DOI] [PubMed] [Google Scholar]
- 64.McKinley L, Alcorn JF, Peterson A, Dupont RB, Kapadia S, Logar A, Henry A, Irvin CG, Piganelli JD, Ray A, Kolls JK. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol 181: 4089–4097, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mitzner W, Tankersley C. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am J Respir Crit Care Med 158: 340–341, 1998. [DOI] [PubMed] [Google Scholar]
- 66.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol 136: 2348–2357, 1986. [PubMed] [Google Scholar]
- 67.Nials AT, Uddin S. Mouse models of allergic asthma: acute and chronic allergen challenge. Dis Models Mech 1: 213–220, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Noble PB, McFawn PK, Mitchell HW. Intraluminal pressure oscillation enhances subsequent airway contraction in isolated bronchial segments. J Appl Physiol 96: 1161–1165, 2004. [DOI] [PubMed] [Google Scholar]
- 69.Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 448: 480–483, 2007. [DOI] [PubMed] [Google Scholar]
- 70.Ochkur SI, Jacobsen EA, Protheroe CA, Biechele TL, Pero RS, McGarry MP, Wang H, O'Neill KR, Colbert DC, Colby TV, Shen H, Blackburn MR, Irvin CC, Lee JJ, Lee NA. Coexpression of IL-5 and eotaxin-2 in mice creates an eosinophil-dependent model of respiratory inflammation with characteristics of severe asthma. J Immunol 178: 7879–7889, 2007. [DOI] [PubMed] [Google Scholar]
- 71.Oettgen HC Regulation of the IgE isotype switch: new insights on cytokine signals and the functions of epsilon germline transcripts. Curr Opin Immunol 12: 618–623, 2000. [DOI] [PubMed] [Google Scholar]
- 72.Padrid P, Snook S, Finucane T, Shiue P, Cozzi P, Solway J, Leff AR. Persistent airway hyperresponsiveness and histologic alterations after chronic antigen challenge in cats. Am J Respir Crit Care Med 151: 184–193, 1995. [DOI] [PubMed] [Google Scholar]
- 73.Pantano C, Ather JL, Alcorn JF, Poynter ME, Brown AL, Guala AS, Beuschel SL, Allen GB, Whittaker LA, Bevelander M, Irvin CG, Janssen-Heininger YM. Nuclear factor-kappaB activation in airway epithelium induces inflammation and hyperresponsiveness. Am J Respir Crit Care Med 177: 959–969, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Peslin R, Fredberg JJ. Oscillation mechanics of the respiratory system. In: Handbook of Physiology Section 3: The Respiratory System, edited by Macklem PT and JM. Bethesda, MD: American Physiological Society, 1986, p. 145–178.
- 75.Petak F, Hantos Z, Adamicza A, Asztalos T, Sly PD. Methacholine-induced bronchoconstriction in rats: effects of intravenous vs. aerosol delivery. J Appl Physiol 82: 1479–1487, 1997. [DOI] [PubMed] [Google Scholar]
- 76.Reynolds JS, Johnson VJ, Frazer DG. Unrestrained acoustic plethysmograph for measuring specific airway resistance in mice. J Appl Physiol 105: 711–717, 2008. [DOI] [PubMed] [Google Scholar]
- 77.Schnyder-Candrian S, Togbe D, Couillin I, Mercier I, Brombacher F, Quesniaux V, Fossiez F, Ryffel B, Schnyder B. Interleukin-17 is a negative regulator of established allergic asthma. J Exp Med 203: 2715–2725, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schuessler TF, Bates JH. A computer-controlled research ventilator for small animals: design and evaluation. IEEE Trans Biomed Eng 42: 860–866, 1995. [DOI] [PubMed] [Google Scholar]
- 79.Southam DS, Ellis R, Wattie J, Inman MD. Components of airway hyperresponsiveness and their associations with inflammation and remodeling in mice. J Allergy Clin Immunol 119: 848–854, 2007. [DOI] [PubMed] [Google Scholar]
- 80.Stephens NL, Morgan G, Kepron W, Seow CY. Changes in cross-bridge properties of sensitized airway smooth muscle. J Appl Physiol 61: 1492–1498, 1986. [DOI] [PubMed] [Google Scholar]
- 81.Tepper RS, Ramchandani R, Argay E, Zhang L, Xue Z, Liu Y, Gunst SJ. Chronic strain alters the passive and contractile properties of rabbit airways. J Appl Physiol 98: 1949–1954, 2005. [DOI] [PubMed] [Google Scholar]
- 82.Thorpe CW, Bates JH. Effect of stochastic heterogeneity on lung impedance during acute bronchoconstriction: a model analysis. J Appl Physiol 82: 1616–1625, 1997. [DOI] [PubMed] [Google Scholar]
- 83.Tomioka S, Bates JH, Irvin CG. Airway and tissue mechanics in a murine model of asthma: alveolar capsule vs. forced oscillations. J Appl Physiol 93: 263–270, 2002. [DOI] [PubMed] [Google Scholar]
- 84.Turner DJ, Myron P, Powell WS, Martin JG. The role of endogenous corticosterone in the late-phase response to allergen challenge in the brown Norway rat. Am J Respir Crit Care Med 153: 545–550, 1996. [DOI] [PubMed] [Google Scholar]
- 85.Uchida DA, Ackerman SJ, Coyle AJ, Larsen GL, Weller PF, Freed J, Irvin CG. The effect of human eosinophil granule major basic protein on airway responsiveness in the rat in vivo. A comparison with polycations. Am Rev Respir Dis 147: 982–988, 1993. [DOI] [PubMed] [Google Scholar]
- 86.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24: 179–189, 2006. [DOI] [PubMed] [Google Scholar]
- 87.Wagers S, Lundblad LK, Ekman M, Irvin CG, Bates JH. The allergic mouse model of asthma: normal smooth muscle in an abnormal lung? J Appl Physiol 96: 2019–2027, 2004. [DOI] [PubMed] [Google Scholar]
- 88.Wagers SS, Haverkamp HC, Bates JH, Norton RJ, Thompson-Figueroa JA, Sullivan MJ, Irvin CG. Intrinsic and antigen-induced airway hyperresponsiveness are the result of diverse physiological mechanisms. J Appl Physiol 102: 221–230, 2007. [DOI] [PubMed] [Google Scholar]
- 89.Wang CG, Almirall JJ, Dolman CS, Dandurand RJ, Eidelman DH. In vitro bronchial responsiveness in two highly inbred rat strains. J Appl Physiol 82: 1445–1452, 1997. [DOI] [PubMed] [Google Scholar]
- 90.Wanner A, Abraham WM, Douglas JS, Drazen JM, Richerson HB, Ram JS. NHLBI Workshop Summary. Models of airway hyperresponsiveness. Am Rev Respir Dis 141: 253–257, 1990. [DOI] [PubMed] [Google Scholar]
- 91.Wenzel SE Asthma: defining of the persistent adult phenotypes. Lancet 368: 804–813, 2006. [DOI] [PubMed] [Google Scholar]
- 92.Whittaker L, Niu N, Temann UA, Stoddard A, Flavell RA, Ray A, Homer RJ, Cohn L. Interleukin-13 mediates a fundamental pathway for airway epithelial mucus induced by CD4 T cells and interleukin-9. Am J Respir Cell Mol Biol 27: 593–602, 2002. [DOI] [PubMed] [Google Scholar]
- 93.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Interleukin-13: central mediator of allergic asthma. Science 282: 2258–2261, 1998. [DOI] [PubMed] [Google Scholar]
- 94.Yiamouyiannis CA, Schramm CM, Puddington L, Stengel P, Baradaran-Hosseini E, Wolyniec WW, Whiteley HE, Thrall RS. Shifts in lung lymphocyte profiles correlate with the sequential development of acute allergic and chronic tolerant stages in a murine asthma model. Am J Pathol 154: 1911–1921, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang W, Gunst SJ. Interactions of airway smooth muscle cells with their tissue matrix: implications for contraction. Proc Am Thoracic Soc 5: 32–39, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhen G, Park SW, Nguyenvu LT, Rodriguez MW, Barbeau R, Paquet AC, Erle DJ. IL-13 and epidermal growth factor receptor have critical but distinct roles in epithelial cell mucin production. Am J Respir Cell Mol Biol 36: 244–253, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zosky GR, Larcombe AN, White OJ, Burchell JT, Janosi TZ, Hantos Z, Holt PG, Sly PD, Turner DJ. Ovalbumin-sensitized mice are good models for airway hyperresponsiveness but not acute physiological responses to allergen inhalation. Clin Exp Allergy 38: 829–838, 2008. [DOI] [PubMed] [Google Scholar]
- 98.Zuyderduyn S, Sukkar MB, Fust A, Dhaliwal S, Burgess JK. Treating asthma means treating airway smooth muscle cells. Eur Respir J 32: 265–274, 2008. [DOI] [PubMed] [Google Scholar]