

Abstract
The nitric oxide/soluble guanylyl cyclase (sGC) signal transduction pathway plays an important role in smooth muscle relaxation and phenotypic regulation. However, the transcriptional regulation of sGC gene expression is largely unknown. It has been shown that sGC expression increases in pulmonary arteries from chronic hypoxia-induced pulmonary hypertensive animals. Since the transcription factor NFATc3 is required for the upregulation of the smooth muscle hypertrophic/differentiation marker α-actin in pulmonary artery smooth muscle cells from chronically hypoxic mice, we hypothesized that NFATc3 is required for the regulation of sGC-α1 expression during chronic hypoxia. Exposure to chronic hypoxia for 2 days induced a decrease in sGC-α1 expression in mouse pulmonary arteries. This reduction was independent of NFATc3 but mediated by nuclear accumulation of the mRNA-stabilizing protein human antigen R (HuR). Consistent with our hypothesis, chronic hypoxia (21 days) upregulated pulmonary artery sGC-α1 expression, bringing it back to the level of the normoxic controls. This response was prevented in NFATc3 knockout and cyclosporin (calcineurin/NFATc inhibitor)-treated mice. Furthermore, we identified effective binding sites for NFATc in the mouse sGC-α1 promoter. Activation of NFATc3 increased sGC-α1 promoter activity in human embryonic derived kidney cells, rat aortic-derived smooth muscle cells, and human pulmonary artery smooth muscle cells. Our results suggest that NFATc3 and HuR are important regulators of sGC-α1 expression in pulmonary vascular smooth muscle cells during chronic hypoxia-induced pulmonary hypertension.
Keywords: human antigen R, nuclear factor of activated T cells isoform c3
the nitric oxide (NO) signal transduction pathway plays an important role in smooth muscle (SM) relaxation and phenotypic regulation, and it has been an intensively investigated field for more than two decades. NO activates soluble guanylyl cyclase (sGC) that subsequently produces guanosine 3′,5′-cyclic monophosphate (cGMP). sGC is a heterodimer composed of two subunits, α and β, of which four types exist (α1, β1, α2, and β2). Cloning and expression experiments have demonstrated that both α- and β-subunits are required for sGC activity (20, 42). However, expression of the β1-subunit is somewhat invariant, whereas alterations in the expression of the α-subunit(s) may be an important physiological control mechanism (69).
The intracellular actions of cGMP are primarily mediated by cGMP-dependent protein kinases (PKG), but several types of cGMP-activated ion channels also appear to be involved (18, 47) in the regulation of the SM relaxation. However, the pathways controlled by cGMP levels are complex due to the existence of several phosphodiesterases (PDE), which hydrolyze cGMP (59).
In addition to its vasodilator action, NO/sGC/PKG signaling also has been shown to antagonize the proliferative responses of growth factors and vasoactive peptides such as endothelin-1 (ET-1) in vascular smooth muscle cells (VSMC) (6, 21). In addition, NO/sGC/PKG signaling has been shown to induce differentiation (contractile phenotype) of VSMC through the upregulation of myosin heavy chain 2, SM α-actin, and SM-calponin (4, 8, 46, 47). PKG also has been reported to inhibit proliferation of SM in pulmonary arteries (10). Consistently, several studies have shown that sGC mRNA, protein, and enzyme activity are upregulated in pulmonary arterial smooth muscle cells (PASMC) from chronic hypoxia (CH)-induced pulmonary hypertensive animals (31, 44, 45, 73). The administration of BAY 63-2521 (activator of sGC) partially reversed pulmonary hypertension and the structural remodeling of the lung vasculature (73). In addition, administration of PDE isoform 1 inhibitor 8-methoxymethyl 3-isobutyl-1-methylxanthine reversed the increased pulmonary arterial pressure and arterial remodeling in two models of pulmonary hypertension (74). These studies suggest that the pulmonary hypertension and arterial remodeling might be even more severe without the upregulation of sGC. However, the transcriptional regulation of sGC gene expression is largely unknown. Only recently has a detailed description of the human sGC-α1 and -β1 (52, 76) and the mouse sGC-α1 (81) promoters been reported. However, considerable information is now available regarding the mechanism of posttranscriptional control of sGC-α1. Among posttranscriptional events, mRNA turnover is emerging as a critical paradigm of gene regulation through the mRNA-stabilizing protein human antigen R (HuR). This member of the embryonic lethal abnormal vision (ELAV) protein family is a ligand for mRNAs containing adenosine/uridine-rich elements located in the 3′-untranslated regions of certain mRNA (for review, see Refs. 15, 30, 70, 79, 86). Unlike other members of the ELAV family (HuB, HuC, and HuD), which are exclusively found in neuronal tissue, HuR is ubiquitously expressed (24, 50). Klöss and colleagues (39, 41) have demonstrated that sGC-α1 expression is posttranscriptionally regulated by HuR (64).
Since we have demonstrated that the transcription factor NFATc3 (nuclear factor of activated T cells isoform c3) is required for upregulation of one of the SM hypertrophic/differentiation markers, SM α-actin, in PASMC from CH-induced pulmonary hypertensive mice (12), we propose that NFATc3 is similarly required for the regulation of sGC-α1 expression during CH. NFATc3 is a member of the NFATc family of transcription factors, along with NFATc1, NFATc2, and NFATc4, that share the property of Ca2+/calcineurin-dependent nuclear translocation (reviewed in Refs. 28, 29, 66). The NFATc3 isoform is specifically implicated in vasculature development (25, 26), maintenance of SM contractile phenotype (22, 58), and regulation of VSMC contractility (1, 57).
We found that sGC-α1 expression in mouse pulmonary arteries has a biphasic response to CH. sGC-α1 expression is initially decreased after 2 days of CH exposure, whereas its expression recovers to normoxic control values after 21 days of CH exposure. The initial decrease is independent of calcineurin/NFATc3. Our results suggest that increased nuclear accumulation of HuR limits the stabilization of sGC-α1 mRNA by cytosolic HuR, which then results in decreased sGC-α1 expression. The recovery in sGC-α1 expression observed after 21 days of CH requires calcineurin/NFATc3, because it is prevented in NFATc3 knockout (KO) and cyclosporin A (CsA)-treated mice. In addition, we found that NFATc3 binds to the mouse sGC-α1 promoter both in vivo and ex vivo. Activation of NFATc3 increases sGC-α1 promoter activity in human embryonic kidney (HEK-293) cells, rat aortic SMC (RAoSMC), and human PASMC. Our results demonstrate that HuR and NFATc3 are important regulators of sGC-α1 expression in PASMC.
EXPERIMENTAL PROCEDURES
All protocols employed in this study were reviewed and approved by the Institutional Animal Care and Use Committee of the University of New Mexico School of Medicine (Albuquerque, NM).
Animals.
Adult male FVBN, NFATc3 KO, and Balb/C wild-type (WT) mice (20–25 g) were used. NFATc3 KO mice were kindly provided by Dr. Laurie Glimcher (Harvard University, Boston, MA) (60). Heterozygous mice were bred to obtain age-matched WT (Balb/C) and KO mice. Importantly, it has been shown that loss of the NFATc3 isoform does not induce a compensatory regulation of other NFATc isoforms (88). Mice were administered 18 mg·kg−1·day−1 of CsA (Calbiochem) in food treats (BioServe) for 2 or 21 days.
Chronic hypoxia exposure.
Animals designated for exposure to CH were housed in a hypobaric chamber with barometric pressure maintained at ∼380 mmHg for 2 or 21 days. The chamber was opened one time per week to provide animals with fresh food, water, and clean bedding. Control animals were housed at ambient barometric pressure (normoxia, ∼630 mmHg). All animals were maintained on a 12:12-h light-dark cycle.
Isolation of intrapulmonary arteries.
Mice were anesthetized with pentobarbital sodium (Sleepaway; 200 mg/kg ip). Lungs were removed and placed in ice-cold HEPES-PSS solution. Intrapulmonary arteries (1st, 2nd, and 3rd order) were isolated and cleaned from the adventitia.
Quantitative real-time PCR.
Intrapulmonary arteries (2nd and 3rd order) were stored in RNAlater (Ambion). Total RNA was isolated using the RNeasy mini kit (Qiagen) following the manufacturer's protocol. Total RNA was reverse transcribed to cDNA using a High-Capacity reverse transcription kit (Applied Biosystems). For real-time detection of target genes mouse sGC-α1, mouse HuR, and reference gene mouse β-actin, TaqMan gene expression assays (Applied Biosystems) were used. PCR was performed using Applied Biosystems 7500 fast real-time PCR system. The normalized gene expression method (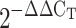 ) for relative quantification was used (48). β-Actin was used as an endogenous control because its expression is not affected by CH, as we (12) previously demonstrated.
) for relative quantification was used (48). β-Actin was used as an endogenous control because its expression is not affected by CH, as we (12) previously demonstrated.
Western blot analysis.
Isolated intrapulmonary arteries (1st to 4th order) were homogenized in lysis buffer (10 mM Tris·HCl, pH 7.4, 1 mM EDTA, 1% IGEPAL, 0.1% sodium deoxycholate, 500 nM sodium orthovanadate, 50 nM NaF, 1 μg/ml pepstatin/leupeptin/aprotinin, and 1 mM PMSF) at 4°C and centrifuged at a relative centrifugal force of 16,000 g for 2 min. Protein concentration was determined in the supernatant using Bradford's method (Bio-Rad) as recommended by the manufacturer. Supernatants (2–20 μg/lane) were resolved by SDS-PAGE, and proteins were transferred to polyvinylidene difluoride membranes. After being blocked for nonspecific binding, the membranes were incubated with primary anti-sGC-α1 antibody (1:100; Cayman) or anti-β-actin antibody (1:5,000; Sigma) at 4°C overnight, washed, and incubated with a peroxidase-conjugated secondary antibody (Pierce) for 1 h at room temperature. Specifically bound antibody was detected by enhanced chemiluminescence detection (ECL; Pierce). Relative content of the antigen protein was evaluated using a GeneGnome imaging system and GeneSnap software (Syngene, Cambridge, UK). Band densities were normalized to total protein loaded per lane as determined by Coomassie blue (Bio-Rad) staining of the membrane (NIH Image software). β-Actin was used as an endogenous control because its expression is not affected by CH, as shown in Fig. 2B and as we (12) previously demonstrated.
sGC mRNA-HuR immunoprecipitation.
Isolated intrapulmonary arteries (1st to 4th order) were homogenized in polysome lysis buffer [100 mM KCl, 5 mM MgCl2, 10 mM HEPES, 0.5% Nonidet P-40, 1 mM DTT, 10 mM vanadyl ribonucleoside complex inhibitor (VRN), 0.2 mM PMSF, Sigma proteinase inhibitor cocktail, and 50 U/ml RNasin]. HuR was immunoprecipitated by incubating the homogenate with anti-HuR antibody (40 μg; 19F12; Santa Cruz Biotechnology) and paramagnetic protein G Dynabeads (Invitrogen) overnight at 4°C (12a). The protein G Dynabeads were washed and incubated with proteinase K (30 μg) and 0.1% SDS at 55°C for 30 min, and coimmunoprecipitated total RNA was purified using the RNeasy mini kit (Qiagen). The negative control was performed in the absence of antibody. RNA (30 ng) was reverse transcribed to cDNA, and sGC-α1 mRNA was quantified by real-time PCR (see Quantitative real-time PCR). The relative change in sGC-α1 mRNA binding to HuR was calculated using the comparative threshold method (ΔΔCT) with input DNA as the reference. CT in the negative control samples was consistently higher than in the presence of primary antibody.
Cell culture.
Tet-inducible HEK-293 cells (Clontech), which constitutively express reverse tetracycline trans-activator protein (rtTA), were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% Tet system approved fetal bovine serum (FBS), 4 mM l-glutamine, 100 U/ml penicillin, 0.1 mg/ml streptomycin, and 0.2 mg/ml G418 (Mediatech). Primary human pulmonary arterial smooth muscle cells (HPASMC) obtained from ScienCell were cultured in ScienCell HPASMC culture medium containing 2% FBS. RAoSMC were obtained as described previously (79) and cultured in DMEM supplemented with 10% FBS, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. HPASMC and RAoSMC were used between passages 2 and 8. All cell types were cultured at 37°C in a humidified atmosphere of 95% O2-5% CO2.
Plasmids.
Mouse sGC-α1 promoter-luciferase reporter plasmid (sGC-luc, pGL3 backbone vector) was kindly provided by Dr. Roberto Vazquez-Padron (University of Miami, Miami, FL) (81). pGL3 luciferase reporter empty plasmid (Promega) was used as control. Tet-inducible-operated EGFP-NFATc3 plasmid (Tet-Op-NFATc3) (22) was used to overexpress NFATc3. NFATc dominant negative plasmid (DN-NFAT, pBluescript backbone vector) was kindly provided by Dr. Mercedes Rincon (University of Vermont, Burlington, VT). TK-Renilla-luciferase reporter plasmid (Renilla-luc; Promega) was used as a transfection efficiency control and to normalize luciferase activity.
Transient transfections.
HEK-293 cells, HPASMC, and RAoSMC were plated at 80% confluency on 24-well plates. The cells were transfected to express exogenous DNA using Superfect (Qiagen) according to the manufacturer's instructions, with a total of 0.5 μg of plasmid DNA and 15 ng of Renilla-luc. To activate the overexpression of NFATc3, we treated HEK-293 cells after transfection with doxycycline (DOX; 2 μg/ml). The next day, HEK-293 cells were treated with ionomycin (Io; 1 μM) for 24 h in the presence or absence of CsA (1 μM). After treatments, cells were harvested for chromatin immunoprecipitation (ChIP) assay or dual-luciferase assay. HPASMC or RAoSMC were FBS deprived for 24 h before treatment with Io with or without CsA. Luciferase activity was determined using the dual-luciferase reporter assay system (Promega) following the manufacture's recommendations.
ChIP assay.
ChIP assay was performed using ChIP-IT Express (Active Motif) according to the manufacturer's instructions. Isolated pulmonary arteries from WT mice exposed to 21 days of CH and Io-treated Tet-Op-NFATc3/ sGC-luc-transfected HEK-293 cells were fixed with 1% formaldehyde in PBS at room temperature for 10 min to cross-link DNA and proteins. The cross-linking reaction was stopped by adding glycine stop-fix solution. Tissues and cells were lysed in lysis buffer containing a protease inhibitor mixture and homogenized on ice. The lysate was sonicated to shear DNA to lengths between 200 and 1,000 bp. A fraction of the sheared chromatin/lysate was DNA purified and used as control in the later PCR analysis (input DNA). The remaining sheared chromatin/lysates were diluted and incubated with polyclonal anti-NFATc3 (Santa Cruz Biotechnology) and with nonimmune antibody as a preimmune negative control. Immunoprecipitated chromatin was captured with protein G magnetic beads. After washes and elution steps, cross-links were reversed at 65°C for 4 h and proteinase K digestion. An aliquot of the immunoprecipitated DNA (NFATc3 and negative antibody) and 1:10 dilution of the input DNA, as suggested by the manufacturer, were amplified by PCR with specific primers (described below) and analyzed on 3% agarose gel.
Putative NFATc binding sites were determined using Patch 1.0 public software (Biobase) and MatInspector (Genomatix) over the sequence of sGC-α1 promoter (GenBank accession no. AY116663). Each putative site was designated with a number.
The DNA obtained by ChIP was analyzed by semiquantitative PCR using the following primers (IDT) designed for the different putative NFATc binding sites. Primer sequences and fragment sizes were as follows: site 1+2, forward 5′-CAG TGT CAG ACC TGA AGA TGC TGTCTA CAC TGA TGT CCC TCT TTC CCA-3′ and reverse 5′-CTA CAC TGA TGT CCC TCT TTC CCA-3′ (104 bp); site 3+4, forward 5′-CTT CAA GAT CCC ACA CTG ACA CCA-3′ and reverse 5′-TCA GAC CAT GAA CGT AAG GCC CAA-3′ (206 bp); site 4, forward 5′-GGC CTT ACG TTC ATG GTC TGA GAA-3′ and reverse 5′-CAT GAA CTT CTC TTA GTT CAG CAG-3′ (68 bp); site 5, forward 5′-GAA TTG CTC TGG GAG TAA GAG AAG C-3′ and reverse 5′-AAG ATG GAA CCA AAC CTC GTA GGT CC-3′ (120 bp); site 5+6, forward 5′-TGC TCT GGG AGT AAG AGA AGC TTT GG-3′ and reverse 5′-ACA GCT ACA GGA ACC AGG GAA ACT-3′ (139 bp); site 7, forward 5′-TGT AGC TGT CTT GGC ATT TCT GGC-3′ and reverse 5′-TCG ACT CAT CAT CGA CTC TGG TCT-3′ (169 bp); and site 8, forward 5′-CCA CCC AAG GAG AAA GAA AGT TCC-3′ and reverse 5′-ACA CAC ACA CAC ACA TAG CAG GGA-3′ (179 bp). Positive and negative primers were purchased from Active Motif for PCR quality control.
Site-directed mutagenesis of sGC promoter.
Site-specific mutation of NFATc binding site 3 in the sGC-α1 promoter was carried out using a QuickChange system (Stratagene) according to the manufacturer's protocol. Primers were designed for the mutated site 3: forward 5′-TGC TTT ACA TTA TGA GTC CCT TTG TTT TTG TAA CCT TTT GGG CCT TAC G-3′ and reverse 5′-CGT AAG GCC CAA AAG GTT ACA AAA ACA AAG GGA CTC ATA ATG TAA AGC A-3′. The mutation in the binding site was confirmed by DNA sequencing.
Immunofluorescence confocal microscopy.
Isolated lungs were fixed with Histochoice (Amrefco). Lungs were cryoprotected with 30% sucrose in PBS, embedded in OCT medium, and frozen. Cryostat sections (10 μm) were permeabilized and blocked for nonspecific binding, and primary antibodies [rabbit polyclonal anti-HuR; 1:100 (Santa Cruz Biotechnology) and anti-α-actin; 1:250 (Sigma)] were prepared in 0.2% gelatin in PBS and applied overnight at 4°C. Secondary antibodies [anti-rabbit Cy5 and anti-mouse Cy3; 1:500 (Jackson ImmunoResearch Laboratories)] were prepared in 0.2% gelatin in PBS and applied for 1 h at room temperature. Nuclei were stained using SYTOX green (1:5,000 in PBS; Molecular Probes). Sections were examined using a ×40 objective on a META Zeiss 510 laser scanning confocal microscope. Specificity of immune staining was confirmed by the absence of fluorescence in tissues incubated with primary or secondary antibodies alone. For scoring of HuR-positive nuclei, multiple fields for each vessel were imaged and counted by two independent observers using MetaMorph software (Universal Imaging). The software was programmed so that individual pixels would appear white instead of yellow if the green nucleic acid stain and red HuR stain colocalized. Thus a cell was considered positive if colocalization (white) was uniformly distributed in the nucleus and negative if no colocalization (green only) was observed (12, 17, 23).
Statistical analysis.
Data are means ± SE. Statistical significance was tested at 95% (P < 0.05) confidence level using unpaired t-test or one-way or two-way ANOVA followed by the Student-Newman-Keuls method.
RESULTS
CH induces an initial decrease in sGC-α1 expression in mouse pulmonary arteries that recovers after 21 days of exposure: role of calcineurin/NFATc3 and HuR.
Recently, we reported the novel finding that NFATc3 is maximally activated in pulmonary arteries from mice exposed for 2 days to CH, remaining active even at 21 days of exposure (12). In the same study, we demonstrated that NFATc3 is required for CH-induced increases in pulmonary arterial wall thickness and associated SM α-actin upregulation. In addition, we have previously shown that NFATc3 contributes to the maintenance of VSMC contractile phenotype (22).
sGC is downregulated during conditions in which VSMC becomes dedifferentiated or is converted to a synthetic phenotype (reviewed in Ref. 54). However, sGC levels and activity have been shown to be upregulated in PASMC from CH-induced pulmonary hypertensive mice and rats (31, 44, 45). The mechanism that leads to increased sGC expression is unexplored. Thus we hypothesized that NFATc3 regulates sGC-α1 expression in mouse pulmonary arteries during CH. To address this hypothesis, we measured sGC-α1 mRNA levels in pulmonary arteries isolated from vehicle (V)- and CsA-treated FVBN mice exposed to normoxia and to 2 and 21 days of CH. CsA is an immunosuppressant that inhibits calcineurin, the Ca2+/calmodulin-dependent phosphatase that dephosphorylates NFATc, allowing its nuclear import (66).
Interestingly, we found that there was an approximately twofold reduction in pulmonary arterial sGC-α1 mRNA levels after 2 days of CH in both V- and CsA-treated mice (Fig. 1) compared with normoxic arteries. These results suggest the decrease is independent of calcineurin/NFATc activation, despite NFATc3 being maximally activated after 2 days of CH (12). Longer exposure to CH, 21 days, induced a significant increase in sGC-α1 mRNA from the levels observed at 2 days (∼3-fold) that was even higher than the levels of sGC-α1 mRNA in normoxic control arteries (1.7-fold). The recovery of sGC-α1 mRNA levels to control values (or higher) was only observed in V-treated mice (Fig. 1). No differences were observed between normoxic V- and CsA-treated mice. These results suggest that the recovery and small but significant increase in sGC-α1 are dependent on calcineurin/NFATc activation.
Fig. 1.

Calcineurin/nuclear factor of activated T cells isoform c3 (NFATc) inhibition prevents chronic hypoxia (CH)-dependent increases in expression of soluble guanylyl cyclase-α1 (sGC-α1) in mouse pulmonary arteries. sGC-α1 mRNA was measured by quantitative real-time PCR in pulmonary arteries from FVBN mice treated with vehicle (V) or cyclosporin A (CsA) and exposed to normoxia (N) or CH for 2 (2d) or 21 days (21d). β-Actin was used as endogenous control. Data are expressed as a fold change from normoxic vehicle (NV) using the equation 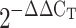 . Values are means ± SE; n = 6. *P < 0.05 vs. NV. &P < 0.05 vs. CH 2d V and CH 21d CsA. #P < 0.05 vs. NV, N CsA, and CH 21d CsA.
. Values are means ± SE; n = 6. *P < 0.05 vs. NV. &P < 0.05 vs. CH 2d V and CH 21d CsA. #P < 0.05 vs. NV, N CsA, and CH 21d CsA.
Since we have previously demonstrated that NFATc3 is the isoform activated by CH (12), we evaluated sGC-α1 mRNA levels in NFATc3 KO and WT littermates exposed to normoxia and to 2 and 21 days of CH. We also measured sGC-α1 protein levels using Western blot analysis in NFATc3 KO and WT littermates exposed to normoxia and 21 days of CH. sGC-α1 mRNA and protein were significantly lower in NFATc3 KO compared with WT mice under normoxic conditions (Fig. 2, A and B). Similar to the previous experiment, sGC-α1 mRNA levels were significantly lower (∼2.5-fold) after 2 days of CH in WT mice compared with normoxic WT (Fig. 2A). However, there was no difference between 2-day CH-exposed NFATc3 KO and normoxic NFATc3 KO mice. These results demonstrate that NFATc3 is necessary for basal sGC-α1 normoxic expression and that the low level of expression in its absence is not further reduced by 2 days of hypoxia.
Fig. 2.

NFATc3 genetic ablation prevents CH-mediated increases in expression of sGC-α1 in mouse pulmonary arteries. Data are fold change in mRNA (A) and protein (B) normalized to β-actin in pulmonary arteries from NFATc3 wild-type (WT) and knockout (KO) mice exposed to N or CH for 2d or 21d. Values are means ± SE; n = 6. *P < 0.05 vs. N WT and CH 21d WT. #P < 0.05 vs. N WT.
Consistent with our hypothesis and CsA experiments, sGC-α1 expression recovered to normoxic levels after 21 days of CH only in pulmonary arteries from CH NFATc3 WT mice (Fig. 2A), suggesting NFATc3 is required for both basal sGC-α1 expression and the recovery phase induced by long-term exposure to CH. Similar results were obtained for protein levels (Fig. 2B). In this strain of mice (Balb/C), we did not observe a further increase in either sGC-α1 mRNA or protein in pulmonary arteries from 21-day CH WT (Balb/C) compared with normoxic mice as observed in V-treated FVBN mice. This is probably due to differences in the genetic backgrounds between FVBN and Balb/C mice. However, both strains responded with an increase in sGC-α1 mRNA levels after the reduction induced by 2 days of CH.
Modulation of mRNA turnover by HuR is an important regulatory pathway of posttranscriptional control of gene expression (for review, see Refs. 15, 30, 70, 79, 86). Cytosolic HuR accumulation has been linked to the mechanism of stabilization of the mRNAs (15). Furthermore, sGC-α1 mRNA is posttranscriptionally regulated by HuR in VSMC (39, 41). To address whether posttranscriptional control of gene expression is involved in CH (2 days)-induced downregulation of sGC-α1, HuR mRNA levels and nuclear localization were determined in isolated pulmonary arteries from the two different strains of mice used in these studies (FVBN and Balb/C). No differences in HuR mRNA were observed in either strain of mouse after exposure to normoxia or to 2 and 21 days of CH (Fig. 3, A and B). However, HuR nuclear localization increased after 2 days of CH exposure and returned to normoxic levels after 21 days of CH (Fig. 4, A–C).
Fig. 3.

CH does not modify human antigen R (HuR) expression in mouse pulmonary arteries. Mouse HuR mRNA normalized to β-actin in FVBN mice treated with V (A) and Balb/C NFATc3 WT mice (B) exposed to N or CH for 2d or 21d. Data are expressed as fold change from N (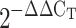 ). Values are means ± SE; n = 6.
). Values are means ± SE; n = 6.
Fig. 4.

Short-term CH (2d) increases HuR nuclear accumulation and decreases HuR/sGC-α1 mRNA coimmunoprecipitation in pulmonary arteries. A: lung sections from mice exposed to N and CH for 2d or 21d were costained with the DNA-binding dye SYTOX (green) and anti-HuR (red) and anti-SM α-actin (blue) antibodies. Representative images show nuclear colocalization of HuR (white) in FVBN mice. Arrows indicate examples of HuR-positive nuclei in pulmonary arteries. Scale bar, 20 μm. Data are summaries of HuR nuclear accumulation in FVBN mice (B) and Balb/C NFATc3 WT mice (C). Values are means ± SE; n = 23–38 arteries from 4 animals/group. *P < 0.05 vs. N. D: HuR was immunoprecipitated (IP) in pulmonary arteries from N- and CH (2d)-exposed NFATc3 WT mice. sGC-α1 mRNA bound to it was amplified by real-time PCR. Input RNA was used as reference control.
To further demonstrate that a reduction in HuR-mediated sGC-α1 mRNA stabilization is the underlying mechanism of CH (2 days)-induced reduction in sGC-α1 expression, we performed immunoprecipitation assays. Intrapulmonary artery lysates from normoxia- and CH (2 days)-exposed NFATc3 WT (Balb/C) mice were immunoprecipitated with anti-HuR antibody or no antibody (no primary control), and coimmunoprecipitated sGC-α1 mRNA was measured by real-time PCR. CH (2 days) significantly reduced the amount of sGC-α1 mRNA coimmunoprecipitated with HuR (Fig. 4D). Together, these results suggest that short-term exposure to CH leads to an increase in nuclear HuR (i.e., decreased cytosolic HuR that binds to sGC-α1 mRNA), which mediates a reduction in sGC-α1 mRNA stability and expression. On the other hand, NFATc3 is required for the recovery phase of sGC-α1 expression in pulmonary arteries exposed to long-term CH.
NFATc binding sites in mouse sGC-α1 promoter sequence.
To characterize the sGC-α1 transcriptional system, in silico analysis of the 5′-flanking region of the Mus musculus sGC-α1 gene [accession no. AY116663 (81)] was performed. Eight potential NFATc binding sites were identified by Patch 1.0 (Biobase) and designated with numbers, as shown in Fig. 5. MatInspector release Professional 7 software detected one site for NFATc, which is identical to site 3 recognized by Patch analysis. These programs use previously described NFATc consensus sequences (GGAA, AAGTGA, TTTTCC, TCAGCA, AGAAATTCC, GGAGCC) (66).
Fig. 5.

Putative NFATc binding sites in the 5′-flanking region sequence of mouse sGC-α1 gene. Putative NFATc binding sites (underlined) were identified using Patch 1.0 public software (Biobase) and MatInspector (Genomatix). The transcription initiation site is indicated with an arrow and designated +1.
To confirm the results of in silico analysis of the mouse sGC-α1 promoter, we performed ChIP assays in isolated pulmonary arteries of three FVBN mice exposed to CH for 3 wk. The DNA samples were analyzed by PCR using primers designed to amplify all the putative binding sites (see ChIP assay in experimental procedures for primer sequences). Unfortunately, in some cases it was not possible to design efficient primers that would amplify individual sites. As a negative control for the PCR, we used negative control primers suggested by the kit's manufacturer designed to amplify a silent DNA region.
Figure 6 A presents the putative binding sites that were positive for NFATc3 binding. Figure 6B is a representation of three experiments showing the amplicon for binding site 3+4, which was clearly enriched in every assay performed, and the control PCR products corresponding to no immune antibody, input DNA, and negative control primers. PCR primers originated the expected product length. The intensity of the bands was reproducibly higher in the DNA immunoprecipitated with NFATc3 antibody than the DNA immunoprecipitated using nonimmunogenic antibody. In contrast, PCR amplifications using negative control primers on the same DNA template did not show any comparative enrichment.
Fig. 6.

NFATc3 binds to several sites in mouse pulmonary arterial sGC-α1 promoter. A: summary of results of chromatin immunoprecipitation (ChIP) assays on isolated pulmonary arteries from FVBN mice exposed to CH for 3 wk. Data show the number of assays that were positive (enrichment) over the total number of assays (animals). B: PCR products from the chromatin/protein complexes immunoprecipitated with NFATc3 antibody or nonimmunogenic antibody (Neg Ab). Nonimmunoprecipitated DNA was used as input DNA.
NFATc activation increases mouse sGC-α1 promoter activity.
Since putative NFATc binding sites were identified in the mouse sGC-α1 promoter by in silico analysis and confirmed with the in vivo ChIP assay, we investigated whether NFATc acts as a trans-activator of the mouse sGC-α1 promoter in RAoSMC, HPASMC, and HEK-293 cells. To test promoter activity, we inserted the 5′-flanking region of sGC-α1 gene into the pGL3 vector upstream of the luciferase gene to yield sGC-luc plasmid.
RAoSMC and HPASMC were transiently transfected with the sGC-luc plasmid, serum deprived for 24 h, and stimulated with 1 μM Io ± 1 μM CsA. NFATc activation with Io induced a significant increase in reporter activity that was prevented by CsA in both sources of SMC (fig. 7, A and B).
Fig. 7.

Ionomycin (Io) increases mouse sGC-α1 promoter activity via NFATc. Rat aortic smooth muscle cells (RAoSMC; A) and human pulmonary artery smooth muscle cells (HPASMC; B) were cotransfected with sGC-luc and Renilla-luc plasmids and treated with 1 μM Io for 24 h ± 1 μM CsA. Values are means ± SE; n = 7. Dual-luciferase activity was measured and expressed as %change of relative luciferase units/relative Renilla units (RLU/ RRU) from basal. Values are means ± SE; n = 6. *P < 0.05 vs. basal.
To address whether NFATc3 is capable of trans-activating mouse sGC-α1 promoter, we transiently transfected HEK-293 cells with Tet-Op-NFATc3 and sGC-luc. To overexpress NFATc3, we added DOX to one-half of the plate. NFATc3 was activated with Io (1 μM) for 24 h in the absence or presence of CsA (1 μM) pretreatment. Consistent with the results from SMC, sGC-α1 promoter activity increased significantly in NFATc3-overexpressing activated HEK-293 cells (Fig. 8A). In addition, the inhibition of the calcineurin/NFATc pathway with CsA prevented this increase. A similar response was obtained in HEK-293 cells that were not overexpressing NFATc3 (see below), although, as expected, basal sGC-α1 promoter activity was lower than in NFATc3 overexpressing cells [relative luciferase units/relative Renilla units (RLU/RRL): overexpressing, 3.24 ± 0.14 vs. nonoverexpressing, 1.65 ± 0.04; n = 6, P < 0.05]. Consistent with these findings, a DN-NFAT plasmid clearly prevented Io-induced increases in reporter activity (Fig. 8B). Together, these results suggest that NFATc3 regulates sGC-α1 promoter activity.
Fig. 8.

NFATc3 activation increases mouse sGC-α1 promoter activity in HEK-293 cells. A: cells overexpressing NFATc3 were cotransfected with WT sGC-luc or sGC promoter mutated at site 3 (sGC-MUT-luc) and Renilla-luc plasmids. NFATc was activated with 1 μM Io for 24 h ± 1 μM CsA. B: NFATc3-overexpressing HEK cells were cotransfected with a dominant negative NFAT plasmid (DN) ± 1 μM Io for 24 h. Total DNA plasmid was maintained constant. Values are means ± SE; n = 6. *P < 0.05 vs. basal. #P < 0.05 vs. sGC-luc.
Every cell type transfected with the empty plasmid of the sGC-luc (pGL3) showed no differences in luciferase activity after stimulation with Io (data not shown).
As previously shown by Vazquez-Padron et al. (81), sGC promoter activity under basal conditions was higher in SMC compared with non-smooth muscle-derived cells such as HEK-293 cells not overexpressing NFATc3 (RLU/RRL: HEK-293, 1.70 ± 0.04; RAoSMC, 10.60 ± 1.70, P < 0.05 vs. HEK-293; HPASMC, 17.40 ± 3.90, P < 0.05 vs. HEK-293, n = 6). In addition, the order in the degree of increase in sGC promoter activity after Io stimulation was HPASMC > RAoSMC > HEK (relative change from basal: HEK-293, 1.5 ± 0.2-fold; RAoSMC, 1.7 ± 0.2-fold, P < 0.05 vs. HEK293 and HPASMC; HPASMC, 2.3 ± 0.4-fold, P < 0.05 vs. RAoSMC and HEK-293; n = 6). These results highlight the importance of sGC for SMC biology.
To confirm the results from the in vivo ChIP, we performed ChIP assays on Io-treated HEK-293 cells overexpressing NFATc3 and transfected with sGC-luc. As described earlier, negative control primers were used to amplify a silent DNA region.
Figure 9 A indicates the putative binding sites that were positive for NFATc3 binding. The results were very consistent with the in vivo ChIP assay shown in Fig. 6. In particular, site 3+4 clearly binds NFATc3 (Fig. 9B). PCR amplifications using negative control primers on these same DNA templates did not show any comparative enrichment. The remaining putative binding sites were not clearly positive and therefore are not considered as potential NFATc3 binding sites at this time.
Fig. 9.

NFATc3 binds to several sites in mouse sGC-α1 promoter transfected in HEK-293 cells. A: summary of results of ChIP assays on HEK-293 cells overexpressing NFATc3 cotransfected with sGC-luc and activated with 1 μM Io for 24 h. Data show the number of assays that were positive (enrichment) over the total number of assays. B: PCR products from the chromatin/protein complexes immunoprecipitated with NFATc3 antibody or nonimmunogenic antibody. Nonimmunoprecipitated DNA was used as input DNA.
A point mutation on NFATc binding site 3 drastically decreases mouse sGC-α1 promoter activity.
NFATc3 binding site 3 (Fig. 5) was chosen based on the fact that it was identified by both MatInspector and Patch and consistently bound NFATc3 in both pulmonary arteries and HEK-293 cells (Figs. 6 and 9). Thus we studied the importance of this site in the regulation of sGC-α1 promoter activity by mutating the site in the sGC-luc plasmid. The new plasmid was designated sGC-MUT-luc. HEK-293 cells were transfected with sGC-MUT-luc and treated with or without 1 μM Io for 24 h in the presence or absence of CsA. To compare the activity of both constructs, we took care to use the same quantity and ratio of plasmid DNA/transfection reagent. Figure 8A shows that sGC-MUT-luc transfected cells had markedly reduced sGC-α1 promoter activity compared with sGC-luc (WT)-transfected cells. In addition, the effect of Io was absent, demonstrating that NFATc binding site 3 is of primary importance in the transcriptional regulation of mouse sGC-α1 gene. The deletion did not introduce any new binding sites as confirmed by sequence analysis with MatInspector and Patch software. Similar results were obtained in SMC (data not shown).
DISCUSSION
This is the first study demonstrating that NFATc3 regulates sGC-α1 expression. In addition, it confirms that mouse sGC-α1 expression is regulated by HuR as has been shown previously in rats (39–41, 64). The analysis of the mouse sGC-α1 promoter showed several putative NFATc binding sites. Mouse sGC-α1 promoter reporter studies have demonstrated that NFATc3 activation enhances promoter activity, which is dependent on calcineurin. NFATc3 clearly binds to at least three regions in the promoter. Interestingly, site-directed mutagenesis of one of the NFATc binding sites in the mouse sGC-α1 promoter drastically decreased both basal reporter activity and NFATc-mediated activation of the promoter. Together, these results demonstrate that NFATc3 is a positive regulator of sGC-α1 expression through direct binding to the promoter of this gene, supporting previous evidence that NFATc3 is an important regulator of VSMC differentiated phenotype.
sGC-α1 expression in pulmonary arteries was initially decreased by CH exposure (2 days), but it recovered to normoxic levels after 21 days. The initial decrease was not dependent on NFATc3, because neither calcineurin/NFATc inhibition with CsA nor genetic ablation of NFATc3 prevented the downregulation. However, the recovery was dependent on NFATc3, because it did not occur in either CsA-treated or NFATc3 KO mice.
Paddenberg et al. (63) have showed that proliferative activity of pulmonary arterial SMC increased by 34% at day 4 in mice held under CH compared with normoxia, leveling off within 3 wk, although arterial wall thickness was increased. Therefore, the initial reduction in sGC-α1 expression may be consistent with other reports showing sGC downregulation under conditions of VSMC dedifferentiation or conversion to a synthetic/proliferative phenotype (reviewed in Ref. 54). Interestingly, sGC downregulation has been reported in the vasculature of different models of systemic hypertension (38, 49, 71), in pulmonary arteries from pulmonary hypertensive fetal lambs (80), and in hypoxia-exposed rat PASMC in culture (27). However, neither these studies nor our study have determined whether the reduction in sGC expression correlates with increased smooth muscle proliferation or dedifferentiation, and this needs to be further investigated. In addition, the mechanism of this downregulation was not previously explored. Both transcriptional (independent of NFATc) as well as posttranscriptional mechanisms could have contributed to the observed downregulation.
It has been demonstrated that sGC-α1 expression is posttranscriptionally modulated by HuR (39–41, 64). The precise mechanisms whereby HuR mediates mRNA stabilization are still poorly understood. HuR is predominantly localized in the nucleus of most unstimulated cells but can translocate to the cytoplasm on cell stimulation, a process that has been linked to the stabilization of many target mRNAs (15). Furthermore, a reduction in sGC-α1 expression in old spontaneously hypertensive rats compared with age-matched controls has been demonstrated to be due to HuR downregulation (40). Our findings are in agreement with these reports. Although no changes were observed in HuR mRNA levels in pulmonary arteries between 2-day CH and normoxic mice, nuclear localization of HuR was increased. Furthermore, less sGC-α1 mRNA coimmunoprecipitated with HuR in pulmonary arteries from 2-day CH- compared with normoxia-exposed mice. These findings suggest that a decrease in HuR-dependent sGC-α1 mRNA stabilization mediates the reduction in its expression after short-term exposure to CH (2 days).
The increased sGC-α1 expression over 2-day CH values after 21 days of hypoxia exposure in both strains of mice is consistent with previous reports showing that in adult animals, long-term CH induces an increase in sGC mRNA, protein, and enzyme activity (2, 3, 13, 16, 31, 36, 43–45, 68, 89). NO/cGMP signaling is not only involved in VSMC relaxation but also is antiproliferative (46). Therefore, it is possible that sGC upregulation offsets the increased PASMC proliferation that occurs during CH-induced pulmonary arterial remodeling (5, 63, 74, 87). Along these lines, we have shown that NFATc3 is involved in CH-induced increases in pulmonary arterial wall thickness with upregulation of SM α-actin (12), suggesting that NFATc3 mediates PASMC hypertrophy rather than proliferation in mice. However, the significance of hypoxia-induced increases in sGC-α1 levels and activity is not completely understood and has to be further investigated. It has been suggested that the upregulation of sGC-α1 induced by hypoxia in the pulmonary vasculature may be a mechanism of physiological adaptation to or modulation of pulmonary hypertension, antagonizing/offsetting the vascular remodeling and increased vasoconstriction (44, 65). In support of this hypothesis, chronic inhibition of PDEs or activation of sGC in hypoxic lungs reverses pulmonary hypertension (14, 51, 74, 87). In addition, chronic inhibition of sGC by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), a selective inhibitor, augments vasoconstriction in hypoxic lungs but not in normoxic control rat lungs (19).
On the contrary, it also has been proposed that CH-induced elevations in vascular production of reactive oxygen species scavenge NO and oxidize sGC (32, 54, 77), thereby reducing NO-dependent activation of sGC and attenuating NO-dependent pulmonary vasodilation (7, 32, 56). Therefore, although the components of the NO pathway are upregulated, they may not be effectively preventing the development of pulmonary hypertension during CH.
Indeed during CH, PASMC exhibit reduced KV channel activity and expression (53, 67, 85) with increased receptor potential channel activity and expression (83, 84), leading to increased intracellular Ca2+ levels and pulmonary vasoconstriction. Increases in intracellular Ca2+ have been linked to SMC hypertrophy and vascular remodeling (62). Along these lines, NFATc3 activation downregulates the expression of KV2.1 and the β-subunit of BK channels, thereby increasing cerebral arterial VSMC contractility (1, 57). Both NFATc and sGC/PKG pathway are linked to SM phenotypic maintenance (20–25) through regulation of the expression of SM myosin heavy chain, SM α-actin, α1-integrin, and caldesmon genes (4, 8, 22, 46, 47, 58, 82). Therefore, NFATc3 regulation of sGC-α1 also may be part of the repertoire of genes that control the maintenance of SM contractile phenotype contributing to the increased vasoreactivity and/or remodeling observed in pulmonary hypertensive animals (33, 34, 55, 67, 78).
On the other hand, it has been demonstrated that the NFATc2 isoform mediates PASMC KV1.5 downregulation and increased proliferation in HPASMC from idiopathic pulmonary hypertensive patients (5). In the same study, CsA reduced monocrotaline-induced pulmonary hypertension and pulmonary arterial remodeling in rats by increasing PASMC KV1.5 and inducing PASMC apoptosis. Therefore, in our study we cannot discard the possibility that early activation of another NFATc isoform linked to PASMC proliferation triggers sGC-α1 downregulation. However, our results are not consistent with this possibility, because CsA, which inhibits calcineurin and therefore all NFATc isoforms, did not prevent 2-day CH-induced sGC-α1 downregulation in pulmonary arteries. In addition, we have previously demonstrated that NFATc3 is the isoform activated in mouse pulmonary arteries exposed to CH and that the same dose of CsA effectively prevents CH-induced NFATc activation (12).
Our in vivo findings in mice are consistent with our molecular findings of sGC-α1 promoter regulation by NFATc3. Little is known about transcriptional regulation of sGC-α1. Vazquez-Padron et al. (81) have elegantly characterized the mouse sGC-α1 promoter, determined sequence homology with other species, and identified the initiation transcription site. A recent publication has implicated NFATc as a negative regulator of the human sGC-α1 and -β1 promoter. Marro et al. (52) showed that deletion of a putative NFATc binding site from these promoters increases activity in human aortic SMC, suggesting a repressor effect of NFATc. However, NFATc binding to these sites was not confirmed by either ChIP or electrophoretic mobility shift assay. In addition, the 0.3-kb fragment of the human sGC-α1 promoter studied by Marro et al. has only 44% homology with the mouse sequence (52) and is very close to the transcription start site (−100 bp). In the present study, the identified NFATc binding site (site 3) in the mouse sGC-α1 promoter present in chromosome 3 region 81950726-81950739 has 78.6% identity with the human sGC-α1 gene upstream regulatory region present in chromosome 4 (156806032-156809745) (61). Site 3 is present at position −955 from the transcription start site, which is a region excluded in the analysis performed by Marro et al. in the human sGC-α1 gene. Our results show that a single nucleotide mutation in this site drastically decreases mouse sGC-α1 promoter activity. The mutation did not generate a new putative computationally identified site. Site 3 does not overlap with any other previously described transcription factor, but it is in close proximity to other NFATc, Sp-1, and NF-κB sites. It has been shown previously that Sp-1 contributes to the expression of the sGC-α1 subunit in resting pituitary cells (35). The other interesting finding from the analysis of sGC-α1 upstream region, performed by Vazquez-Padron et al. (81), is the presence of p300/cAMP response element binding protein (CBP) binding sites conserved between rodents and human. p300/CBP forms a complex with several transcription factors, including NFATc, regulating transcription through chromatin acetylation. It has been shown that p300/CBP can influence different physiological processes, including but not limited to cell growth, proliferation, and differentiation (9). Future studies are needed to address the NFATc transcriptional cofactors, such as AP-1 or GATA, that also are upregulated by CH (37, 75), along with NF-κB, Sp-1, serum response factor (SRF), or p300/CBP (11), all of which have putative binding sites described in the mouse sGC-α1 promoter (81) and have been implicated in phenotypic modulation of VSMC.
Another important finding derived from our studies, confirming data previously shown by Vazquez-Padron et al. (81), is that sGC promoter activity under basal conditions is higher in SMC compared with non-smooth muscle-derived cells. In addition, sGC promoter responded to NFATc activation greatly in HPASMC > RAoSMC > HEK, highlighting the importance of sGC and NFATc for SMC biology.
In conclusion, NFATc3 promotes mouse sGC-α1 transcription in pulmonary arteries under basal and long-term exposure to CH. In addition, short-term exposure to CH increases HuR nuclear localization, decreasing sGC-α1 expression independently of NFATc3. This study further supports NFATc3 as an important regulator of smooth muscle gene transcription in pulmonary hypertension.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant R01-HL-088151, the American Lung Association, and a Postdoctoral Fellowship from Ministerio de Educación y Ciencia, Spain (to Sergio de Frutos).
Acknowledgments
We thank Dr. Thomas Resta for helpful discussions and Dr. Nora Perrone-Bizzozero for helping with the RIP assays.
REFERENCES
- 1.Amberg GC, Rossow CF, Navedo MF, Santana LF. NFATc3 regulates Kv2.1 expression in arterial smooth muscle. J Biol Chem 279: 47326–47334, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Archer SL, Tolins JP, Raij L, Weir EK. Hypoxic pulmonary vasoconstriction is enhanced by inhibition of the synthesis of an endothelium derived relaxing factor. Biochem Biophys Res Commun 164: 1198–1205, 1989. [DOI] [PubMed] [Google Scholar]
- 3.Barer G, Emery C, Stewart A, Bee D, Howard P. Endothelial control of the pulmonary circulation in normal and chronically hypoxic rats. J Physiol 463: 1–16, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boerth NJ, Dey NB, Cornwell TL, Lincoln TM. Cyclic GMP-dependent protein kinase regulates vascular smooth muscle cell phenotype. J Vasc Res 34: 245–259, 1997. [DOI] [PubMed] [Google Scholar]
- 5.Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, Hashimoto K, Bonnet SN, Michelakis ED. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci USA 104: 11418–11423, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouallegue A, Daou GB, Srivastava AK. Nitric oxide attenuates endothelin-1-induced activation of ERK1/2, PKB, and Pyk2 in vascular smooth muscle cells by a cGMP-dependent pathway. Am J Physiol Heart Circ Physiol 293: H2072–H2079, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Brennan LA, Steinhorn RH, Wedgwood S, Mata-Greenwood E, Roark EA, Russell JA, Black SM. Increased superoxide generation is associated with pulmonary hypertension in fetal lambs: a role for NADPH oxidase. Circ Res 92: 683–691, 2003. [DOI] [PubMed] [Google Scholar]
- 8.Brophy CM, Woodrum DA, Pollock J, Dickinson M, Komalavilas P, Cornwell TL, Lincoln TM. cGMP-dependent protein kinase expression restores contractile function in cultured vascular smooth muscle cells. J Vasc Res 39: 95–103, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci 114: 2363–2373, 2001. [DOI] [PubMed] [Google Scholar]
- 10.Chiche JD, Schlutsmeyer SM, Bloch DB, de la Monte SM, Roberts J, Filippov G, Janssens SP, Rosenzweig A, Bloch KD. Adenovirus-mediated gene transfer of cGMP-dependent protein kinase increases the sensitivity of cultured vascular smooth muscle cells to the antiproliferative and pro-apoptotic effects of nitric oxide/cGMP. J Biol Chem 273: 34263–34271, 1998. [DOI] [PubMed] [Google Scholar]
- 11.Cummins E, Taylor C. Hypoxia-responsive transcription factors. Pflügers Arch 450: 363–371, 2005. [DOI] [PubMed] [Google Scholar]
- 12.de Frutos S, Spangler R, Alo D, Gonzalez Bosc LV. NFATc3 mediates chronic hypoxia-induced pulmonary arterial remodeling with alpha -actin up-regulation. J Biol Chem 282: 15081–15089, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12a.Deschênes-Furry J, Belanger G, Mwanjewe J, Lunde JA, Parks RJ, Perrone-Bizzozero N, Jasmin BJ. The RNA-binding protein HuR binds to acetylcholinesterase transcripts and regulates their expression in differentiating skeletal muscle cells. J Biol Chem 280: 25361–25368, 2005. [DOI] [PubMed] [Google Scholar]
- 13.Dinh-Xuan A Endothelial modulation of pulmonary vascular tone. Eur Respir J 5: 757–762, 1992. [PubMed] [Google Scholar]
- 14.Dumitrascu R, Weissmann N, Ghofrani HA, Dony E, Beuerlein K, Schmidt H, Stasch JP, Gnoth MJ, Seeger W, Grimminger F, Schermuly RT. Activation of soluble guanylate cyclase reverses experimental pulmonary hypertension and vascular remodeling. Circulation 113: 286–295, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Figueroa A, Cuadrado A, Fan J, Atasoy U, Muscat GE, Munoz-Canoves P, Gorospe M, Munoz A. Role of HuR in skeletal myogenesis through coordinate regulation of muscle differentiation genes. Mol Cell Biol 23: 4991–5004, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fike CD, Kaplowitz MR, Thomas CJ, Nelin LD. Chronic hypoxia decreases nitric oxide production and endothelial nitric oxide synthase in newborn pig lungs. Am J Physiol Lung Cell Mol Physiol 274: L517–L526, 1998. [DOI] [PubMed] [Google Scholar]
- 17.Filosa JA, Nelson MT, Gonzalez Bosc LV. Activity-dependent NFAT3 nuclear accumulation in pericytes from cortical parenchymal microvessel. Am J Physiol Cell Physiol 293: C1797–C1805, 2007. [DOI] [PubMed] [Google Scholar]
- 18.Finn JT, Grunwald ME, Yau KW. Cyclic nucleotide-gated ion channels: an extended family with diverse functions. Annu Rev Physiol 58: 395–426, 1996. [DOI] [PubMed] [Google Scholar]
- 19.Fouty B, Komalavilas P, Muramatsu M, Cohen A, McMurtry IF, Lincoln TM, Rodman DM. Protein kinase G is not essential to NO-cGMP modulation of basal tone in rat pulmonary circulation. Am J Physiol Heart Circ Physiol 274: H672–H678, 1998. [DOI] [PubMed] [Google Scholar]
- 20.Friebe A, Koesling D. Regulation of nitric oxide-sensitive guanylyl cyclase. Circ Res 93: 96–105, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest 83: 1774–1777, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gonzalez Bosc LV, Layne J, Nelson MT, Hill-Eubanks DC. Nuclear factor of activated T-cells and serum response factor cooperatively regulate an α-actin intronic enhancer. J Biol Chem 280: 26113–26120, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Gonzalez Bosc LV, Wilkerson MK, Bradley KN, Eckman DM, Hill-Eubanks DC, Nelson MT. Intraluminal pressure is a stimulus for NFATc3 nuclear accumulation—role of calcium, endothelium-derived nitric oxide, and cGMP-dependent protein kinase. J Biol Chem 279: 10702–10709, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Good PJ A conserved family of elav-like genes in vertebrates. Proc Natl Acad Sci USA 92: 4557–4561, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graef IA, Chen F, Chen L, Kuo A, Crabtree GR. Signals transduced by Ca2+/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell 105: 863–875, 2001. [DOI] [PubMed] [Google Scholar]
- 26.Graef IA, Chen F, Crabtree GR. NFAT signaling in vertebrate development. Curr Opin Genet Dev 11: 505–512, 2001. [DOI] [PubMed] [Google Scholar]
- 27.Hassoun PM, Filippov G, Fogel M, Donaldson C, Kayyali US, Shimoda LA, Bloch KD. Hypoxia decreases expression of soluble guanylate cyclase in cultured rat pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol 30: 908–913, 2004. [DOI] [PubMed] [Google Scholar]
- 28.Hill-Eubanks DC, Gomez MF, Stevenson AS, Nelson MT. NFAT regulation in smooth muscle. Trends Cardiovasc Med 13: 56–62, 2003. [DOI] [PubMed] [Google Scholar]
- 29.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 17: 2205–2232, 2003. [DOI] [PubMed] [Google Scholar]
- 30.Hollams EM, Giles KM, Thomson AM, Leedman PJ. mRNA stability and the control of gene expression: implications for human disease. Neurochem Res 27: 957–980, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Jernigan NL, Resta TC. Chronic hypoxia attenuates cGMP-dependent pulmonary vasodilation. Am J Physiol Lung Cell Mol Physiol 282: L1366–L1375, 2002. [DOI] [PubMed] [Google Scholar]
- 32.Jernigan NL, Resta TC, Walker BR. Contribution of oxygen radicals to altered NO-dependent pulmonary vasodilation in acute and chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 286: L947–L955, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Jernigan NL, Walker BR, Resta TC. Endothelium-derived reactive oxygen species and endothelin-1 attenuate NO-dependent pulmonary vasodilation following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 287: L801–L808, 2004. [DOI] [PubMed] [Google Scholar]
- 34.Jernigan NL, Walker BR, Resta TC. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 295: L515–L529, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang Y, Stojilkovic SS. Molecular cloning and characterization of alpha1-soluble guanylyl cyclase gene promoter in rat pituitary cells. J Mol Endocrinol 37: 503–515, 2006. [DOI] [PubMed] [Google Scholar]
- 36.Johns RA, Linden JM, Peach MJ. Endothelium-dependent relaxation and cyclic GMP accumulation in rabbit pulmonary artery are selectively impaired by moderate hypoxia. Circ Res 65: 1508–1515, 1989. [DOI] [PubMed] [Google Scholar]
- 37.Kallio PJ, Okamoto K, O'Brien S, Carrero P, Makino Y, Tanaka H, Poellinger L. Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J 17: 6573–6586, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klöss S, Bouloumie A, Mulsch A. Aging and chronic hypertension decrease expression of rat aortic soluble guanylyl cyclase. Hypertension 35: 43–47, 2000. [PubMed] [Google Scholar]
- 39.Klöss S, Furneaux H, Mulsch A. Post-transcriptional regulation of soluble guanylyl cyclase expression in rat aorta. J Biol Chem 278: 2377–2383, 2003. [DOI] [PubMed] [Google Scholar]
- 40.Klöss S, Rodenbach D, Bordel R, Mulsch A. Human-antigen R (HuR) expression in hypertension: downregulation of the mRNA stabilizing protein HuR in genetic hypertension. Hypertension 45: 1200–1206, 2005. [DOI] [PubMed] [Google Scholar]
- 41.Klöss S, Srivastava R, Mulsch A. Down-regulation of soluble guanylyl cyclase expression by cyclic AMP is mediated by mRNA-stabilizing protein HuR. Mol Pharmacol 65: 1440–1451, 2004. [DOI] [PubMed] [Google Scholar]
- 42.Koesling D Studying the structure and regulation of soluble guanylyl cyclase. Methods 19: 485–493, 1999. [DOI] [PubMed] [Google Scholar]
- 43.Le Cras TD, Xue C, Rengasamy A, Johns RA. Chronic hypoxia upregulates endothelial and inducible NO synthase gene and protein expression in rat lung. Am J Physiol Lung Cell Mol Physiol 270: L164–L170, 1996. [DOI] [PubMed] [Google Scholar]
- 44.Li D, Zhou N, Johns RA. Soluble guanylate cyclase gene expression and localization in rat lung after exposure to hypoxia. Am J Physiol Lung Cell Mol Physiol 277: L841–L847, 1999. [DOI] [PubMed] [Google Scholar]
- 45.Li D, Laubach VE, Johns RA. Upregulation of lung soluble guanylate cyclase during chronic hypoxia is prevented by deletion of eNOS. Am J Physiol Lung Cell Mol Physiol 281: L369–L376, 2001. [DOI] [PubMed] [Google Scholar]
- 46.Lincoln TM, Wu X, Sellak H, Dey N, Choi CS. Regulation of vascular smooth muscle cell phenotype by cyclic GMP and cyclic GMP-dependent protein kinase. Front Biosci 11: 356–367, 2006. [DOI] [PubMed] [Google Scholar]
- 47.Lincoln TM, Dey N, Sellak H. cGMP-dependent protein kinase signaling mechanisms in smooth muscle: from the regulation of tone to gene expression. J Appl Physiol 91: 1421–1430, 2001. [DOI] [PubMed] [Google Scholar]
- 48.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25: 402–408, 2001. [DOI] [PubMed] [Google Scholar]
- 49.Lopez-Farre A, Rodriguez-Feo JA, Garcia-Colis E, Gomez J, Lopez-Blaya A, Fortes J, de AR, Rico L, Casado S. Reduction of the soluble cyclic GMP vasorelaxing system in the vascular wall of stroke-prone spontaneously hypertensive rats: effect of the alpha1-receptor blocker doxazosin. J Hypertens 20: 463–470, 2002. [DOI] [PubMed] [Google Scholar]
- 50.Ma WJ, Cheng S, Campbell C, Wright A, Furneaux H. Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J Biol Chem 271: 8144–8151, 1996. [DOI] [PubMed] [Google Scholar]
- 51.MacLean MR, Johnston ED, McCulloch KM, Pooley L, Houslay MD, Sweeney G. Phosphodiesterase isoforms in the pulmonary arterial circulation of the rat: changes in pulmonary hypertension. J Pharmacol Exp Ther 283: 619–624, 1997. [PubMed] [Google Scholar]
- 52.Marro ML, Peiro C, Panayiotou CM, Baliga RS, Meurer S, Schmidt HHHW, Hobbs AJ. Characterization of the Human alpha1beta1 soluble guanylyl cyclase promoter: key role for NF-kappaB(p50) and CCAAT-binding factors in regulating expression of the nitric oxide receptor. J Biol Chem 283: 20027–20036, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mauban JRH, Remillard CV, Yuan JXJ. Hypoxic pulmonary vasoconstriction: role of ion channels. J Appl Physiol 98: 415–420, 2005. [DOI] [PubMed] [Google Scholar]
- 54.Munzel T, Daiber A, Ullrich V, Mulsch A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler Thromb Vasc Biol 25: 1551–1557, 2005. [DOI] [PubMed] [Google Scholar]
- 55.Nagaoka T, Morio Y, Casanova N, Bauer N, Gebb S, McMurtry I, Oka M. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 287: L665–L672, 2004. [DOI] [PubMed] [Google Scholar]
- 56.Nakanishi K, Tajima F, Nakamura A, Yagura S, Ookawara T, Yamashita H, Suzuki K, Taniguchi N, Ohno H. Effects of hypobaric hypoxia on antioxidant enzymes in rats. J Physiol 489: 869–876, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nieves-Cintron M, Amberg GC, Nichols CB, Molkentin JD, Santana LF. Activation of NFATC3 down-regulates the beta 1 subunit of large conductance, calcium-activated K+ channels in arterial smooth muscle and contributes to hypertension. J Biol Chem 282: 3231–3240, 2006. [DOI] [PubMed] [Google Scholar]
- 58.Ohkawa Y, Hayashi K, Sobue K. Calcineurin-mediated pathway involved in the differentiated phenotype of smooth muscle cells. Biochem Biophys Res Commun 301: 78–83, 2003. [DOI] [PubMed] [Google Scholar]
- 59.Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res 100: 309–327, 2007. [DOI] [PubMed] [Google Scholar]
- 60.Oukka M, Ho IC, de la Brousse FC, Hoey T, Grusby MJ, Glimcher LH. The transcription factor NFAT4 is involved in the generation and survival of T cells. Immunity 9: 295–304, 1998. [DOI] [PubMed] [Google Scholar]
- 61.Ovcharenko I, Nobrega MA, Loots GG, Stubbs L. ECR browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res 32: W280-W286, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Owens GK, Rabinovitch PS, Schwartz SM. Smooth muscle cell hypertrophy versus hyperplasia in hypertension. Proc Natl Acad Sci USA 78: 7759–7763, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paddenberg R, Stieger P, von Lilien AL, Faulhammer P, Goldenberg A, Tillmanns H, Kummer W, Braun-Dullaeus R. Rapamycin attenuates hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy in mice. Respir Res 8: 15, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Priviero FBM, Zemse SM, Teixeira CE, Webb RC. Oxidative stress impairs vasorelaxation induced by the soluble guanylyl cyclase activator BAY 41-2272 in spontaneously hypertensive rats. Am J Hypertens 22: 493–499, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rabinovitch M, Gamble W, Nadas AS, Miettinen OS, Reid L. Rat pulmonary circulation after chronic hypoxia: hemodynamic and structural features. Am J Physiol Heart Circ Physiol 236: H818–H827, 1979. [DOI] [PubMed] [Google Scholar]
- 66.Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol 15: 707–747, 1997. [DOI] [PubMed] [Google Scholar]
- 67.Remillard CV, Yuan JX. High altitude pulmonary hypertension: role of K+ and Ca2+ channels. High Alt Med Biol 6: 133–146, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Resta TC, Gonzales RJ, Dail WG, Sanders TC, Walker BR. Selective upregulation of arterial endothelial nitric oxide synthase in pulmonary hypertension. Am J Physiol Heart Circ Physiol 272: H806–H813, 1997. [DOI] [PubMed] [Google Scholar]
- 69.Ritter D, Taylor JF, Hoffmann JW, Carnaghi L, Giddings SJ, Zakeri H, Kwok PY. Alternative splicing for the alpha1 subunit of soluble guanylate cyclase. Biochem J 346: 811–816, 2000. [PMC free article] [PubMed] [Google Scholar]
- 70.Ross J Control of messenger RNA stability in higher eukaryotes. Trends Genet 12: 171–175, 1996. [DOI] [PubMed] [Google Scholar]
- 71.Ruetten H, Zabel U, Linz W, Schmidt HHHW. Downregulation of soluble guanylyl cyclase in young and aging spontaneously hypertensive rats. Circ Res 85: 534–541, 1999. [DOI] [PubMed] [Google Scholar]
- 73.Schermuly RT, Stasch JP, Pullamsetti SS, Middendorff R, Muller D, Schluter KD, Dingendorf A, Hackemack S, Kolosionek E, Kaulen C, Dumitrascu R, Weissmann N, Mittendorf J, Klepetko W, Seeger W, Ghofrani HA, Grimminger F. Expression and function of soluble guanylate cyclase in pulmonary arterial hypertension. Eur Respir J 32: 881–891, 2008. [DOI] [PubMed] [Google Scholar]
- 74.Schermuly RT, Pullamsetti SS, Kwapiszewska G, Dumitrascu R, Tian X, Weissmann N, Ghofrani HA, Kaulen C, Dunkern T, Schudt C, Voswinckel R, Zhou J, Samidurai A, Klepetko W, Paddenberg R, Kummer W, Seeger W, Grimminger F. Phosphodiesterase 1 upregulation in pulmonary arterial hypertension: target for reverse-remodeling therapy. Circulation 115: 2331–2339, 2007. [DOI] [PubMed] [Google Scholar]
- 75.Semenza GL Perspectives on oxygen sensing. Cell 98: 281–284, 1999. [DOI] [PubMed] [Google Scholar]
- 76.Sharina IG, Martin E, Thomas A, Uray KL, Murad F. CCAAT-binding factor regulates expression of the beta1 subunit of soluble guanylyl cyclase gene in the BE2 human neuroblastoma cell line. Proc Natl Acad Sci USA 100: 11523–11528, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stasch JP, Schmidt PM, Nedvetsky PI, Nedvetskaya TY, AKHS, Meurer S, Deile M, Taye A, Knorr A, Lapp H, Muller H, Turgay Y, Rothkegel C, Tersteegen A, Kemp-Harper B, Muller-Esterl W, Schmidt HH. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest 116: 2552–2561, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stenmark KR, McMurtry IF. Vascular remodeling versus vasoconstriction in chronic hypoxic pulmonary hypertension: a time for reappraisal? Circ Res 97: 95–98, 2005. [DOI] [PubMed] [Google Scholar]
- 79.Travo P, Barrett G, Burnstock G. Differences in proliferation of primary cultures of vascular smooth muscle cells taken from male and female rats. Blood Vessels 17: 110–116, 1980. [DOI] [PubMed] [Google Scholar]
- 80.Tzao C, Nickerson PA, Russell JA, Gugino SF, Steinhorn RH. Pulmonary hypertension alters soluble guanylate cyclase activity and expression in pulmonary arteries isolated from fetal lambs. Pediatr Pulmonol 31: 97–105, 2001. [DOI] [PubMed] [Google Scholar]
- 81.Vazquez-Padron RI, Pham SM, Pang M, Li S, Aïtouche A. Molecular dissection of mouse soluble guanylyl cyclase alpha1 promoter. Biochem Biophys Res Commun 314: 208–214, 2004. [DOI] [PubMed] [Google Scholar]
- 82.Wada H, Hasegawa K, Morimoto T, Kakita T, Yanazume T, Abe M, Sasayama S. Calcineurin-GATA-6 pathway is involved in smooth muscle-specific transcription. J Cell Biol 156: 983–991, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang J, Shimoda LA, Sylvester JT. Capacitative calcium entry and TRPC channel proteins are expressed in rat distal pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol 286: L848–L858, 2004. [DOI] [PubMed] [Google Scholar]
- 84.Wang J, Weigand L, Lu W, Sylvester JT, Semenza L, Shimoda LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 98: 1528–1537, 2006. [DOI] [PubMed] [Google Scholar]
- 85.Wang J, Weigand L, Wang W, Sylvester JT, Shimoda LA. Chronic hypoxia inhibits Kv channel gene expression in rat distal pulmonary artery. Am J Physiol Lung Cell Mol Physiol 288: L1049–L1058, 2005. [DOI] [PubMed] [Google Scholar]
- 86.Wang W, Fan J, Yang X, Furer-Galban S, Lopez de Silanes I, von Kobbe C, Guo J, Georas SN, Foufelle F, Hardie DG, Carling D, Gorospe M. AMP-activated kinase regulates cytoplasmic HuR. Mol Cell Biol 22: 3425–3436, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wharton J, Strange JW, Moller GMO, Growcott EJ, Ren X, Franklyn AP, Phillips SC, Wilkins MR. Antiproliferative effects of phosphodiesterase type 5 inhibition in human pulmonary artery cells. Am J Respir Crit Care Med 172: 105–113, 2005. [DOI] [PubMed] [Google Scholar]
- 88.Wilkins BJ, De Windt LJ, Bueno OF, Braz JC, Glascock BJ, Kimball TF, Molkentin JD. Targeted disruption of NFATc3, but not NFATc4, reveals an intrinsic defect in calcineurin-mediated cardiac hypertrophic growth. Mol Cell Biol 22: 7603–13, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xue C, Johns RA. Upregulation of nitric oxide synthase correlates temporally with onset of pulmonary vascular remodeling in the hypoxic rat. Hypertension 28: 743–753, 1996. [DOI] [PubMed] [Google Scholar]