

Abstract
Gene microarray analyses represent potentially effective means for high-throughput gene expression profiling in nonhuman primates. In the companion article we emphasize effective experimental design based on the in vivo physiology of the rhesus macaque, whereas this article emphasizes considerations for gene annotation and data interpretation using gene microarray platforms from Affymetrix®. Initial annotation of the rhesus genome array was based on Affymetrix® human GeneChips®. However, annotation revisions improve the precision with which rhesus transcripts are identified. Annotation of the rhesus GeneChip® is under continuous revision with large percentages of probesets under multiple annotation systems having undergone multiple reassignments between March 2007 and November 2008. It is also important to consider that quantitation and comparison of gene expression levels across multiple chips requires appropriate normalization. External corroboration of microarray results using PCR-based methodology also requires validation of appropriate internal reference genes for normalization of expression values. Many tools are now freely available to aid investigators with microarray normalization and selection of internal reference genes to be used for independent corroboration of microarray results.
Keywords: Macaca Mulatta, Microarray, GeneChip® Rhesus Macaque Genome Array
1. Introduction
Gene microarrays are comprised of single strands of complementary DNA (cDNA) or oligomers for genes of interest immobilized in locations called features [1, 2] bonded to a solid substrate in a systematic arrangement (array) [3, 4]. An individual microarray construction is commonly referred to as a “chip” (e.g., microarray chip, array chip, gene chip etc.). Microarray technology is based on the principle that in a mixture of thousands of nucleic acid species, labeled species (targets) applied in solution preferentially hybridize with immobilized complementary sequences (probes) identified by a specific feature [5]. For review, see [6].
Affymetrix® GeneChips® (Affymetrix, Inc., Santa Clara, CA) have been the most prominent commercially available microarrays [7, 8]. The Affymetrix® GeneChip® Rhesus Macaque Genome Array is a single-labeled high-density oligonucleotide array [9] where the entire monkey genome is represented (52,024 rhesus probe-sets, representing >20,000 genes)[10, 11]. This means that researchers interrogate expression of all mRNA transcripts from a given sample (thousands of genes) simultaneously. In general, RNA from the sample of interest is extracted, synthesized into a cDNA template, from which labeled cRNA is produced and then hybridized to the microarray. By measuring the quantity of hybridization on each feature the abundance of the corresponding RNA transcript in the sample can be determined from the signal intensity of the labeled target. This signal intensity correlates directly to the degree of hybridization occurring at a feature (typically representing a gene) of choice. As simple as this may sound, there are several potential pitfalls associated with this relatively new methodology, especially when profiling gene expression in rhesus macaques. The submitted companion article [12] focuses on experimental design considerations, when planning to perform gene profiling in the rhesus macaque (Macaca mulatta). In the present article we focus two other important considerations when performing gene microarray studies: normalization of signal intensities and annotation of nucleotide sequences used on the microarray. By carefully addressing each of these issues one can optimize the physiological relevance of the differential gene expression results, and gain more meaningful insights into the mechanism that underlie normal and pathological human physiology.
2. Overall microarray considerations
Among the multiple microarray designs, dual-labeled and single-labeled microarrays are currently widely available. In the current article we focus on use of Affymetrix® GeneChip® single-labeled high-density oligonucleotide microarrays. The sequence of steps for conducting microarray experiments may be described as follows: 1) Data acquisition. i.e., array hybridization and image analysis. 2) Image analysis, sometimes referred to as preprocessing, and mainly addresses background correction for each array. 3) Normalization, sometimes called preprocessing, typically algorithm-based comparative methods aimed background correction and removal of non-experimental variation within and between microarrays. 4) Quantification of differential gene expression, which is typically the statistical testing for differences in normalized signal intensities for specified genes [7, 8, 13]. The literature discussing each of these steps is vast. However, in the current article we restrict discussion to methodologies for normalizing expression signals across microarrays within an experiment and methods for detecting differential gene expression under multiple experimental conditions.
Affymetrix® GeneChip® microarrays interrogate genes with the use of several probe pairs, oligomers that are representative of sequences spread throughout the gene. In order to distinguish non-specific hybridization from probe binding, each probe pair consists of a perfect match (PM) sequence, and a mismatch (MM), which possesses one mismatched base pair located at the center of the sequence (http://www.affymetrix.com/). Hybridization signal intensity data is subject to multiple sources of variation ranging from the microarray manufacturing process to preparation of the biological sample, and can stem from factors such as differences in RNA quality or quantity, hybridization conditions, scanning efficiency and so on [14] Normalization procedures are designed to account for these technical variations in hybridization results by balancing signal intensities across experimental factors while maintaining signal intensity differences due to the conditions under investigation [15].
The Affymetrix® Microarray Suite 5.0 software (MAS 5.0; [16]) provides one of the simplest approaches to normalization [17] using trimmed means to scale each microarray to a reference array so that all microarrays have the same mean intensity [18]. However, MAS 5.0 is sub-optimal where there are large chip-to-chip differences in probe level intensity within the data set [19]. The DNA-Chip (dChip®) algorithm [20] (http://biosun1.harvard.edu/complab/dchip/) was developed from the premise that “rank invariant set” approaches [21, 22] using non-linear smooth curves could be used to normalize the summarized gene-level intensity data. The dChip® algorithms normalize each array with reference to a baseline array which has the nearest global intensity to the average of the group.
An additional normalization method, Robust multi-array analysis (RMA) [23, 24], uses probe-level quantile normalization [25] in multiple arrays. A comparison of four differentially distributed raw GeneChip® data sets using MAS 5.0, dChip® and RMA for each indicated that RMA showed more symmetrical density distribution compared to those shown by MAS 5.0 and dChip® [15].
Whereas RMA combines background adjustments and normalization using PM values only, Gene Chip RMA (GCRMA) [26] can use PM intensities only or combinations of PM-MM intensities [27], and may compensate for lost accuracy using MM-based nonspecific hybridization adjustments lost to RMA. A comparative performance analysis of five different normalizing algorithms, MAS 5.0, dChip®, PerfectMatch [28], RMA and GCRMA, to test the capacity of each algorithm to accurately model differential expression between two arrays, stated in conclusion that RMA and GCRMA most precisely modeled expression changes on the Affymetrix® GeneChip®, with the GCRMA performing better for weakly-expressed genes and RMA for strongly-expressed ones [29, 30].
Affymetrix® recommends use of the Probe Logarithmic Intensity Error (PLIER) algorithm; this is a model-based signal estimator which builds upon RMA and MAS 5.0 signal detection analysis by also including parameters that account for systematic differences in intensity between features [31, 32]. PLIER can use MM probes, but without MM data, it behaves similarly to RMA [33]. In a comparison with dChip®, MAS 5.0, Probe Profiler PCA (http://www.corimbia.com/Pages/ProbeProfiler.htm) and RMA, PLIER appeared superior to the other algorithms in avoiding false positives with poorly performing probesets [34]. Even more recently, a global rank-invariant set normalization (GRSN) post-processing tool has been proposed [35] based on the general idea of rank-invariant genes presented by Li and Wong [20] to reduce systematic distortions in microarray data produced by MAS 5.0, RMA or dChip® preprocessing.
Investigators using microarray experiments for primate studies are routinely presented with issues of sample size limitation. The literature concerning proposed methods for sample size and power analysis calculations [36] is extensive [37-41]. In terms of experimental design, if the aim of the study is to identify more than two-fold differences in expression between conditions, then experiments with three samples per condition have been considered adequate, with six samples per condition allowing for meaningful permutation tests and less conservative multiple-comparison corrections to p-values and false discovery rates. The suggested minimum for meaningful clustering is 20 samples with at least five groups [42]. A small fraction of the thousands of genes in a microarray experiment are typically differentially expressed and measured intensities among many different genes may be correlated. Thus use of nominal significance levels without multiplicity adjustments could lead to high incidences of false positive findings [43]. These types of errors are addressed using statistical approaches in which significance levels are determined based on family-wise error rate (FWER) [44] and the false discovery rate (FDR) [45].
More than 50 methodological proposals for processing Affymetrix® GeneChip® data have been published and there is debate regarding the best methods of integrating PM and MM hybridization intensities into an assembled signal for each gene [24]. BioConductor [46, 47] is a collection of open source software packages using the programming language R (http://www.r-project.org/) designed to support the analysis of biological data. BioConductor has more than 200 packages representing analytical and annotation tools for normalization and expression summary [33]. An “affycomp” package, is available as part of the BioConductor project (http://www.bioconductor.org). This web tool (http://affycomp.biostat.jhsph.edu) was made available for developers to benchmark their microarray preprocessing procedures [24]. Additional computational tools for microarray preprocessing, normalization and quantification of differential gene expression [13] are freely available and widespread. More recent studies evaluating performance of multiple combinations of preprocessing and gene ranking algorithms using Affymetrix® GeneChip® recommend additional combinations of analytical methods for enhancing detection specificity and sensitivity [48].
3. Methodology
3.1. Tissue collection (brain)
All of the animal-based studies described here were approved by the Institutional Animal Care and Use Committee at the Oregon National Primate Research Center (ONPRC). To reduce any potential effect of circadian rhythms on patterns of gene expression, tissue samples were collected during a short time-frame, during the late morning to early afternoon. The main body of the hippocampus (HPC) and amygdala (AMD) were collected by dissection and then frozen in liquid nitrogen, followed by storage at -80°C. The medial basal hypothalamus, comprising the arcuate nucleus (MBH), was removed and immersed in RNAlater (http://www.ambion.com) and stored at 4°C. The use of this RNA stabilization agent allowed additional time for further microdissection of the arcuate nucleus from the surrounding hypothalamic tissue. We note that RNAlater will result in tissue hardening, which can be advantageous for more precise dissection. However, hardening is also accompanied by tissue shrinkage, so optimization of the time of storage should be observed. In addition, it is important to note the maximum recommended storage times at various temperatures are also provided in the product literature. Once the entire bilateral extent of the MBH was dissected it was also stored at -80°C. The other brain regions were not subdivided into smaller nuclei for this initial effort, although it is recognized that enrichment by excision of homogenous areas has its advantages.
3.2. RNA extraction and characterization
Dissected brain regions were homogenized using a PowerGen rotor-stator homogenizer (Fisher Scientific, Pittsburgh, PA). Total RNA was isolated from the homogenates using RNeasy Mini kits (http://www.qiagen.com) according to manufacturer's instructions. Samples were lysed and homogenized in a denaturing buffer containing 1% β-mercaptoethanol and guanidine isothiocyanate to ensure inactivation of RNAses. The RNA was then stabilized with ethanol and bound to a silica matrix. Washes were accomplished by microcentrifugation and the purified RNA was eventually eluted with water. The concentration of RNA was measured by spectroscopy, with an expected A260/A280 ratio close to 2, denoting an acceptably pure nucleic acid sample. Qualitative assessment of the RNA was also performed using the Bioanalyzer 2100 (Agilent Technologies, http://www.agilent.com), which utilizes microfluidic technology, and so requires sparingly small sample mass. The output allows visualization of the major 18S and 28S RNA peaks, thereby providing additional information on molecular weight and critically, the degree of RNA degradation. The optimal use of microarray analysis is predicated on the use of high quality RNA.
3.3. Affymetrix® GeneChip® microarrays
Reflecting the time line in development of Affymetrix® GeneChip® microarrays, our earlier studies of circadian and seasonal gene expression in the adrenal gland (see accompanying article [12]) utilized the human HG_U133A microarray platform. In the subsequent study examining the effect of the phase of the menstrual cycle on gene expression in sub-regions of the primate brain, we utilized the Affymetrix® HG_U133 Plus 2.0 microarray platform. Wang et al. [49] evaluated the utility of human GeneChips® [50, 51] for assessing gene expression patterns in non-human primates (NHP). They aligned expressed sequence tags (EST) [52, 53] for NHP probe sequences to identify inter-species conserved (ISC) probesets, and found that ISC probesets expressed higher interspecies reproducibility than overall expressed probesets. Appropriate normalization methods could be leveraged to improve interspecies correlation, and RMA normalization was recommended over dChip® and MAS 5.0 for improving interspecies reproducibility for both expressed and ISC probesets [49].
More recently, in our hormone replacement study we [12] took advantage of the commercial release of the Affymetrix® Rhesus Macaque Genome Array. Following washes, the microarrays were assessed for gross systematic problems and the data were given a first pass assessment utilizing spike-in exogenous controls (Bacillus subtilis, non-eukaryotic RNA) built into the microarray. By utilizing the 3′/5′ ratios for the control genes ACTB and GAPDH, the relative quality (intactness) of the RNA can be assessed. Digitized images from microarray scans are used for the initial creation of the data set, which is subject to normalization and analysis (see below).
Because of our increased use of the Affymetrix® GeneChip® Rhesus Macaque Genome Array (rhesus GeneChip®), a more in-depth description of its design and continuing evolution follows. The rhesus GeneChip® was designed in close collaboration with Dr. Robert Norgren of the University Nebraska Medical Center, to increase specificity of macaque genome interrogation above levels possible using human-specific chips. The rhesus GeneChip® design [10] leverages the homology of expressed sequences represented on the GeneChip® Human Genome U133 Plus 2.0 array to annotate the Baylor School of Medicine's rhesus macaque whole-genome shotgun-assembly of the U133 GeneChip® [54], using updated probesets to assess expression of specified genes.
A layout of general probeset design assignments for the rhesus GeneChip® are categorized in Table 1. As with the design of prior GeneChips®, prefixes, suffixes and codes assigned to probesets by Affymetrix® are combined to create descriptors, which categorize probesets according to their design and according to gene clusters used to interpret prospective sequence function [55].
Table 1.
Quantity distribution and design descriptions of probe sets used to construct the GeneChip® Rhesus Macaque Genome Array.
| Total # | Design description |
|---|---|
| 2060 | Mmu.xxxx (EST/mRNA PSRs derived from genome-anchored clusters) |
| 43942 | MmugDNA.xxxx (U133-based orthologous gDNA PSRs) |
| 989 | MmunewRS.xxxx (New human RefSeq-based orthologous gDNA PSRs) |
| 4943 | MmuSTS.xxxx (Rhesus last exon STS-based PSRs) |
| 80 | MmuAffx.xxxx (EST/mRNA PSRs derived from D2 clusters) |
| 6 | MmuMitochon.xxxx (Rhesus mitochondrial-based PSRs) |
| 4 | MmurRNA.xxxx (Rhesus ribosomal RNA PSRs) |
| 59 | RPTR-Mmu-xxxx (Affymetrix reporter genes) |
| 683 | NC-00xxxx and AY588945 (Viral pathogens) |
| 13 | AFFX-Mmu-actin or gapdh or ef1a (Rhesus, 5′ M and 3′ controls) |
| 24 | AFFX-Mmu-xxxx (Affymetrix other controls) |
Information obtained from Affymetrix® NetAffx™ Technical support in November and December 2008.
Examples of specific probeset IDs are listed in the legends of Figures 1 – 3. In these IDs the descriptor construction can be explained as follows: The “Mmu” prefix refers to “Macaca mulatta”, “gDNA” to “genomic DNA” and “AFFX” typically to Affymetrix® controls. However, AFFX in the middle of the probeset ID indicates a de novo cluster. Identical numbers differing only in digits after the decimal point (e.g., 2662.1 and 2662.2) represent two sub clusters. Codes used to further refine the descriptors include sequence tagged sites (STS), short sequences traditionally detected using two PCR primers [56], and expressed sequence tags (EST) [52, 53], small segments of DNA generated by sequencing either one or both ends of an expressed gene. A “newRS” is an updated sequence used to replace an old MmugDNA based on a human U133 sequence.
Fig. 1.
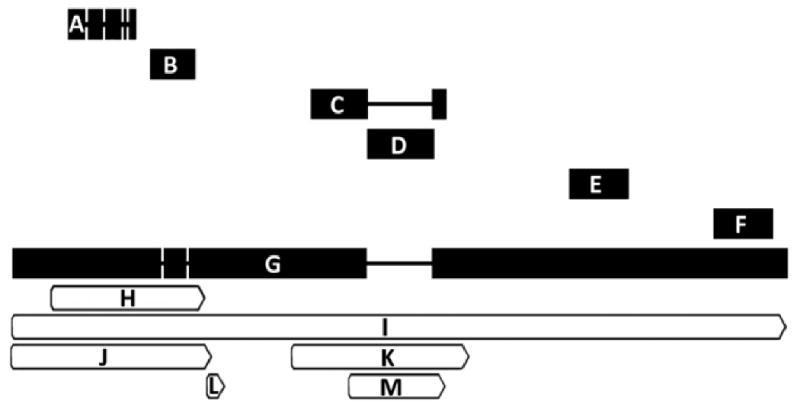
Alignment of probesets currently featured under NetAffx™ query “GABRB3”. Probesets are ordered from 5′ to 3′ with respect to the M. mulatta GABRB3 mRNA sequence listed under NCBI accession number XM_00109005 with reference annotation for the protein coding sequence (CDS) and STS sites for the GABRB3 gene. Alignments were made using Geneious software (Biomatters, Ltd) version 4.5.4 for windows. For items A-G, thick horizontal bar length is proportional to sequence length (base pairs) shown in parentheses below. Spaces connected by thin lines represent nucleotide sequence gaps. Bars for items H-M are shown relative to endpoints on the XM_001109005 sequence (G). A = MmugDNA.33250.1.S1_at (521 bp); B = MmugDNA.24108.1.S1_at (360 bp); C = MmuSTS.4813.1.S1_at (567 bp); D = MmugDNA.23997.1.S1_at (549 bp); E = MmugDNA.2520.1.S1_at (470 bp); F = MmugDNA.2522.1.S1_at (478 bp); G = XM_001109005 (5838 bp); H = GABRB3 CDS: Protein ID = “XP_001109005.1. db_xref = GI:109080423 & GeneID:711754; I = GABRB3 gene: db_xref = GeneID:711754; J = GABRB3 STS: db_xref = UniSTS:484873; K = GABRB3 STS: db_xref = UniSTS:277264; L = GABRB3 STS: db_xref = UniSTS:90465; M = GABRB3 STS: db_xref = UniSTS:49473. Note that the gap induced by MmugDNA.23997.1.S1_at (D) indicates non-alignment with the reference sequence and may explain the low expression values observed for this probeset (Table 3).
Fig. 3.
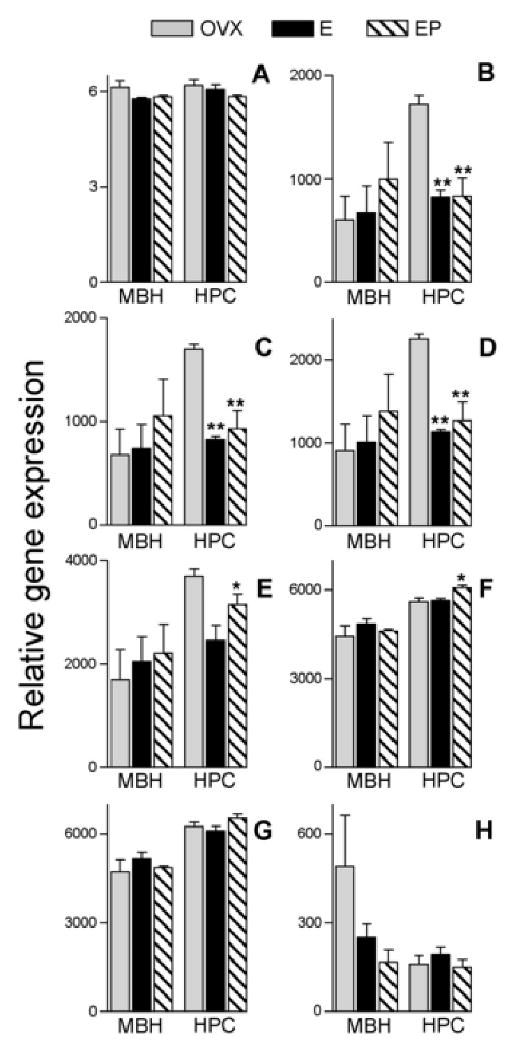
Mean and SEM of Antilogarithms to RMA-normalized rhesus macaque GeneChip® data for probesets currently featured under NetAffx™ query “ACTB”. Data are from ovariectomized females receiving hormone replacement. Probesets are ordered from 5′ to 3′ with respect to alignment on the M. mulatta β-actin mRNA sequence listed under RefSeq transcript ID NM_001033084 (see Figure 2); A = MmunewRS.18.1.S1_at; B = AFFX-Mmu-actin-5_at; C = AFFX-Mmu-actin-M_at; D = AFFX-Mmu-actin-M_x_at; E = MmunewRS.624.1.S1_s_at; F = AFFX-Mmu-actin-3_s_at; G = MmugDNA.28776.1.S1_s_at; H = MmurRNA.1.1.S1_at. MBH = arcuate nucleus of the medial basal hypothalamus, HPC = hippocampus, OVX = ovariectomized controls, E = Estrogen replacement, EP = Estrogen + Progesterone replacement. Asterisks represent significant differences in expression relative to the OVX group and show results of Newman-Keuls post hoc test following one-way ANOVA conducted on log2 transformed RMA-normalized values within each tissue. *p< 0.05, **p< 0.01.
Suffixes such as “A” and “S” represent the strandedness of the sequence derived from the cluster that is represented, “A” refers to antisense strands and “S” to sense strands relative to the orientation of the cluster. Both strands are represented in unique cases where the true orientation of the EST (gene) cannot be determined. Regarding “a”, “s” and “x” suffixes: About 10 to 20% of the probesets represent multiple sequences; “a” indicates that the sequence may be shared by members of the same gene family. In “a” cases, all 11 PM probes exactly match all the gene family member-sequences. “s” means there is uncertainty regarding whether sequence is in the same gene family, but the regions interrogated by the probes are identical to all other sequences. Lastly, “x” is a default used where there are multiple similar sequences, but probes that match all of them cannot be found. In these cases, probes can match any or all of these similar sequences. Some probes are allowed to be “polymorphic” with (usually) single base substitutions. Any probeset without an “a”, “s”, or “x” encoding is unique amongst the input sequences according to the Affymetrix® cross-hybridization model.
As a tool for examining gene expression, the rhesus GeneChip® represents a significant improvement over the use of the human GeneChip® for rhesus macaque gene expression studies [11]. Quality ratings, such as annotation grades for transcript assignments, are provided by NetAffx™ based on the perceived reliability of the source material and alignment specificity of individual probes within the probeset. Rhesus GeneChip® annotation updates are posted every four months, in July, November, and March, and can be accessed through the NetAffx™ analysis center website (http://www.affymetrix.com/analysis/index.affx). However, some probeset identifiers are more actively updated than others. For example, although overall percentages of probesets annotated with Entrez Gene, RefSeq transcript ID or Ensembl identifiers have remained between 40 - 70% over the last 2 years, the percentage of UniGene ID assignments decreased from more than 70% in early 2007 to less than 1% by November of the same year. Since then, the percentage of UniGene ID assignments Rhesus GeneChip® probesets has increased with every annotation update (Table 2). Comparatively, when the Human U133 array design was released, 36.2 % of the 44,199 probesets were EST-only probesets. Two years later, only 20.7 percent remained unassigned to a specific mRNA. In the case of mouse MOE430 array, 44.2 % of EST-only probeset assignments remain unassigned in March 2003 versus only 17 % unassigned in 2006. For the Rat 230 array, 81.5 % of the probesets were EST-based in March 2003 versus 51.1 % at the end of 2004 [55].
Table 2.
Summary of chronological NetAffx™ annotation updates to the GeneChip® Rhesus Macaque Genome Array.
| NetAffx build # | Annotation date | UniGene ID* | SwissProt ID* | Entrez Gene ID | RefSeq transcript ID | Ensembl ID |
|---|---|---|---|---|---|---|
| 22 | March 2007 | 71.9 | 52.0 | 66.6 | 65.7 | 42.8 |
| 23 | July 2007 | 2.2 | 18.4 | 56.6 | 54.4 | 39.8 |
| 24 | November 2008 | 0.6 | 12.5 | 54.9 | 54.9 | 40.5 |
| 25 | March 2008 | 2.1 | 19.9 | 54.7 | 54.6 | 40.3 |
| 26 | July 2008 | 37.4 | 13.4 | 54.6 | 54.5 | 58.6 |
| 27 | November 2008 | 37.4 | 20.6 | 52.2 | 52.2 | 40.6 |
Numbers listed under identifier IDs show the percentage of total probe sets on the Rhesus Macaque Genome Array annotated using the respective identifier. Data are listed according to the annotation date of the NetAffx™ build #. The Unigene, SwissProt, Entrez Gene, RefSeq transcript and Ensembl IDs represent annotation systems with varying degrees of redundancy.
Prospective non-redundant identifiers showing chronological changes in overall annotation assignment profiles (de-annotation and re-annotation) during a period of just less than 2 years. Annotation builds were obtained from “Current NetAffx Annotation Files” and “Archived NetAffx™ Annotation Files” sections located at: http://www.affymetrix.com/products_services/arrays/specific/rhesus_macaque.affx#1_4
Because transcript sequencing efforts are maturing rapidly, annotation analysis is a major feature of current microarray analysis. The goal of updating annotations is to ensure that probesets are associated with functional genes. The information integrated from multiple sequence, protein and cluster databases used in the public domain to associate a probeset with a given gene is provided through NetAffx™ (http://www.affymetrix.com/analysis/index.affx). To select an appropriate probeset for expression analysis, it is necessary to both validate the probeset-to-mRNA association supplied through NetAffx™, and the mRNA gene name association provided by public domain databases, such as UniGene and Entrez Gene [57]. Multiple probesets assigned to the same gene may detect cases of alternative splicing or use of alternative polyadenylation sites [58]. Although Netaffx™ annotation updates make use of new information to correct incomplete or erroneous records, interpretation of transcripts measured by probesets can be confounded by the constantly evolving mRNA sequence record in the public domain. As a result, current NetAffx™ annotations may differ significantly from the annotations assigned when the array was originally designed or from those assigned in prior annotation updates (Tables 2 and 3).
Table 3.
Relative expression of probesets for GABA-A receptor subunit β3 (GABRB3).
| Annotation Grade | Relative Gene Expression | ||||
|---|---|---|---|---|---|
| Probeset ID | Spring 2008 | Spring 2009 | MBH | HPC | AMD |
| MmuSTS.4813.1.S1_at | A | A | 2337 | 3586 | 3416 |
| MmugDNA.2520.1.S1_at | A | A | 1052 | 1293 | 1535 |
| MmugDNA.2522.1.S1_at | A | A | 958 | 1803 | 1896 |
| MmugDNA.24108.1.S1_at | A | B | 218 | 353 | 366 |
| MmugDNA.33250.1.S1_at | B | B | 60 | 65 | 79 |
| MmugDNA.23997.1.S1_at | A | A | 16 | 24 | 27 |
Relative gene expression values from three different brain regions. Each value represents the mean from twelve microarrays, and was obtained from globally scaled (MAS 5.0) analysis of probe sets on the GeneChip® Rhesus Macaque Genome Array, identified using NetAffx™ query “Gabrb3”. Changes in annotation grade reflect some changes in NetAffx™ annotation. MBH = arcuate nucleus of the medial basal hypothalamus, HPC = hippocampus, AMD = amygdala.
For example, we used Affymetrix® probeset ID numbers to search the rhesus GeneChip® for oligonucleotide signal intensities associated with genes for γ-aminobutyric acid (GABA) receptor subunits. Probeset ID assignments were referenced and searched using the Affymetrix® online Netaffx™ Analysis Center software query function at https://www.affymetrix.com/analysis/netaffx/index.affx. Using data from the gene encoding the GABAA receptor β3 subunit (GABRB3) as a descriptive example: multiple GABRB3-annotated Affymetrix® probesets align with a well described NCBI representative of the rhesus macaque GABRB3 mRNA sequence (Figure 1), but show dramatic differences in hybridization with macaque cDNA (Table 3). Such results may lead to disparate interpretations of gene expression levels; however, each of these probesets has undergone annotation revisions which are not uniform across all identifiers (Table 4). Thus without further corroboration, interpretation of the results will be dependent on annotations available at the time of analysis.
Table 4.
Chronological annotation of two GABRB3 probe sets.
| Probeset ID | NetAffx build # | date | UniGene ID | Swissprot ID | Entrez Gene ID | RefSeq transcript ID | Ensembl ID |
|---|---|---|---|---|---|---|---|
| MmugDNA.24108.1.S1_at | 22 | 3/07 | --- | Q3LUA0 | --- | --- | --- |
| 23 | 7/07 | --- | Q3LUA0 | 711754 |
XM_001108947 XM_001109005 XM_001109060 XM_001109118 |
ENSMMUG00000004314 | |
| 24 | 11/07 | --- | --- | 711754 |
XM_001108947 XM_001109005 XM_001109060 XM_001109118 |
ENSMMUG00000004314 | |
| 25 | 3/08 | --- | Q3LUA0 | 711754 |
XM_001108947 XM_001109005 XM_001109060 XM_001109118 |
ENSMMUG00000004314 | |
| 26 | 7/08 | Mmu.16343 | --- | 711754 |
XM_001108947 XM_001109005 XM_001109060 XM_001109118 |
ENSMMUG00000004314 | |
| 27 | 11/08 | --- | --- | --- | --- | ENSMMUG00000004314 | |
| MmugDNA.24108.1.S1_at | 22 | 3/07 | Hs.302352 |
P28472 Q3LUA0 Q68CS5 |
2562 |
NM_021912 NM_000814 |
ENSG00000166206 |
| 23 | 7/07 | --- | Q3LUA0 | 711754 |
XM_001108947 XM_001109005 XM_001109060 XM_001109118 |
ENSMMUG00000004314 | |
| 24 | 11/07 | --- | --- | 711754 |
XM_001108947 XM_001109005 XM_001109060 XM_001109118 |
ENSMMUG00000004314 | |
| 25 | 3/08 | --- | Q3LUA0 | 711754 |
XM_001108947 XM_001109005 XM_001109060 XM_001109118 |
ENSMMUG00000004314 | |
| 26 | 7/08 | Mmu.16343 | --- | 711754 |
XM_001108947 XM_001109005 XM_001109060 XM_001109118 |
ENSMMUG00000004314 | |
| 27 | 11/08 | Mmu.7293 | --- | 707956 | XR_011809 | ENSMMUG00000004314 |
The table shows annotation assignments according to identifier. Data are arranged chronologically by NetAffx™ build #, and demonstrate differences in identifier redundancy. Probe sets were identified using NetAffx™ query “Gabrb3”. Data obtained from “Current NetAffx Annotation Files” and “Archived NetAffx Annotation Files” sections located at: http://www.affymetrix.com/products_services/arrays/specific/rhesus_macaque.affx#1_4
Rather than using microarrays strictly for gene discovery, we have been using gene-by-gene interrogation to screen for potential genes to be used as normalizers in PCR applications. There is mounting consensus that the expression of normalizers labeled as “housekeeping” or “structural” genes vary across experimental conditions, and that lack of regulation should be verified before use [59]. Here we illustrate considerations for this selection process using well-annotated β-actin probesets from the rhesus GeneChip®.
Each of our experimental replicates is represented by one microarray. Using single or multiple microarrays, analysis using Affymetrix® Microarray Analysis Suite (MAS 5.0) can be used to determine background expression levels. We used MAS 5.0 globally scaled expression values to provide conservative means of assessing whether a target sequence is expressed (above background) or not expressed (below background) [60], under the experimental conditions represented by the hybridized material. Once we determined that a target sequence is expressed (MAS 5.0 analysis), we used RMA normalization before looking for expression differences according to experimental condition. In effect, we partitioned the strengths of the two algorithms, using the PM - MM algorithms of MAS 5.0 to screen out genes likely to be non-expressing, and using RMA normalization to account for differences in chip quality or measurement conditions.
To illustrate the process of screening through variations in expression profiling results related to annotation technicalities, we discuss data from our hormone replacement study using the extremely well characterized β-actin (ACTB) gene which includes Affymetrix® 5′ and 3′ control assignments [61]. Seven of the eight probesets retrieved under a February 2009 NetAffx™ rhesus GeneChip® query for ACTB align with sequence data in the public domain such as the M. mulatta β-actin mRNA sequence listed under RefSeq transcript ID NM_001033084 (Figure 2). Despite this alignment, care must be taken regarding which probesets are included in analysis. For example, using RMA analysis only, one may consider the probeset “MmunewRS.18.1.S1_at” (Figure 3A) to show no variation in expression according to tissue or treatment, thus indicating that β-actin may be a suitable gene for normalization during the PCR corroboration process. Note however, that the relative expression values for this probeset are low compared to expression values of other probesets (Figure 3B-H). In fact, MAS 5.0 analysis (data not shown) indicates that the target sequence for this MmunewRS.18.1.S1_at is not expressed under any of the experimental conditions used in the study (all samples “absent”). Therefore, sequences represented by this probeset would be unsuitable for use in corroboration.
Fig. 2.
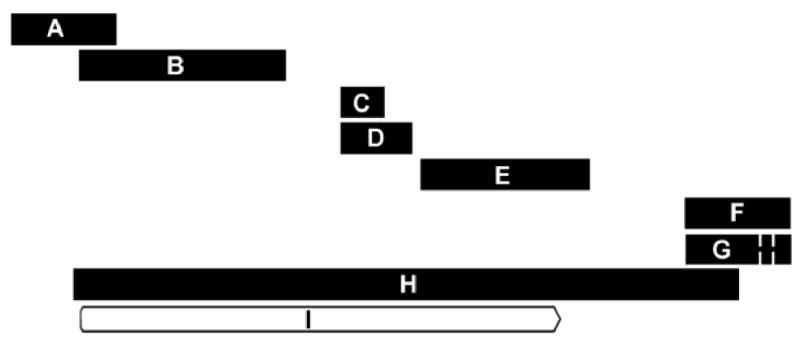
Alignment of probesets currently featured under NetAffx™ query “ACTB”. Probesets are ordered from 5′ to 3′ with respect to the M. mulatta β-actin (ACTB) mRNA sequence listed under RefSeq transcript ID NM_001033084 and the reference annotation for the protein coding sequence (CDS) of the β-actin gene. Alignments were made using Geneious software (Biomatters, Ltd) version 4.5.4 for Windows. Thick horizontal bar length is proportional to sequence length (base pairs) shown in parentheses below. Spaces connected by thin lines represent nucleotide sequence gaps. A = MmunewRS.18.1.S1_at (246 bp); B = AFFX-Mmu-actin-5_at (483 bp); C = AFFX-Mmu-actin-M_at (100 bp); D = AFFX-Mmu-actin-M_x_at (169 bp); E = MmunewRS.624.1.S1_s_at (403 bp); F = AFFX-Mmu-actin-3_s_at (247 bp); G = MmugDNA.28776.1.S1_s_at (245 bp); H = NM_001033084 (1584 bp); I = ACTB CDS: Protein ID= “NP_001028256.1”, db_xref = GI:74316002 &“GeneID:574285.
Potential differences in tissue expression, as well as treatment-related changes in β-actin expression may be discerned from the remaining probesets. Differences in the HPC expression appear more pronounced in the probesets interrogating 5′ portions of the ACTB reference sequence (Figures 2 and 3). Although the “MmurRNA.1.1.S1_at” probeset in Figure 3H is an A-grade annotation [55], it appears to interrogate a portion of the rhesus genome outside the realm covered by the other seven probesets (Figure 2). Here we show an example of both tissue and treatment-related differential regulation of a gene commonly used for corroborative normalization, highlighting the importance of evaluating probeset-specific expression data regarding conditions of use.
4. Interpretation of data
4.1. Identification of regulated genes
To identify regulated genes, we have used the GeneSifter Analysis package (http://www.genesifter.net/), a web-based service, which allows RMA normalization and subsequent analysis for treatment effects. Currently, the software package can perform pairwise, multi-group analysis (1-way and 2-way ANOVA, balanced design only) and cluster analysis. We employed a balanced design with the same number (four) of individual samples in each of three treatments (three phases of menstrual cycle or hormone replacement) and using three brain regions (MBH, HPC, AMD). To analyze the effect of treatment by brain region, we opted for a 2-way analysis of variance. We varied the fold-change threshold depending upon the number of genes of interest we sought to explore, but usually set the significance level at p < 0.05. Initial attempts with MAS 5.0 usually produced many more candidate genes than RMA, as previously observed [62]. Moreover, we also confirm the observation that Bonferroni adjustments for false discovery rate, is very conservative and hence used the Benjamini-Hochberg technique for false discovery correction [45, 63] as incorporated in the GeneSifter software package. Similar to other analysis packages, the GeneSifter program also has additional tools for the further organization of significantly regulated genes. This includes organization by gene ontology, KEGG and principal components analyses.
4.2. Corroboration of gene expression using quantitative real-time RT-PCR
Quantitative real-time RT-PCR (qPCR) [64, 65] is a popular method for independently validating gene expression data for the most important genes in a microarray study [66-68]. In cases where microarray annotation is suspect, we perform reverse transcriptase PCR on cDNA generated from rhesus macaque RNA and sequence the resulting PCR products to obtain macaque-specific mRNA sequences. These sequences are then used to design primers and TaqMan® probes for the real-time assay, using Primer Express® software (Applied Biosystems, Foster City, CA). Quantitation of mRNA expression is then analyzed using a TaqMan® PCR Core Reagent Kit reagents with the 7900HT Fast Real-Time PCR thermal cycler and Sequence Detection Systems software version 2.2.1 (Applied Biosystems) with thermal cycler conditions of 2 min at 50°C, 10 min at 95°C and 40 cycles of 15s at 95°C (DNA melting) and 1 min at 60°C (primer annealing/extension).
Although qPCR is a commonly accepted method for quantifying gene expression results, the gene expression data obtained need to be normalized against a stably-expressed gene for accurate interpretation [69]. Many of the genes previously adopted as standards for normalization of gene expression data, have recently been shown to vary widely depending on experimental conditions [70, 71]. Therefore, it is imperative that the stability of prospective reference genes is verified under each of the specific experimental conditions under study, and only the most appropriate ones selected for use as normalizers [72-74]. Again we emphasize the importance of organismal biology considerations when evaluating use of internal reference genes for normalization. For example Lemos et al. (supplemental table [75]) found the commonly used internal reference gene GAPDH to show rhythmic circadian expression variation in rhesus macaques. The current trend is to select the most stably expressed reference gene from groups of candidate genes [76, 77]. However, we have found simultaneous use of multiple internal reference genes to be effective when comparing brain regions differentially affected by multiple hormone treatments (Noriega et al. submitted[78]) and we anticipate that normalization using multiple reference genes may become more common in the near future.
Three approaches for assessing gene expression stability using qPCR data are currently widely used and are available in separate software algorithm bundles named geNorm, NormFinder and BestKeeper. “geNorm” [79] calculates gene stability values using average pairwise variation between all genes under consideration. The least stable gene is then removed, new pairwise variation comparisons are made and the next least stable gene is removed. This sequential elimination process is re-iterated until the most stable gene or combination of genes is identified [80]. “NormFinder” [81] was developed using a model-based estimation approach for expression variation proposed to allow more robust and precise measures of gene expression stability than the pairwise comparison approach [82]. We have found this useful in that it allows the user to account for experimental groupings, as well as facilitating evaluations of intra- and inter-group expression variation for each gene. The BestKeeper [83] algorithm generates descriptive statistics of candidate normalizers based on their threshold crossing point [84] values obtained from qPCR analysis. BestKeeper computes descriptive statistics allowing the user to rank expression stability according to standard deviation or coefficient of variation based on crossing point [85].
Once appropriate internal reference genes are identified, their expression data can be used to normalize overall expression data in the experiment. Additional software for qPCR normalization, such as “qpcrNorm” (http://cgi.uc.edu/cgi-bin/kzhang/QpcrNorm.cgi/), is also integrated in packages available from the BioConductor project (http://www.bioconductor.org/).
4.3. Corroboration of gene expression using semi-quantitative RT-PCR
Although use is declining and it is not as quantitative as qPCR, traditional semi-quantitative RT-PCR using digital image analysis of agarose gels [86] can often be used as an inexpensive and practical method of corroborating gene array findings. The advantage of this procedure is that it does not require specialized equipment, other than that found in a typical molecular biology laboratory, and does not depend on costly supplies (e.g., labeled real-time probes). However, the RT-PCR requires use of electrophoresis gels, and construction of dilution curves to ensure that the sample image intensity falls within the linear limits of the gel image intensity profiles.
Whenever possible we design primers against targeted sequences common to the rhesus macaque and human transcripts. The macaque sequences can be obtained from the Human Genome Sequencing Center at Baylor College of Medicine (http://www.hgsc.bcm.tmc.edu/projects/rmacaque/). Resulting PCR products are then resolved by electrophoresis on 2% agarose gels with ethidium bromide and photographed under ultraviolet light. Gel bands are analyzed using NIH Image-J software 1.37v (Bethesda, MD, http://rsb.info.nih.gov/nih-image).
4.4 Translation corroboration techniques
Protein measures can be used to assess whether a change in gene expression truly translates to regulation of downstream protein levels. In the past, our laboratory has relied upon in situ hybridization histochemistry (ISH) and immunocytochemistry (ICC) to explore relationships between mRNA expression and protein expression respectively in the primate brain [87-89]. These techniques offer the additional advantage of addressing correlations between gene and protein expression changes in specific anatomically intact areas [90]. Western blotting for the measure of specific proteins levels may be used with samples from nonhuman primates [91].
5. Closing remarks
Gene profiling in the rhesus macaque represent a powerful new approach in our quests for gaining insights into the genetic causes of normal and abnormal human physiology and behavior. However, there are potential pitfalls with this new technology, which can affect validity of the results. These can be largely overcome by careful consideration of appropriate experimental design [12], together with appropriate data analysis.
Acknowledgments
This work was supported by National Institutes of Health grants: AG029612, HD029186 and RR000163. All of the gene microarray assays were performed in the Affymetrix® Microarray Core of the Gene Microarray Shared Resource at OHSU.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kendrew J, editor. The Encyclopedia of Molecular Biology. Blackwell Science Ltd; 1994. 1994. [Google Scholar]
- 2.Raychaudhuri S, Sutphin PD, Chang JT, Altman RB. Trends Biotechnol. 2001;19:189–193. doi: 10.1016/s0167-7799(01)01599-2. [DOI] [PubMed] [Google Scholar]
- 3.Schena M, Shalon D, Davis RW, Brown PO. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 4.DeRisi J, Penland L, Brown PO, Bittner ML, Meltzer PS, Ray M, Chen Y, Su YA, Trent JM. Nat Genet. 1996;14:457–460. doi: 10.1038/ng1296-457. [DOI] [PubMed] [Google Scholar]
- 5.Brown PO, Botstein D. Nat Genet. 1999;21:33–37. doi: 10.1038/4462. [DOI] [PubMed] [Google Scholar]
- 6.Phimister B. Nat Genet. 1999;21 doi: 10.1038/11880. [DOI] [PubMed] [Google Scholar]
- 7.Ehrenreich A. Appl Microbiol Biotechnol. 2006;73:255–273. doi: 10.1007/s00253-006-0584-2. [DOI] [PubMed] [Google Scholar]
- 8.Lee NH, Saeed AI, Hilario E, Mackay J, editors. Protocols for Nucleic Acid Analysis by Nonradioactive Probes. Vol. 353. Humana Press Inc; Totowa, NJ: 2007. pp. 265–296. [Google Scholar]
- 9.Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Horton H, Brown EL. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- 10.Affymetrix GeneChip® Rhesus Macaque Genome Array Data Sheet. 2005 www.affymetrix.com/support/technical/datasheets/rhesus_datasheet.pdf.
- 11.Duan F, Spindel ER, Li YH, Norgren RB., Jr BMC Genomics. 2007;8:61. doi: 10.1186/1471-2164-8-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urbanski HF, Noriega NC, Lemos DR, Kohama SG. Methods. doi: 10.1016/j.ymeth.2009.05.009. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinhoff C, Vingron M. Brief Bioinform. 2006;7:166–177. doi: 10.1093/bib/bbl002. [DOI] [PubMed] [Google Scholar]
- 14.Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP. Nucleic Acids Res. 2002;30:e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Do JH, Choi DK. Mol Cells. 2006;22:254–261. [PubMed] [Google Scholar]
- 16.Hubbell E, Liu WM, Mei R. Bioinformatics. 2002;18:1585–1592. doi: 10.1093/bioinformatics/18.12.1585. [DOI] [PubMed] [Google Scholar]
- 17.Bolstad BM, Collin F, Simpson KM, Irizarry RA, Speed TP. Int Rev Neurobiol. 2004;60:25–58. doi: 10.1016/S0074-7742(04)60002-X. [DOI] [PubMed] [Google Scholar]
- 18.Affymetrix. Affymetrix Microarray Suite Users Guide, Version 5. 2001. [Google Scholar]
- 19.Zhang Q, Ushijima R, Kawai T, Tanaka H. Chem-Bio Informatics Journal. 2004;4:56–72. [Google Scholar]
- 20.Li C, Wong WH. Proc Natl Acad Sci U S A. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schadt EE, Li C, Su C, Wong WH. J Cell Biochem. 2000;80:192–202. [PubMed] [Google Scholar]
- 22.Schadt EE, Li C, Ellis B, Wong WH. J Cell Biochem Suppl. 2001 37:120–125. doi: 10.1002/jcb.10073. [DOI] [PubMed] [Google Scholar]
- 23.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 24.Irizarry RA, Wu Z, Jaffee HA. Bioinformatics. 2006;22:789–794. doi: 10.1093/bioinformatics/btk046. [DOI] [PubMed] [Google Scholar]
- 25.Bolstad BM, Irizarry RA, Astrand M, Speed TP. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 26.Wu Z, Irizarry RA. Nat Biotechnol. 2004;22:656–658. doi: 10.1038/nbt0604-656b. author reply 658. [DOI] [PubMed] [Google Scholar]
- 27.Liu WM, Li R, Sun JZ, Wang J, Tsai J, Wen W, Kohlmann A, Williams PM. J Theor Biol. 2006;243:273–278. doi: 10.1016/j.jtbi.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 28.Zhang L, Miles MF, Aldape KD. Nat Biotechnol. 2003;21:818–821. doi: 10.1038/nbt836. [DOI] [PubMed] [Google Scholar]
- 29.Zhijin W, Irizarry RA. Nat Biotechnol. 2004;6:656–658. doi: 10.1038/nbt0604-656b. [DOI] [PubMed] [Google Scholar]
- 30.Zhijin W, Irizarry R, Gentleman RC, Martinez-Murillo F, Spencer F. J Am Stat Assoc. 2004;99:909–917. [Google Scholar]
- 31.Affymetrix. Gene signal estimates from exon arrays. 2005. [Google Scholar]
- 32.Affymetrix. Technical note: Guide to Probe Logarithmic Intensity Error (PLIER) Estimation. 2005 http://www.affymetrix.com/support/technical/technotes/plier_technote.pdf.
- 33.Okoniewski MJ, Miller CJ. PLoS Comput Biol. 2008;4:e6. doi: 10.1371/journal.pcbi.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seo J, Hoffman EP. BMC Bioinformatics. 2006;7:395. doi: 10.1186/1471-2105-7-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pelz CR, Kulesz-Martin M, Bagby G, Sears RC. BMC Bioinformatics. 2008;9:520. doi: 10.1186/1471-2105-9-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lerman J. Can J Anaesth. 1996;43:184–191. doi: 10.1007/BF03011261. [DOI] [PubMed] [Google Scholar]
- 37.Lee ML, Whitmore GA. Stat Med. 2002;21:3543–3570. doi: 10.1002/sim.1335. [DOI] [PubMed] [Google Scholar]
- 38.Zien A, Fluck J, Zimmer R, Lengauer T. J Comput Biol. 2003;10:653–667. doi: 10.1089/10665270360688246. [DOI] [PubMed] [Google Scholar]
- 39.Gadbury GL, Page GP, Edwards J, Kayo T, Prolla TA, Weindruch R, Permana PA, Mountz JD, Allison DB. Stat Methods Med Res. 2004;13:325–338. [Google Scholar]
- 40.Muller P, Parmigiani G, Robert C, Rousseau J. J Am Stat Assoc. 2004;99:990–1001. [Google Scholar]
- 41.Wang SJ, Chen JJ. J Comput Biol. 2004;11:714–726. doi: 10.1089/cmb.2004.11.714. [DOI] [PubMed] [Google Scholar]
- 42.Reimers M. Addict Biol. 2005;10:23–35. doi: 10.1080/13556210412331327795. [DOI] [PubMed] [Google Scholar]
- 43.Tsai CA, Wang SJ, Chen DT, Chen JJ. Bioinformatics. 2005;21:1502–1508. doi: 10.1093/bioinformatics/bti162. [DOI] [PubMed] [Google Scholar]
- 44.Westfall PH, Young SS. Resampling-based multiple testing: Examples and methods for p-value adjustment. John Wiley & Sons; 1993. [Google Scholar]
- 45.Benjamini Y, Hochberg Y. Journal of the Royal Statistical Society B. 1995;57:289–300. [Google Scholar]
- 46.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.BioConductor. http://www.bioconductor.org.
- 48.Kadota K, Nakai Y, Shimizu K. Algorithms Mol Biol. 2009;4:7. doi: 10.1186/1748-7188-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Z, Lewis MG, Nau ME, Arnold A, Vahey MT. BMC Bioinformatics. 2004;5:165. doi: 10.1186/1471-2105-5-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Affymetrix Technical Note: Design and Performance of the GeneChip® Human Genome U133 Plus 2.0 and Human Genome U133A 2.0, Arrays. 2003 http://www.affymetrix.com/support/technical/technotes/hgu133_p2_technote.pdf.
- 51.Affymetrix GeneChip® Human Genome Arrays. 2004 http://www.affymetrix.com/support/technical/datasheets/human_datasheet.pdf.
- 52.Adams MD, Kelley JM, Gocayne JD, Dubnick M, Polymeropoulos MH, Xiao H, Merril CR, Wu A, Olde B, Moreno RF, et al. Science. 1991;252:1651–1656. doi: 10.1126/science.2047873. [DOI] [PubMed] [Google Scholar]
- 53.Boguski MS, Lowe TM, Tolstoshev CM. Nat Genet. 1993;4:332–333. doi: 10.1038/ng0893-332. [DOI] [PubMed] [Google Scholar]
- 54.Affymetrix Array Design for the GeneChip® Human Genome U133 Set. http://www.affymetrix.com/support/technical/technotes/hgu133_design_technote.pdf.
- 55.Affymetrix Transcript Assignment for NetAffx™ Annotations Revision 2.3. 2006 www.affymetrix.com/support/technical/whitepapers/Transcript_Assignment_whitepaper.pdf.
- 56.Green ED, Green P. PCR Methods Appl. 1991;1:77–90. doi: 10.1101/gr.1.2.77. [DOI] [PubMed] [Google Scholar]
- 57.Affymetrix Sleuthing With the Affymetrix NetAffx™ Website: Identifying and Examining Probe Sets and Their Genomic Context. http://www.affymetrix.com/support/technical/whitepapers/Sleuthing_NetAffx_whitepaper.pdf.
- 58.Stalteri MA, Harrison AP. BMC Bioinformatics. 2007;8:13. doi: 10.1186/1471-2105-8-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ahn K, Huh JW, Park SJ, Kim DS, Ha HS, Kim YJ, Lee JR, Chang KT, Kim HS. BMC Mol Biol. 2008;9:78. doi: 10.1186/1471-2199-9-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu WM, Mei R, Di X, Ryder TB, Hubbell E, Dee S, Webster TA, Harrington CA, Ho MH, Baid J, Smeekens SP. Bioinformatics. 2002;18:1593–1599. doi: 10.1093/bioinformatics/18.12.1593. [DOI] [PubMed] [Google Scholar]
- 61.Affymetrix Performance and Validation of the GeneChip® Human Genome U133 Set (Technical Note) http://www.affymetrix.com/support/technical/technotes/hgu133_performance_technote.pdf.
- 62.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Benjamini Y, Hochberg Y. Journal of Educational and Behavioral Statistics. 2000;25:60–83. [Google Scholar]
- 64.Gibson UE, Heid CA, Williams PM. Genome Res. 1996;6:995–1001. doi: 10.1101/gr.6.10.995. [DOI] [PubMed] [Google Scholar]
- 65.Heid CA, Stevens J, Livak KJ, Williams PM. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- 66.Mutch DM, Berger A, Mansourian R, Rytz A, Roberts MA. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-12-preprint0009. preprint0009.0001-0009.0029. [DOI] [PubMed] [Google Scholar]
- 67.Mutch DM, Berger A, Mansourian R, Rytz A, Roberts MA. BMC Bioinformatics. 2002;3:17. doi: 10.1186/1471-2105-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wurmbach E, Yuen T, Sealfon SC. Methods. 2003;31:306–316. doi: 10.1016/s1046-2023(03)00161-0. [DOI] [PubMed] [Google Scholar]
- 69.Bustin SA, Nolan T. J Biomol Tech. 2004;15:155–166. [PMC free article] [PubMed] [Google Scholar]
- 70.Zhong H, Simons JW. Biochem Biophys Res Commun. 1999;259:523–526. doi: 10.1006/bbrc.1999.0815. [DOI] [PubMed] [Google Scholar]
- 71.Glare EM, Divjak M, Bailey MJ, Walters EH. Thorax. 2002;57:765–770. doi: 10.1136/thorax.57.9.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dheda K, Huggett JF, Bustin SA, Johnson MA, Rook G, Zumla A. Biotechniques. 2004;37:112–114. 116, 118–119. doi: 10.2144/04371RR03. [DOI] [PubMed] [Google Scholar]
- 73.Dheda K, Huggett JF, Chang JS, Kim LU, Bustin SA, Johnson MA, Rook GA, Zumla A. Anal Biochem. 2005;344:141–143. doi: 10.1016/j.ab.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 74.Sellars MJ, Vuocolo T, Leeton LA, Coman GJ, Degnan BM, Preston NP. J Biotechnol. 2007;129:391–399. doi: 10.1016/j.jbiotec.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 75.Lemos DR, Downs JL, Urbanski HF. Mol Endocrinol. 2006;20:1164–1176. doi: 10.1210/me.2005-0361. [DOI] [PubMed] [Google Scholar]
- 76.Allen D, Winters E, Kenna PF, Humphries P, Farrar GJ. J Dermatol Sci. 2008;49:217–225. doi: 10.1016/j.jdermsci.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 77.Barsalobres-Cavallari CF, Severino FE, Maluf MP, Maia IG. BMC Mol Biol. 2009;10:1. doi: 10.1186/1471-2199-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Noriega NC, Kryger SG, Eghlidi DH, Garyfallou VT, Kohama SG, Urbanski HF. Brain Res. doi: 10.1016/j.brainres.2009.10.011. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.geNorm Software http://medgen.ugent.be/∼jvdesomp/genorm/
- 80.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. research0034.0031-0034.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.NormFinder Software http://www.mdl.dk/publicationsnormfinder.htm
- 82.Andersen CL, Jensen JL, Orntoft TF. Cancer Res. 2004;64:5245–5250. doi: 10.1158/0008-5472.CAN-04-0496. [DOI] [PubMed] [Google Scholar]
- 83.BestKeeper software http://gene-quantification.com/bestkeeper.html
- 84.Rasmussen R, editor. Rapid Cycle Real-Time PCR: Methods and Applications. Springer-Verlag Press; Heidelberg: 2001. pp. 21–34. [Google Scholar]
- 85.Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Biotechnol Lett. 2004;26:509–515. doi: 10.1023/b:bile.0000019559.84305.47. [DOI] [PubMed] [Google Scholar]
- 86.Marone M, Mozzetti S, De Ritis D, Pierelli L, Scambia G. Biol Proced Online. 2001;3:19–25. doi: 10.1251/bpo20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bethea CL, Brown NA, Kohama SG. Endocrinology. 1996;137:4372–4383. doi: 10.1210/endo.137.10.8828498. [DOI] [PubMed] [Google Scholar]
- 88.Garyfallou VT, Kohama SG, Urbanski HF. Brain Res. 1996;716:22–28. doi: 10.1016/0006-8993(95)01545-0. [DOI] [PubMed] [Google Scholar]
- 89.Kohama SG, Garyfallou VT, Urbanski HF. Brain Res Mol Brain Res. 1998;53:328–332. doi: 10.1016/s0169-328x(97)00282-9. [DOI] [PubMed] [Google Scholar]
- 90.Rowe WB, Blalock EM, Chen KC, Kadish I, Wang D, Barrett JE, Thibault O, Porter NM, Rose GM, Landfield PW. J Neurosci. 2007;27:3098–3110. doi: 10.1523/JNEUROSCI.4163-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Duce JA, Podvin S, Hollander W, Kipling D, Rosene DL, Abraham CR. Glia. 2008;56:106–117. doi: 10.1002/glia.20593. [DOI] [PubMed] [Google Scholar]