

Abstract
One of the most significant obstacles for systematic delivery of nano-payloads is the foreign particle clearance by the mononuclear phagocyte system (MPS). The majority of biocompatible nano-payloads with charged groups on their surface cannot fully evade the clearance by MPS during systemic circulation. For safe and effective targeted nano-drug delivery in vivo, we describe a novel approach for evading the macrophage clearance. We demonstrate that neutral and hydrophilic materials can effectively evade the macrophage uptake and also quickly degrade into bioeliminable fragments. We show that there is no opsonization effect and no toxic effect on living cells. In addition, the payloads are stable in an aqueous environment and they can release drugs in a cellular environment. These results suggest that the unique properties of this kind of payloads may make them useful for designing new drug delivery systems.
The development of payloads for drugs (small molecules, proteins, DNAs and RNAs) has been significantly impacted by the use of nanometer-scale materials. 1-5 It is envisioned that nano-sscale payloads can improve the stability and solubility of hydrophobic drugs in physiological environments; can guide payloads to specific sites in organs or cells by tagging with bio-recognition markers; alter protein binding affinity and drug bio-distribution in the body by surface modification; change drug pharmacokinetics and pharmaco-dynamics; and decrease immunogenicity.
Despite these clear advantages, nanometer sized materials are targets for removal from the circulation by the mononuclear phagocyte system (MPS). The rate and extent of removal is determined by the physico-chemical properties of the materials used. 6 Large, hydrophobic or charged materials are rapidly cleared from systemic circulation by the MPS and are sequestered in the liver, the kidney or the spleen. Thus the design of biocompatible nanoparticle matrices is a critical step in the optimization of nanoplatform (NP)-based drug delivery.
The most commonly used synthetic degradable polymers are hydrophobic materials. They find widespread use as biodegradable materials for tissue implant and implant drug release. Nevertheless, their inherent hydrophobicity limits their utility as flexible intravenous drug delivery systems. 7-13 First, the bulk hydrophobic moieties of the polymers slow the enzymatic degradation, prolong the time needed to achieve effective local concentrations of the drug/pro-drug and significantly narrows the optimal therapeutic window. 7-10 Persistence of the nanoplatform in the body enhances the probability of coincidental and unintentional adverse effects. Besides, the long-time degradation of these hydrophobic polymers dramatically alters local pH, physiology / pathophysiology and may lead to unwanted degradation of the drug within payloads. 7,12,13 Second, The MPS clearance has to be taken into account during payload design. Post-synthetic modification of these hydrophobic nanoparticles with hydrophilic moieties that makes the nanoparticle soluble in physiological environments may cause MPS clearance.11 It has been observed that hydrophobic quantum dots coated with charged ligands and PolyEthyleneGlycols (PEG) could not completely avoid capture by macrophages. 14 The large fraction of the nanoparticles that accumulate at the liver indicates that the surface charge is a dominating factor in the clearance, contravening modification with PEGs and control of nanoparticle size (30nm). All of the above considerations suggest that neutral, hydrophilic polymers could be optimal payload materials for drug delivery.
Evidence exists for the use of neutral, hydrophilic matrices that reduce protein opsonization in the bloodstream and eliminate initiation of immunogenic responses, thereby increasing circulation life times and minimizing premature clearance by macrophages. 15, 16 Embedding the active constituents in such a biocompatible matrix separates the drugs from the biological environment, further reducing coincidental interactions that may stimulate adverse MPS responses. In addition to immunological considerations, the size of the nanoplatform dictates its ability to readily pass through the renal glomerula,, avoiding entrapment in the circulation and kidney. 6
It has been shown that polyacrylamide-based polymers provide an alternative for modifying the hydrophobicity of drugs. 15, 16 Neutral hydrophilic homo-polymerized N-(2-hydroxypropyl) methacrylamide (HPMA) has been used successfully as a co-polymer in a variety of clinical trials. This hydrophilic alternative imparts higher molecular weight (extended half-life in the circulation) and has improved a variety of therapeutic measures. However, as currently used, HMPA is simply a tag that enhances the half-life of the drug/pro-drug by hindering renal elimination mechanisms. This simple ‘linker’ approach does not provide separation of the drug from the physiological environment or provide any of the attendant benefits that come with embedding of the therapeutic moiety within a polymer matrix. Moreover, HPMA-linked hydrophobic drugs tend to accumulate in the liver despite being a potential candidate for renal clearance. 17 Many of these complications are obviated by encapsulation in a hydrophilic matrix. 18 Previously published data support the idea that potentially toxic materials may be successfully encapsulated in polyacrylamide-based hydrogels while retaining their therapeutic mode of action such as production of singlet oxygen for the destruction of cancer cells. 19-24 However, conventional polyacrylamide hydrogels do not degrade in biological environments and tend to persist in the body. While the free radical co-polymerization of acrylamide with degradable cross-linkers can produce partially degradable hydrogels, the main chains of the polymer backbone do not readily dissociate in solution and thus the molecular weight of the degraded nanoparticle is maintained in the range of small to medium-sized proteins, i.e., thousands of kD. These degraded nanoparticles are robust and are not readily excreted. Precise control of the polymer to optimize delivery of the drug to the target while providing for degradation and renal or fecal elimination would minimize toxic potential and enhance the therapeutic usefulness of the platform. The molecular design presented here for new degradable polyacrylamide-based nanoparticles combines both biodegradable and biocompatible characteristics into novel hydrogels that specifically avoid uptake by macrophages and have a greater potential for excretion from the body (Scheme 1). Therefore, our proposed payload design criteria are: (i) neutral and hydrophilic polymers; (ii) nano-platforms that are degradable through chemical or enzyme hydrolysis; (iii) non-toxic hydrogels with high potential for bio-elimination; (iv) a small size, i.e. less than 30 nm, to avoid capture by macrophages; (v) a matrix that facilitates incorporation of drugs; (vi) in situ synthesis without further modification; and (vii) ready, tunable release of drug from the matrix.
Scheme 1.

The design of bioeliminable nano-hydrogels for drug delivery
Replacing the routine free radical polymerization approach with that of chain transfer polymerization enables “living control” of the polyacrylamide chain length. 25, 26 This is particularly advantageous in view of the short chain polymer’s good potential for bio-elimination from the body. Another attractive feature of polyacrylamide-based hydrogels is that acrylamide monomers can copolymerize with degradable cross linkers in situ to form degradable nanoparticles in a single step. The size of the polyacrylamide-based nanoparticle can also be easily modulated by this synthetic approach. Furthermore, polyacrylamide can easily copolymerize with other functional monomers to introduce targeting tags for guided drug delivery. All these considerations suggest that the designed degradable polyacrylamide-based hydrogels would be a novel class of safe payloads for in vitro study and eventually for in vivo use. The nanoparticles described here take advantage of short polyacrylamide chains (<20KDa) that are assembled into nanohydrogels by degradable cross linkers. The degradable nanoparticles enable several distinct features compared to traditional ones: 1. The hydrophilicity, size and neutral surface significantly reduce capture by macrophages. 2. The degraded products are sufficiently small so that they may be eliminated from cells and hence tissues, organs, etc. 3. Various drugs can be easily encapsulated within the inert matrix and thus one can avoid their chemical toxicity to normal cells and the triggering of MPS responses, when they are introduced into a biological environment. 4. These matrices degrade easily and release drugs upon entering The targeted cells.
The synthesis of the short chain polymer and its degradable hydrogels are described in detail in the experimental section. Briefly, 3-mercapto-1-propanol mediated chain transfer polymerization of acrylamide monomers was optimized first in aqueous solution in order to assure the synthetic process. The results indicated that the monomer to chain transfer reagent optimization ratio is 250:1. The obtained polyacrylamide molecular weight was 15KDa, monitored by gel permeation chromatography (GPC) (Figure 1S), which is a reasonable size for the polyacrylamide that could be eliminated from cells.7 By varying the monomer to chain transfer reagent ratio, the polyacrylamide chain can be precisely controlled between 8 KDa and 800 KDa. Based on the above results, acrylamide monomers copolymerize with degradable glycerol dimethacrylate cross linkers to assemble the degradable hydrogels in a hexane/water microemulsion system. The apparent molecular weight of the neutral and hydrophilic NPs is 1.4 MDa as measured by GPC (Figure 1), and they are approximately 20 nm in diameter as measured by scanning electron microscopy (SEM) (Figure 2S). These nanoparticles were incubated in PBS buffer to evaluate their stability. It is interesting to note that the degradable NPs were very stable in neutral aqueous solution for two months (Figure 3S). This suggests that these NPs can circulate in the blood stream over a long time period without significant leaching of the drug, resulting in reduced drug-related toxicity. However, the synthesized nano-hydrogels are easily degraded to pieces with apparent molecular weight of 24 KDa (Figure 1) as measured by GPC, under hydrolyzing conditions. The resultant molecular weight of the degraded product allows renal clearance, so that, after cross-linker degradation, the degraded products can readily be excreted from the body.
Figure 1.

GPC of nano-hydrogel (blue) and its degraded product (black).
The toxicity of the polymer used for drug delivery is of high concern. Cytotoxicity of degradable and non-degradable nano-particles to rat C6 glioma cells was tested (Figure 4S). The cells were seeded in 96-well plates at a concentration of 4.0×104 cells/100 μl per well. After 12 h of incubation, each well was added with NPs in a volume of 100 μl. The plates were incubated for 72 h at 37 °C, and the cellular viability was evaluated by the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) assay kit. Each polymer concentration was tested in eight average measurements. The degradable and non-degradable nano-hydrogels had no effect on rat C6 glioma cells up to 5 mg/ml. Cellular viability remained approximately 100% (±5%) and 93% (±5%), for non-degradable and degradable polyacrylamide nanoparticles, respectively, after 3day incubation.
It is interesting to find out if the synthesized nanohydrogels can be uptaken by macrophages. The nano-hydrogel clearance by a mouse macrophage RAW cell line was evaluated with propidium iodide (PI) encapsulated degradable NPs. PI is a cell impermeable nuclear staining dye and is generally excluded from viable cells. If the degradable PI-encapsulated NPs are uptaken by macrophages, one would expect that the degraded NPs release PI to stain the nuclei. The PI-encapsulated degradable NPs were incubated with macrophages in serum containing medium for 2h. After incubation, the cells were washed with medium for several times to remove any absorbed NPs. The bright field and calcein AM stain showed that the cells were healthy. We did not observe any PI nuclear staining on the nuclei of macrophage cells (Figure 2). The result also indicated that there is no opsonization effect for these polyacrylamide based nanoparticles. The nanoparticles did not associate with proteins and there was no particle uptaken by macrophages through endocytosis. However, for amine-functionalized polyacrylamide nanoparticles, the charged particles were internalized by macrophages.27 In comparison, the results showed that the small, neutral and hydrophilic nano-particles can evade macrophage clearance.
Figure 2.

Nano-hydrogel uptaken by macrophages. PI encapsulated degradable NPs were incubated with macrophage cells for 2 hours. Then the cells were washed with medium and were excited at 568 nm (PI excitation). No PI staining was observed. This means that these PI encapsulated NPs were stable in the biological environment, thus were not taken up by macrophages. (a). bright field image; (b). Calcein AM fluorescence image; (c). with excitation at PI excitation wavelength. The scale bar is 30 μm
Nanohydrogel degradation was further evaluated in vitro, within cells. Because our nanometer-scale hydrogels were not taken up by macrophages during our experiments, we therefore transferred the PI-containing degradable and non-degradable particles into macrophages by the gene gun method in order to investigate the degradation of NPs. Confocal imaging showed that for the non-degradable NPs, after washing with medium, no nucleus PI staining was observed (Figure 5S). This means that the non-degradable NPs were not degraded in the cytoplasm of the cells, thus preventing the encapsulated PI from staining the nucleus. To confirm that the nanoparticles were actually inside these cells without degradation, free PI was added to the same cell culture medium. The free PI staining of these cells’ nuclei confirmed that the non-degradable NPs did go through the cell membrane but did not degrade within the cells after the gene-gun delivery. This further supports our thesis that the NP matrix can effectively prevent the PI from interacting with the nucleus. For the case of degradable PI-containing NPs, they were also delivered into cells by gene-gun, and then the cells were washed with medium and incubated for 20 minutes. Bright field visualization showed both NP containing cells and normal cells in the same field of view (Figure 3a). Unlike for the non-degradable NPs, the confocal imaging showed that the degradable NP containing cells can be visualized by excitation at the PI excitation wavelength (568nm), while NP-free normal cells (controls) cannot be visualized by the PI excitation (Figure 3b). This means that the degradable nano-particles indeed underwent hydrolysis by various enzymes in the cytoplasm of the macrophage cells and then quickly released PI and stained the nucleus. Figure 3 also shows that no nucleus staining was observed for the cells without nano-particles. This demonstrates that the nuclear staining was only caused by the nanohydrogel degradation. These results therefore show that our designed degradable nano-platform did degrade after being introduced into cells.
Figure 3.
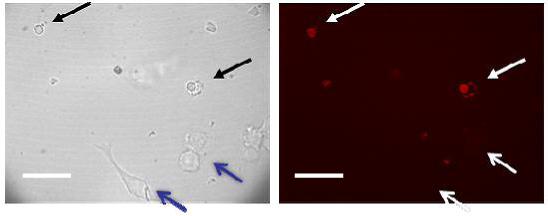
Nanohydrogel in vitro degradation within macrophages. PI encapsulating degradable NPs were introduced into macrophage cells by the gene gun method. (a) Bright field visualization of NP loaded cells (black arrows) and normal cells (blue arrows). (b) NP loaded cells can be visualized by excitation at the PI excitation wavelength (568nm). This means that the degradable NPs were indeed hydrolyzed within cells and the released PI thus stained the nuclei. The scale bar is 40 μm.
Our experiments supported the thesis that the small, neutral and hydrophilic polyacrylamide-based hydrogels could avoid the capture of macrophages and have less cellular toxicity than charged NPs, such as silica NPs, as well as minimal macrophage uptake. In order to avoid the accumulation of non-degradable polyacrylamide-based NPs in the body, degradable short chain polacrylamides were assembled into degradable NPs with degradable crosslinkers. These payloads are stable in aqueous environments for at least two months; however, they can quickly degrade under cytoplasmic conditions, within an hour, so as to rapidly release drugs once they enter cells. The low molecular weight of the final degraded products can be precisely controlled by the synthetic approach. These short-chain assembled NPs should result in the removal of the degraded polymers from the body. This work thus provides a feasible alternative proof-of-concept approach for the development of novel biodegradable materials and it can be extended to other materials of interest. Further research will focus on investigating peptide guided targeting and the in vivo bio-elimination of the synthesized nanoparticles.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the National Cancer Institute (NIH) UIP contract N01-CO-37123 (RK).
REFERENCES
- (1).Silva GA. Nature Reviews Neuroscience. 2006;7:65–74. doi: 10.1038/nrn1827. [DOI] [PubMed] [Google Scholar]
- (2).Ferrari M. Nature Reviews Cancer. 2005;5:161–171. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- (3).Nayak S, Lyon LA. Angew. Chem. Int. Ed. 2005;44:7686–7708. doi: 10.1002/anie.200501321. [DOI] [PubMed] [Google Scholar]
- (4).Allen TM, Cullis PR. Science. 2004;303:1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- (5).Sahoo SK, Labhasetwar V. Drug Discovery Today. 2003;8:1112–1120. doi: 10.1016/s1359-6446(03)02903-9. [DOI] [PubMed]
- (6).Moghimi SM, Hunter AC, Murray JC. Pharmacol Rev. 2001;53:283–318. [PubMed] [Google Scholar]
- (7).Zhu G, Mallery SR, Schwendeman SP. Nature Biotechnology. 2000;18:52–57. doi: 10.1038/71916. [DOI] [PubMed] [Google Scholar]
- (8).Jain RA. Biomaterials. 2000;21:2475–2490. doi: 10.1016/s0142-9612(00)00115-0. [DOI] [PubMed] [Google Scholar]
- (9).Anderson JM, Shive MS. Advanced Drug Delivery Reviews. 1997;28:5–24. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
- (10).Ha C-S, Gardella JA., Jr. Chem. Rev. 2005;105:4205–4232. doi: 10.1021/cr040419y. [DOI] [PubMed] [Google Scholar]
- (11).Harris JM, Chess RB. Nature Reviews Drug Discovery. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- (12).Fu K, Pack DW, Klibanov AM, Langer R. Pharm. Res. 2000;17:100–106. doi: 10.1023/a:1007582911958. [DOI] [PubMed] [Google Scholar]
- (13).Wang C, Ge Q, Ting D, Nguyen D, Shen H, Chen J, Eisen HN, Heller J, Langer R, Putnam D. Nature Materials. 2004;3:190–196. doi: 10.1038/nmat1075. [DOI] [PubMed] [Google Scholar]
- (14).Gao X, Cui Y, Levenson RM, Chung LWK, Nie S. Nature Biotechnology. 2004;22:969–976. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- (15).Duncan R. Nature Reviews Drug Discovery. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- (16).Duncan R. Nature Reviews Cancer. 2006;6:688–701. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]
- (17).Duncan R, Seymour LCW, Scarlett L, Lloyd JB, Rejmanova P, Kopecek J. Biochimica et Biophysica Acta. 1986;880:62–71. doi: 10.1016/0304-4165(86)90120-0. [DOI] [PubMed] [Google Scholar]
- (18).Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Advanced Materials. 2006;18:1345–1360. [Google Scholar]
- (19).nucleus Gao D, Agayan RR, Xu H, Philbert MA, Kopelman R. Nano Letters. 2006;6:2383–2386. doi: 10.1021/nl0617179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Monson E, Brasuel M, Philbert MA, Kopelman R. Biomedical Photonics Handbook. 2003;59:1–14. [Google Scholar]
- (21).Buck SM, Koo Y-E, Park E, Xu H, Philbert MA, Brasuel MA, Kopelman R. Current Opinion in Chemical Biology. 2004;8:540–546. doi: 10.1016/j.cbpa.2004.08.011. [DOI] [PubMed] [Google Scholar]
- (22).Sumner JP, Westerberg NM, Stoddard AK, Hurst TK, Cramer M, Thompson RB, Fierke CA, Kopelman R. Biosensors and Bioelectronics. 2006;21:1302–1308. doi: 10.1016/j.bios.2005.04.023. [DOI] [PubMed] [Google Scholar]
- (23).Tang W, Xu H, Kopelman R, Philbert MA. Photochemistry and Photobiology. 2005;81:242–249. doi: 10.1562/2004-05-24-RA-176.1. [DOI] [PubMed] [Google Scholar]
- (24).Kopelman R, Koo Y-E, Philbert MA, Moffat BA, Ramachandra RG, McConville P, Hall DE, Chenevert TL, Bhojani MS, Buck SM, Rehemtulla A, Ross BD. Journal of Magnetism and Magnetic Materials. 2005;293:404–410. [Google Scholar]
- (25).Henriquez C, Bueno C, Lissi EA, Encinas MV. Polymer. 2003;44:5559–5561. [Google Scholar]
- (26).Endo K, Sawada T. Colloid and Polymer Science. 2001;279:1058–1063. [Google Scholar]
- (27).Schneider RJ. Ph.D. Thesis. University of Michigan; Ann Arbor, MI: 2005. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.