

Abstract
Background
The 5 subunits of native pentameric C-reactive protein (CRP) are dissociated to generate monomeric form of CRP (mCRP) in some in vitro conditions, both physiological and non-physiological, and also in vivo. Many bioactivities of mCRP generated by urea-treatment of CRP and of mCRP generated by mutating the primary structure of CRP have been reported. The bioactivities of mCRP generated by spontaneous dissociation of CRP are largely unexplored.
Methods
We purified mCRP generated by spontaneous dissociation of CRP and investigated the binding of mCRP to enzymatically-modified low-density lipoprotein (E-LDL).
Results
mCRP was approximately 60 times more potent than CRP in binding to E-LDL. In the presence of the small-molecule compound phosphoethanolamine (PEt), at 37°C, the binding of mCRP to E-LDL was enhanced <2-fold, while the binding of CRP to E-LDL was enhanced >10-fold. In contrast, PEt inhibited the binding of both CRP and mCRP to pneumococcal C-polysaccharide, another phosphocholine-containing ligand to which CRP and mCRP were found to bind. We have not investigated yet whether PEt alters the structure of CRP at 37°C.
Conclusions
Combined data suggest that the targeting of CRP with the aim to monomerize CRP in vivo may be an effective approach to capture modified forms of LDL.
Keywords: C-reactive protein, Monomeric C-reactive protein, Phosphoethanolamine, Pneumococcal C-polysaccharide, Enzymatically-modified low-density lipoprotein
1. Introduction
Throughout this paper, the abbreviation CRP represents “native pentameric C-reactive protein”. CRP is composed of 5 identical, non-covalently associated, 23 kDa-subunits. Some of the biologically significant binding specificities of CRP include substances containing phosphocholine (PCh) and phosphoethanolamine (PEt), such as pneumococcal C-polysaccharide (PnC) and damaged cells of the myocardial infarcts. There are 5 PCh-binding sites in CRP, one located on each subunit. Calcium ions are required for the binding of CRP to PCh and PEt [1–7].
Although CRP is secreted by hepatocytes in the form of pentameric molecules, the 5 subunits of CRP are dissociated to generate monomeric form of CRP (mCRP) in some in vitro conditions, both physiological and non-physiological, and also in vivo [1,8,9]. Four in vitro conditions for the generation of mCRP have been described. 1. mCRP can be generated by exposing CRP to protein denaturants such as urea and heat [10]. 2. Recombinant mCRP can be generated by mutating CRP amino acids Cys36 and Cys97 to alanine so that the expressed CRP does not pentamerize [11,12]. 3. mCRP can also be generated by spontaneous dissociation of CRP when CRP is stored in the absence of calcium [13–16]. Also, in the absence of calcium, both CRP and mCRP are susceptible to proteolytic degradation [17,18]. 4. Under physiological conditions, mCRP is generated when CRP is associated with a cell membrane or liposome [19–21]. This mechanism of mCRP generation has been considered to explain the production of mCRP in vivo [20,21]. The presence of mCRP has been demonstrated in normal vascular tissue, atherosclerotic lesions, damaged brain tissue, diseased kidney, urine, and in other inflammatory conditions including inflamed tissues in animal studies [22–29]. These demonstrations suggest that mCRP is a major tissue form of CRP, and therefore, it is important to investigate the functions of mCRP.
CRP has been shown to bind to enzymatically-modified low-density lipoprotein (E-LDL) which is an atherogenic form of LDL and a constituent of atherosclerotic lesions [30–34]. The PCh-binding site of CRP participates in the binding of CRP to E-LDL because PCh has been shown to inhibit binding of CRP to E-LDL. We have shown recently that PEt, which, like PCh, presumably also blocks the PCh-binding site of CRP, does not inhibit binding of CRP to E-LDL; instead, PEt enhances the binding of CRP to E-LDL [34, 35]. The capacity of CRP to bind to E-LDL is a beneficial function of CRP because, by doing so, CRP can prevent formation of macrophage foam cells [34,35]. mCRP exhibits bioactivities different from the bioactivities of CRP [15,36–39]. However, like CRP, recombinant mCRP has been shown to bind to E-LDL [14]. The PCh and cholesterol moieties of E-LDL molecules serve as the ligands for the binding of CRP and mCRP to E-LDL [21,32,34].
The reported bioactivities of mCRP, including the E-LDL-binding activity, are mostly based on the investigations using either recombinant mCRP or mCRP generated by urea-treatment of CRP. Little is known about the bioactivities of purified mCRP generated by spontaneous dissociation of CRP [38, 39]. While investigating CRP-E-LDL interactions [34], we routinely observed that the stored CRP was more efficient than freshly-purified CRP in binding to E-LDL. Gel filtration analysis of the stored preparations of purified CRP revealed the presence of mCRP. In this study, we purified mCRP from the stored CRP preparations and investigated the binding of purified mCRP to E-LDL.
2. Materials and methods
2.1. Purification of CRP and mCRP
As described previously [40], CRP was purified from pleural fluid by 3 steps: a Ca++-dependent affinity chromatography on a PCh-conjugated Sepharose column (Pierce, Rockford, IL), followed by anion-exchange chromatography on a MonoQ column (GE Healthcare, Piscataway, NJ), and gel filtration on a Superose12 column (10/300 GL, GE Healthcare) using the Biologic Duo Flow Protein Purification System (BioRad, Hercules, CA). Purified CRP was stored in 10 mmol/l Tris-HCl, 150 mmol/l NaCl, pH 7.2 (TBS) at −80°C. The duration of storage was at least 8 weeks at −80°C followed by 2 weeks at 4°C. We called this preparation of CRP as “stored CRP”.
Stored CRP was centrifuged and subjected to gel filtration again on the Superose12 column to remove any mCRP which might have formed upon storage of CRP. The column was eluted with TBS and the fractions containing CRP were collected, diluted in appropriate Ca2+-containing buffer as described below, and then used in the assays the same day. After the re-purification procedure, we called this preparation of CRP as “freshly-purified CRP”.
During the re-purification of CRP, the mCRP-containing fractions were also collected, diluted in appropriate Ca2+-containing buffer as described below, and then used in the assays. Thus, mCRP, that we report here, was generated by spontaneous dissociation of stored CRP.
2.2. Preparation of E-LDL
E-LDL was prepared as described previously, with some modifications [31, 34, 41]. First, native LDL was isolated from normal human plasma according to a published method [42]. Native LDL was dialyzed against 5 mmol/l Veronal containing 150 mmol/l NaCl, pH 7.3. Next, native LDL (5 mg/ml) was treated with plasmin (0.1 U/ml; Roche, Indianapolis, IN) and EDTA (0.5 mmol/l) and incubated for 24 h at 37°C with shaking. Cholesterol esterase (40 μg/ml; Roche) was added next and incubated for 24 h at 37°C with shaking. Then, once again, plasmin and cholesterol esterase were added together and incubated overnight at 37°C with shaking. Finally, the mixture was centrifuged at 10,000 rpm for 5 min and the supernatant was stored at 4°C in aliquots. The concentration of protein in the E-LDL preparations was measured using a protein assay kit (Bio-Rad).
2.3. E-LDL-binding assay
E-LDL-binding assays were performed as described previously [34]. Microtiter wells were coated with 10 μg/ml of E-LDL diluted in TBS overnight at 4°C. The unreacted sites in the wells were blocked with TBS containing 0.5% gelatin. CRP and mCRP diluted in TBS containing 0.1% gelatin, 0.02% Tween-20 and 2 mmol/l CaCl2 (TBS-Ca) were added in duplicate wells. After incubation for 3 h at 37°C, the wells were washed with TBS-Ca. Immunoaffinity-purified polyclonal rabbit anti-CRP antibody (1 μg/ml) was used to detect bound CRP. Immunoaffinity-purified polyclonal rabbit anti-CRP antibody was purified from the rabbit anti-human CRP antiserum (Sigma-Aldrich, St. Louis, MO) by affinity chromatography on a CRP-conjugated agarose column prepared by using the AminoLink Immobilization kit (Pierce) [7]. HRP-conjugated goat anti-rabbit IgG (Pierce) were used as the secondary antibody. Color was developed, and the absorbance was read at 405 nm in the microtiter plate reader (Molecular Devices, Sunnyvale, CA). To determine the effects of PEt (Sigma-Aldrich, P0503) on the binding of CRP and mCRP to E-LDL, CRP and mCRP were added to E-LDL-coated wells in the presence of 10 mmol/l PEt diluted in TBS-Ca buffer.
2.4. PnC-binding assay
Binding activity of CRP and mCRP for PCh was evaluated by using PnC (Statens Serum Institut, Copenhagen) as described previously [34]. Briefly, microtiter wells were coated with PnC (10 μg/ml) in TBS. After blocking, serial dilutions of CRP in TBS containing 0.1% BSA, 0.02% Tween-20 and 5 mmol/l CaCl2 were added to the wells and incubated for 3 h at 37°C. Immunoaffinity-purified polyclonal rabbit anti-CRP antibody (1 μg/ml) was used to detect bound CRP. HRP-conjugated goat anti-rabbit IgG were used as the secondary antibody. Color was developed, and the absorbance was read at 405 nm in the microtiter plate reader. In some assays, an anti-CRP monoclonal antibody (HD2.4) was used to detect PnC-bound CRP [7]. To determine the effects of PEt on the binding of CRP and mCRP to PnC, CRP and mCRP were added to PnC-coated wells in the presence of 10 mmol/l PEt prepared in TBS containing 0.1% BSA, 0.02% Tween-20 and 5 mmol/l CaCl2.
3. Results
3.1. The binding of stored CRP to E-LDL is more efficient than that of freshly-purified CRP
We first compared the binding of stored CRP and freshly-purified CRP to E-LDL using the E-LDL-binding assay (Fig. 1). CRP from both preparations bound to E-LDL in a dose-dependent manner, but with different efficiency. For equivalent binding of CRP to E-LDL, 10 μg/ml of freshly-purified CRP was required compared to 1.73±0.4 μg/ml of stored CRP. Based on the data obtained from 4 assays, we calculated that 80–90% less of stored CRP was required compared to freshly-purified CRP for equivalent binding to E-LDL. Because it has been shown earlier that the storage of CRP under certain conditions causes its dissociation into mCRP and that mCRP also binds to E-LDL [13–16], we hypothesized that the increased binding of stored CRP to E-LDL might be due to the presence of mCRP in stored CRP preparations.
Fig. 1.
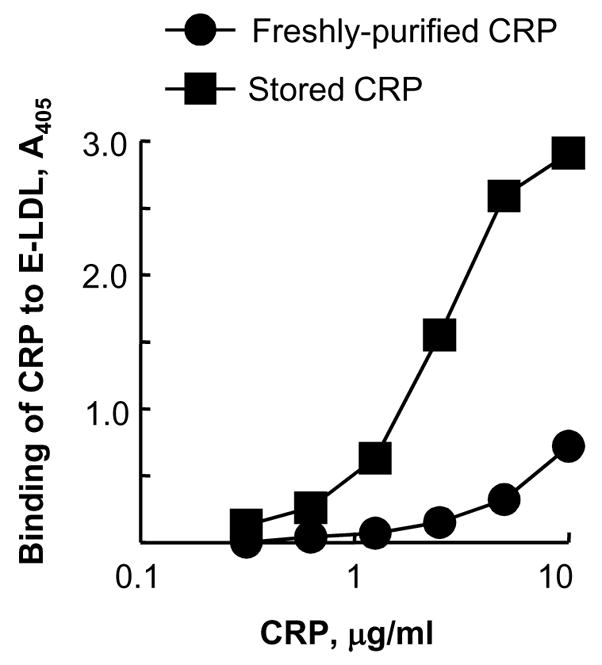
Stored CRP is more potent than freshly-purified CRP in binding to E-LDL. CRP was added to microtiter wells coated with E-LDL. Bound CRP was detected by using anti-CRP antibody. A representative of four experiments using four different preparations of CRP is shown.
3.2. Purification of mCRP
To test the hypothesis that the increased binding of stored CRP to E-LDL was due to the presence of mCRP, we performed gel filtration chromatography of stored CRP. For comparison, we performed gel filtration chromatography of freshly-purified CRP. The elution profiles of freshly-purified CRP and stored CRP from the gel filtration chromatography column are shown in Figs. 2A and 2B. The elution profile of stored CRP showed two peaks, indicating the presence of mCRP. The first peak corresponded with the elution profile of freshly-purified CRP. The molecular weight of the protein in the second peak was calculated to be approximately 31 kDa. Next, we used reducing SDS-PAGE (Fig. 2C), to determine the molecular weight of the subunits of freshly-purified CRP and the protein present in the second peak (labelled as mCRP in Fig. 2B). The apparent molecular weights for both were found to be approximately 27.5 kDa. The degradation products (Fig. 2C, lane 3) appearing after electrophoresis reflected the unstable nature of mCRP under electrophoresis conditions. We concluded that the protein present in the second peak was mCRP generated from spontaneous dissociation of CRP (further confirmed by the results of the PnC-binding assays; see below), and used it as purified mCRP.
Fig. 2.

Purification of mCRP. (A) Gel filtration chromatography of freshly-purified CRP. A representative of 5 chromatograms from a Superose12 column is shown. (B) Gel filtration chromatography of stored CRP. A representative of 5 chromatograms from a Superose12 column is shown. The arrows point to the elution volumes of molecular weight marker proteins. The first peak contained CRP while the second peak contained mCRP. (C) CRP and mCRP were subjected to 10–20% SDS-PAGE under reducing conditions. A representative Coomassie blue-stained gel is shown. Lane 1, molecular weight marker; Lane 2, freshly-purified CRP (10 μg); Lane 3, purified mCRP (10 μg).
3.3. The binding of purified mCRP to E-LDL is more efficient than that of CRP: Effects of phosphoethanolamine
In the experiments shown in Figures 3 and 4, we used purified mCRP and freshly-purified CRP. In the E-LDL-binding assay (Fig. 3A), mCRP bound to E-LDL more efficiently than did CRP. The amount of mCRP required for 50% of maximal binding to E-LDL was 60.5±27.7 times (98.2%) less than the required amount of CRP. Prompted by our previous finding that PEt enhances the binding of CRP to E-LDL [34], we next determined whether PEt also had an effect on the binding of mCRP to E-LDL. In the presence of 10 mmol/l PEt, the binding of mCRP to E-LDL was slightly enhanced at all concentrations of mCRP (Fig. 3B). In the presence of PEt, the amount of mCRP required for 50% of maximal binding of mCRP to E-LDL was 2.3±1.2 times (56.5%) less compared to that required in the absence of PEt.
Fig. 3.

Both mCRP and PEt-bound CRP are more potent than CRP in binding to E-LDL. CRP and mCRP were added to E-LDL-coated wells. Bound CRP and mCRP were detected by using anti-CRP antibody. (A) Comparison of the binding of CRP and mCRP to E-LDL. A representative of three experiments is shown. (B) Increasing concentration of mCRP, with and without 10 mmol/l PEt, was added to E-LDL-coated wells. A representative of three experiments is shown. (C) Increasing concentration of CRP and mCRP, with and without 10 mmol/l PEt, was added to E-LDL-coated wells. A representative of two experiments is shown. (D) Relative E-LDL-binding capacity of CRP and mCRP, in the absence and presence of PEt. Values on the x-axis were derived from the experiment shown in Fig. 3C, and represent the relative concentration of CRP and mCRP, in the absence and presence of PEt, required for their equivalent binding to E-LDL.
Fig. 4.
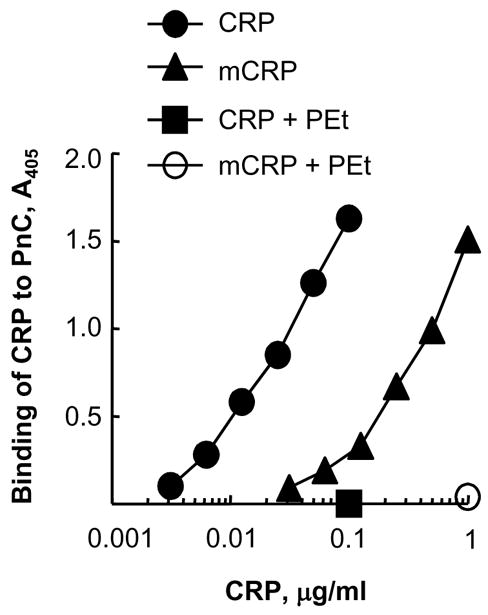
PEt inhibits the binding of CRP and mCRP to PnC. Increasing concentration of CRP and mCRP, with and without 10 mmol/l PEt, was added to PnC-coated wells. Bound CRP and mCRP were detected by using polyclonal anti-CRP antibody. A representative of 5 experiments is shown.
Next, we compared the binding of CRP and mCRP to E-LDL, in the presence and absence of PEt, in a single assay (Fig. 3C). PEt again enhanced the binding of both CRP and mCRP to E-LDL. From the data shown in Figure 3C, we determined the concentration of CRP and mCRP, in the presence and absence of PEt, required for equivalent binding to E-LDL (Fig. 3D). CRP was required at a concentration of 9.4 μg/ml, CRP in the presence of PEt was required at a concentration of 0.92 μg/ml, mCRP was required at a concentration of 0.23 μg/ml, and mCRP in the presence of PEt was required at a concentration of 0.16 μg/ml. Thus, relatively, CRP was least efficient in binding to E-LDL while PEt-bound mCRP was most efficient. We also found that a temperature of 37°C was critical for PEt-dependent enhancement of the binding of CRP to E-LDL; PEt did not enhance the binding of CRP to E-LDL when the E-LDL-binding assays were performed at room temperature (data not shown).
3.4. PEt inhibits the binding of CRP and mCRP to PnC
We performed PnC-binding assay to investigate whether mCRP retained the PCh-binding activity of CRP and, if so, to determine the effects of PEt on the binding of mCRP to PnC. Both CRP and mCRP bound to PnC in a dose-dependent manner (Fig. 4). However, the avidity of the binding of mCRP to PnC was much lower than that of CRP. For comparable binding to PnC, 59.7±7.8 times more mCRP was required than the required amount of CRP. Identical results were obtained when an anti-CRP monoclonal antibody HD2.4 was used to detect mCRP bound to PnC (data not shown). These results showed that the HD2.4 epitope of CRP was present on mCRP generated by spontaneous dissociation of CRP, and also provided information on the specificity of the HD2.4 antibody for future use. Combined data also suggested structural similarities between mCRP and CRP subunits in the pentamer. As expected, PEt inhibited the binding of both CRP and mCRP to PnC, indicating that mCRP retained the PEt-binding activity of CRP and that the function of PEt to enhance the binding of CRP and mCRP to E-LDL was selective for E-LDL.
4. Discussion
In this study, we purified mCRP generated by spontaneous dissociation of CRP, and investigated the binding of mCRP to E-LDL. Our major findings were: 1. mCRP was approximately 60 times more potent than CRP in binding to E-LDL. 2. In the presence of the small-molecule compound PEt, at 37°C, the binding of mCRP to E-LDL was enhanced less than 2-fold, while the binding of CRP to E-LDL was enhanced more than 10-fold. 3. mCRP generated by spontaneous dissociation of CRP retained the PCh-binding and PEt-binding activities of CRP. 4. The PEt-mediated enhancement in the binding of mCRP and CRP to E-LDL was selective for E-LDL because PEt inhibited the binding of both CRP and mCRP to PnC, another PCh-containing ligand to which CRP and mCRP were found to bind.
Investigating the functions of mCRP has implications because mCRP has been found to be generated in vivo. Even a mechanism for the processing of CRP into mCRP in physiological conditions has been proposed, that is, when CRP is associated with a cell membrane, it is converted to mCRP [20,21]. In the arterial wall, mCRP has been shown to localize with LDL and macrophages in the atherosclerotic lesions [12]. Not only mCRP, but CRP has also been found deposited and localized with LDL and macrophages in atherosclerotic lesions in humans and experimental animal models [31,43–45]. However, it is important to note that in the studies where CRP had been found deposited in atherosclerotic lesions, polyclonal anti-CRP antibodies were used which did not differentiate between CRP and mCRP. Many polyclonal anti-CRP antibodies, which have been examined for their mCRP specificity to date, have been shown to cross-react with mCRP [46].
It has been reported previously that recombinant mCRP also binds to E-LDL and it has been suggested that the capacity of mCRP to bind to modified forms of LDL is a beneficial function of mCRP [12,14]. Accordingly, mCRP has been found to decrease the uptake of oxidized LDL by macrophages and it has been proposed that the interaction of mCRP with oxidized LDL may contribute to retardation of the foam cell formation by reducing the aggressive macrophage response to oxidized LDL [14]. Using ApoE−/− murine model of atherosclerosis, mCRP has been shown to be atheroprotective [12,47]. The functions of mCRP have not yet been investigated in the Apob100/100Ldlr−/− mouse model of atherosclerosis, a model in which CRP has been shown to be atheroprotective [48].
The mechanism of action of PEt in enhancing the binding of CRP to E-LDL is not known, but it is reasonable to assume that PEt modifies CRP in a way similar to storage-induced modification of CRP. Several effects of PEt, model liposomes, and cell membranes on the integrity of CRP have been described recently [20,21,49,50]. Data obtained from the studies on Limulus CRP suggested that the binding of CRP to liposomes and bacterial membranes causes hyper-oligomerization of CRP [49]. The binding of PEt to Limulus SAP-like pentraxin was shown to alter the structure of the Ca2+-binding site of Limulus SAP-like pentraxin; upon binding PEt, each subunit of the pentraxin bound an additional calcium ion. In the crystal structure of PEt-bound Limulus SAP-like pentraxin, the ethanolamine group was poorly defined, suggesting structural and binding variability of this group [50]. In addition to these effects of PEt and membranes on the structural integrity of CRP, the binding of membranes to CRP has been shown to induce monomerization of CRP, and thereby exposing an otherwise hidden cholesterol-binding site of CRP [20,21]. We have not investigated yet whether PEt induces monomerization of CRP at 37°C.
We found that PEt had minimal effect on the binding of CRP to E-LDL if CRP was already monomerized. The minimal enhancing effect of PEt that was seen on mCRP may be due to possible PEt-mediated separation of self-aggregates of mCRP. Alternatively, mCRP, when alone, may be binding to only a certain population of E-LDL molecules in the E-LDL preparations. PEt, probably by inducing a structural alteration of CRP, may confer mCRP the ability to bind to those E-LDL molecules also to which mCRP cannot bind alone. It is less likely that our mCRP was contaminated with some CRP and the effect of PEt on mCRP was due to the CRP contaminant in purified mCRP preparations. Although mCRP by itself was efficient in binding to E-LDL, the beneficial effect of PEt would be to abolish mCRP’s PCh-binding activity while retaining the E-LDL-binding activity.
Using mice, it has been shown that the half-life of mCRP in the circulation is <5 min. mCRP is rapidly cleared from the circulation and migrates to tissues [51]. These findings indicate that the transport of mCRP from circulation to various sites in the body is faster than CRP. We report here that mCRP is much more potent than CRP in binding to E-LDL. Taken together, our data suggest that the targeting of CRP with the aim to monomerize CRP in vivo may be an effective approach to capture modified atherogenic forms of LDL.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (R01HL071233 to A.A.).
Abbreviations
- CRP
pentameric C-reactive protein
- mCRP
monomeric C-reactive protein
- LDL
low-density lipoprotein
- E-LDL
enzymatically-modified LDL
- PCh
phosphocholine
- PEt
phosphoethanolamine
- PnC
pneumococcal C-polysaccharide
- TBS
10 mmol/l Tris-HCl, 150 mmol/l NaCl, pH 7.2
- TBS-Ca
TBS containing 0.1% gelatin, 0.02% Tween-20 and 2 mmol/l CaCl2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Singh SK, Suresh MV, Voleti B, Agrawal A. The connection between C-reactive protein and atherosclerosis. Ann Med. 2008;40:110–120. doi: 10.1080/07853890701749225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Black S, Kushner I, Samols D. C-reactive protein. J Biol Chem. 2004;279:48487–48490. doi: 10.1074/jbc.R400025200. [DOI] [PubMed] [Google Scholar]
- 3.Peisajovich A, Marnell L, Mold C, Du Clos TW. C-reactive protein at the interface between innate immunity and inflammation. Expert Rev Clin Immunol. 2008;4:379–390. doi: 10.1586/1744666X.4.3.379. [DOI] [PubMed] [Google Scholar]
- 4.Shrive AK, Cheetham GMT, Holden D, et al. Three dimensional structure of human C-reactive protein. Nature Struct Biol. 1996;3:346–354. doi: 10.1038/nsb0496-346. [DOI] [PubMed] [Google Scholar]
- 5.Volanakis JE, Wirtz KWA. Interaction of C-reactive protein with artificial phosphatidylcholine bilayers. Nature. 1979;281:155–157. doi: 10.1038/281155a0. [DOI] [PubMed] [Google Scholar]
- 6.Schwalbe RA, Dahlbäck B, Coe JE, Nelsestuen GL. Pentraxin family of proteins interacts specifically with phosphorylcholine and/or phosphorylethanolamine. Biochemistry. 1992;31:4907–4915. doi: 10.1021/bi00135a023. [DOI] [PubMed] [Google Scholar]
- 7.Agrawal A, Simpson MJ, Black S, Carey MP, Samols D. A C-reactive protein mutant that does not bind to phosphocholine and pneumococcal C-polysaccharide. J Immunol. 2002;169:3217–3222. doi: 10.4049/jimmunol.169.6.3217. [DOI] [PubMed] [Google Scholar]
- 8.Schwedler SB, Filep JG, Galle J, Wanner C, Potempa LA. C-reactive protein: a family of proteins to regulate cardiovascular function. Am J Kidney Dis. 2006;47:212–222. doi: 10.1053/j.ajkd.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 9.Boncler M, Watala C. Regulation of cell function by isoforms of C-reactive protein: a comparative analysis. Acta Biochim Pol. 2009;56:17–31. [PubMed] [Google Scholar]
- 10.Potempa LA, Siegel JN, Fiedel BA, Potempa RT, Gewurz H. Expression, detection and assay of a neoantigen (neo-CRP) associated with a free, human C-reactive protein subunit. Mol Immunol. 1987;24:531–541. doi: 10.1016/0161-5890(87)90028-9. [DOI] [PubMed] [Google Scholar]
- 11.Khreiss T, József L, Potempa LA, Filep JG. Conformational rearrangement in C-reactive protein is required for proinflammatory actions on human endothelial cells. Circulation. 2004;109:2016–2022. doi: 10.1161/01.CIR.0000125527.41598.68. [DOI] [PubMed] [Google Scholar]
- 12.Ji SR, Wu Y, Potempa LA, Liang YH, Zhao J. Effect of modified C-reactive protein on complement activation: a possible complement regulatory role of modified or monomeric C-reactive protein in atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2006;26:935–941. doi: 10.1161/01.ATV.0000206211.21895.73. [DOI] [PubMed] [Google Scholar]
- 13.Wang HW, Wu Y, Chen Y, Sui SF. Polymorphism of structural forms of C-reactive protein. Int J Mol Med. 2002;9:665–671. [PubMed] [Google Scholar]
- 14.Ji SR, Wu Y, Potempa LA, Qiu Q, Zhao J. Interactions of C-reactive protein with low-density lipoproteins: implications for an active role of modified C-reactive protein in atherosclerosis. Int J Biochem Cell Biol. 2006;38:648–661. doi: 10.1016/j.biocel.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 15.Taylor KE, van den Berg CW. Structural and functional comparison of native pentameric, denatured monomeric and biotinylated C-reactive protein. Immunology. 2007;120:404–411. doi: 10.1111/j.1365-2567.2006.02516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hakobyan S, Harris CL, van den Berg CW, et al. Complement factor H binds to denatured rather than to native pentameric C-reactive protein. J Biol Chem. 2008;283:30451–30460. doi: 10.1074/jbc.M803648200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kinoshita CM, Ying SC, Hugli TE, et al. Elucidation of a protease-sensitive site involved in the binding of calcium to C-reactive protein. Biochemistry. 1989;28:9840–9848. doi: 10.1021/bi00451a044. [DOI] [PubMed] [Google Scholar]
- 18.Suresh MV, Singh SK, Agrawal A. Interaction of calcium-bound C-reactive protein with fibronectin is controlled by pH: in vivo implications. J Biol Chem. 2004;279:52552–52557. doi: 10.1074/jbc.M409054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang HW, Sui SF. Dissociation and subunit rearrangement of membrane-bound human C-reactive proteins. Biochem Biophys Res Comm. 2001;288:75–79. doi: 10.1006/bbrc.2001.5733. [DOI] [PubMed] [Google Scholar]
- 20.Ji SR, Wu Y, Zhu L, et al. Cell membranes and liposomes dissociate C-reactive protein (CRP) to form a new, biologically active structural intermediate: mCRPm. FASEB J. 2007;21:284–294. doi: 10.1096/fj.06-6722com. [DOI] [PubMed] [Google Scholar]
- 21.Ji SR, Ma L, Bai CJ, et al. Monomeric C-reactive protein activates endothelial cells via interaction with lipid raft microdomains. FASEB J. 2009;23:1806–1816. doi: 10.1096/fj.08-116962. [DOI] [PubMed] [Google Scholar]
- 22.Rees RF, Gewurz H, Siegel JN, Coon J, Potempa LA. Expression of a C-reactive protein neoantigen (neo-CRP) in inflamed rabbit liver and muscle. Clin Immunol Immunopathol. 1988;48:95–107. doi: 10.1016/0090-1229(88)90160-2. [DOI] [PubMed] [Google Scholar]
- 23.Diehl EE, Haines GK, III, Radosevich JA, Potempa LA. Immunohistochemical localization of modified C-reactive protein antigen in normal vascular tissue. Am J Med Sci. 2000;319:79–83. doi: 10.1097/00000441-200002000-00002. [DOI] [PubMed] [Google Scholar]
- 24.Schwedler SB, Guderian F, Dämmrich J, Potempa LA, Wanner C. Tubular staining of modified C-reactive protein in diabetic chronic kidney disease. Nephrol Dial Transplant. 2003;18:2300–2307. doi: 10.1093/ndt/gfg407. [DOI] [PubMed] [Google Scholar]
- 25.Kiefer CR, McKenney JB, Trainor JF, Snyder LM. Pulse pressure-driven neutral lipid accumulation and correlative proinflammatory markers of accelerated atherogenesis. Atherosclerosis. 2005;183:17–24. doi: 10.1016/j.atherosclerosis.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 26.Slevin M, Matou-Nasri S, Turu M, et al. Modified C-reactive protein is expressed by stroke neovessels and is a potent activator of angiogenesis in vitro. Brain Pathol. 2009 doi: 10.1111/j.1750-3639.2008.00256.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agrawal A, Bhattacharya S. Possible role of C-reactive protein in detoxication of mercury. Indian J Exp Biol. 1990;28:638–641. [PubMed] [Google Scholar]
- 28.Ruleva NY, Lyukova TK, Tarabarko NV, Komolov IS, Domogatskii SP. Structure of C-reactive protein excreted in urine during acute rejection episodes. Bull Exp Biol Med. 2003;135:250–252. doi: 10.1023/a:1024180813651. [DOI] [PubMed] [Google Scholar]
- 29.Wetterö J, Nilsson L, Jonasson L, Sjöwall C. Reduced serum levels of autoantibodies against monomeric C-reactive protein (CRP) in patients with acute coronary syndrome. Clin Chim Acta. 2009;400:128–131. doi: 10.1016/j.cca.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 30.Bhakdi S, Dorweiler B, Kirchmann R, et al. On the pathogenesis of atherosclerosis: enzymatic transformation of human low density lipoprotein to an atherogenic moiety. J Exp Med. 1995;182:1959–1971. doi: 10.1084/jem.182.6.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhakdi S, Torzewski M, Klouche M, Hemmes M. Complement and atherogenesis: binding of CRP to degraded, nonoxidized LDL enhances complement activation. Arterioscler Thromb Vasc Biol. 1999;19:2348–2354. doi: 10.1161/01.atv.19.10.2348. [DOI] [PubMed] [Google Scholar]
- 32.Taskinen S, Kovanen PT, Jarva H, Meri S, Pentikainen MO. Binding of C-reactive protein to modified low-density-lipoprotein particles: identification of cholesterol as a novel ligand for C-reactive protein. Biochem J. 2002;367:403–412. doi: 10.1042/BJ20020492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Biró A, Thielens NM, Cervenák L, Prohászka Z, Füst G, Arlaud GJ. Modified low density lipoproteins differentially bind and activate the C1 complex of complement. Mol Immunol. 2006;44:1169–1177. doi: 10.1016/j.molimm.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 34.Singh SK, Suresh MV, Prayther DC, Moorman JP, Rusinol AE, Agrawal A. C-reactive protein-bound enzymatically modified low-density lipoprotein does not transform macrophages into foam cells. J Immunol. 2008;180:4316–4322. doi: 10.4049/jimmunol.180.6.4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agrawal A. Therapeutic potential of phosphoethanolamine-bound C-reactive protein in atherosclerosis. Future Lipidol. 2008;3:599–602. doi: 10.2217/17460875.3.6.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heuertz RM, Schneider GP, Potempa LA, Webster RO. Native and modified C-reactive proteins bind different receptors on human neutrophils. Int J Biochem Cell Biol. 2005;37:320–335. doi: 10.1016/j.biocel.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 37.Boguslawski G, McGlynn PW, Potempa LA, Filep JG, Labarrere CA. Conduct unbecoming: C-reactive protein interactions with a broad range of protein molecules. J Heart Lung Transplant. 2007;26:705–713. doi: 10.1016/j.healun.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 38.Agrawal A, Singh SK, Thompson JA, Hammond DJ, Rusinol AE. Requirements for the binding of C-reactive protein to oxidized low-density lipoprotein. FASEB J. 2009;23:1006.2. [Google Scholar]
- 39.Hammond DJ, Singh SK, Suresh MV, Pangburn MK, Agrawal A. Requirements for the binding of C-reactive protein to complement factor H. J Immunol. 2009;182:134.60. [Google Scholar]
- 40.Singh SK, Suresh MV, Prayther DC, Moorman JP, Rusinol AE, Agrawal A. Phosphoethanolamine-complexed C-reactive protein: a pharmacological-like macromolecule that binds to native low-density lipoprotein in human serum. Clin Chim Acta. 2008;394:94–98. doi: 10.1016/j.cca.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Torzewski M, Suriyaphol P, Paprotka K, et al. Enzymatic modification of low-density lipoprotein in the arterial wall: a new role for plasmin and matrix metalloproteinases in atherogenesis. Arterioscler Thromb Vasc Biol. 2004;24:2130–2136. doi: 10.1161/01.ATV.0000144016.85221.66. [DOI] [PubMed] [Google Scholar]
- 42.Brown AJ, Leong SL, Dean RT, Jessup W. 7-Hydroperoxycholesterol and its products in oxidized low density lipoprotein and human atherosclerotic plaque. J Lipid Res. 1997;38:1730–1745. [PubMed] [Google Scholar]
- 43.Torzewski M, Rist C, Mortensen RF, et al. C-reactive protein in the arterial intima: role of C-reactive protein receptor-dependent monocyte recruitment in atherogenesis. Arterioscler Thromb Vasc Biol. 2000;20:2094–2099. doi: 10.1161/01.atv.20.9.2094. [DOI] [PubMed] [Google Scholar]
- 44.Sun H, Koike T, Ichikawa T, et al. C-reactive protein in atherosclerotic lesions: its origin and pathophysiological significance. Am J Pathol. 2005;167:1139–1148. doi: 10.1016/S0002-9440(10)61202-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reynolds GD, Vance RP. C-reactive protein immunohistochemical localization in normal and atherosclerotic human aortas. Arch Pathol Lab Med. 1987;111:265–269. [PubMed] [Google Scholar]
- 46.Samberg NL, Bray RA, Gewurz H, Landay AL, Potempa LA. Preferential expression of neo-CRP epitopes on the surface of human peripheral blood lymphocytes. Cell Immunol. 1988;116:86–98. doi: 10.1016/0008-8749(88)90212-2. [DOI] [PubMed] [Google Scholar]
- 47.Schwedler SB, Amann K, Wernicke K, et al. Native C-reactive protein increases whereas modified C-reactive protein reduces atherosclerosis in apolipoprotein E-knockout mice. Circulation. 2005;112:1016–1023. doi: 10.1161/CIRCULATIONAHA.105.556530. [DOI] [PubMed] [Google Scholar]
- 48.Kovacs A, Tornvall P, Nilsson R, Tegnér J, Hamsten A, Björkegren J. Human C-reactive protein slows atherosclerosis development in a mouse model with human-like hypercholesterolemia. Proc Natl Acad Sci USA. 2007;104:13768–13773. doi: 10.1073/pnas.0706027104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harrington JM, Chou HT, Gutsmann T, et al. Membrane activity of a C-reactive protein. FEBS Lett. 2009;583:1001–1005. doi: 10.1016/j.febslet.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 50.Shrive AK, Burns I, Chou HT, Stahlberg H, Armstrong PB, Greenhough TJ. Crystal structures of Limulus SAP-like pentraxin reveal two molecular aggregations. J Mol Biol. 2009;386:1240–1254. doi: 10.1016/j.jmb.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 51.Motie M, Schaul KW, Potempa LA. Biodistribution and clearance of 125I-labeled C-reactive protein and 125I-labeled modified C-reactive protein in CD-1 mice. Drug Metab Disp. 1998;26:977–981. [PubMed] [Google Scholar]