

Abstract
Biogenic amine transporters for serotonin, norepinephrine and dopamine (SERT, NET and DAT respectively), are the key players terminating transmission of these amines in the central nervous system by their high-affinity uptake. They are also major targets for many antidepressant drugs. Interestingly however, drugs targeted to a specific transporter do not appear to be as clinically efficacious as those that block two or all three of these transporters. A growing body of literature, reviewed here, supports the idea that promiscuity among these transporters (the uptake of multiple amines in addition to their “native” transmitter) may account for improved therapeutic effects of dual and triple uptake blockers. However, even these drugs do not provide effective treatment outcomes for all individuals. An emerging literature suggests that “non-traditional” transporters such as organic cation transporters (OCT) and the plasma membrane monoamine transporter (PMAT) may contribute to the less than hoped for efficacy of currently prescribed uptake inhibitors. OCT and PMAT are capable of clearing biogenic amines from extracellular fluid and may serve to buffer the effects of frontline antidepressants, such as selective serotonin reuptake inhibitors. In addition, polymorphisms that occur in the genes encoding the transporters can lead to variation in transporter expression and function (e.g. the serotonin transporter linked polymorphic region; 5-HTTLPR) and can have profound effects on treatment outcome. This may be accounted for, in part, by compensatory adaptations in other transporters. This review synthesizes the existing literature, focusing on serotonin to illustrate and revive a model for the rationale design of improved antidepressants.
Keywords: serotonin transporter, norepinephrine transporter, dopamine transporter, organic cation transporter, plasma membrane monoamine transporters, antidepressants
1. Introduction
Biogenic amine transporters, including the serotonin, norepinephrine and dopamine transporters (SERT, NET and DAT respectively) are pivotal players regulating neurotransmission by the high-affinity uptake of serotonin (5-HT), norepinephrine (NE) and dopamine (DA) released from nerve terminals. While today this statement can be made in an almost cavalier fashion, tremendous advances made in our understanding of biogenic amine transporters since the initial discovery that [3H]NE could be taken up into organs containing sympathetic nerves (Whitby et al., 1961) should not be understated. For example, until relatively recent times (within the last two decades), it was believed that these transporters resided statically in the intrasynaptic plasma membrane of nerve terminals, where they served to faithfully take up neurotransmitter upon its release into the synaptic cleft. With the advent of molecular biological techniques came the cloning of these transporters (Blakely et al., 1991; Hoffman et al., 1991; Pacholczyk et al., 1991; Giros and Caron, 1993) and an explosion in our understanding of how these important proteins work. We now know that these transporters are not static and can be dynamically regulated (for reviews see, Torres et al., 2003b; Blakely et al., 2005; Mandela and Ordway, 2006; Miranda and Sorkin, 2007), including being trafficked in and out of the plasma membrane in response to numerous and diverse stimuli, and by a host of receptor triggered signal transduction pathways (Blakely et al., 1998; Ramamoorthy and Blakely, 1999; Daws et al., 2000; and for reviews see, Blakely and Bauman, 2000; Robinson, 2002; Zahniser and Sorkin, 2004; Steiner et al., 2008). We know that they can form oligomers (Torres et al., 2003a; Chen and Reith, 2008; and for review see, Sitte et al., 2004), that they can operate in different modes (alternating access or channel mode) (Galli et al., 1996; Kahlig et al., 2005) and that polymorphisms in these transporters can determine their expression and activity level (Lesch et al., 1996; and for reviews see Bannon et al., 2001; Bönisch and Brüss, 2006; Murphy and Lesch, 2008). Because these transporters are primary sites of action for both psychotherapeutic drugs, such as those used for the treatment of depression (fluoxetine or “Prozac”) and ADHD (methylphenidate or “Ritalin”), as well as for abused drugs such as 3,4-methylenedioxmethamphetamine (MDMA) and cocaine (Blakely et al., 1998; Ramamoorthy and Blakely, 1999; Dutta et al., 2002; and for reviews see, Amara and Sonders, 1998; Schenk, 2002; Gether et al., 2006; Iversen 2006; Fleckenstein et al., 2007), the relationship between polymorphisms in these transporters and drug response has come under intense scrutiny.
One aspect of biogenic amine transporter function that has received relatively less attention is that of their “promiscuous nature”. The idea of transporter promiscuity heralds back to the 1960s when several groups provided evidence that 5-HT could be accumulated by catecholaminergic neurons (Bertler et al., 1964; Burgen and Iverson, 1965, Lichtensteiger et al., 1967; Fuxe and Ungerstedt, 1967). Uptake of 5-HT into rat brain slices was defined by at least two processes. These were termed “uptake-1” and “uptake-2”. Uptake-1, the “SERT”, had a high affinity but low capacity to take up [3H]5-HT and uptake-2, then thought to be the “NET”, had low affinity but high capacity to take up [3H]5-HT into brain slices (Burgen and Iverson, 1965; Shaskan and Snyder, 1970). Conclusions drawn from such studies were that 5-HT, even in relatively low concentrations, may enter catecholaminergic neurons in significant amounts. This line of investigation was given additional impetus with the discovery that many antidepressants were inhibitors of both NE and 5-HT uptake (Glowinski and Axelrod, 1966; Ross and Renyi, 1969). These early discoveries form the basis of this review, which describes recent advances in our understanding of biogenic amine transporter promiscuity and implications for the development of therapeutics for psychiatric disorders.
2. Transporter Location Sets the Scene for Transporter Promiscuity
The concept of paracrine or volume transmission (Fuxe and Agnati, 1991; Fuxe et al., 2007) as the primary mode for monoamine neurotransmission is now well entrenched in our current thinking. In contrast to wiring transmission, where there exists “quasi contact” between neurons, volume transmission is mediated by diffusion of neurotransmitter through the extracellular matrix to its target (for detailed review and historical detail see Fuxe et al., 2007). Ultrastructural studies reveal SERT, NET and DAT to be located in the plasma membrane outside the synaptic cleft. They are located in the perisynaptic area and sometimes along axons and dendrites (Sur et al., 1996; Nirenberg et al., 1996 & 1997, Zhou et al., 1998, Pickel and Chan 1999; Schroeter et al., 2000; Miner et al., 2000 & 2003; Liprando et al., 2004); therefore they are positioned such that neurotransmitter must reach these transporters by volume transmission (see Figure 1).
Figure 1.
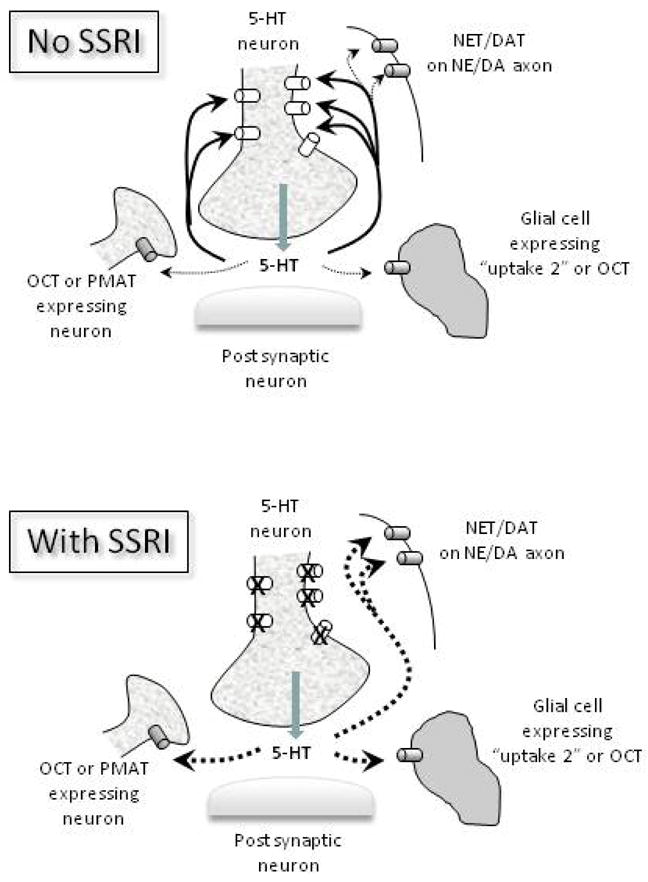
Schematic representation of volume (paracrine) transmission for 5-HT and how it may influence the contribution of SERTs (open cylinders) and other plasma membrane transporters (NET, DAT, OCT, PMAT) (shaded cylinders) in mediating uptake of 5-HT in brain. The top panel shows that in the absence of uptake blockers, uptake of 5-HT is mediated primarily by the high affinity-low capacity SERT (dark solid arrows), with only minor contribution from other plasma membrane transporters (light dashed arrows). The lower panel shows that when an SSRI is present, 5-HT uptake continues via low affinity-high capacity transporters for 5-HT located either on neighboring neurons or axons (NET, DAT, OCT, PMAT) or glial cells (OCT and “uptake 2” transporters) (dark dashed arrows). Similar mechanisms of 5-HT uptake may occur when SERT expression or function is genetically reduced.
An elegant study by Bunin and Wightman (1998) provides functional support for these anatomical findings. Using fast scan cyclic voltammetry to measure 5-HT release in brain slices, they were able to use mathematical models to estimate synaptic concentrations of 5-HT. This was determined to be in the range of 6 mM, a concentration that greatly exceeds the affinity of 5-HT receptors and the SERT for 5-HT. These investigators calculated that 5-HT must diffuse greater than 20 μm from its release site for extrasynaptic concentrations to diminish to the nanomolar range, a range typical for receptor affinity values calculated using in vitro methods. Notably the distance that 5-HT is able to diffuse from its release site implies that 5-HT is able to interact with many extrasynaptic elements and not just those receptors and transporters specific to 5-HT neurons (Figure 1). Similar synaptic concentration and diffusion distances have been estimated for other neurotransmitters such as glutamate and DA (Clements 1996; Cragg and Rice, 2004).
3. What Can Affinity Values Tell Us?
These new estimates of synaptic neurotransmitter concentration bring an interesting twist to the way we think about biogenic amine transporters in terms of their affinity (Km) and maximal capacity to transport (Vmax) various substrates. For example, in vitro preparations made from homogenates, synaptosomes or transfected cell lines provide an excellent means by which to estimate Km and Vmax values, particularly in terms of the relative selectivity of a transporter for various substrates. These systems allow transporters to be studied in relative isolation and typically result in Km values in the nanomolar range for the “native” substrate (e.g. DA for DAT). Interestingly, however, when affinity values for transporters have been estimated using in vivo approaches, as afforded by voltammetry and chronoamperometry, the range shifts to the micromolar level (Wightman and Zimmerman, 1990; Zahniser et al., 1999). For example, Zahniser and colleagues determined the in vivo affinity value (denoted KT to distinguish it from Km values derived in vitro) of DAT for DA in striatum to be 12 μM, a value greatly exceeding the range reported for Km values determined using in vitro methods (0.1–0.4 μM) (See Table 1 in Nicholson, 1995). Even when corrected for volume fraction (α), which reduces bias in the estimate due to diffusion, the KT value was 2.5 μM, still 6-fold higher than Km values determined using in vitro methods. Likewise, estimates for the KT of SERT for 5-HT are consistently higher (e.g. 1.0 – 2.0 μM) than Km values derived in vitro (e.g. 0.01–0.5 μM) (see Codd and Walker 1987; Shaskan and Snyder, 1970; Masson et al., 1999; Montañez et al., 2003, Daws et al., 2005). Since these in vivo KT estimates have already been corrected for diffusion, a plausible explanation for the inflated affinity values in vivo (i.e. greater KT value or lower affinity compared to Km value) could be the presence of other transporters, low affinity-high capacity transporters, skewing the affinity value upward (see also Daws et al., 2005); a reasonable assertion given these measures of neurotransmitter clearance are made in the heterogeneous environment of intact brain where volume transmission is occurring.
Of course, many factors regulate extracellular fluid levels of neurotransmitter, including release rate, amount of transmitter released, rate of degradation, rate of diffusion, genetic influence on these processes and so on. Based on the strong evidence for biogenic amine transmission to occur via volume transmission (discussed below), the question is not will any given neurotransmitter reach a heterogeneous population of transporters, but rather, what factors influence how much will reach any given transporter? - The focus of this review.
4. Functional Evidence for Promiscuous Transport
4.1. Heterologous Uptake of Dopamine and Norepinephrine
Existence of heterologous uptake of DA by the NET is arguably the most comprehensively documented to date (see Carboni and Silvagni, 2004 for additional review). Several groups have independently shown that extracellular DA is increased following blockade of the NET. For example, NET blockade in prefrontal cortex leads to a significantly greater increase in extracellular DA than blockade of DAT (Carboni et al., 2006), consistent with the greater density of NET relative to DAT in this brain region (Raiteri et al., 1977; Moll et al., 2000). In another study using DAT knockout (KO) mice, Morón and co-workers (2002) showed the NET blocker, nisoxetine, inhibited 40% of DA uptake into synaptosomes prepared from the nucleus accumbens. In contrast, and consistent with the very sparse expression of NET in caudate, NET blockers failed to inhibit DA uptake in this region (Carboni et al., 1990; Jones et al., 1998; Morón et al., 2002).
Evidence for DA uptake by the SERT has also come from studies in KO mice. Cocaine, which is a non-selective biogenic amine transporter inhibitor, was found to increase extracellular DA in the striatum of DAT KOs but not in striatum of DAT/SERT double KO mice, suggesting that DA is taken up by the SERT (Shen et al., 2004). Similarly, in rats treated with the neurotoxin 6-hydroxydopamine (6-OHDA) to lesion striatal DA terminals, L-3,4-dihydroxyphenylalanine (L-DOPA) derived DA was increased by the selective serotonin reuptake inhibitor (SSRI) fluoxetine (Kannari, et al., 2006). While these data support the idea of uptake of DA by striatal SERT, there may also be a contribution by NET. At the doses of fluoxetine used in the Kannari et al., (2006) study, NET would also be blocked. However, NET is sparsely expressed in striatum (Tejani-Butt, 1992) and so any contribution of DA clearance by NET would likely be small by comparison.
Uptake of NE by the SERT has also been documented, again using gene KO technology, this time of the NET. Vizi and coworkers (2004) showed that although [3H]NE uptake was greatly retarded in mice lacking the NET, uptake was still measurable (12.6% and 33.5% in hippocampus and cortex respectively). This residual uptake could be essentially abolished by co-incubation of synaptosomes with the SSRI, citalopram. Similarly, release of [3H]NE was eliminated by greater than 90% in synaptosomes from NET KO mice when citalopram was present during loading (Vizi et al., 2004). These data provide compelling evidence that serotonergic varicosities can accumulate and release NE.
4.2. Heterologous Uptake of Serotonin
The DAT was first shown to be capable of taking up and releasing 5-HT by Stamford et al., 1990 and later Jackson and Wightman (1995). More recently, other groups have demonstrated DAT-mediated uptake of 5-HT in striatum when extracellular 5-HT was elevated by exogenous application or by administration of an SSRI (Callaghan et al., 2005; Zhou et al., 2005). Moreover, 5-HT and DA could be co-released from DA terminals in striatum (Zhou et al., 2005). The DAT also appears to be a compensatory mechanism for 5-HT uptake in SERT KO mice (Pan et al., 2001; Zhou et al., 2002) as well as mice with a double KO of SERT and monoamine oxidase A (MAOA) (Mössner et al., 2006). Similarly, the NET can take up 5-HT. For example, when 5-HT terminals in the dentate gyrus of hippocampus are eliminated by treatment with the 5-HT neurotoxin 5,7-dihydroxytryptamine, 5-HT clearance can be inhibited by the NET blocker, desipramine, but not by a SSRI (Daws et al., 1998). In intact rats, when extracellular 5-HT concentration is driven higher by exogenous application, DMI more potently inhibits 5-HT clearance than the SSRI, fluvoxamine (Daws et al., 2005).
Consistent with transporter promiscuity, more recent studies have revealed inhibitory actions of alcohol on both 5-HT and DA clearance in SERT and DAT KO mice respectively (Daws et al., 2006; Mathews et al., 2006). Clearly alcohol is acting on transporters other than, or in addition to SERT and DAT to inhibit the uptake of these biogenic amines. While the sites of action mediating these effects of alcohol remain unclear, what is evident is that pharmacologic and genetic manipulations can unmask transport systems “unfaithful” to their neurotransmitter.
The body of work described thus far underscores the tremendous redundancy and plasticity of uptake systems, particularly the SERT, NET and DAT, which are closely linked to a host of psychiatric disorders including depression, anxiety and addiction. However, the critical question remains. Does promiscuous uptake of neurotransmitter have biological and therapeutic relevance? The following sections seek to answer this question by focusing on mechanisms for 5-HT uptake and revisiting established models for treatment strategies for depression.
5. Biological Significance of Transporter Promiscuity for Serotonin
While there is no doubt that the SERT is the “prime-mover” of active 5-HT uptake from extracellular fluid, as just discussed, substantial evidence for uptake of 5-HT by other transporters, including the NET and DAT has amassed over the past 5 decades (e.g. Burgen and Iverson, 1965; Shaskan and Snyder,1970; Jackson and Wightman, 1995; Daws et al., 1998 and 2005; Pan et al., 2001; Zhou et al., 2002; Callaghan et al., 2005; Zhou et al., 2005). The studies discussed herein suggest that these alternative mechanisms for 5-HT transport play a larger role when extracellular levels of 5-HT are very high and/or when SERT function/expression is compromised or eliminated (e.g. genetically or pharmacologically). These findings are particularly intriguing given growing evidence that a mutation in the promoter region of the human SERT gene, which leads to reduced SERT expression, is associated with increased risk for emotional disorders and alcoholism (Feinn et al., 2005; Rausch, 2005; Wurtman, 2005; Munafò et al., 2008). Moreover, they raise important considerations for the use of SSRIs and other therapeutics used in the treatment of disorders such as depression, anxiety and alcoholism. For example, patients who do not respond well to SSRIs may have a greater relative amount of functioning NET, (or other transporter capable of 5-HT uptake), than do responders. One could speculate that greater NET (or other transporter) activity, by taking up 5-HT, would serve to reduce the overall ability of SSRI treatment to elevate extracellular 5-HT, presumably one of the first critical steps in achieving therapeutic benefits. Taken together with the rapidly expanding literature describing interactions between genetically defined expression of the SERT (e.g. serotonin transporter linked polymorphic region, 5-HTTLPR polymorphism) (e.g. Collier et al., 1996; Smits et al., 2004), environment (e.g. Caspi et al., 2003) and response to drug treatment (e.g. Eichhammer et al., 2003; Smits et al., 2004; Serretti et al., 2007), the design of a biological system with built in functional redundancies makes evolutionary sense in order to maintain homeostasis of serotonergic neurotransmission. As discussed in subsequent sections, consideration of these alternative mechanisms for 5-HT uptake may lead to the development of improved therapeutics for numerous disorders.
5.1 Polymorphisms of SERT
Of growing interest to clinical researchers is the relationship between SERT expression and propensity for disease. In particular, the relationship between carriers of the low expressing (s) variant of the 5-HTTLPR, drug abuse and mental disorders, has received enormous attention in recent years; however the neurochemical underpinnings remain poorly understood. Given carriers of one or two copies of the s allele constitute more than half of the population, the relationship between SERT expression and drug efficacy remains an area of intense research. As discussed, a common feature of drugs used to treat depression and related disorders is that they produce an acute increase in levels of extracellular 5-HT, even though therapeutic efficacy takes some weeks to emerge. This delay is thought to be due to the time needed for compensatory desensitization of 5-HT1A autoreceptors (Blier et al., 1987; Gardier et al., 1996; Artigas et al., 1996) and likely other adaptive changes. Today the most widely prescribed treatment for depression and related disorders are the SSRIs, such as fluoxetine (Prozac®), which act at the SERT. In this regard, the low expressing variant of the human 5-HTTLPR creates a conundrum. For example, SERT+/− mice, which express half as many SERTs as wildtype mice, have elevated levels of 5-HT. Like these mice, if we assume that individuals carrying the s allele have lower SERT expression and elevated extracellular 5-HT basally, it is curious and somewhat paradoxical then, that these individuals are not less prone to, or even protected from such disorders. In fact, when combined with stressful life events, these individuals are more likely to have episodes of depression (e.g. Caspi et al., 2003; Wilhelm et al., 2006).
Of course, whether extracellular concentrations of 5-HT are increased in low expressing variants of the SERT remains unknown. Where humans and non-human primates are concerned, the only clues available to date come from studies reporting cerebral spinal fluid levels of 5-hydroxyindolacetic acid (5-HIAA, the metabolite of 5-HT), an indirect measure of extracellular 5-HT in brain (Williams et al.,2001; Bennett et al., 2002; Lesch, 2005). The results have been variable, but appear to be dependent upon both 5-HTTLPR genotype and environment in which the individual is raised. Clearly there is complex interplay between genes and environment and the ultimate outcome on 5-HT neurotransmission. Manipulations of the murine SERT are helping to unravel these mysteries.
5.2 Lessons Learned from SERT+/− and −/− Mice
SERT mutant mice provide an excellent model system to study regulation of 5-HT clearance by novel transporters. SERT mutant mice do have elevated levels of extracellular 5-HT, at least in striatum and frontal cortex where microdialysis measures have been made (Shen et al., 2004; Mathews et al., 2004; and see Murphy and Lesch, 2008 for review). Given that increased extracellular fluid 5-HT is linked to therapeutic efficacy of many frontline antidepressants, this observation has raised the question, why then, would medications designed to elevate levels of 5-HT in an already elevated state, be expected to lessen signs of depression? A likely explanation is that it is not the basal level of 5-HT that determines therapeutic response but rather the change in extracellular 5-HT, which follows immediately after treatment, that is important in initiating the cascade of events leading to antidepressant effect, regardless of basal 5-HT level. This idea is supported by findings that 5-HTT mutant mice are normal in a range of neurological and behavioral tests (Holmes et al., 2002 and 2003), including tests for depression, however, they do respond differently from wildtype mice to challenge with antidepressant drugs (although this does vary according to background strain of the mouse) (Holmes et al., 2002). Similarly, humans with the low expressing form of the 5-HTTLPR (s/l and s/s) are normal in a large range of neurological tests, however they do exhibit an exaggerated response to emotional/stressful stimuli (Hariri and Holmes, 2006; Munafò et al., 2008 for reviews) and interestingly, are often resistant to treatment with SSRIs (Rausch et al., 2002; Rausch, 2005; Serretti et al., 2007 for reviews).
Can transporter promiscuity account, at least in part, for the apparent paradoxes summarized above? Published literature indicates it can. For example, using in vivo chronoamperometry to measure clearance of 5-HT applied exogenously into the CA3 region of hippocampus in concentrations up to 8 micromolar, Montañez and co-workers (2003) reported that 5-HT clearance, as predicted, was slower in SERT−/− and SERT+/− mice than wildtype mice. Consistent with these data and a diminished ability to recapture released 5-HT, others using microdialysis, reported increased extracellular 5-HT in SERT+/− and SERT−/− mice (Shen et al., 2004; Mathews et al., 2004). However, in vivo chronoamperometry studies, have more recently revealed that when 5-HT signal amplitudes produced by exogenous application of 5-HT exceed ~ 20 μM, 5-HT clearance rate between wildtype and SERT−/− mice becomes indistinguishable (see Daws and Toney, 2007). While the apparent maximal velocity (Vmax) for 5-HT clearance was not different between genotypes, there was a marked shift to the right in the concentration effect curve in SERT mutant mice indicative of a low affinity, but high capacity transporter mechanism for clearing 5-HT (Daws and Toney, 2007). These data are also in keeping with the ideas discussed in section 3.0; that KT values determined using in vivo approaches are inflated (i.e. lower affinity) compared to those determined using in vitro approaches, in part due the presence of multiple transporters for any given neurotransmitter.
As discussed earlier, many factors can influence clearance of neurotransmitter in extracellular fluid, including enzymatic breakdown. Monoamine-oxidase (MAO)-A and MAO-B break down 5-HT. It is worth noting that the level of MAO-A and MAO-B do not differ among SERT genotypes (Mathews et al., 2004), and so cannot account for shifts in clearance rate reported among SERT+/+, +/− and −/− mice as a function of increasing extracellular 5-HT concentration.
An interesting caveat then, is that SERT+/− and SERT−/− mice do have significantly higher levels of extracellular 5-HT, consistent with a reduced ability to recapture released 5-HT (Shen et al., 2004; Mathews et al., 2004). These data at first lend themselves to the interpretation that uptake of 5-HT by another transporter does not fully compensate for reduction or loss of SERT to maintain “normal” 5-HT neurotransmission. However, there are several important considerations:
The concentration of extracellular 5-HT in striatum, according to zero-net flux microdialysis measurements (corrected for extraction fraction), is in the order of 3, 9 and 18 nM in SERT+/+, +/− and −/− mice respectively. In frontal cortex concentrations are lower; ~ 1.5, 3.5 and 14 nM for SERT+/+, +/− and −/− mice respectively (Mathews et al., 2004).
As discussed in section 2, current estimates for synaptic concentration of 5-HT in brain are around 6 mM (Bunin and Wightman, 1998). Serotonin is estimated to diffuse greater than 20 μm away from the synaptic cleft, where the concentration of 5-HT falls to the micromolar and nanomolar range (Bunin and Wightman, 1998 & 1999). Thus, the “low” micromolar amounts of 5-HT required to reach Vmax for 5-HT clearance measured using in vivo chronoamperometry fit well within the current framework of our knowledge (Daws and Toney, 2007, and see Figure 2).
Based on these estimates, it is somewhat surprising that extracellular 5-HT, measured by in vivo microdialysis, is increased by only 6 to 10-fold in SERT−/− mice and not much higher; for example, 1000-fold higher would shift the concentration of extracellular 5-HT from a nanomolar to micromolar range. Of course, the fact that recordings were made in different brain regions (striatum and frontal cortex for microdialysis and CA3 region of hippocampus for chronoamperometry), may also contribute to these differences (Mathews et al., 2004; Daws and Toney, 2007). That notwithstanding it is plausible that extracellular 5-HT does not reach the micromolar range because of the presence of alternate mechanisms for 5-HT transport. Based on the premise that these alternate transporters have low affinity, but high capacity to take up 5-HT, their affinity for 5-HT imposes a threshold; i.e. a set concentration of 5-HT must be reached in the extracellular matrix before these transporters contribute significantly to uptake (Figure 2). Following this line of reasoning, microdialysis studies (Mathews et al., 2004) indicate a threshold in the order of 14–18 nM. Based on chronoamperometry data (Daws and Toney, 2007), the threshold appears to be in the range of 10–15 micromolar. It is worth keeping in mind the dissimilarities between these sampling techniques, (summarized in Table 1). Compared to chronoamperometry, microdialysis samples from a considerably larger pool of neurotransmitter (probes ranging 1–3 mm in length and 250–350 μm in diameter) and with relatively long sampling intervals (5–20 min). Electrodes used for chronoamperometry are markedly smaller (carbon fiber recording tip in the order of 50–300 μm long and 5–30 μm in diameter) and sampling rates are in the millisecond range. Thus, the electrochemical approaches of chronoamperometry and other forms of voltammetry afford sampling from a neurotransmitter pool only microns away from neural terminals (Borland and Michael, 2007; and Table 1). Given the differences between these sampling techniques, it is reasonable to conclude that data obtained from both microdialysis studies and chronoamperometry studies support this “threshold” hypothesis and that the absolute molar value of this threshold varies somewhat based on the technique applied. Moreover it is possible that different thresholds for different transporters may exist and so would be dependent on regional distribution of each transporter in brain as well as their activity state.
Figure 2.
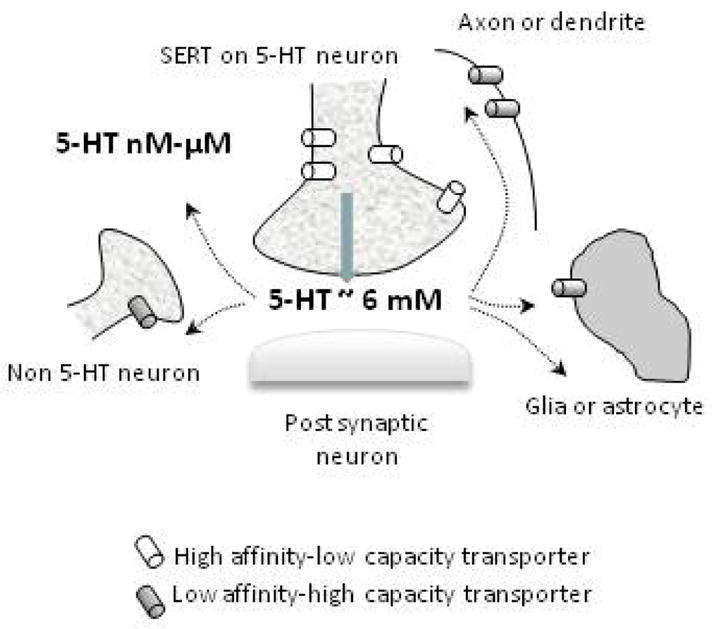
Keeping the brake on extracellular serotonin. Synaptic concentrations of 5-HT are estimated to reach the millimolar range. When SERT expression and function is intact, uptake of 5-HT is mediated primarily by the SERT (high affinity-low capacity). This activity serves to help constrain extrasynaptic concentrations of 5-HT in the micromolar-nanomolar range. When SERT expression or function is compromised (e.g. pharmacologically, genetically), low affinity-high capacity transporters for 5-HT, such as the NET and DAT, help to prevent extrasynaptic serotonin from remaining in high micromolar to millimolar ranges (see text for details).
Table 1.
Comparison of microdialysis and chronoamperometry for recording biogenic amine neurotransmitter in vivo.
| MICRODIALYSIS | CHRONOAMPEROMETRY | |
|---|---|---|
| Recording dimensions | ~1000–3000 μm long 200–350 μm diameter |
~50–300 μm long 5–30 μm diameter |
| Neurotransmitter Pool Sampled | Large Surface area of probe* ~0.66 to 3.39 mm2 |
Small Surface area of carbon fiber electrode* ~0.81 × 10−3 to 0.06 mm2 |
| Sampling Time | Minutes | Milliseconds |
| Ability to Decipher Basal Neurotransmitter Concentration | Good | Poor |
| Ability to Decipher Release from Uptake | Poor | Good |
| Ability to Measure Kinetics of Uptake | No | Yes |
based on equation for surface area of a cylinder with recording area on bottom dimension only πr2 + (2πr)h, where r is the radius and h is the length of the cylinder.
6. Old Ideas, New Discoveries
Promiscuous uptake of biogenic amine transporters is not a new idea, as evidenced by the pioneering work of several groups in the 1960s and 1970s (Bertler et al., 1964; Lichtensteiger et al., 1967; Fuxe and Ungerstedt, 1967; Shaskan and Snyder, 1970). However, it took until the 1990s and 2000s for this notion to become accepted as physiologically significant and of potential impact to the design of therapeutics such as antidepressants. In 2004, Schildkraut and Mooney proposed the extraneuronal monoamine transporter (uptake 2) hypothesis as a means to speed antidepressant action. Their hypothesis was based on their own work as well as a thorough review of the literature, which turned up the consistent finding that treatment with antidepressant drugs that block the NET led to a gradual increase in normetanephrine, the O-methylated metabolite of NE (Schildkraut and Mooney, 2004). Normetanephrine is a potent inhibitor of uptake 2 (Burgen and Iversen, 1965; Männistö PT and Kaakkola, 1999). Thus, uptake of NE by the extraneuronal monoamine transporter can be reduced by the presence of normetanephrine leading to further increases in extracellular NE. Schildkraut and Mooney (2004) proposed the hypothesis that drugs that increase normetanephrine, or that block the extraneuronal transporter (uptake 2), will reduce the latency to clinical efficacy of antidepressants, such as desipramine, which block NE uptake. While evidence supporting this hypothesis is growing (Mooney et al., 2008), debate continues regarding the identity of “uptake 2”. Some schools of thought believe “uptake 2” is the organic cation transporter (OCT), specifically the OCT3 subtype, while others dispute this idea. The main reason for this disagreement lies in the location of these transporters. “Uptake 2” is thought to be located exclusively on glia (Burgen and Iversen, 1965; Hendley et al., 1970; Henn and Hamberger, 1971; Streich et al, 1996; Grüdemann et al., 1998), whereas OCT3 is located predominantly on neurons (Wu et al., 1998; Kristufek et al., 2002; Schmitt et al., 2003; Vialou et al., 2004). It seems likely that “uptake 2” may more accurately describe a group of transporters, including the OCTs and plasma membrane monoamine transporters (PMAT), which have low affinity, but high capacity to transport biogenic amines relative to their “parent”, high affinity-low capacity transporters the SERT, NET and DAT. Indeed, current consensus is that “uptake 1” encompasses these Na+ and Cl− dependent high affinity plasma membrane transporters, while “uptake 2” encompasses Na+ and Cl− independent high capacity transporters for the biogenic amines.
6.1 Organic Cation Transporters
OCTs were first cloned in 1994 by Gründemann and colleagues. They belong to the SLC22 family of transporters and are widely distributed in brain. There are 3 subtypes of OCTs (OCT 1, 2 & 3). OCT3 is densely expressed in brain compared to OCT1. OCT2 expression levels in brain are very low (Gorboulev et al., 1997; Gründemann et al., 1997). OCT3 expression is particularly rich in cerebellum, subfornical organ, dorsal raphe, hypothalamic nuclei as well as cortex and hippocampus (Wu et al., 1998; Amphoux et al., 2006). Importantly, OCTs are capable of taking up 5-HT, NE and DA as well as amines such as histamine (Busch et al., 1996 & 1998; Koepsell and Endou, 2004; Amphoux et al., 2006; Gasser et al., 2006; for reviews see Koepsell et al., 2003 and 2007). Vialou and co-workers (2004), using OCT3 deficient mice, showed that OCT3 is critical for the regulation of salt intake (in the form of hypertonic saline solution or low sodium chow). This study was the first to provide physiological evidence for the importance of OCTs in central nervous system function. Salt-sensitive hypertension may be linked to dysfunction of OCTs and perhaps contributes to comorbid hypertension, heart disease and depressive disorders (Esler and Parati, 2004; Critchley et al., 2005). Amphoux et al. (2006) recently found that HEK293 cells transfected with rOCT showed MDMA-sensitive inhibition of [3H]1-methyl-4-phenylpyridium ion (MPP+) uptake, raising the possibility that some of MDMA’s effects are mediated via OCTs. Similarly, others have shown that methamphetamine can block DA transport via OCT3 (Wu et al., 1998; Nakayama et al., 2007).
Although their affinity is much lower for 5-HT than that of the SERT, OCTs have a high capacity to transport 5-HT (Busch et al., 1996). For example, Feng and co-workers (2005) reported that perfusion of the OCT blocker, decynium 22 (D-22), via a dialysis probe in the medial hypothalamus produced robust and dose-dependent increases in dialysate 5-HT. The magnitude of this effect (~200–650% increase, depending on dose) is remarkably comparable to that reported for the SSRI, fluoxetine, delivered by the same route to the hypothalamus (~400%, increase in extracellular 5-HT) (Maswood et al., 1999).
Reports that OCT1 (Chen et al., 2001) and OCT3 (Schmitt et al., 2003; Baganz et al., in press) expression are increased in brains of SERT+/− and SERT KO mice support the idea that, like DAT, OCTs may compensate at least partially, for reduced or absent SERT expression. It is conceivable therefore, that transport of 5-HT by OCT in primate and human carriers of the low functioning variant of the 5-HTTLPR may constrain extracellular 5-HT to relatively “normal” levels. Given that corticosterone blocks OCT (Wu et al., 1998; Arndt et al., 2001; Gasser et al., 2006), it is tempting to speculate that if OCTs are increased in individuals carrying the low expressing variant of the 5HTTLPR, this “imbalance” in transporter expression may contribute to the susceptibility of these individuals to early life stress and depression in adulthood (see Caspi et al., 2003).
6.2 Plasma Membrane Monoamine Transporter
The PMAT, also known as equilibrative nucleoside transporter 4 (ENT4), was cloned and characterized by the Wang group (Engel et al., 2004; Engel and Wang, 2005). PMAT is not homologous with SERT, NET or DAT but shares approximately 20% homology with the ENT family (SLC29). PMAT favors uptake of 5-HT and DA over other monoamines and in cell expression systems the transport efficiency (Km × Vmax) for 5-HT and DA falls only marginally short of that for SERT and DAT (Engel et al., 2004). The apparent affinities (Km) of PMAT for 5-HT and DA are 2–3 orders of magnitude lower than those of SERT and DAT. However, the Vmax values of PMAT-mediated 5-HT or DA transport are 2–3 orders of magnitude higher. Together, these data suggest that PMAT is a low affinity but high capacity transporter for 5-HT and DA (Engel et al., 2004). PMAT is expressed widely throughout brain but is most richly expressed in forebrain cortex, olfactory tubercle, hippocampus, cerebellum and epithelial cells of the choroid plexus. Notably, PMAT is expressed on neurons and not on astrocytes (Dahlin et al., 2007; Vialou et al., 2007). While studies of PMAT expression and function in mice deficient in SERT, NET or DAT remain to be investigated, the possibility exists, that like OCT3, PMAT offers a compensatory alternative for uptake of biogenic amines. Importantly, the regional distribution of both PMAT and OCT3 in brain overlaps largely with that for SERT, NET and DAT. These transporters are therefore anatomically positioned to play a significant role in maintaining homeostasis of biogenic amine neurotransmission.
7. “Old Dog, New Tricks”: New Vistas for Therapeutics
The over 40-year-old monoamine hypothesis of depression, postulating a functional deficiency of noradrenergic or serotonergic transmission in brain, stemmed from studies examining the acute pharmacologic actions of tricyclic antidepressants (TCAs) and MAO inhibitors (MAOIs) (Prange, 1964; Schildkraut, 1965; Coppen, 1967; Lapin and Oxenkrug, 1969; Carlsson et al., 1969; and see Frazer, 1997 for review). It has been proposed that drugs that augment the increase in extracellular 5-HT and/or NE that follows treatment with SSRIs, tricyclic and related antidepressants, may speed therapeutic efficacy. Indeed, a shift back to less selective compounds is the current trend (Skolnick et al., 2006). First generation antidepressants, such as the tricyclic antidepressants, although reported to have dual action to inhibit uptake of biogenic amines by the SERT and NET according to in vitro analyses, were found in vivo to affect primarily NE uptake (for review see Frazer, 1997). Second generation antidepressants, which include the SSRIs, were developed to be highly selective for one uptake mechanism. While these antidepressants generally had a much improved side-effect profile compared to TCAs, antidepressant efficacy was not greatly increased (Frazer, 1997). Indeed, a recent meta-analysis reported that SSRIs were no more effective than placebo in a significant majority of depressed patients (Kirsch et al., 2008). More recently, clinical use of antidepressant drugs with dual activity, to block the SERT and NET, has increased. However, again they are not fully satisfactory because of their inability to reduce the delay in onset of therapeutic effect.
Of course a role for DA and the DAT in depression should not be overlooked, as recently reviewed by Montgomery (2008). A pre-clinical study by Kobayashi and colleagues (2008) provides an elegant illustration of this. They found that antidepressants that increased extracellular DA and NE were more efficacious in reducing time spent immobile in the forced swim test (a test for antidepressant efficacy) than antidepressants that increased 5-HT. The converse was true for suppression of marble burying behavior (Kobayashi et al., 2008). Their data, along with others (e.g. Cryan et al., 2005), support the idea that different biogenic amine neurotransmitter systems mediate distinct aspects of depression-like behaviors. Importantly, studies such as these underscore the importance of considering multiple systems in the treatment of affective disorders. Indeed in humans, deficiencies in 5-HT, NE and DA appear to underlie the pathology of major depressive disorder (Nutt, 2006 for review). This has led to the emergence of triple uptake inhibitors, those which block the SERT, NET and DAT (for review see Chen and Skolnick, 2007; Sulzer and Edwards, 2005). So far, this newest class of antidepressants appears to be superior in terms of shortest time to onset of therapeutic benefit. Given what is now known about promiscuity among these transporters, this is perhaps not a surprising finding; one that could potentially be improved upon by consideration of the less well recognized players in regulating biogenic amine neurotransmission, the OCT and PMAT.
OCTs have been the focus of considerable research with respect to their role in peripheral function (e.g. as regulators of the interstitial concentrations of endogenous compounds, drugs and xenobiotics) (Koepsell et al., 2003), but until very recently their role in brain has received little attention. A growing literature points to OCT3 in particular, as a target for augmentation of therapeutic response. For example, Kitaichi and colleagues (2005) tested the antidepressant efficacy of the tricyclic antidepressant, imipramine, in mice treated with antisense to decrease OCT3 expression. Antisense treatment not only decreased immobility in the forced swim test, but augmented the decrease in immobility induced by imipramine. Consistent with these findings, a recent study showed that short term treatment of mice with corticosterone, a blocker of OCT, reduced immobility in the forced swim test. Notably, corticosterone did so to a greater extent than desmethylimipramine (DMI), a NET blocker (and TCA), given via the same route and dose and for the same duration (Stone and Lin, 2008). While it cannot be ruled out that this action of corticosterone is mediated by other targets (e.g. mineralocorticoid receptors), blockade of glucocorticoid receptors did not abolish the antidepressant-like effect of DMI in the forced swim test (Stone and Lin, 2008). Similarly, in a separate study, mice subjected to acute footshock 2–4 min. prior to the tail suspension test (another test for antidepressant activity), spent less time immobile than control mice (Swiergiel et al., 2008). The footshock resulted in a dramatic increase in plasma corticosterone. Thus, the antidepressant-like effect of acute stress might also be explained by acute blockade of OCT3 by corticosterone. In another study, treatment with antisense against OCT3 augmented methamphetamine-induced hyperactivity (Kitaichi et al., 2005), suggesting that OCT blockade also promotes dopamine signaling. Together these data strongly suggest that reduced OCT3 expression, or OCT blockade, enhances monoaminergic neurotransmission. As such, OCTs may represent a novel target for the development of treatments for disorders including depression and drug abuse. Studies from our own group support this notion. We found OCT3 expression and function to be increased in mice with constitutive deficiencies in SERT. Blockade of OCT3 with decynium-22 produced antidepressant-like effects (reduced immobility time in the tail suspension test) in SERT+/− and SERT−/− mice, but had little effect in SERT+/+ mice with a full complement of SERT (Baganz et al., in press).
While studies exploring the potential for PMAT as a therapeutic target are just beginning, evidence already favors this transporter as an excellent candidate. Work from Wang’s group shows that the SSRIs fluoxetine, sertraline, citalopram, fluvoxamine and paroxetine inhibit PMAT with IC50 values 3–4 fold greater than those of SERT (Zhou et al., 2007). Therefore, residual uptake of 5-HT by PMAT after treatment with SSRI may help to explain treatment resistance in certain individuals and suggests that like OCT3, PMAT may be a target for the development of adjunctive or first line antidepressants.
7.1 A Working Model: Considerations
As discussed in this review, focusing on 5-HT transport, several groups have provided evidence for uptake of 5-HT by multiple transporters, that is transporters with a high affinity for 5-HT but with low capacity to transport, and low affinity transporters with high a high capacity to transport the same substrate (Bertler et al., 1964; Burgen and Iverson, 1965, Lichtensteiger et al., 1967; Fuxe and Ungerstedt, 1967; Butler et al., 1988). An elegant study by Butler and colleagues, using the olfactory bulbectomy (OB) model of depression in rats, provided some of the first evidence for low affinity but high capacity transporters in the therapeutic efficacy of antidepressants (Butler et al., 1988). They studied [3H]5-HT uptake into synaptosomes prepared from cortex and amygdala. Importantly, to decipher high affinity and low affinity uptake components they studied 5-HT over two concentration ranges (see Table 2). The first, 0.01–0.5 μM, was inhibited by the SERT blocker, sertraline. The second range, 0.1 – 2.0 μM, was not inhibited by sertraline, consistent with this uptake being mediated by a low affinity – high capacity transporter for 5-HT. Vmax and Km values for high and low affinity uptake of [3H]5-HT into synaptosomes from sham operated rats are shown in Table 2. Compared to sham operated rats, OB rats showed enhancement of both high and low affinity uptake of [3H]5-HT into synaptosomes prepared from cortex and amygdala. This was reflected as an increase in Vmax for both high and low affinity 5-HT uptake, with no change in Km. This enhanced 5-HT uptake could be normalized by chronic treatment (3 weeks, daily i.p. injection) with the SERT blocker, sertraline (10 mg/kg), or the NET blocker, DMI (10 mg/kg). In the case of low affinity 5-HT uptake, Vmax in the OB rats was the only parameter influenced by these antidepressant treatments. In contrast, chronic treatment with sertraline increased the Km value for 5-HT uptake in OB rats, again consistent with some contribution of a low affinity-high capacity transporter for 5-HT (Butler et al., 1988).
Table 2.
Vmax and Km values for [3H]5-HT uptake into synaptosomes prepared from cortex and amygdala of sham operated male Sprague-Dawley rats as a function of 5-HT concentration (from Butler et al., 1988).
| 5-HT concentration (μM) | Vmax (pmoles/mg protein) | Km (μM) |
|---|---|---|
| 0.01–0.5 | 35.70 ± 9.13 | 0.80 ± 0.43 |
| 0.1–2.0 | 266.00 ± 37.00 | 4.30 ± 0.24 |
An especially interesting aspect of this study is that 5-HT uptake could be normalized with equivalent efficiency by chronic treatment with either a SERT blocker or a NET blocker (Butler et al., 1988). This underscores the fact, that although the focus of this review has been on 5-HT, depression and related disorders are complex disorders, involving multiple neurotransmitter systems, including, but not limited to, 5-HT, NE and DA. Indeed, numerous studies, utilizing a variety of approaches have provided evidence for a multiplicity of functionally distinct uptake sites, which also vary widely in their affinities for antidepressant drugs (e.g. Hrdina, 1981; Hughes and Stanford, 1998). For example, fluoxetine (Prozac) appears to stand alone from other SSRIs in that it is the least selective of the SSRIs and profoundly influences catecholaminergic neurotransmission as well as serotonergic function (for review see Stanford, 1996). This complexity makes a role for the new “players”, OCTs and PMAT potentially all the more important in mediating antidepressant response. These transporters are capable of taking up 5-HT, NE and DA, and so their presence likely buffers the effect of all currently prescribed biogenic amine transport blockers. To illustrate this point, 5-HT will be used as an example.
7.1.1. Implications for Treatment Resistance: SSRIs as an example
Buffering of 5-HT uptake by OCTs or PMAT may help explain the greater incidence of treatment resistance to SSRIs, particularly that which occurs in carriers of the s allele of the 5-HTTLPR. Based on our own data generated using high-speed chronoamperometry (Daws and Toney, 2007), together with literature discussed in this review, in particular that of Butler and colleagues (1988), we have developed a model to illustrate the potential therapeutic utility of non-SERT uptake mechanisms in SSRI resistant patients. This is illustrated in Figure 3. For the sake of simplicity and clarity, SERT−/− mice are used to depict the “treatment resistant” cohort in this figure. However, it must be emphasized that the basis of treatment resistance remains largely unknown. It is known however, that there is a strong association between treatment resistance to SSRIs and humans carrying the low expressing variant of the SERT (Rausch et al 2002; Rausch, 2005; Serretti et al., 2007 for reviews). Thus, there is a rationale basis for predicting that a constitutive reduction in SERT expression is related to SSRI treatment resistance.
Figure 3.

Model predicting the efficacy of uptake blockers to inhibit 5-HT clearance from extracellular fluid in brain as a function of extracellular 5-HT concentration and genetically defined expression level of SERT. The left panel shows 5-HT clearance rate and predicted effects of uptake blockers in wildtype (SERT+/+) mice and the right panel for mice lacking SERT (SERT−/−). The solid black line represents a polynomial function showing that clearance of 5-HT is mediated by at least two processes, a high affinity/low capacity transporter (SERT), and likely a combination of low affinity/high capacity transporters for 5-HT (OCT, PMAT, NET, DAT). The efficacy of drugs to inhibit 5-HT clearance is depicted as the distance of the symbols below the polynomial function; solid circles = SSRI, open squares = blocker of OCT, PMAT, NET or DAT. See the text for a detailed description of the model.
In Figure 3 the polynomial function (solid black line) represents hypothetical clearance profiles measured using in vivo chronoamperometry for 5-HT in SERT+/+ mice (left panel) and SERT−/− mice (right panel) and is based on existing literature (see discussion in section 5.2; Mathews et al., 2004; Daws et al., 2007). Although not shown graphically, it is hypothesized that results for SERT+/− mice would be intermediate. Shown on the unshaded side (on the left of each figure) is 5-HT clearance rate mediated primarily by a high affinity, low capacity transporter, in this case, the SERT. Because SERT−/− mice lack SERT, this portion of the curve reflects very slow rates of 5-HT clearance compared to SERT+/+ mice. When the extracellular concentration of 5-HT is “low”, the slow rate of 5-HT clearance in SERT−/− mice is due largely to diffusion of 5-HT away from the recording electrode as well as some active uptake by low affinity-high capacity transporters of 5-HT such as OCT, PMAT, NET and DAT (see Figure 1). Shaded areas on the right side of each figure show 5-HT clearance rates when the extracellular concentration of 5-HT is “high”. Here the profiles for 5-HT clearance are quite similar between SERT+/+ and SERT−/− mice (see also Daws and Toney, 2007). Based on existing literature, clearance of high extracellular concentrations of 5-HT is very likely mediated by one or more low affinity-high capacity transporters for 5-HT – OCT, PMAT, NET or DAT; which of these transporters contributes predominantly to 5-HT clearance is predicted to depend on their expression level in brain regions relative to that of SERT. Based on evidence that OCT3 (Baganz et al., in press), and perhaps other non-SERT transporters, are upregulated in brain of SERT mutant mice, the ability of blockers of these transporters to inhibit 5-HT clearance is likely to be greater in SERT mutant mice than in SERT+/+ mice. This idea is illustrated by the open squares. The squares represent the change in rate of 5-HT clearance after administration of a blocker of OCT, PMAT, NET or DAT. The model predicts inhibition of 5-HT clearance even when the extracellular concentration of 5-HT is relatively “low”. However, the magnitude of this inhibition is predicted to increase with increasing concentration of extracellular 5-HT (shown by the increasing distance of the squares below the polynomial function, black line). In contrast, in SERT+/+ mice, blockers of these transporters will have either no, or very modest inhibitory effects on 5-HT clearance when extracellular 5-HT is “low” due to the presence of the high affinity SERT. When extracellular 5-HT rises, the model predicts that significant inhibition of 5-HT clearance by blockers of OCT, PMAT, NET or DAT in SERT+/+ mice will be revealed. The magnitude of this inhibition will not be as great as that in SERT mutants because (1) these transporters will not be functionally upregulated as in SERT mutant mice and importantly, (2) the effect of these blockers to inhibit 5-HT uptake will be buffered by the presence SERT. This idea is shown by the solid circles. The solid circles represent the expected effect of an SSRI to inhibit 5-HT clearance in SERT+/+ and SERT−/− mice. In SERT−/− mice the circles fall on the polynomial function indicating that regardless of extracellular 5-HT levels, an SSRI will not affect 5-HT clearance. This result is wholly consistent with the absence of SERTs in these mice, the primary molecular target of SSRIs. In contrast, blockade of SERT in SERT+/+ mice will lead to marked inhibition of 5-HT clearance. It is predicted that this will be particularly pronounced at lower concentrations of 5-HT and that the magnitude of this effect will diminish, and perhaps even disappear, as the extracellular concentration of 5-HT increases, due to buffering of uptake by low affinity-high capacity transporters for 5-HT – OCT, PMAT, NET and DAT. The model predicts that in SERT heterozygote (+/−) mice the patterns of 5-HT clearance and inhibition of 5-HT clearance by blockers of these transporters will be intermediate between SERT+/+ and SERT−/− mice but will more closely resemble those in SERT−/− mice. Moreover, the magnitude of effect will be dependent on the relative ratio of SERTs to OCT, PMAT, NET or DAT and as such, will vary with brain region expression of each transporter.
While further studies are needed to validate this model, it provides a rationale basis for resistance to treatment with SSRIs in certain populations. For example, as discussed there is a strong association between those individuals carrying the low expressing variant of the 5-HTTLPR and treatment resistance to SSRIs. Moreover, chronic blockade of SERT with SSRIs may also lead to upregulation of other transporters capable of clearing 5-HT from extracellular fluid. This model may therefore also provide a basis for diminished therapeutic efficacy of SSRIs after long term use (Byrne and Rothschild, 1998). No doubt many factors contribute to treatment resistance. For now at least, development of broader spectrum antidepressant drugs (e.g. triple uptake blockers) appears to be a promising approach (Skolnick et al., 2006; Chen and Skolnick, 2007). According to the model for high and low affinity uptake mechanisms for the biogenic amines as it relates to therapeutic potential, it appears we now have two legitimate new players to factor into our thinking, OCTs and PMAT.
8. Concluding Remarks
The focus of this review has centered on 5-HT, but could be applied to any of the biogenic amine neurotransmitters. Clearly there are converging lines of evidence that novel mediators of 5-HT (and other biogenic amines) uptake may be critically important in human disease. OCT and PMAT provide an exciting new direction for research efforts directed toward discovery and design of improved therapeutics for depression. In addition, the relatively high affinity of abused drugs such as methamphetamine and MDMA for OCTs point to these transporters as significant players in mediating addiction. Similarly, many hormones, including corticosterone, have high affinity for OCTs suggesting that these transporters for the biogenic amines may play a much more important role in maintaining normal neural function than previously thought. Exploration of the role of “non-traditional” transporters for biogenic amine transmitters in brain will undoubtedly uncover distinct and unanticipated functions for this versatile and pleiotropic transporter family.
Acknowledgments
Supported in part from USPHS grant MH64489, NARSAD and the Alcoholic Beverage Medical Research Foundation. Dr. Glenn M. Toney is gratefully acknowledged for his comments on this manuscript.
Abbreviations
- DA
dopamine
- DAT
dopamine transporter
- KO
knockout
- MDMA
3,4-methylenedioxymethamphetamine
- NE
norepinephrine
- NET
norepinephrine transporter
- OCT
organic cation transporter
- PMAT
plasma monoamine transporter
- SSRI
selective serotonin reuptake inhibitor
- 5-HT
serotonin, 5-hydroxytrytamine
- SERT/5-HTT
serotonin transporter
- 5-HTTLPR
serotonin transporter linked polymorphic region
- +/+
wildtype
- +/−
heterozygote
- −/−
null; knockout
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amara SG, Sonders MS. Neurotransmitter transporters as molecular targets for addictive drugs. Drug Alcohol Depend. 1998;51:87–96. doi: 10.1016/s0376-8716(98)00068-4. [DOI] [PubMed] [Google Scholar]
- Amphoux A, Vialou V, Drescher E, Brüss M, Mannoury La Cour C, Rochat C, Millan MJ, Giros B, Bönisch H, Gautron S. Differential pharmacological in vitro properties of organic cation transporters and regional distribution in rat brain. Neuropharmacology. 2006;50:941–952. doi: 10.1016/j.neuropharm.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Arndt P, Volk C, Gorboulev V, Budiman T, Popp C, Ulzheimer-Tueber I, Akhoundova A, Koppatz S, Bamberg E, Nagel G, Koepsell H. Interaction of cations, anions, and weak base quinine with rat renal cation transporter rOCT2 compared with rOCT1. Am J Physiol Renal Physiol. 2001;281:F454–F468. doi: 10.1152/ajprenal.2001.281.3.F454. [DOI] [PubMed] [Google Scholar]
- Artigas F, Romero L, de Montigny C, Blier P. Acceleration of the effect of selected antidepressant drugs in major depression by 5-HT1A antagonists. Trends Neurosci. 1996;19:378–383. doi: 10.1016/S0166-2236(96)10037-0. [DOI] [PubMed] [Google Scholar]
- Baganz NL, Horton RE, Calderon AS, Owens WA, Munn JL, Watts LT, Koldzic-Zivanovic N, Jeske NA, Koek W, Toney GM, Daws LC. Organic cation transporter 3: Keeping the brake on extracellular serotonin in serotonin transporter deficient mice. Proc Nat Acad Sci USA. doi: 10.1073/pnas.0800466105. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannon MJ, Michelhaugh SK, Wang J, Sacchetti P. The human dopamine transporter gene: gene organization, transcriptional regulation, and potential involvement in neuropsychiatric disorders. Eur Neuropsychopharmacol. 2001;11:449–455. doi: 10.1016/s0924-977x(01)00122-5. [DOI] [PubMed] [Google Scholar]
- Bennet AJ, Lesch KP, Heils A, Long JC, Lorenz JG, Shoaf ES, Champoux M, Suomi SJ, Linnoila MC, Higley JD. Early experience and serotonin transporter gene variation interact to influence primate CNS function. Mol Psychiatry. 2002;7:118–122. doi: 10.1038/sj.mp.4000949. [DOI] [PubMed] [Google Scholar]
- Bertler A, Falck B, Owman C. Studies on 5-hydroxytryptamine stores in pineal gland of rat. Acta Physiol Scand Suppl. 1964;239:1–18. doi: 10.1111/j.1748-1716.1964.tb04118.x. [DOI] [PubMed] [Google Scholar]
- Blakely RD, Berson HE, Fremeau RT, Jr, Caron MG, Peek MM, Prince HK, Bradley CC. Cloning and expression of a functional serotonin transporter from rat brain. Nature. 1991;354:66–70. doi: 10.1038/354066a0. [DOI] [PubMed] [Google Scholar]
- Blakely RD, Ramamoorthy S, Schroeter S, Qian Y, Apparsundaram S, Galli A, DeFelice LJ. Regulated phosphorylation and trafficking of antidepressant-sensitive serotonin transporter proteins. Biol Psychiatry. 1998;44:169–178. doi: 10.1016/s0006-3223(98)00124-3. [DOI] [PubMed] [Google Scholar]
- Blakely RD, Bauman AL. Biogenic amine transporters: regulation in flux. Curr Opin Neurobiol. 2000;10:328–336. doi: 10.1016/s0959-4388(00)00088-x. [DOI] [PubMed] [Google Scholar]
- Blakely RD, DeFelice LJ, Galli A. Biogenic amine neurotransmitter transporters: just when you thought you knew them. Physiology. 2005;20:225–231. doi: 10.1152/physiol.00013.2005. [DOI] [PubMed] [Google Scholar]
- Blier P, deMontigny C, Chaput Y. Modifications of the serotonin system by antidepressant treatments: Implications for the therapeutic response in major depression. J Clin Psychopharmacol. 1987;7(suppl):24S–35S. [PubMed] [Google Scholar]
- Bönisch H, Brüss M. The norepinephrine transporter in physiology and disease. Handb Exp Pharmacol. 2006;175:485–524. doi: 10.1007/3-540-29784-7_20. [DOI] [PubMed] [Google Scholar]
- Borland LM, Michael AC. An introduction to electrochemical methods in neuroscience. In: Michael AC, Borland LM, Michael AC, Simon SA, Nicolelis MAL, editors. Electrochemical Methods in Neuroscience for Methods and New Frontiers in Neuroscience Series. Boca Raton, Fl: CRC Press; 2007. pp. 1–15. [Google Scholar]
- Bowden CL, Koslow SH, Hanin I, Maas JW, Davies JM, Robins E. Effects of amitriptyline and imipramine on brain amine neurotransmitter metabolites in cerebrospinal fluid. Clin Pharmacol Ther. 1985;37:316–324. doi: 10.1038/clpt.1985.46. [DOI] [PubMed] [Google Scholar]
- Bunin MA, Wightman RM. Quantitative evaluation of 5-hydroxytryptamine (serotonin) neuronal release and uptake: and investigation of extrasynaptic transmission. J Neurosci. 1998;18:4854–4860. doi: 10.1523/JNEUROSCI.18-13-04854.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunin MA, Wightman RM. Paracrine neurotransmission in the CNS: Involvement of 5-HT. Trends Neurosci. 1999;22:377–382. doi: 10.1016/s0166-2236(99)01410-1. [DOI] [PubMed] [Google Scholar]
- Burgen ASV, Iversen LL. The inhibition of noradrenaline uptake by sympathomimetic amines in the rat isolated heart. Br J Pharmacol. 1965;25:34–49. doi: 10.1111/j.1476-5381.1965.tb01754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch AE, Quester S, Ulzheimer JC, Gorboulev V, Akhoundova A, Waldegger S, Lang F, Koepsell H. Monoamine neurotransmitter transport mediated by the polyspecific cation transporter rOCT1. FEBS Lett. 1996;395:153–156. doi: 10.1016/0014-5793(96)01030-7. [DOI] [PubMed] [Google Scholar]
- Busch AE, Karbach U, Miska D, Gorboulev V, Akhoundova A, Volk C, Arndt P, Ulzheimer JC, Sonders MS, Bauman C, Waldegger S, Lang F, Koepsell H. Human neurons express the polyspecific cation transporter hOCT2, which translocates monoamine neurotransmitters, amantadine and memantine. Mol Pharmacol. 1998;54:342–352. doi: 10.1124/mol.54.2.342. [DOI] [PubMed] [Google Scholar]
- Butler J, Tannian M, Leonard BE. The chronic effects of desipramine and sertraline on platelet and synaptosomal 5HT uptake in olfactory bulbectomized rats. Prog Neuro-Psychopharmacol & Biol Psychiat. 1988;12:585–594. doi: 10.1016/0278-5846(88)90004-8. [DOI] [PubMed] [Google Scholar]
- Byrne SE, Rothschild AJ. Loss of antidepressant efficacy during maintenance therapy: possible mechanisms and treatments. J Clin Psychiatry. 1998;59:279–288. doi: 10.4088/jcp.v59n0602. [DOI] [PubMed] [Google Scholar]
- Callaghan PD, Irvine RJ, Daws LC. Differences in the in vivo dynamics of neurotransmitter release and serotonin uptake after acute para-methoxyamphetamine and 3,4-methylenedioxymethamphetamine revealed by chronoamperometry. Neurochem Int. 2005;47:350–361. doi: 10.1016/j.neuint.2005.04.026. [DOI] [PubMed] [Google Scholar]
- Carboni E, Tanda GL, Frau R, Di Chiara G. Blockade of the noradrenaline carrier increases extracellular dopamine concentrations in the prefrontal cortex: evidence that dopamine is taken up in vivo by noradrenergic terminals. J Neurochem. 1990;55:1067–1070. doi: 10.1111/j.1471-4159.1990.tb04599.x. [DOI] [PubMed] [Google Scholar]
- Carboni E, Silvagni A. Dopamine reuptake by norepinephrine neurons: exception or rule? Crit Rev Neurobiol. 2004;16:121–128. doi: 10.1615/critrevneurobiol.v16.i12.130. [DOI] [PubMed] [Google Scholar]
- Carboni E, Silvagni A, Vacca C, Di Chiara G. Cumulative effect of norepinephrine and dopamine carrier blockade on extracellular dopamine increase in the nucleus accumbens shell, bed nucleus of stria terminalis and prefrontal cortex. J Neurochem. 2006;96:473–481. doi: 10.1111/j.1471-4159.2005.03556.x. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Corrodi H, Fuxe K, Hokfelt T. Effect of antidepressant drugs on the depletion of intraneuronal brain 5-hydroxytryptamine stores caused by 4-methyl-a-ethyl-meta-tyramine. Eur J Pharmacol. 1969;5:357–366. doi: 10.1016/0014-2999(69)90113-7. [DOI] [PubMed] [Google Scholar]
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- Chen JJ, Li Z, Pan H, Murphy DL, Tamir H, Koepsell H, Gershon MD. Maintenance of serotonin in the intestinal mucosa and ganglia of mice that lack the high-affinity serotonin transporter: Abnormal intestinal motility and the expression of cation transporters. J Neurosci. 2001;21:6348–6361. doi: 10.1523/JNEUROSCI.21-16-06348.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Reith ME. Substrates dissociate dopamine transporter oligomers. J Neurochem. 2008;105:910–920. doi: 10.1111/j.1471-4159.2007.05195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Skolnick P. Triple uptake inhibitors: therapeutic potential in depression and beyond. Expert Opin Investig Drugs. 2007;16:1365–1377. doi: 10.1517/13543784.16.9.1365. [DOI] [PubMed] [Google Scholar]
- Clements JD. Transmitter time course in the synaptic cleft: Its role in central synaptic function. Trends Neurosci. 1996;19:163–171. doi: 10.1016/s0166-2236(96)10024-2. [DOI] [PubMed] [Google Scholar]
- Codd EE, Walker RF. Platelet serotonin uptake: methods and strain differences. Biol Psychiatry. 1987;44:169–178. doi: 10.1016/0165-1781(87)90051-5. [DOI] [PubMed] [Google Scholar]
- Collier DA, Stober G, Li T, Heils A, Catalano M, Di Bella D, Arranz MJ, Murray RM, Vallada HP, Bengel D, Muller CR, Roberts GW, Smeraldi E, Kirov G, Sham P, Lesch KP. A novel function of polymorphism within the promoter of the serotonin transporter gene: Possible role in susceptibility to affective disorders. Mol Psychiatry. 1996;1:453–460. [PubMed] [Google Scholar]
- Coppen A. The biochemistry of affective disorders. Br J Psychiatry. 1967;113:1237–1264. doi: 10.1192/bjp.113.504.1237. [DOI] [PubMed] [Google Scholar]
- Cragg SJ, Rice ME. DAncing past the DAT at a DA synapse. Trends Neurosci. 2004;27:270–277. doi: 10.1016/j.tins.2004.03.011. [DOI] [PubMed] [Google Scholar]
- Critchley HD, Taggart P, Sutton PM, Holdright DR, Batchvarov V, Hnatkova K, Malik M, Dolan RJ. Mental stress and sudden cardiac death: asymmetric midbrain activity as a linking mechanism. Brain. 2005;128:75–85. doi: 10.1093/brain/awh324. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Valentino RJ, Lucki I. Assessing substrates underlying the behavioral effects of antidepressants using the modified rat forced swimming test. Neurosci Behav Rev. 2005;29:547–569. doi: 10.1016/j.neubiorev.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Dahlin A, Xia L, Kong W, Hevner R, Wang J. Expression and immunolocalization of the plasma membrane monoamine transporter in the brain. Neuroscience. 2007;146:1193–1211. doi: 10.1016/j.neuroscience.2007.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daws LC, Toney GM, Gerhardt GA, Frazer A. In vivo chronoamperometric measures of extracellular serotonin clearance in rat dorsal hippocampus: Contributions of serotonin and norepinephrine transporters. J Pharmacol Exp Ther. 1998;286:967–976. [PubMed] [Google Scholar]
- Daws LC, Gould GG, Teicher SD, Gerhardt GA, Frazer A. 5-HT1B receptor- mediated regulation of serotonin clearance in rat hippocampus in vivo. J Neurochem. 2000;75:2113–2122. doi: 10.1046/j.1471-4159.2000.0752113.x. [DOI] [PubMed] [Google Scholar]
- Daws LC, Montañez S, Owens WA, Gould GG, Frazer A, Toney GM, Gerhardt GA. Transport mechanisms governing clearance in vivo revealed by high-speed chronoamperometry. J Neurosci Meth. 2005;143:49–62. doi: 10.1016/j.jneumeth.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Daws LC, Montañez S, Munn JL, Owens WA, Baganz NL, Boyce-Rustay JM, Millstein R, Wiedholz LM, Murphy DL, Holmes A. Ethanol inhibits clearance of brain serotonin by a serotonin transporter independent mechanism. J Neurosci. 2006;26:6431–6438. doi: 10.1523/JNEUROSCI.4050-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daws LC, Toney GM. Voltammetric Methods to Study Kinetics and Mechanisms for Serotonin Clearance In Vivo. In: Michael AC, Borland LM, Michael AC, Simon SA, Nicolelis MAL, editors. Electrochemical Methods in Neuroscience for Methods and New Frontiers in Neuroscience Series. Boca Raton, Fl.; CRC Press: 2007. pp. 63–81. [Google Scholar]
- Dutta AK, Zhang S, Kolhathar R, Reith ME. Dopamine transporter as target for drug development of cocaine dependence medications. Eur J Pharmacol. 2003;479:93–106. doi: 10.1016/j.ejphar.2003.08.060. [DOI] [PubMed] [Google Scholar]
- Eichhammer P, Langguth B, Wiegand R, Kharraz A, Frick U, Hajak G. Allelic variation in the serotonin transporter promoter affects the neuromodulatory effects of a selective serotonin transporter reuptake inhibitor (SSRI) Psychopharmacology. 2003;166:294–297. doi: 10.1007/s00213-002-1370-1. [DOI] [PubMed] [Google Scholar]
- Engel K, Zhou M, Wang J. Identification and characterization of a novel monoamine transporter in the human brain. J Biol Chem. 2004;279:50042–50049. doi: 10.1074/jbc.M407913200. [DOI] [PubMed] [Google Scholar]
- Engel K, Wang J. Interaction of organic cations with a newly identified plasma membrane monoamine transporter. Mol Pharmacol. 2005;68:1397–1407. doi: 10.1124/mol.105.016832. [DOI] [PubMed] [Google Scholar]
- Esler M, Parati G. Is essential hypertension sometimes a psychosomatic disorder? J Hypertension. 2004;22:873–876. doi: 10.1097/00004872-200405000-00003. [DOI] [PubMed] [Google Scholar]
- Feinn R, Nellissery M, Kranzler HR. Meta-analysis of the association of a functional serotonin transporter promoter polymorphism with alcohol dependence. Am J Med Genet B Neuropsychiatr Genet. 2005;133:79–84. doi: 10.1002/ajmg.b.30132. [DOI] [PubMed] [Google Scholar]
- Feng N, Mo B, Johnson PL, Orchinik M, Lowry CA, Renner KJ. Local inhibition of organic cation transporters increases extracellular serotonin in the medial hypothalamus. Brain Res. 2005;1063:69–76. doi: 10.1016/j.brainres.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Fleckenstein AE, Volz TJ, Riddle EL, Gibb JW, Hanson GR. New insights into the mechanism of action of amphetamines. Annu Rev Pharmacol Toxicol. 2007;47:681–698. doi: 10.1146/annurev.pharmtox.47.120505.105140. [DOI] [PubMed] [Google Scholar]
- Frazer A. Pharmacology of antidepressants. J Clin Psychopharmacol. 1997;17:2S–18S. doi: 10.1097/00004714-199704001-00002. [DOI] [PubMed] [Google Scholar]
- Fuxe K, Ungerstedt U. Localization of 5-hydroxtryptamine uptake in rat brain after intraventricular injection. J Pharm Pharmacol. 1967;19:335–337. doi: 10.1111/j.2042-7158.1967.tb08097.x. [DOI] [PubMed] [Google Scholar]
- Fuxe K, Agnati LF. Two principle modes of electrochemical communication in the brain: Volume versus wiring transmission. In: Fuxe K, Agnati LF, editors. Volume transmission in the brain: Novel mechanisms of neuronal transmission. New York: Raven Press; 1991. pp. 1–9. [Google Scholar]
- Fuxe K, Dahlström A, Höistad M, Marcellino D, Jansson A, Rivera A, Diaz-Cabiale Z, Jacobsen K, Tinner-Staines B, Hagman B, Leo G, Staines W, Guidolin D, Kehr J, Genedani S, Belluardo N, Agnati LF. From the Golgi-Cajal mapping to the transmitter-based characterization of the neuronal networks leading to two modes of brain communication: Wiring and volume transmission. Brain Res Rev. 2007;55:17–54. doi: 10.1016/j.brainresrev.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Galli A, Blakely RD, DeFelice LJ. Norepinephrine transporters have channel modes of conduction. Proc Natl Acad Sci USA. 1996;93:8671–8676. doi: 10.1073/pnas.93.16.8671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardier AM, Malagié L, Trillat AC, Jacquot C, Artigas F. Role of 5-HT1A autoreceptors in the mechanism of action of serotonergic antidepressant drugs: recent findings from in vivo microdialysis studies. Fundam Clin Pharmacol. 1996;10:16–27. doi: 10.1111/j.1472-8206.1996.tb00145.x. [DOI] [PubMed] [Google Scholar]
- Gasser PJ, Lowry CA, Orchinik M. Corticosterone-sensitive monoamine transport in the rat dorsomedial hypothalamus: Potential role for organic cation transporter 3 in stress-induced modulation of monoaminergic neurotransmission. J Neurosci. 2006;26:8758–8766. doi: 10.1523/JNEUROSCI.0570-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gether U, Andersen PH, Larsson OM, Schousboe A. Neurotransmitter transporters: molecular function of important drug targets. Trends Pharmacol Sci. 2006;27:375–383. doi: 10.1016/j.tips.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Giros B, Caron MG. Molecular characterization of the dopamine transporter. Trends Pharmacol Sci. 1993;14:43–49. doi: 10.1016/0165-6147(93)90029-j. [DOI] [PubMed] [Google Scholar]
- Glowinski J, Axelrod J. Effects of drugs on the disposition of H-3-norepinephrine in the rat brain. Pharmacol Rev. 1966;18:775–785. [PubMed] [Google Scholar]
- Gorboulev V, Ulzheimer JC, Akhoundova A, Ulzheimer-Tueber I, Karbach U, Quester S, Baumann C, Lang F, Busch AE, Koepsell H. Cloning and characterization of two human polyspecific organic cation transporters. DNA Cell Biol. 1997;16:871–881. doi: 10.1089/dna.1997.16.871. [DOI] [PubMed] [Google Scholar]
- Gründemann D, Gorboulev V, Gambaryan S, Vehyl M, Koepsell H. Drug excretion mediated by a new prototype of polyspecific transporter. Nature. 1994;372:549–552. doi: 10.1038/372549a0. [DOI] [PubMed] [Google Scholar]
- Gründemann D, Babin-Ebell J, Martel F, Örding N, Schmidt A, Schömig E. Primary structure and functional expression of the apical organic cation transporter from kidney epithelial LLC-PK1 cells. J Biol Chem. 1997;272:10408–10413. doi: 10.1074/jbc.272.16.10408. [DOI] [PubMed] [Google Scholar]
- Gründemann D, Schechinger B, Rappold GA, Schömig E. Molecular identification of the corticosterone-sensitive extraneuronal catecholamine transporter. Nat Neurosci. 1998;1:349–352. doi: 10.1038/1557. [DOI] [PubMed] [Google Scholar]
- Hariri AR, Holmes A. Genetics of emotional regulation: The role of the serotonin transporter in neural function. Trends Cog Sci. 2006;10:182–191. doi: 10.1016/j.tics.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Hendley ED, Taylor KM, Snyder SH. 3H-normetanephrine uptake in rat brain slices. Relationship to extraneuronal accumulation of norepinephrine. Eur J Pharmacol. 1970;12:167–169. doi: 10.1016/0014-2999(70)90062-2. [DOI] [PubMed] [Google Scholar]
- Henn FA, Hamberger A. Glial cell function: uptake of transmitter substances. Proc Natl Acad Sci USA. 1971;68:2686–2690. doi: 10.1073/pnas.68.11.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman BJ, Mezey E, Brownstein MJ. Cloning of a serotonin transporter affected by antidepressants. Science. 1991;254:579–580. doi: 10.1126/science.1948036. [DOI] [PubMed] [Google Scholar]
- Holmes A, Yang RJ, Murphy DL, Crawley JN. Evaluation of antidepressant-related behavioral responses in mice lacking the serotonin transporter. Neuropsychopharmacology. 2002;27:914–923. doi: 10.1016/S0893-133X(02)00374-3. [DOI] [PubMed] [Google Scholar]
- Holmes A, Li Q, Murphy DL, Gold E, Crawley JN. Abnormal anxiety-related behavior in serotonin transporter null mutant mice: the influence of genetic background. Genes Brain Behav. 2003;2:365–380. doi: 10.1046/j.1601-1848.2003.00050.x. [DOI] [PubMed] [Google Scholar]
- Hrdina PD. Pharmacological characterization of [3H] desipramine binding in rat cerebral cortex. Prog Neuropsychopharmacol. 1981;5:553–557. doi: 10.1016/0364-7722(81)90045-x. [DOI] [PubMed] [Google Scholar]
- Hughes ZA, Stanford SC. Evidence from microdialysis and synaptosomal studies of rat cortex for noradrenergic uptake sites with different sensitivities to SSRIs. Br J Pharmacol. 1998;124:1141–1148. doi: 10.1038/sj.bjp.0701947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iversen L. Neurotransmitter transporters and their impact on the development of psychopharmacology. Br J Pharmacol. 2006;147:S82–88. doi: 10.1038/sj.bjp.0706428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson BP, Wightman RM. Dynamics of 5-hydroxytryptamine released from dopamine neurons in caudate putamen of the rat. Brain Res. 1995;674:163–166. doi: 10.1016/0006-8993(95)00019-m. [DOI] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Wightman RM, Caron MG. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J Neurosci. 1998;18:1979–1986. doi: 10.1523/JNEUROSCI.18-06-01979.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlig KM, Binda R, Khoshbouei H, Blakely RD, McMahon DG, Javitch JA, Galli A. Amphetamine induces dopamine efflux through a dopamine transporter channel. Proc Natl Acad Sci USA. 2005;102:3495–3500. doi: 10.1073/pnas.0407737102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannari K, Shen H, Arai A, Tomiyama M, Baba M. Reuptake of L-DOPA-derived extracellular dopamine in the striatum with dopaminergic denervation via serotonin transporters. Neurosci Lett. 2006;402:62–65. doi: 10.1016/j.neulet.2006.03.059. [DOI] [PubMed] [Google Scholar]
- Kirsch I, Deacon BJ, Huedo-Medina TB, Scoboria A, Moore TJ, Johnson BT. Initial severity and antidepressant benefits: a meta-analysis of data submitted to the Food and Drug Administration. PLoS Med. 2008;5:260–268. doi: 10.1371/journal.pmed.0050045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitaichi K, Fukuda M, Nakayama H, Aoyama N, Ito Y, Fujimoto Y, Takagi K, Takagi K, Hasegawa T. Behavioral changes following antisense oligonucleotide-induced reduction of organic cation transporter-3 in mice. Neurosci Lett. 2005;382:195–200. doi: 10.1016/j.neulet.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Hayashi E, Shimamura M, Konoshita M, Murphy NP. Neurochemical responses to antidepressants in the prefrontal cortex of mice and their efficacy in preclinical models of anxiety-like and depression-like behavior: a comparative and correlational study. Psychopharmacology. 2008;197:567–580. doi: 10.1007/s00213-008-1070-6. [DOI] [PubMed] [Google Scholar]
- Koepsell H, Schmitt BM, Gorboulev V. Organic cation transporters. Rev Physiol Biochem Pharmacol. 2003;150:36–90. doi: 10.1007/s10254-003-0017-x. [DOI] [PubMed] [Google Scholar]
- Koepsell H, Endou H. The SLC22 drug transporter family. Pflugers Arch. 2004;477:666–676. doi: 10.1007/s00424-003-1089-9. [DOI] [PubMed] [Google Scholar]
- Koepsell H, Lips K, Volk C. Polyspecific organic cation transporters: Structure, function, physiological roles, and biopharmaceutical implications. Pharmaceutical Res. 2007;24:1227–1251. doi: 10.1007/s11095-007-9254-z. [DOI] [PubMed] [Google Scholar]
- Kristufek D, Rudorfer W, Pifl C, Huck S. Organic cation transporter mRNA and function in the rat superior cervical ganglion. J Physiol. 2002;543:117–134. doi: 10.1113/jphysiol.2002.021170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapin IP, Oxenkrug GF. Intensification of the central serotonergic processes as a possible determinant of the thymoleptic effect. Lancet. 1969;1:132–136. doi: 10.1016/s0140-6736(69)91140-4. [DOI] [PubMed] [Google Scholar]
- Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, Benjamin J, Muller CR, Hamer DH, Murphy DL. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996;274:1527–1531. doi: 10.1126/science.274.5292.1527. [DOI] [PubMed] [Google Scholar]
- Lesch KP. Alcohol dependence and gene × environment interaction in emotion regulation: Is serotonin the link? Eur J Pharmacol. 2005;526:113–124. doi: 10.1016/j.ejphar.2005.09.027. [DOI] [PubMed] [Google Scholar]
- Lichtensteiger W, Mutzner U, Langemann H. Uptake of 5-hydroxytryptamine and 5-hydroxytryptophan by neurons of the central nervous system normally containing catecholamines. J Neurochem. 1967;14:489–497. doi: 10.1111/j.1471-4159.1967.tb09548.x. [DOI] [PubMed] [Google Scholar]
- Liprando LA, Miner LH, Blakely RD, Lewis DA, Sesack SR. Ultrastructural interactions between terminals expressing the norepinephrine transporter and dopamine neurons in the rat and monkey ventral tegmental area. Synapse. 2004;52:233–244. doi: 10.1002/syn.20023. [DOI] [PubMed] [Google Scholar]
- Mandela P, Ordway GA. The norepinephrine transporter and its regulation. J Neurochem. 2006;97:310–333. doi: 10.1111/j.1471-4159.2006.03717.x. [DOI] [PubMed] [Google Scholar]
- Männistö PT, Kaakkola S. Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev. 1999;51:593–628. [PubMed] [Google Scholar]
- Masson J, Sagné C, Hamon M, El Mestikawy S. Neurotransmitter transporters in the central nervous system. Pharmacol Rev. 1999;51:439–464. [PubMed] [Google Scholar]
- Maswood S, Truitt W, Hotema M, Caldarola-Pastuszka M, Uphouse L. Estrous cycle modulation of extracellular serotonin in mediobasal hypothalamus: role of the serotonin transporter and terminal autoreceptors. Brain Res. 1999;831:146–154. doi: 10.1016/s0006-8993(99)01439-0. [DOI] [PubMed] [Google Scholar]
- Mathews TA, Fedele DE, Coppelli FM, Avila AM, Murphy DL, Andrews AM. Gene dose-dependent alterations in extraneuronal serotonin but not dopamine in mice with reduced serotonin transporter expression. J Neurosci Meth. 2004;140:169–181. doi: 10.1016/j.jneumeth.2004.05.017. [DOI] [PubMed] [Google Scholar]
- Mathews TA, John CE, Lapa GB, Budygin EA, Jones SR. No role of the dopamine transporter in acute ethanol effects on striatal dopamine dynamics. Synapse. 2006;60:288–294. doi: 10.1002/syn.20301. [DOI] [PubMed] [Google Scholar]
- Miner LH, Schroeter S, Blakely RD, Sesack SR. Ultrastructural localization of the serotonin transporter in superficial and deep layers of the rat prelimbic prefrontal cortex and its spatial relationship to dopamine terminals. J Comp Neurol. 2000;427:220–234. doi: 10.1002/1096-9861(20001113)427:2<220::aid-cne5>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Miner LH, Schroeter S, Blakely RD, Sesack SR. Ultrastructural localization of the norepinephrine transporter in superficial and deep layers of the rat prelimbic prefrontal cortex and its spatial relationship to probable dopamine terminals. J Comp Neurol. 2003;466:478–494. doi: 10.1002/cne.10898. [DOI] [PubMed] [Google Scholar]
- Miranda M, Sorkin A. Regulation of receptors and transporters by ubiquitination: new insights into surprisingly similar mechanisms. Mol Interv. 2007;7:157–167. doi: 10.1124/mi.7.3.7. [DOI] [PubMed] [Google Scholar]
- Moll GH, Mehnert C, Wicker M, Bock N, Rothenberger A, Ruther E, Huether G. Age-associated changes in the densities of presynaptic monoamine transporters in different regions of the rat brain from early juvenile life into late adulthood. Brain Res Dev Brain Res. 2000;119:251–257. doi: 10.1016/s0165-3806(99)00182-0. [DOI] [PubMed] [Google Scholar]
- Montañez S, Owens WA, Gould GG, Murphy DL, Daws LC. Exaggerated effects of fluvoxamine in heterozygote serotonin transporter knockout mice. J Neurochem. 2003;86:210–219. doi: 10.1046/j.1471-4159.2003.01836.x. [DOI] [PubMed] [Google Scholar]
- Montgomery SA. The under-recognized role of dopamine in the treatment of major depressive disorder. Int Clin Psychopharmacol. 2008;23:63–69. doi: 10.1097/YIC.0b013e3282f2b3cb. [DOI] [PubMed] [Google Scholar]
- Mooney JJ, Samson JA, Hennen J, Pappalardo K, McHale N, Alpert J, Koutsos M, Schildkraut JJ. Enhanced norepinephrine output during long-term desipramine treatment: a possible role for the extraneuronal monoamine transporter (SLC22A3) J Psychiat Res. 2008;42:605–611. doi: 10.1016/j.jpsychires.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morón JA, Brockington A, Wise RA, Rocha MA, Hope BT. Dopamine uptake through the norepinephrine transporter in brain regions with low levels of the dopamine transporter: evidence from knock-out mouse lines. J Neurosci. 2002;22:389–395. doi: 10.1523/JNEUROSCI.22-02-00389.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mössner R, Simantov R, Marx A, Lesch KP, Seif I. Aberrant accumulation of serotonin in dopamine neurons. Neurosci Lett. 2006;401:49–54. doi: 10.1016/j.neulet.2006.02.081. [DOI] [PubMed] [Google Scholar]
- Munafò MR, Brown SM, Hariri AR. Serotonin transporter (5-HTTLPR) genotype and amygdala activation: A meta analysis. Biol Psychiatry. 2008;63:852–857. doi: 10.1016/j.biopsych.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DL, Lesch KP. Targeting the murine serotonin transporter: insights into human neurobiology. Nat Rev Neurosci. 2008;9:85–96. doi: 10.1038/nrn2284. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Kitaichi K, Ito Y, Hashimoto K, Takagi K, Yokoi T, Takagi K, Ozaki N, Yamamoto T, Hasegawa T. The role of organic cation transporter-3 in methamphetamine disposition and its behavioral response in rats. Brain Res. 2007;1184:260–269. doi: 10.1016/j.brainres.2007.09.072. [DOI] [PubMed] [Google Scholar]
- Nicholson C. Interaction between diffusion and Michaelis-Menten uptake of dopamine after iontophoresis in striatum. Biophys J. 1995;68:1699–1715. doi: 10.1016/S0006-3495(95)80348-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirenberg MJ, Vaughan RA, Uhl GR, Kuhar MJ, Pickel VM. The dopamine transporter is localized to dendritic and axonal plasma membranes of nigrostriatal dopaminergic neurons. J Neurosci. 1996;16:436–447. doi: 10.1523/JNEUROSCI.16-02-00436.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirenberg MJ, Chan J, Pohorille A, Vaughan RA, Uhl GR, Kuhar MJ, Pickel VM. The dopamine transporter: Comparative ultrastructure of dopaminergic axons in limbic and motor compartments of the nucleus accumbens. J Neurosci. 1997;17:6899–6907. doi: 10.1523/JNEUROSCI.17-18-06899.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt DJ. The role of dopamine and norepinephrine in depression and antidepressant treatment. J Clin Psychiatry. 2006;67(Suppl 6):3–8. [PubMed] [Google Scholar]
- Pacholczyk T, Blakely RD, Amara SG. Expression and cloning of a cocaine-and antidepressant-sensitive human noradrenaline transporter. Nature. 1991;350:350–354. doi: 10.1038/350350a0. [DOI] [PubMed] [Google Scholar]
- Pan Y, Gembom E, Peng W, Lesch KP, Mossner R, Simantov R. Plasticity in serotonin uptake in primary neuronal cultures of serotonin transporter knockout mice. Brain Res Dev Brain Res. 2001;126:125–129. doi: 10.1016/s0165-3806(00)00145-0. [DOI] [PubMed] [Google Scholar]
- Pickel VM, Chan J. Ultrastructural localization of the serotonin transporter in limbic and motor compartments of the nucleus accumbens. J Neurosci. 1999;19:7356–7366. doi: 10.1523/JNEUROSCI.19-17-07356.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prange AJ., Jr The pharmacology and biochemistry of depression. Dis Nerv Syst. 1964;25:217–221. [PubMed] [Google Scholar]
- Ramamoorthy S, Blakely RD. Phosphorylation and sequestration of serotonin transporters differentially modulated by psychostimulants. Science. 1999;285:763–766. doi: 10.1126/science.285.5428.763. [DOI] [PubMed] [Google Scholar]
- Raiteri M, Del Carmine R, Bertolini A, Levi G. Effects of sympathomimetic amines on the synaptosomal transport of noradrenaline, dopamine and 5-hydroxytryptramine. Eur J Pharmacol. 1977;41:133–143. doi: 10.1016/0014-2999(77)90202-3. [DOI] [PubMed] [Google Scholar]
- Rausch JL, Johnson ME, Fei YJ, Li JQ, Shendarkar N, Hobby HM, Ganapathy V, Leibach FH. Initial conditions of serotonin transporter kinetics and genotype: Influence on SSRI treatment trial outcome. Biol Psychiatry. 2002;51:723–732. doi: 10.1016/s0006-3223(01)01283-5. [DOI] [PubMed] [Google Scholar]
- Rausch JL. Initial conditions of psychotropic drug response: Studies of serotonin transporter long promoter region (5-HTTLPR), serotonin transporter efficiency, cytokine and kinase gene expression relevant to depression and antidepressant outcome. Prog Neuropsychopharmacol Biol Psychiatr. 2005;29:1046–1061. doi: 10.1016/j.pnpbp.2005.03.011. [DOI] [PubMed] [Google Scholar]
- Robinson MB. Regulated trafficking of neurotransmitter transporters: common notes but different melodies. J Neurochem. 2002;80:1–11. doi: 10.1046/j.0022-3042.2001.00698.x. [DOI] [PubMed] [Google Scholar]
- Ross SB, Renyi AL. Inhibition of the uptake of tritiated 5-hydroxytryptamine in brain tissue. Eur J Pharmacol. 1969;7:270–277. doi: 10.1016/0014-2999(69)90091-0. [DOI] [PubMed] [Google Scholar]
- Schenk JO. The functioning neuronal transporter for dopamine: kinetic mechanisms and effects of amphetamines, cocaine and methylphenidate. Prog Drug Res. 2002;59:111–131. doi: 10.1007/978-3-0348-8171-5_4. [DOI] [PubMed] [Google Scholar]
- Schildkraut JJ. The catecholamine hypothesis of affective disorders: A review of supporting evidence. Am J Psychiatry. 1965;122:509–522. doi: 10.1176/ajp.122.5.509. [DOI] [PubMed] [Google Scholar]
- Schildkraut JJ, Mooney JJ. Toward a rapidly acting antidepressant: the normetanephrine and extraneuronal monoamine transporter (uptake 2) hypothesis. Am J Psychiatry. 2004;161:909–911. doi: 10.1176/appi.ajp.161.5.909. [DOI] [PubMed] [Google Scholar]
- Schmitt A, Mössner R, Gossmann A, Fishcer IG, Gorboulev V, Murphy DL, Koepsell K, Lesch KP. Organic cation transporter capable of transporting serotonin is up-regulated in serotonin transporter-deficient mice. J Neurosci Res. 2003;71:701–709. doi: 10.1002/jnr.10521. [DOI] [PubMed] [Google Scholar]
- Schroeter S, Apparsundaram S, Wiley RG, Miner LH, Sesack SR, Blakely RD. Immunolocalization of the cocaine- and antidepressant-sensitive l-norepinephrine transporter. J Comp Neurol. 2000;420:211–232. [PubMed] [Google Scholar]
- Serretti A, Kato M, De Ronchi D, Kinoshita T. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with selective serotonin reuptake inhibitor efficacy in depressed patients. Mol Psychiatry. 2007;12:247–257. doi: 10.1038/sj.mp.4001926. [DOI] [PubMed] [Google Scholar]
- Shaskan EG, Snyder SH. Kinetics of serotonin accumulation into slices from rat brain: Relationship to catecholamine uptake. J Pharmacol Exp Ther. 1970;175:404–418. [PubMed] [Google Scholar]
- Shen HW, Hagino Y, Kobayashi H, Shinohara-Tanaka K, Ikeda K, Yamamoto H, Yamamoto T, Lesch KP, Murphy DL, Hall FS, Uhl GR, Sora I. Regional differences in extracellular dopamine and serotonin assessed by in vivo microdialysis in mice lacking dopamine and/or serotonin transporters. Neuropsychopharmacology. 2004;29:1790–1799. doi: 10.1038/sj.npp.1300476. [DOI] [PubMed] [Google Scholar]
- Sitte HH, Farhan H, Javitch JA. Sodium-dependent neurotransmitter transporters: oligomerization as a determinant of transporter function and trafficking. Mol Interv. 2004;4:38–47. doi: 10.1124/mi.4.1.38. [DOI] [PubMed] [Google Scholar]
- Skolnick P, Krieter P, Tizzano J, Basile A, Popik P, Czobor P, Lippa A. Preclinical and clinical pharmacology of DOV 216, 303, a “triple” reuptake inhibitor. CNS Drug Rev. 2006;12:123–134. doi: 10.1111/j.1527-3458.2006.00123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits KM, Smits LJ, Schouten JS, Stelma FF, Nelemans P, Prins MH. Influence of SERTPR and STin2 in the serotonin transporter gene on the effect of selective serotonin reuptake inhibitors in depression: a systematic review. Mol Psychiatry. 2004;9:433–441. doi: 10.1038/sj.mp.4001488. [DOI] [PubMed] [Google Scholar]
- Stamford JA, Kruk ZL, Millar J. Striatal dopamine terminals release serotonin after 5- HTP pretreatment: in vivo voltammetric data. Brain Res. 1990;515:173–180. doi: 10.1016/0006-8993(90)90593-z. [DOI] [PubMed] [Google Scholar]
- Stanford SC. Prozac: panacea or puzzle? Trends Pharmacol Sci. 1996;17:150–154. doi: 10.1016/0165-6147(96)81591-4. [DOI] [PubMed] [Google Scholar]
- Steiner JA, Carneiro AM, Blakely RD. Going with the flow: Trafficking-dependent and - independent regulation of serotonin transport. Traffic. 2008;9:1393–1402. doi: 10.1111/j.1600-0854.2008.00757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone EA, Lin Y. An anti-immobility effect of exogenous corticosterone in mice. Eur J Pharmacol. 2008;580:135–142. doi: 10.1016/j.ejphar.2007.10.045. [DOI] [PubMed] [Google Scholar]
- Streich S, Brüss M, Bönisch H. Expression of the extraneuronal monoamine transporter (uptake 2) in human glioma cells. Naunyn Schmeidebergs Arch Pharmacol. 1996;353:328–333. doi: 10.1007/BF00168636. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Edwards RH. Antidepressants and the monoamine masquerade. Neuron. 2005;46:1–2. doi: 10.1016/j.neuron.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Sur C, Betz H, Schloss P. Immunocytochemical detection of the serotonin transporter in rat brain. Neuroscience. 1996;73:217–231. doi: 10.1016/0306-4522(96)00030-9. [DOI] [PubMed] [Google Scholar]
- Swiergiel AH, Leskov IL, Dunn AJ. Effects of chronic and acute stressors and CRF on depression-like behavior in mice. Behav Brain Res. 2008;186:32–40. doi: 10.1016/j.bbr.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Tejani-Butt SM. [3H]Nisoxetine: A radioligand for quantitation of norepinephrine uptake sites by autoradiography or by homogenate binding. J Pharmacol Exp Ther. 1992;260:427–436. [PubMed] [Google Scholar]
- Torres GE, Carneiro A, Seamans K, Fiorentini C, Sweeney A, Yao WD, Caron MG. Oligomerization and trafficking of the human dopamine transporter. Mutational analysis identifies critical domains important for the functional expression of the transporter. J Biol Chem. 2003a;278:2731–2739. doi: 10.1074/jbc.M201926200. [DOI] [PubMed] [Google Scholar]
- Torres GE, Gainetdinov RR, Caron MG. Plasma membrane monoamine transporters: structure, regulation and function. Nat Rev Neurosci. 2003b;4:13–25. doi: 10.1038/nrn1008. [DOI] [PubMed] [Google Scholar]
- Vialou V, Amphoux A, Zwart R, Giros B, Gautron S. Organic cation transporter 3 (Slc22a3) is implicated in salt-intake regulation. J Neurosci. 2004;24:2846–2851. doi: 10.1523/JNEUROSCI.5147-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialou C, Balasse L, Dumas S, Giros B, Gautron S. Neurochemical characterization of pathways expressing plasma membrane monoamine transporter in the rat brain. Neuroscience. 2007;144:616–622. doi: 10.1016/j.neuroscience.2006.09.058. [DOI] [PubMed] [Google Scholar]
- Vizi E, Zsilla G, Caron MG, Kiss JP. Uptake and release of norepinephrine by serotonergic terminals in norepinephrine transporter knock-out mice: Implications for the action of selective serotonin reuptake inhibitors. J Neurosci. 2004;24:7888–7894. doi: 10.1523/JNEUROSCI.1506-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitby LG, Axelrod J, Weil-Malherbe H. The fate of 3H-norepinephrine in animals. J Pharmacol Exp Ther. 1961;132:193–201. [PubMed] [Google Scholar]
- Wightman RM, Zimmerman JB. Control of dopamine extracellular concentration in rat striatum by impulse flow and uptake. Brain Res Brain Res Rev. 1990;15:135–144. doi: 10.1016/0165-0173(90)90015-g. [DOI] [PubMed] [Google Scholar]
- Wilhelm K, Mitchell PB, Niven H, Finch A, Wedgwood L, Scimone A, Blair IP, Parker G, Schofield PR. Life events, first depression onset and the serotonin transporter gene. Br J Psychiatry. 2006;188:210–215. doi: 10.1192/bjp.bp.105.009522. [DOI] [PubMed] [Google Scholar]
- Williams RB, Marchuk DA, Gadde KM, Barefoot JC, Grichnik K, Helms MJ, Kuhn CM, Lewis JG, Schanberg SM, Stafford-Smith M, Suarez EC, Clary GL, Svenson IK, Siegler IC. Central nervous system serotonin function and cardiovascular responses to stress. Psychosom Med. 2001;63:300–305. doi: 10.1097/00006842-200103000-00016. [DOI] [PubMed] [Google Scholar]
- Wu X, Kekuda R, Huang W, Fei YJ, Leibach FH, Chen J, Conway SJ, Ganapathy V. Identity of the organic cation transporter OCT3 as the extraneuronal monoamine transporter (uptake 2) and evidence for the expression of the transporter in brain. J Biol Chem. 1998;273:32776–32786. doi: 10.1074/jbc.273.49.32776. [DOI] [PubMed] [Google Scholar]
- Wurtman RJ. Genes, stress and depression. Metabolism. 2005;54:16–19. doi: 10.1016/j.metabol.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Zahniser NR, Larson GA, Gerhardt GA. In vivo dopamine clearance in rate in rat striatum: regulation by extracellular dopamine concentration and dopamine transporter inhibitors. J Pharmacol Exp Ther. 1999;289:266–277. [PubMed] [Google Scholar]
- Zahniser NR, Sorkin A. Rapid regulation of the dopamine transporter: role in stimulant addiction. Neuropharmacology. 2004;47:S80–91. doi: 10.1016/j.neuropharm.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Zhou FC, Tao-Cheng JH, Segu L, Patel T, Wang Y. Serotonin transporters are located on the axons beyond the synaptic junctions: anatomical and functional evidence. Brain Res. 1998;805:241–254. doi: 10.1016/s0006-8993(98)00691-x. [DOI] [PubMed] [Google Scholar]
- Zhou FC, Lesch K-P, Murphy DL. Serotonin uptake into dopamine neurons via dopamine transporters: a compensatory alternative. Brain Res. 2002;942:109–119. doi: 10.1016/s0006-8993(02)02709-9. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Lang Y, Salas R, Zhang L, De Biasi M, Dani JA. Corelease of dopamine and serotonin from striatal dopamine terminals. Neuron. 2005;46:65–74. doi: 10.1016/j.neuron.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Zhou M, Engel K, Wang J. Evidence for significant contribution of a newly identified monoamine transporter (PMAT) to serotonin uptake in the human brain. Biochem Pharmacol. 2007;72:147–154. doi: 10.1016/j.bcp.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]