

Abstract
Tiopronin monolayer-protected silver nanoparticles with different core sizes (average diameter = 2, 5, 20 nm) were prepared by using different mole ratios of silver nitrate/tiopronin. Ligands on the silver particles were partially displaced by fluorescein-labeled thiolate single-stranded oligonucleotides or their complementary unlabeled oligonucleotides through ligand exchange. The fluorophores on silver particles showed a surfaceenhanced fluorescence (SEF) dependent on the size of metallic cores. The particles could be coupled through hybridizations of oligonucleotides bound on the particles. The coupled particles were aggregated due to multiple displacements of oligonucleotides on each particle, resulting in stronger SEF. The dye-labeled oligonucleotides were assembled on the silver islands on the solid substrate, and the complementary oligonucleotide-displaced particles were coupled via oligonucleotide hybridization. The couplings between particles and islands resulted in an obvious fluorescence enhancement.
Introduction
Fluorescence detection is a useful technique for biological assays.1,2 Although the fluorescence is highly sensitive, it is always crucial to increase sensitivity to detect lower concentration of the target molecule.3-6 Numerous methods have been developed,7,8 including surface-enhanced fluorescence on metal nanostructures (SEF),9 where fluorescence enhancement occurs when a fluorophore is localized near the surface of metallic nanoparticles.10,11 SEF is believed to happen through the coupling of the fluorophore with radiating plasmon from the metallic particles.12 However, fluorescence is also quenched competitively by the metal when the fluorophore is close to the metal core. The maximum enhancement for the fluorophore occurs at about 10 nm from the metal surface.9 Examinations from Mie theory show that small particles quench fluorescence because the absorption dominates over the scattering. Conversely, large particles enhance fluorescence because the scattering component is the dominant factor.12a In addition, it has been found that the quenching of excited fluorophores on the metal nanoparticle is also dependent on core size.12c Therefore, the size of the metal core can be regarded as an important factor affecting enhancement and quenching of the fluorophore on the particle. In this study, we verified this theoretical viewpoint through experiment.
We hope to develop a sensitive approach for DNA detection using the SEF principle. Herein, the silver particles were prepared by a modified Brust method.13 The fluorophore-labeled oligonucleotides, which had 23 bases and a ca. 8 nm length, were bound to the silver particles. The distance from the fluorophore to the metal core was estimated to be about 4-5 nm. At this distance, quenching became negligible but the enhancement dominated the emission.9b To prevent the self-quenching of fluorophores, only a limited number of fluorophores were displaced on the particles.14 In this study, organic monolayer-protected metallic nanoparticles were utilized because the oligonucleotide displacement could be controlled quantitatively through the mole ratio of thiolate oligonucleotide/organic ligand on the particle.13,15,16N-(2-Mercaptopropionyl)glycine (abbreviated as tiopronin) functioned as a ligand in the preparation of particle, and the tiopronin-coated particles displayed good chemical stability and solubility in water.17 The core size was controlled by the mole ratio of silver salt/tiopronin in the preparation. The thiolate fluorescein-labeled single-stranded oligonucleotides and their complementary unlabeled oligonucleotides were bound to the tiopronin-coated silver particles through ligand exchanges.
It is believed that there is an enhanced electromagnetic field near the metallic core and that SEF is induced when the fluorophore is localized in this electromagnetic field.9-11 The fields overlap and become denser when metallic nanoparticles are in close proximity to each other.18 Hence, in addition to the individual particles, the oligonucleotide-bound particles were also coupled through oligonucleotide hybridizations (Scheme 1) to investigate the SEF. In addition, the oligonucleotide-displaced particles were coupled with complementary oligonucleotides capped on the immobilized silver particles as islands on the glass slide (Scheme 2) to investigate SEF caused by the coupled particles on the solid substrate.
SCHEME 1.
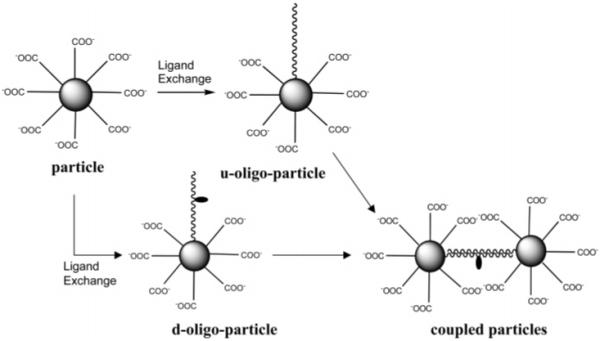
Oligonucleotide Displacements on Particles by Ligand Exchanges and Particle Couplings by Oligonucleotide Hybridizations
SCHEME 2.
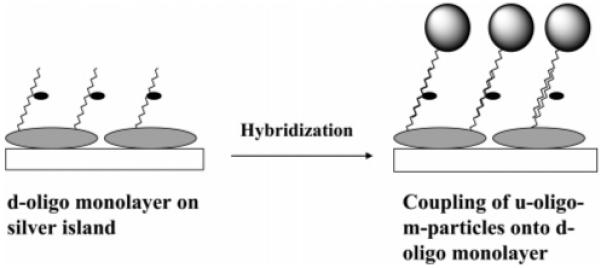
Couplings of Silver Particle with Silver Islands on a Solid Substrate by Oligonucleotide Hybridizations
Experimental Section
All reagents (Aldrich) and spectroscopic grade solvents (Fisher, Aldrich) were used as received. Oligonucleotides were synthesized by the Biopolymer Laboratory of the University of Maryland at Baltimore, and one of them was fluorescein labeled (Scheme 3). RC dialysis membrane (MWCO 8000) was available from Spectrum Laboratories, Inc. Nanopure water (>18.0 MΩ), purified using the Millipore Milli-Q gradient system, was used in all experiments.
SCHEME 3.

Oligonucleotide Structures
Tiopronin Metallic Nanoparticles and Their Coupling by Hybridization
Tiopronin-coated silver nanoparticles were prepared by using a modified Brust reaction17,19,20 with moleratios of tiopronin/silver nitrate ) 6/1 (small particle, abbreviated as s-particle), 1/1 (mid-size particle, abbreviated as m-particle), and 1/6 (large particle, abbreviated as l-particle), respectively, in methanol. The products were precipitated from solution. After removing the solution by filtration, the solid residuals were washed with methanol. The particles (1 mg/mL) and tiopronin (10 mM) were co-dissolved in water and the solution was stirred for 24 h for annealing of the particles. The solvents were removed under vacuum and the residuals were washed thoroughly with methanol. The solid particles were then dissolved in water and dialyzed against water.
Ligands on the particles were displaced partially by thiolate dye-labeled single-stranded oligonucleotides (d-oligo) or unlabeled single-stranded oligonucleotides (u-oligo) by stirring in 10 mM KCl solution with mole ratios of oligonucleotide/tiopronin ) 1/1 for 72 h at room temperature.16 Uncapped oligonucleotides were removed by dialysis against water. The d-oligo and u-oligo bound particles were abbreviated as d-oligoparticles and u-oligo-particles, respectively.
The coupling between d-oligo-particles and u-oligo-particles occurred by oligonucleotide hybridization. The d-oligo- and u-oligo-particles were mixed by a mole ratio of 1/1 containing 1 mg of particles/mL in 10 mM KCl solution at room temperature for 24 h. The coupled particles were then purified by dialysis against water.
Surface Couplings of Oligonucleotide Displaced Particles on Solid Substrate
The silver islands on glass substrates were prepared as described earlier.9c The plasmon absorbances of silver islands were near 0.2. The thiolate oligonucleotides were assembled on the island surfaces by dipping the slides in a 1 mM oligonucleotide aqueous solution for 24 h. The slides were washed thoroughly with water. The oligonucleotide monolayers on the glass substrates were hybridized by the complementary oligonucleotide-displaced particles by dipping the slides into a 10 mM KCl solution containing 1 mg of particles/mL for 72 h. The slides were washed with water.
Spectra
Absorption spectra were monitored with a Hewlett-Packard 8453 spectrophotometer. Fluorescence spectra of samples in solution were recorded with a Cary Eclipse Fluorescence Spectrophotometer, while fluorescence spectra on the surface were recorded with a SLM 8000 Spectrofluorometer in front face under excitation at 514 nm with a mode-locked argon ion laser.9c Transmission electron micrographs (TEM) were taken with a side-entry Philips electron microscope at 120 keV. Samples were cast from water solutions onto standard carboncoated (200-300 Å) Formvar films on copper grids (200 mesh) by placing a droplet of a 1 mg/mL aqueous sample solution on the grids.
Results and Discussion
The tiopronin-coated particles were generated through reduction of silver sulfur salts formed from silver nitrate and tiopronin by NaBH4.17 Ligands were expected to pack densely on the metallic cores.14 The particles were annealed in 10 mM tiopronin aqueous solution for 24 h and then dialyzed against water. This gentle annealing procedure was used by Murray et al. in their work with alkanethiol-coated Au particles.21a Schaaff et al. also observed a narrowing of the dispersity of alkanethiol-coated Au particles using a similar procedure with heating.21b Because the NaBH4 was used in excess in the preparation, the ligands on the particles were expected to be deprotonated and the pH was measured at 7.5 in 10 mM KCl aqueous solution.
The size of metallic cores was controlled by the mole ratios of AgNO3/tiopronin in the preparation. The particles could be outlined by TEM images (Figure 1). Size distributions of these silver cores were obtained from at least 200 individual particles,using Scion Image Beta Release 2 (available at www.scioncor-p.com). The particles had average diameters of 2 nm (AgNO3/tiopronin = 1/6, s-particle), 5 nm (AgNO3/tiopronin = 1/1, m-particle), and 20 nm (AgNO3/tiopronin = 6/1, l-particle), respectively, according to their histograms. Although the particles had been annealed, it was still difficult to achieve good monodispersity of the silver particles.17 In fact, the thiol-coated silver particles often demonstrated a broadening in the core size distribution (polydispersity).15 However, the size difference could be determined clearly from their TEM images. Over 80% of particles had their diameters in a range of 2 ± 0.6 nm for s-particles, 5 ± 3 nm for m-particles, and 20 ± 6 nm for l-particles, implying that they could be used qualitatively in core size-dependent measurements.
Figure 1.
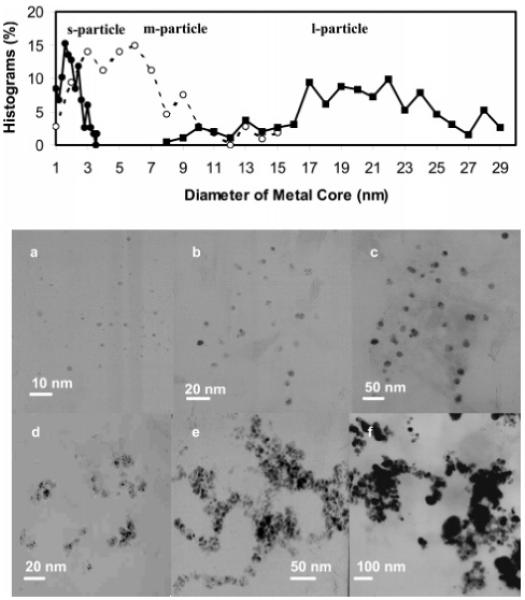
TEM images of (a) s-particles (2 nm), (b) m-particles (5 nm), (c) l-particles (20 nm), (d) s-aggregates, (e) m-aggregates, and (f) l-aggregates.
The tiopronin-coated particles showed different absorbance spectra depending on their core size (Figure 2).20 The s-particle displayed a spectrum of metallic crystal with an absorbance monotonic decrease from high to low energy wavelengths without a plasmon band, while the m- and l-particles displayed plasmon absorbances at 405 and 411 nm, respectively. The broadened and red-shifted extinction spectrum of l-particles overlapped that of m-particles. Similar effects have already been reported.20b These results were consistent with observations from TEM images.
Figure 2.

Absorbance spectra of s-, m-, and l-particles in water.
Tiopronin ligands could be displaced by thiolate dye-labeled single-stranded oligonucleotides (d-oligo) or unlabeled complementary single-stranded oligonucleotides (u-oligo).22 The ligand exchanges were slow and inefficient due to steric hindrance of bulky oligonucleotides. Similar to the s-particle, the oligonucleotide-bound small particle (d-oligo-s-particle for d-oligo and u-oligo-s-particle for u-oligo) did not exhibit a plasmon absorbance (Figure 3). The oligonucleotide displacements on the midsize (d-oligo-m-particle for d-oligo and u-oligo-m-particle for u-oligo) and large particles (d-oligo-l-particle for d-oligo and u-oligo-l-particle for u-oligo) led to small blue shifts of their plasmon wavelengths to 398 and 407 nm, respectively. The plasmon wavelength of the metal particle is known to be sensitive to the ligand composition on the metal core and display a blue or red shift in the ligand exchange.23,24 Although the binding of oligonucleotides on the citrate-coated Au or Ag particle generally resulted in a red-shift,23 it was reasonable here for the organic monolayer-coated silver particle to display a small blue-shift when the ligands on the particle were displaced partially by the oligonucleotides.
Figure 3.

Absorbance spectra of d-oligo-s-, -m-, and -l-particles in 10 mM KCl solution.
The d-oligo-particles displayed emission maximum at 518 nm upon excitation at 450 nm (Figure 4), a 10 nm blue-shift from that of the free d-oligo. The fluorescence shoulder at 550nm for d-oligo-particles was probably due to the interaction of the immobilized fluorophore and metallic core.
Figure 4.

Fluorescence spectra of d-oligo-s-, -m-, and -l-particles in 10 mM KCl solution upon excitation at 450 nm. A mixture of d-oligo and m-particle at the same concentrations worked as reference.
The average oligonucleotide number bound on each particle could be estimated by a standard linear calibration curve.25 Acyanide solution was used to dissolve the silver core and release the capped ligands on the particle.26 Standard curves were measured with known concentrations of fluorophore-labeled oligonucleotides, using identical amounts of buffer solution and cyanide concentration. After the particles were dissolved, the fluorescence maximums (518 nm) were converted to molar concentrations of the fluorescein-labeled oligonucleotide by interpolation from a standard linear calibration curve. When the concentrations of particles were 2 × 10-2 (d-oligo-s-particle), 5 × 10-2 (d-oligo-m-particle), and 6 × 10-2 mg/mL (d-oligo-l-particle), the concentrations of fluorescein-labeled oligonucleotides were about 4.2 × 10-6, 1.7 × 10-6, and 2.0 × 10-6 M, respectively. It was known that the ligands on the edges of metal cores readily displaced in the ligand exchange. The small particles, which had more defect sites (edges, vertexes) on the core surface, led to greater displacements of bulky oligonucleotides onto the metal core.27
By using mixtures of free d-oligo and corresponding particles with the same concentrations as controls, the fluorescence of d-oligo-particles was discovered to progressively alter with the core size in an order of 0.6, 1.9, and 3.5 times for the small, mid-size, and large particle, respectively. This result reveals that the SEF became stronger with an increase of particle size. In other words, fluorescence enhancement was core size-dependent. Such enhancement was ascribed to at least two principal factors: (1) increases of excitation rate from the local enhanced field by interaction of an incident light with the metal and (2) a higher quantum yield from an increase of the intrinsic decay rate of the fluorophore.11
The extinction properties of metal particles can be expressed by a combination of both absorption (CA) and scattering (CS) factors when the particles are spherical and have comparable sizes to the incident wavelength,9a,12a,b
| (1) |
where k1) 2πn1/λ0 is the wavevector of the incident light in medium 1 and α is the polarizability of the sphere of radius r,
| (2) |
and ∊m is the complex dielectric constant of metal. The first term represents the cross section due to absorption and the second term represents the cross section due to scattering. We expected CA to cause quenching and CS to cause enhancement.12b The quenching term increases as the r3 factor, and the enhancement term increases as the r6 factor. Examinations of CA and CS calculated from Mie theory show that small metal particles are expected to quench fluorescence because the absorption dominates over the scattering, while larger size nanostructures are expected to enhance fluorescence because the scattering component is dominant over the absorption.12a Hence, the d-oligo-s-particle displayed only quenching but the d-oligo-m-particle and the d-oligo-l-particle displayed fluorescence enhancements.
Fluorescence enhancement induced by silver particles in solution was lower than that observed on silver islands on a solid substrate (>10 times),27 which was probably due to three factors: particle size, particle coupling, and distance of the fluorophore from the metal surface. The largest particles in this study had an average diameter near 20 nm, much smaller than that of the islands.12 When the particles were immobilized as islands on the solid substrate, the distances among the particles were close and the enhanced fields overlapped.18 We expect this is another factor leading to stronger SEF in this experiment. In addition, the single-stranded oligonucleotide was flexible in solution and competitive quenching became stronger when the fluorophore approached the silver core. Although the enhanced efficiencies were weaker in this study, SEF has been clearly demonstrated to be a core size-dependent phenomenon.
The tiopronin-coated particles were also displaced by u-oligo to yield u-oligo-particles. Under the analogous displacement conditions with the d-oligo-particles, the u-oligo-particles behaved similar to the d-oligo-particles, in terms of displacement number. The u-oligo-particles could be coupled with the d-oligoparticles through oligonucleotide hybridizations in 10 mM KCl solution. It was known that the particles with the core diameters of 2 and 5 nm possessed chemical compositions of Ag96-(ligand)53 and Ag1082(ligand)453,16b respectively. For the s- and m-oligo-particles, these compositions were Ag96 (ligand)51.4- (oligo)1.6 and Ag1082 (ligand)445 (oligo)8 as estimated from the concentrations of oligonucleotides and particles. We could not find the exact composition of the l-particle from the literature, but there is no doubt that the oligo-l-particle should have multidisplaced oligonucleotides on each particle. Therefore, the couplings resulted in aggregation of particles. The aggregated particles could be outlined by TEM images (Figure 1d-f). The aggregates of small particles (s-aggregates, Figure 1d) exhibited metallic cores in close proximity to the individual s-particles. The metallic cores of aggregated mid-size (m-aggregates, Figure 1e) and large particles (l-aggregates, Figure 1f) were difficult to determine individually because of their compact aggregations.
The aggregates displayed absorbance spectra dependent on particle size (Figure 5). The s-aggregates displayed a similar spectrum to that of the individual s-particles without the plasmon band, suggesting that the small particles could not be aggregated compactly.28 In other words, the oligonucleotide chains were too long for the compact couplings between the coupled small particles. The m- and l-aggregates showed plasmon wavelengths at 395 and 397 nm, respectively, about 10 nm blue shifts from the individual particles.29,30 These observations were consistent with those from the TEM images. The l-aggregates displayed an obvious broadening absorbance and a characteristic longitudinal shoulder at 575 nm,26 showing that the l-particles could result in a more compact aggregation than the m-aggregateswhen the same length of oligonucleotide chain was used to separate the coupled particles.29
Figure 5.

Absorbance spectra of s-, m-, and l-aggregates in 10 mM KCl solution.
The aggregates displayed a fluorescence maximum at 518 nm (Figure 6) upon excitation at 450 nm, close to that of individual particles. Compared to corresponding individual particles coated with oligonucleotides at the same concentrations as controls, the fluorescence of l-aggregates and m-aggregates was enhanced 3.0 and 2.5 times (Figure 6), respectively, but remained almost unchanged for the s-aggregates. The large particles were expected to have denser enhanced fields, so it was suggested that the l-aggregates exhibited the stronger overlapped fields. This result implied that fluorescence enhancement by the aggregation of particles also depends on their core sizes. The double-stranded oligonucleotides between the coupled particles became rigid after hybridization and were expected to weaken the competitive quenching by the metallic cores. The s-aggregate did not have an obvious fluorescence enhancement, indicating that the plasmons of metal particles were crucial for SEF.
Figure 6.

Fluorescence spectra of s-, m-, and l-aggregates in 10 mM KCl solution upon excitation at 450 nm.
In addition to the coupling of silver particles in solution, we also studied SEF by coupling the particles on solid substrate. Silver islands on glass slides, which were prepared by chemical reduction, displayed a plasmon absorbance at 440 nm, indicating that these were nanoscale particles immobilized on the solid substrate.9 The island samples used in this study had absorbances of ca. 0.2. The d- and u-oligo were bound respectively on the islands. Such bindings were verified by the red shifts of the plasmon absorbance from 440 to 467 and 482 nm, respectively. It was uncertain why the wavelength shifts for u-oligo and d-oligo were different, but the large fluorophore on the d-oligo probably led to less assembly relative to the u-oligo.
The d- and u-oligos on silver islands were coupled by the u- and d-oligo-m-particles (abbreviated as d-oligo-u-oligo-m-particle and u-oligo-d-oligo-m-particle), respectively, through oligonucleotide hybridizations in 10 mM KCl solution. The midsize particles were selected because the small particles could not work well to strengthen SEF and the large particles were expected to meet stronger steric hindrances in the couplings. The absorbance change could not be monitored reasonably for the particle-couplings due to the strong absorbance from the islands. Figure 7 shows the emission spectra of u-oligo capped silver islands before and after coupling, on which the coupled system displayed a significant fluorescein emission over the background. Measurements on silver islands were carried out in front face geometry upon excitation at 514 nm and observed through a 530 nm long wave pass filter. We also did a control experiment by dipping the d-oligo assembled slide into the d-oligo-m-particle solution. The surface fluorescence did not change before and after such treatments, implying that the particles were coupled by the oligonucleotide hybridizations, rather than other possible interactions.
Figure 7.

Surface fluorescence spectra of u-oligo on the silver islands before and after coupling by d-oligo-m-particles upon excitation at 514 nm.
The d-oligo-u-oligo-m-particle displayed a fluorescence intensity enhancement of 70% relative to that before coupling (Figure 8), indicating that the coupling of silver particles on islands can enhance fluorescence. However, this enhancement was lower than that of the coupled particles in solution. A possible explanation for this is low coverage of coupled particles on the islands due to strong steric hindrances.
Figure 8.

Surface fluorescence spectra of d-oligo on the silver islands before and after coupling by u-oligo-m-particles upon excitation at 514 nm.
Our previous results revealed that silver islands on a solid substrate could increase surface fluorescence by about 1 order of magnitude.9 These papers describe further improvement of enhancement efficiency by a particle-fluorophore-particle sandwich structure using overlap of the enhanced field by particle couplings. This result could also provide a novel approach to gate the DNA hybridization assay.
Conclusions
Fluorescence enhancement on silver surfaces has been well documented in the recent literature.9,11 There continues to be interest in improving the brightness of fluorescence in solution. This is an extremely difficult task because the effect depends not only on the location of the labeled fluorophore but also on the size and shape of the particle. Theoretical predictions have limited use when one faces experimental difficulties such as precipitation or fluorescence quenching at close proximity to the metal. Therefore, only a limited number of reports on enhanced emission in solution have been published.22 We presented results based on a prediction that enhancement should be dependent on the size of the particle. Tiopronin-protected silver nanoparticles were prepared by varying mole ratios of tiopronin/silver nitrate. Their metallic cores had average diameters of 2, 5, and 20 nm, respectively. Ligands on the particles were partially displaced by thiolate single-stranded oligonucleotides through ligand exchanges. Fluorescence of fluorescein-labeled oligonucleotides could be enhanced relative to uncapped oligonucleotides depending on the metal core sizes. The particles were coupled through oligonucleotide hybridization, which resulted in aggregation because of multiple displacements of oligonucleotides on each particle. The aggregated particles also displayed fluorescence enhancement dependent on the metal core size. Large particles had high enhancement efficiency. Mid-size particles displaced by dye-labeled oligonucleotides were also coupled to the unlabeled complementary oligonucleotides assembled on the silver islands on the solid substrate. Surface fluorescence increased by coupling the particle, indicating that the particle-dye-particle sandwich structure on the solid substrate could work as an efficient approach to improve sensitivity in biological detection assay. When using the mixture of free d-oligo and l-particle in solution as a control, the l-aggregates led to a fluorescence enhancement by 1 order of magnitude (3.5 times for the individual particles and 3 times after aggregation), which was close to that of the silver islands on the solid substrate.
Acknowledgment
This research was supported by a grant from NIH, HG-02655, and NCRR, RR-08119.
References and Notes
- (1).(a) Lakowicz JR. In: Emerging biomedical application of time-resolved florescence spectroscopy, Topic in Fluorescence spectroscopy; Vol. 4, Probe Design and Chemical Sensing. Lakowicz JR, editor. Plenum Press; New York: 1994. [Google Scholar]; (b) Lakowicz JR, Soper S, Thompson RB, editors. Proceedings of Advances in fluorescence sensing technology IV; SPIE: San Jose, CA. 1999. [Google Scholar]
- (2).(a) Scheller FW, Schubert F, Fedrowitz J, editors. Frontiers in Biosensorics. I. Fundamental Aspects. Birkhauser Verlag; Berlin, Germany: 1997. [Google Scholar]; (b) Scheller FW, Schubert F, Fedrowitz J, editors. Frontiers in Biosensorics. II. Practical Applications. Birkhauser Verlag; Berlin, Germany: 1997. [Google Scholar]
- (3).(a) Ulman A. Chem. Rev. 1996;96:1533. doi: 10.1021/cr9502357. [DOI] [PubMed] [Google Scholar]; (b) Taton TA, Mucic RC, Mirkin CA, Letsinger RL. J. Am. Chem. Soc. 2000;122:6305. [Google Scholar]; (c) He L, Musick MD, Nicewarner SR, Salinas FG, Benkovic SJ, Natan MJ, Keating CD. J. Am. Chem. Soc. 2000;122:9071. [Google Scholar]; (d) Albrecht MG, Creighton JA. J. Am. Chem. Soc. 1977;99:5215. [Google Scholar]; (e) Nam J-M, Park S-J, Mirkin CA. J. Am. Chem. Soc. 2002;124:3820. doi: 10.1021/ja0178766. [DOI] [PubMed] [Google Scholar]; (f) Wang G, Zhang J, Murray RW. Anal. Chem. 2002;74:4320. doi: 10.1021/ac0257804. [DOI] [PubMed] [Google Scholar]
- (4).(a) Kneipp K, Kneipp H, Itzkan I, Dasari RR, Feld MS. Chem. Rev. 1999;99:2957. doi: 10.1021/cr980133r. [DOI] [PubMed] [Google Scholar]; (b) Leopold N, Lendl BA. J. Phys. Chem. B. 2003;107:5723. [Google Scholar]; (c) Mulvaney SP, Musick MD, Keating CD, Natan MJ. Langmuir. 2003;19:4784. [Google Scholar]; (d) Goulet PJG, Pieczonka NPW, Aroca RF. Anal. Chem. 2003;75:1918. doi: 10.1021/ac034067r. [DOI] [PubMed] [Google Scholar]
- (5).(a) Ho W. J. Phys. Chem. B. 2002;117:11033. [Google Scholar]; (b) Xu HX, Kall M. Phys. Rev. Lett. 2002;89:246802. doi: 10.1103/PhysRevLett.89.246802. [DOI] [PubMed] [Google Scholar]; (c) Tian ZQ, Ren B, Wu DY. J. Phys. Chem. B. 2002;106:9463. [Google Scholar]; (d) Lee SJ, Kim K. Chem. Commun. 2003;2:212. doi: 10.1039/b210217j. [DOI] [PubMed] [Google Scholar]
- (6).Lakowicz JR. Principles of Fluorescence Spectroscopy. 2nd ed. Kluwer Academic/Plenum Publishers; New York: 1999. [Google Scholar]
- (7).(a) Gosling JPA. Cell. 1990;36:1408. [Google Scholar]; (b) Walker NJ. Science. 2002;296:557. doi: 10.1126/science.296.5567.557. [DOI] [PubMed] [Google Scholar]
- (8).(a) Kronick MN. J. Immunol. Methods. 1986;92:1. doi: 10.1016/0022-1759(86)90496-5. [DOI] [PubMed] [Google Scholar]; (b) Daehne S, Resch-Genger U, Wolfbeis OS, editors. Near-Infrared Dyes for High Technology Applications. Kluwer Academic Publishers; New York: 1998. [Google Scholar]; (c) Casay GA, Shealy DB, Patonay G. Near-infrared fluorescence probes. In: Lakowicz JR, editor. Topics in Fluorescence Spectroscopy; Vol. 4, Probe Design and Chemical Sensing. Plenum Press; New York: 1994. pp. 183–222. [Google Scholar]; (d) Lövgren T, Pettersson K. Time-resolved fluoroimmunoassay, advantages and limitations. In: Van Dyke K, Van Dyke R, editors. Fluorescence Immunoassay and Molecular Applications. CRC Press; New York: 1990. pp. 234–250. [Google Scholar]
- (9).(a) Lakowicz JR. Anal. Biochem. 2005;337:171. doi: 10.1016/j.ab.2004.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lakowicz JR. Anal. Biochem. 2001;298:1. doi: 10.1006/abio.2001.5377. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gryczynski I, Malicka J, Shen YB, Gryczynski Z, Lakowicz JR. J. Phys. Chem. B. 2002;106:2191. doi: 10.1021/jp013013n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Malicka J, Gryczynski I, Fang J, Kusba J, Lakowicz JR. Anal. Biochem. 2003;315:160. doi: 10.1016/S0003-2697(02)00710-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Gersten JI, Nitzan A. Surf. Sci. 1985;158:165. [Google Scholar]
- (10).(a) Kummerlen J, Leitner A, Brunner H, Aussenegg FR, Wokaun A. Mol. Phys. 1993;80:1031. [Google Scholar]; (b) Sokolov K, Chumanov G, Cotton TM. Anal. Chem. 1998;70:3898. doi: 10.1021/ac9712310. [DOI] [PubMed] [Google Scholar]; (c) Antunes PA, Constantino CJL, Aroca RF, Duff J. Langmuir. 2001;17:2958. [Google Scholar]; (d) Kamat PV. J. Phys. Chem. B. 2002;106:7729. [Google Scholar]; (e) Kulakovich O, Strekal N, Yaroshevich A, Maskevich S, Gaponenko S, Nabiev I, Woggon U, Artemyev M. Nano Lett. 2002;2:1449. [Google Scholar]
- (11).(a) Geddes CD, Cao H, Gryczynski I, Gryczynski Z, Fang JY, Lakowicz JR. J. Phys. Chem. A. 2003;107:3443. doi: 10.1021/jp022040q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Geddes CD, Parfenov A, Roll D, Fang JY, Lakowicz JR. Langmuir. 2003;19:6236. doi: 10.1021/la020930r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Parfenov A, Gryczynski I, Malicka J, Geddes CD, Lakowicz JR. J. Phys. Chem. B. 2003;107:8829. doi: 10.1021/jp022660r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Kreibig U, Vollmer M. Optical Properties of Metal Clusters. Springer-Verlag; Berlin and Heidelberg, Germany: 1995. [Google Scholar]; (b) Kerker M, Blatchford CG. Phys. Rev. B. 1982;26:4082. [Google Scholar]; (c) Zhang J, Whitesell JK, Fox MA. J. Phys. Chem. B. 2003;107:605. [Google Scholar]
- (13).Templeton AC, Wuelfing WP, Murray RW. Acc. Chem. Res. 2000;33:27. doi: 10.1021/ar9602664. [DOI] [PubMed] [Google Scholar]
- (14).Aguila A, Murray RW. Langmuir. 2000;16:5949. [Google Scholar]
- (15).(a) Templeton AC, Hostetler MJ, Kraft CT, Murray RW. J. Am. Chem. Soc. 1998;120:1906. [Google Scholar]; (b) Hostetler MJ, Templeton AC, Murray RW. Langmuir. 1999;15:3782. [Google Scholar]; (c) Hostetler MJ, Green SJ, Stokes JJ, Murray RW. J. Am. Chem. Soc. 1996;118:4212. [Google Scholar]
- (16).Ingram RS, Hostetler MJ, Murray RW. J. Am. Chem. Soc. 1997;119:9175. [Google Scholar]
- (17).(a) Huang T, Murray RW. J. Phys. Chem. B. 2001;105:12498. [Google Scholar]; (b) Huang T, Murray RW. Langmuir. 2002;18:7077. [Google Scholar]
- (18).Lakowicz JR. Anal. Biochem. 2001;298:1. doi: 10.1006/abio.2001.5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Brust M, Walker M, Bethell D, Schiffrin DJ, Whyman R. J. Chem. Soc., Chem. Commun. 1994:801. [Google Scholar]
- (20).(a) Hostetler MJ, Wingate JE, Zhong C-J, Harris JE, Vachet RW, Clark MR, Londono JD, Green SJ, Stokes JJ, Wignall GD, Glish GL, Porter MD, Evans ND, Murray RW. Langmuir. 1998;14:17. [Google Scholar]; (b) Kim HS, Ryu JH, Jose B, Lee BG, Ahn BS, Kang YS. Langmuir. 2001;17:5817. [Google Scholar]
- (21).(a) Hicks JF, Miles DT, Murray RW. J. Am. Chem. Soc. 2002;124:13322. doi: 10.1021/ja027724q. [DOI] [PubMed] [Google Scholar]; (b) Schaaff TG, Shafigullin MN, Khoury JT, Vezmar I, Whetten RI. J. Phys. Chem. 2001;105:8785. [Google Scholar]
- (22).Zhang J, Malicka J, Gryczynski I, Lakowicz JR. Anal. Biochem. 2004;330:81. doi: 10.1016/j.ab.2004.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Tokareva I, Hutter E. J. Am. Chem. Soc. 2004;126:15784. doi: 10.1021/ja046779k. [DOI] [PubMed] [Google Scholar]
- (24).Hu J, Zhang J, Liu F, Kittredge K, Whitesell JK, Fox MA. J. Am. Chem. Soc. 2001;123:1464. [Google Scholar]
- (25).Zhao Z, Shen T, Xu H. Spectrochim. Acta. 1989;45A:1113. [Google Scholar]
- (26).(a) Demers LM, Mirkin CA, Mucic RC, Reynolds RA, III, Letsinger RL, Elghanian R, Viswanadham G. Anal. Chem. 2000;72:5535. doi: 10.1021/ac0006627. [DOI] [PubMed] [Google Scholar]; (b) Taton TA, Mucic RC, Mirkin CA, Letsinger RL. J. Am. Chem. Soc. 2000;122:6305. [Google Scholar]; (c) Storhoff JJ, Lazarides AA, Mirkin CA, Letsinger RL, Mucic RC, Schatz GC. J. Am. Chem. Soc. 2000;122:4640. [Google Scholar]; (d) Marinakos SM, Novak JP, Brousseau LC, House AB, Edeki EM, Feldhaus JC, Feldheim DC. J. Am. Chem. Soc. 1999;121:8518. [Google Scholar]
- (27).Donkers RL, Song Y, Murray RW. Langmuir. 2004;20:4703. doi: 10.1021/la0497494. [DOI] [PubMed] [Google Scholar]
- (28).Malicka J, Gryczynski I, Lakowicz JR. Anal. Chem. 2003;75:4408. doi: 10.1021/ac020739m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).(a) Zamborini FP, Leopold MC, Hicks JF, Kulesza PJ, Malik MA, Murray RW. J. Am. Chem. Soc. 2002;124:8958. doi: 10.1021/ja025965s. [DOI] [PubMed] [Google Scholar]; (b) Hicks JF, Seok-Shon Y, Murray RW. Langmuir. 2002;18:2288. [Google Scholar]
- (30).(a) Esumi K, Matsushima Y, Torigoe K. Langmuir. 1995;11:3285. [Google Scholar]; (b) Link S, Mohamed MB, El-Sayed MA. J. Phys. Chem. B. 1999;103:3073. [Google Scholar]