

Abstract
Nitroxides can undergo one- or two-electron reduction reactions to hydroxylamines or oxammonium cations, respectively, which themselves are interconvertible, thereby providing redox metabolic actions. 4-Hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl (tempol) is the most extensively studied nitroxide. It is a cell membrane-permeable amphilite that dismutates superoxide catalytically, facilitates hydrogen peroxide metabolism by catalase-like actions, and limits formation of toxic hydroxyl radicals produced by Fenton reactions. It is broadly effective in detoxifying these reactive oxygen species in cell and animal studies. When administered intravenously to hypertensive rodent models, tempol caused rapid and reversible dose-dependent reductions in blood pressure in 22 of 26 studies. This was accompanied by vasodilation, increased nitric oxide activity, reduced sympathetic nervous system activity at central and peripheral sites, and enhanced potassium channel conductance in blood vessels and neurons. When administered orally or by infusion over days or weeks to hypertensive rodent models, it reduced blood pressure in 59 of 68 studies. This was accompanied by correction of salt sensitivity and endothelial dysfunction and reduced agonist-evoked oxidative stress and contractility of blood vessels, reduced renal vascular resistance, and increased renal tissue oxygen tension. Thus, tempol is broadly effective in reducing blood pressure, whether given by acute intravenous injection or by prolonged administration, in a wide range of rodent models of hypertension.
I. Introduction
A. Development of Knowledge Concerning Nitroxides
The biological activity of nitroxides was recognized in 1964 by Emmerson and Howard-Flanders who reported that nitroxides sensitized bacteria to the lethal effects of radiation (Emmerson and Howard-Flanders, 1964, 1965). This finding sparked interest in their therapeutic potential. In 1965, McConnell and Griffith demonstrated that nitroxides are “free radicals” and paramagnetic “spin labels.” They showed further that nitroxides could be linked stably and covalently to proteins and other agents as biomarkers for molecules of interest such as poly-L-lysine, bovine serum albumin, hemoglobin, or catalase (Griffith and McConnell, 1966; Grebenshchikov et al., 1972). Early studies of nitroxide synthesis and action were described by Rozantsev, Swartz, and coworkers (Rozantsev, 1970; Chumakov et al., 1972, 1974; Grebenshchikov et al., 1972; Rozantsev and Sholle, 1979; Rozantsev and Zhdanov, 1987; Kocherginsky and Swartz, 1995).
In the 1990s Schnackenberg, Welch, and Wilcox reported that intravenous, intraperitoneal, or per os administration of 4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl (tempol)1 to hypertensive rat models led to a reduction in blood pressure (BP) and lipid peroxidation (Schnackenberg et al., 1998; Schnackenberg and Wilcox, 1999). They reported that the acute antihypertensive response to nitroxides was related to their in vitro superoxide dismutase (SOD)-mimetic activity (Patel et al., 2006) and dependent on potentiating the effects of nitric-oxide synthase (NOS) and on inhibition of the sympathetic nervous system (SNS) by actions that included activation of ATP-dependent potassium (KATP) channels (Chen et al., 2007a), whereas the long-term response to tempol entailed correction of salt sensitivity (Welch et al., 2005b), renal hypoxia (Welch and Wilcox, 2001; Welch et al., 2003, 2005a), and renal vasoconstriction (Kawada et al., 2002; Wang et al., 2003b, 2004b, 2006b). They showed further that local microperfusion of tempol into the interstitium of the kidney of the spontaneously hypertensive rat (SHR) model of oxidative stress restored NO signaling between the macula densa and afferent arteriole (Welch and Wilcox, 2001) and that systemic infusion of tempol improved the efficacy with which the kidney used oxygen for tubular sodium (Na+) transport and thereby increased the renal cortical pO2 (Welch et al., 2005a).
Fink, Xu, and coworkers first demonstrated the NO-independent effects of tempol to reduce SNS activity (Xu et al., 2001, 2002) and related this to the antihypertensive response (Xu et al., 2004) via activation of large-conductance, Ca2+-activated potassium (BK) channels (Xu et al., 2005, 2006).
Nishiyama and coworkers reported that activated renal sympathetic nerves directly whereas local neural application of tempol prevented nerve firing (Shokoji et al., 2003, 2004; Majid et al., 2005). With Majid, he reported that NOS blockade in the dog unexpectedly enhanced the natriuresis and diuresis in response to tempol (Majid and Nishiyama, 2002) and related this result to enhanced generation of ROS in the kidney after NOS blockade (Majid et al., 2004).
These studies laid the foundation for an explosion of scientific interest in nitroxides as agents to reduce ROS and BP. These are the subject of this review. The larger field of the role of ROS in hypertension and aging has been extensively reviewed (Cai and Harrison, 2000; Wilcox and Welch, 2001; Himmelfarb et al., 2002; Wilcox, 2002, 2003, 2005; Cai et al., 2003; Touyz, 2003, 2004; Himmelfarb, 2004; Modlinger et al., 2004; Wilcox and Gutterman, 2005; Harrison et al., 2007; Lambeth, 2007; Lambeth et al., 2007).
In this review we describe the published experience of the BP-lowering actions of nitroxides such as tempol. The emphasis is placed on dose, delivery, responsiveness, and mechanisms of action. We do not consider the larger field of organ protection by tempol. Studies with tempol are of importance both because of the potential role of tempol as a therapeutic agent to reduce ROS and BP and because of the insight these studies yield into the roles of ROS in hypertension.
B. Biochemistry of Nitroxides
Nitroxides share a reducible nitroxide (•N–O) group as part of a six- or five-member carbon ring. Some examples discussed in this review from the very large family of nitroxides are represented in Fig. 1. Tempol is a cell membrane-permeable amphilite nitroxide. It is a redox cycling agent that can metabolize superoxide anion ( ) and many other ROS (Krishna et al., 1992, 1996a, 1998; Li et al., 2006). Tempol is among the most potent of the nitroxides in protecting cells and tissues from the damaging effects of ROS (Krishna et al., 1998; Li et al., 2006). The action of nitroxides to metabolize ROS is ascribed primarily to cyclic one- or two-electron transfer among three oxidation states: the oxammonium cation, the nitroxide, and the hydroxylamine (Fig. 2A). Nitroxides undergo a very rapid, one-electron reaction in vivo to the corresponding hydroxylamine (Swartz, 1990; Okajo et al., 2006), which has antioxidant activity (Krishna et al., 1992, 1998; Wu et al., 1997; Hahn et al., 2000). Hydroxylamines can be converted to nitroxides by hydrogen peroxide (H2O2) or other oxidants such as transition metals (Dikalov et al., 1998). Indeed, incubation of tempol hydroxylamine (tempol-H) with H2O2 in the presence of cytochrome c oxidase (Chen et al., 1989) yields radical tempol (Moore et al., 1992). Nitroxides can be converted to the corresponding oxammonium compounds by hypervalent heme (Krishna et al., 1992) and thereafter can undergo fast one-electron reactions to the nitroxide or by interaction with NADPH can undergo two-electron reactions to the hydroxylamine. These reactions contribute to the pro-oxidant and potentially adverse effects of nitroxides (Israeli et al., 2005). A rapid exchange between the nitroxide, hydroxylamine, and oxammonium cation species confers recycling and catalytic activity on nitroxides (Krishna et al., 1992). This interaction among the nitroxide species has been reviewed recently (Soule et al., 2007). Tempol is rapidly converted to tempol-H in tissues but does not undergo significant further metabolism over several hours (Hyodo et al., 2006).
Fig. 1.

Some examples of six- and five-member ring nitroxide compounds. [Reprinted from Patel K, Chen Y, Dennehy K, Blau J, Connors S, Mendonca M, Tarpey M, Krishna M, Mitchell JB, Welch WJ, and Wilcox CS (2006) Acute antihypertensive action of nitroxides in the spontaneously hypertensive rat. Am J Physiol Regul Integr Comp Physiol 290:R37–R43. Copyright © 2006 American Physiological Society. Used with permission.]
Fig. 2.
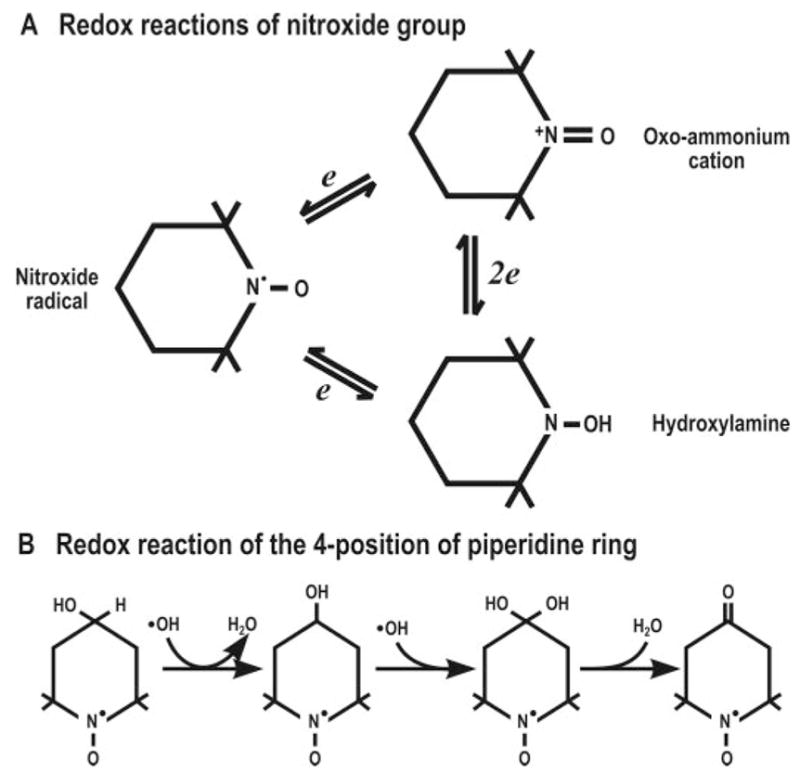
A, oxidized and reduced nitroxide forms, and their intercon-version. [Reprinted from Soule BP, Hyodo F, Matsumoto K, Simone NL, Cook JA, Krishna MC, and Mitchell JB (2007) The chemistry and biology of nitroxide compounds. Free Radic Biol Med 42:1632–1650. Copyright © 2007 Elsevier Limited. Used with permission.] B, conversion of tempol to tempone by reaction of the 4-position of the piperidine ring with hydroxyl radical. [Reprinted from Saito K, Takeshita K, Ueda J, and Ozawa T (2003) Two reaction sites of a spin label, TEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl), with hydroxyl radical. J Pharm Sci 92: 275–280. Copyright © 2003 Wiley-Liss, Inc., a subsidiary of John Wiley & Sons, Inc. Used with permission.]
Saito et al. demonstrated that hydroxyl radical (•OH) interacts both with the nitroxide group and with the 4-position of the piperidine ring of tempol to form 4-oxo-2,2,6,6-tetramethylpiperidine-N-oxyl (tempone) with the appearance of a new triplet electron paramagnetic resonance (EPR) signal (Saito et al., 2003) (Fig. 2B). However, at physiological levels of pH, this reaction accounts for only approximately 10% of the reduction of •OH by tempol (Deffner and Schimmack, 1976; Saito et al., 2003). This reaction is also rapidly reversible because tempone was metabolized in cells (Kroll et al., 1999) or in mice to tempol over 10 min (Kroll and Borchert, 1999; Kroll et al., 1999).
C. Interaction with Reactive Oxygen Species
Nitroxides metabolize to H2O2 by a catalytic action and are thereby termed “SOD mimetics” (Chateauneuf et al., 1988; Samuni et al., 1988, 1990a,b, 2002; Krishna et al., 1992, 1996a; Damiani et al., 1999a; Zhang et al., 1999; Samai et al., 2007; Van Dyke et al., 2007). The catalytic nature of this reaction was challenged by results of stop-flow kinetics (Weiss et al., 1993). In contrast, a detailed EPR study concluded that nitroxides exert apparent catalytic activity above stoichiometric scavenging of (Krishna et al., 1996a). Tempol is effective in metabolizing generated in solutions of xanthine plus xanthine oxidase (Patel et al., 2006) or in cells stimulated by angiotensin (Ang) II (Luo et al., 2007).
The conversion of nitroxides to the hydroxylamine occurs principally intracellularly and is reversible (Onishi and Morales, 1976; Nothiglaslo and Bobst, 1991; Bobko et al., 2007). This reaction is facilitated by ascorbate (Marx et al., 2000) in erythrocytes (Saphier et al., 2003) and the liver (Keana et al., 1987). Ascorbate is oxidized by tempol to dehydroascorbate at a rate that is diffusion limited (Champion et al., 2004; Vislisel et al., 2007). Ascorbate is the preferred reductant in erythrocytes because incubation of human erythrocytes with tempol over 2.5 h depleted 80% of intracellular ascorbate, without measurable effects on glutathione or α-tocopherol (May et al., 1998). Bobko et al. (2007) reported that the bimolecular rate constants of ascorbate-induced reduction are higher for six-member nitroxides than for five-member ring nitroxides. Tetraethyl-substituted imidazoline nitroxides are the most resistant to reduction by ascorbate.
However, nitroxides also can be reduced by glutathione (Finkelstein et al., 1984; Khramtsov et al., 1989; Schafer and Buettner, 2001; Kuppusamy et al., 2002; Glebska et al., 2003; Bobko et al., 2007). In the presence of thiols, reacted with nitroxides to yield a N-hydroxy-N-hydroperoxyl intermediate that decomposed rapidly to the hydroxylamine and a compound believed to be sulfenyl hydroperoxide (Finkelstein et al., 1984). The latter reduced two additional nitroxide molecules to account for the unusual 3:1 stoichiometry of this reaction (Finkelstein et al., 1984).
Ascorbate can convert the nitroxide oxammonium cation rapidly to the hydroxylamine, whereas the nitroxide radical facilitates the dismutation of the ascorbate free radical. These reactions underlie a synergistic antioxidant effect of nitroxides and ascorbate (Bobko et al., 2007), which is facilitated further by scavenging of the ascorbate radical by glutathione. Clearly, there are extensive interactions between nitroxides, ascorbate, and glutathione.
Nitroxides such as tempol also metabolize, detoxify, or prevent the formation or action of a wide range of other ROS. These include H2O2 by a catalase-like action (Krishna et al., 1996b, 1998; Wu et al., 1997; Samuni et al., 2001), which can involve the metabolism of H2O2 by the oxammonium cation (Krishna et al., 1996b) or the hydroxylamine (Dikalov et al., 1998) and interaction with hemeproteins (Krishna et al., 1996b). Nitroxides were shown to possess both catalytic and stoichiometric effects in metabolizing H2O2 (Krishna et al., 1998). Nitroxides metabolized or prevented the generation of •OH (Anastassopoulou and Rakintzis, 1984; Charloux et al., 1995; Wu et al., 1997; Risso-de Faverney et al., 2000; Zeltcer et al., 2002), singlet oxygen (Yoshino et al., 2002), peroxyl radicals (Offer and Samuni, 2002; Gadjeva et al., 2005), nitroxyl anion (Wink et al., 1998; Bai et al., 2001; Hewett et al., 2005), peroxynitrite (ONOO•) (Carroll et al., 2000; Cuzzocrea et al., 2001; El-Remessy et al., 2003; Fernandes et al., 2005; Song et al., 2007; Van Dyke et al., 2007), nitrogen dioxide generated by myeloperoxidase radicals (Borisenko et al., 2004; Dabrowska et al., 2005), and peroxidation products of lipids (Nilsson et al., 1989; Schnackenberg and Wilcox, 1999; Gadjeva et al., 2005) or phospholipids (Manevich et al., 2002). They prevented tissue damage by oxidizing reduced transition metals, including ferrous (Samuni et al., 1991b; Charloux et al., 1995; Zeltcer et al., 1997, 2002; Udassin et al., 1998;Risso-de Faverney et al., 2000; Glebska et al., 2001; Mehta et al., 2004; Murakami et al., 2005, 2006a,b,c; Nouri et al., 2007) and cuprous ions (Damiani et al., 1994; Zeltcer et al., 1997; Burlando and Viarengo, 2005; Murakami et al., 2006b, 2007; Persichini et al., 2006) or cadmium or chromium (Lewinska et al., 2008), thereby decreasing the availability of the reduced species for Fenton reactions (Monti et al., 1996; Glebska et al., 2001).
Tempol has been shown to protect lipids (Samuni and Barenholz, 1997; Samuni et al., 1997, 2000), DNA (Samuni et al., 1991a; Damiani et al., 1999b, 2000b), or proteins (Damiani et al., 2000a) from oxidative damage. Tempol interacted with other antioxidants to promote their ability to reduce oxidized lipids (Champion et al., 2004). Nitroxides prevented oxidative damage in many cellular or organ systems, for example, in the skin after UV radiation (Damiani et al., 2006; Shen et al., 2006), in cells after x-irradiation (Hahn et al., 1992b, 2000; Sasaki et al., 1998), or in tissues after incubation in a high glucose-containing medium (Xia et al., 2006).
Tempol has complicated effects on ONOO−. Tempol prevented ONOO− from nitrating phenol or tyrosine residues (Carroll et al., 2000) but increased nitrosation of phenol (Fernandes et al., 2005). Tempol decreased protein-3-nitrotyrosine formation while increasing the yield of protein nitrocysteine (Fernandes et al., 2005). Studies in solutions and cells implicated the oxammonium form of tempol in the oxidation of ONOO− to NO. Thus, hydroxyl or carbonate radicals, derived from ONOO−, oxidized tempol to the oxammonium cation that itself was reduced back to tempol while oxidizing further ONOO− to O2 and NO (Bonini et al., 2002). Thereafter NO reacted with nitrogen dioxide derived from ONOO− to produce the nitrosating species, dinitrogen trioxide (Bonini et al., 2002).
Li et al. (2006) recently compared the IC50 values (potency) of nitroxides in protecting lipids from peroxidation by •OH [assessed from malondialdehyde (MDA) formation in tissue extracts stimulated with Fe2+ and ascorbic acid], in protecting cells from damage by H2O2 (assessed from red blood cell hemolysis by H2O2), and in enhancing metabolism (assessed from formazan generation by the addition of nitroblue tetrazolium to zymosan A-stimulated leukocytes). Among eight 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) derivatives, 4–bis-TEMPO was the most potent. Interestingly, tempol had a lower IC50 value in the MDA assay for •OH (0.8 ± 0.2 μmol/l) than in the hemolysis assay for H2O2 (10.7 ± 0.2 μmol/l) or in the SOD assay for (326 ± 18 μmol/l). The authors concluded that the rank order of nitroxide scavenging of ROS was •OH > H2O2 > . This finding supports the rather weak SOD-mimetic action of tempol reported previously by Weiss et al. (1993). However, this conclusion is not secure because nitroxides were not tested against equimolar concentrations of ROS. Nevertheless, kinetic EPR studies in vitro demonstrated that the rate constant for the reaction of nitroxides with •OH (109 M−1·s−1) was much higher than that for the reaction of nitroxides with in the presence of cysteine (103–104 M−1·s−1) (Takeshita et al., 2002). The rate of reaction of nitroxides with •OH was almost diffusion-limited (Takeshita et al., 2002).
The multiple antioxidant actions of nitroxides have two consequences. First, a functional response to a nitroxide (e.g., a fall in BP) should not be assumed to relate to metabolism of a single ROS. Second, the ability of nitroxides to inhibit three or more sequential sites in an oxidative chain (for example , H2O2, and •OH) may underlie their efficacy in diverse models of oxidative stress.
These biochemical reactions have been widely studied in animal models, tissues, or cells. For example, the addition of tempol to aortas harvested from mice with oxidative stress reduced the lucigenin-enhanced chemiluminescence signal for vascular but enhanced transiently the luminol signal for vascular H2O2 (Chen et al., 2007b). More prolonged incubation of endothelial cells (ECs) with tempol reduced the dihydrorhodamine signal for H2O2. This result indicated that an increase in vascular H2O2 after tempol was a transient effect of metabolism of to H2O2. The H2O2 was later metabolized to O2 and H2O as a consequence of the catalase-mimetic effects of tempol. The addition of tempol to hepatoma cells blocked •OH signaling (Burlando and Viarengo, 2005). Tempol protected bacteria (Skórko-Glonek et al., 1999) and the stomach (Samuni et al., 1999) from the damaging ability of iron to generate •OH. The hydroxylamine was not effective.
D. Pro-Oxidant Actions
High concentrations of tempol (10−4–10−2 M) can have pro-oxidant effects in vascular smooth muscle and endothelial cells (VSMCs) (Alpert et al., 2004; May et al., 2005). The paradoxical pro-oxidant effects of high concentrations of manganese superoxide dismutase (Mn-SOD) (Omar and McCord, 1990), Cu/Zn-SOD (Omar et al., 1990), or tempol (Offer et al., 2000) have been ascribed to the dual ability of to both terminate and initiate lipid peroxidation (Nelson et al., 1994; Paller and Eaton, 1995; McCord and Edeas, 2005). These findings may account for the ability of low concentrations of tempol to protect cells from oxidant damage by paraquat, whereas very high concentrations of tempol of 10 mmol/l enhanced toxicity (Samai et al., 2007). The pro-oxidant action of tempol in ECs has been inconsistent and can be prevented by coincubation with antioxidants, for example, ascorbate (May et al., 2005).
A special feature of nitroxides is their conversion to the highly oxidizing oxammonium species (Goldstein et al., 2003), whose reduction to the hydroxylamine contributes to their pro-oxidant actions (Israeli et al., 2005).
E. Structure-Activity Relationships
The nitroxide moiety has been found to be essential for full antioxidant activity, whereas substitution at the 4-position affects potency (Samuni et al., 1988, 1990a; Krishna et al., 1998; Samuni and Barenholz, 2003; Anzai et al., 2006; Li et al., 2006). Extensive studies by Mitchell, Krishna, and colleagues using physiochemical methods coupled with EPR established that the one-electron redox cycling of six-member ring nitroxides such as tempol was enhanced by their ability to undergo reversible “boat-and-chair” conformational change (Krishna et al., 1996b). This was not possible with five-member ring nitroxides, which may account for their lesser biological activity (Patel et al., 2006). Krishna et al. (1998) reported a detailed structure-activity analysis of 58 nitroxides for protection against H2O2-induced cytotoxicity or ionizing radiation. Protection against H2O2 depended on the ring size, oxidation state (nitroxides > hydroxylamines > amines), and redox midpoint potentials (lowest potentials were most effective). A basic side chain enhanced radiation protection by facilitating the accumulation of the drug at the site of damage.
F. Metabolism and Pharmacokinetics
Nitroxides are stable organic free radicals without significant plasma protein binding (Okajo et al., 2006). The presence of a single unpaired electron on the radical yields unique insights into their pharmacokinetics because this species is detected by magnetic resonance imaging (MRI) via shortening of the relaxation time (T1) or by a characteristic spectrum on EPR (Hyodo et al., 2006; Swartz et al., 2007). These signals are lost after bioreduction of the nitroxide to the diamagnetic hydroxylamine (Yamaguchi et al., 1984). Tempol is converted to the hydroxylamine in liver microsomes, principally by NADPH and cytochrome c (Iannone et al., 1989a,b) but also can be reduced by sulfhydryl groups on proteins (Couet et al., 1985) or by ascorbate in the cell cytosol (Eriksson et al., 1987). Isolated keratinocytes use thioredoxin reductase to reduce tempol (Kroll et al., 1999).
The half-time (t1/2) for the loss of the nitroxide EPR signal in blood in vivo is dose-dependent and very variable. The t1/2 for the loss of signal from a fixed dose has been used to provide a measure of the rate of reduction of the nitroxide and thereby the redox state of the system. The t1/2 in conscious rats or mice after acute intravenous injection was short (Komarov et al., 1994) with a mean residence time for radical tempol in the inferior vena caval blood, liver, and kidneys of 0.25 to 15 min (Kamataria et al., 2002). Ueda et al. (2003) reported a t1/2 for radical tempol in the kidneys and livers of rats of 15 and 31 s after systemic injection, whereas a more prolonged t1/2 of 148 and 278 s, respectively, was seen ex vivo in organ homogenates. The rate of tempol reduction in an organ was related to ROS production (Turrens, 2003). The t1/2 for tempol reduction in the kidneys of rats was shorter than that in the liver both in vivo (Kamataria et al., 2002; Ueda et al., 2003) and in cell homogenates and correlated with the greater mitochondrial density in the kidneys (Ueda et al., 2003). Hepatic reduction depended on the metabolic rate of the liver. Thus, the t1/2 for tempol of 39 s in the liver of rats in vivo was reduced by 20% after ingestion of glucose, which was related to the development of mitochondrial oxidative stress because it was prevented by inhibition of mitochondrial function with sodium azide (Tada et al., 2001). Likewise, the reduction of tempol in the kidney depends on renal function. The administration of doxorubicin (Adriamycin) (Oteki et al., 2005) or puromycin (Ueda et al., 2002) to rats caused renal damage and proteinuria and prolonged the EPR decay of the tempol signal over the kidneys, indicating a diminished renal reducing ability. Subcellular fractionation revealed that the reducing activity of the kidneys was located primarily in mitochondria (Kamataria et al., 2002), whereas the liver also contained significant reducing actions in microsomes and cytosol (Ueda et al., 2003).
The rate of reduction of tempol has been used to assess the oxygenation or redox state of tissues (Mikuni and Tatsuta, 1998). The decay of the MRI or EPR signal after loading with tempol was reduced by hypoxia in vitro (Chen et al., 1989; Iannone et al., 1989b; Miura et al., 1990; Nakajima et al., 2002) and in vivo (Miura et al., 1992) and in neoplastic tissues, which are significantly hypoxic (Hyodo et al., 2006). The rate of reduction of tempol was increased in the livers of rats given ascorbic acid or glutathione (Tada et al., 2004), in the brains of rats fed vitamin E, vitamin C, or the free radical scavenging compound idebenone (Zs-Nagy, 1990; Matsumoto et al., 1998), or in cells deficient in glucose-6-phosphate dehydrogenase (Branca et al., 1988; Samuni et al., 2004). The time constant for the decay of the relaxation signal in a tissue was related to the initial reduction of the tempol radical to the hydroxylamine. The decay constant for tempol after injection into the mouse was 0.32 min−1 in the leg and 1.2 to 1.5 min−1 in the kidney (Hyodo et al., 2006).
The rate of reduction of six-member ring nitroxides in the presence of a reducing agent such as ascorbate has been shown to be 100-fold faster than that of five-member ring nitroxides (Samuni et al., 1990a; Nothiglaslo and Bobst, 1991). After intraperitoneal injection into mice, the oxidized (radical nitroyl) forms of six-member ring nitroxides such as tempol or 4-amino-2,2,6,6,-tetromethylpiperiodine-N-oxyl (tempamine) were reduced to 10% of peak values within 5 to 10 min, whereas the five-member ring nitroxides such as 3-carbamoyl-PROXYL (3-CP) and 3-aminomethyl-PROXYL remained at 10% or more of peak values for 30 to 60 min (Hahn et al., 1998). Thus, tempamine was considered to be an excellent redox probe, whereas 3-CP was recommended for EPR imaging (Matsumoto et al., 2004).
Takechi et al. (1997) used a continuous blood sampling technique for an in vivo EPR study in the rat to determine the composite pharmacokinetic parameters of a range of nitroxides after intravenous bolus injection (Takechi et al., 1997). A rapid initial distribution phase was followed by a plasma clearance phase whose t1/2 depended on the physical chemistry characteristics of the probe. Lipid-soluble agents had lower plasma clearance values, perhaps because of a greater volume of distribution. The decay of intravenously injected nitroxide radicals in mice had a biphasic curve with an initial rapid decay that was attributed to reduction to the hydroxylamine followed by a slow decay attributed to excretion (Matsumoto et al., 2004). The cationic nitroxide 4-trimethylammonium-2,2,6,6-tetramethylpiperidine-1-oxyl iodide (CAT-1) had a triphasic decay (Matsumoto et al., 2004). Both free radical and hydroxylamine species were excreted into the urine. Reduction to the hydroxylamine and reoxidation to the nitroxide soon reached equilibrium (Matsumoto et al., 2004).
Tempol has been found to enter cells rapidly and to be widely distributed in the body. It reacted with cellular in the cytoplasm and in the mitochondria (van der Poel et al., 2006). Tempol penetrated the blood-brain barrier (Behringer et al., 2002) and accumulated in the brain (Matsumoto et al., 1998). Tempol penetrated intact skin (Herrling et al., 2002) where it accumulated in the lipid compartment of the stratum corneum (Li et al., 2001). It was distributed rapidly into the aqueous humor (Zamir et al., 1999) and diffused through cartilage into the underlying bone (Fischer et al., 1995).
The reduction of nitroxides occurs principally within cells, accounting for the much slower rate of reduction of hydrophilic than lipophilic nitroxides by intact cells or bacteria (Jung et al., 1998). Using erythrocytes as a test system, Gwoździński and coworkers concluded that cell membrane passage of tempol was limited by diffusion (Gwoździński, 1985), which was affected by SH– groups (Gwoździński et al., 1983; Gwoździński, 1985), adenine nucleotides (Jozwiak et al., 1983), and ionizing radiation (Gwoździński, 1986). Negatively charged or amphilite nitroxides such as tempol were concentrated in hydrophobic microdomains of cell membranes (Timoshin and Ruuge, 1994). Positively changed nitroxides such as CAT-1 had very little penetration into cells (Samuni et al., 2001; Okajo et al., 2006) unless they were incorporated into liposomes (Matsumoto et al., 2005). Consequently, tempol, but not CAT-1, protected cells against H2O2-induced DNA damage (Samuni et al., 2001). Negatively charged nitroxides or probes with carboxyl moieties, such as carboxy-TEMPO and carboxy-PROXYL did enter cells, albeit slowly, via an anion transporter that was inhibited by 4-acetamido-4′-isothiocyanostilbene-2,2′-disulfonic acid (Ross and McConnell, 1975; Pikula et al., 1994; Okajo et al., 2006). After intravenous injection, tempol produced additional EPR signals in the bile that were attributed to hepatic uptake and biliary excretion, whereas the highly hydrophilic CAT-1 was not present in bile. One study demonstrated that the blood levels of membrane-permeable nitroxides were replenished by an active enterohepatic recirculation (Hahn et al., 1998), but this finding was not confirmed in another study (Okajo et al., 2006). After oral administration to the rabbit, the great majority of tempol in the plasma or aqueous humor was in the reduced form (Sasaki et al., 1998).
The t1/2 for decay of the tempol radical in the blood pool of mice after intraperitoneal injection was approximately 50 min (Hahn et al., 1992a), which was much longer than the t1/2 of 1 min after intravenous injection or of 5 min after intramuscular injection (Kuppusamy et al., 1998). The t1/2 after subcutaneous injection was prolonged by coinjection with polynitroxyl-albumin (Kuppusamy et al., 1998). Tempol-H given at a very high dose of 1.45 mmol/kg i.p. to mice provided an early whole-body EPR peak within 1 to 2 min, demonstrating some rapid oxidation to tempol, but this was <10% of the signal produced by tempol itself (Hahn et al., 2000). An equilibrium was reached after 10 min. Thereafter, the two signals decayed at similar rates, largely because of renal excretion.
The t1/2 for reduction of tempol was greatly increased by NO (Nakajima et al., 2002). Studies in hepatic microsomes (Nakajima et al., 2002) and cell lines (Samuni et al., 2004) showed that NO donors reduced both the reduction of nitroxides and the reoxidation of hydroxylamines, thereby limiting redox recycling perhaps by inhibition of mitochondrial function by NO (Wolin et al., 1999).
In addition to rapid and reversible redox reduction of nitroxides to hydroxylamines, nitroxide probes also were reduced by enzymic one-electron reduction reactions (Okajo et al., 2006). Liver microsomes were shown to metabolize tempol from a six- to a five-member ring in the presence of Fe+ (Yin et al., 2003, 2004) or to sterically hindered secondary amines (Kroll and Borchert, 1999).
A slow-release formulation of tempol has been provided by incorporation into fluoroalkyl double-ended polyethylene glycol (Rf-PEG) micelles (Prabhutendolkar et al., 2006).
G. Modified Nitroxides
Nitroxides have been joined covalently to other compounds via the 4′-site. Tempol has been linked to drugs such as chlorpromazine to study the pharmacokinetics of the drug (Feldman et al., 1975), to agents such as acyl-coenzyme A to incorporate tempol into the mitochondrial membrane wherein acyl-coenzyme A interacts with a specific ADP carrier protein (Devaux et al., 1975), to agents such as serum albumin to prolong the duration of tempol in the plasma (Li et al., 2002), and to therapeutic agents to reduce their oxidative actions (Alayash, 1999; Buehler et al., 2000, 2004).
II. Mechanistic Basis of the Blood Pressure-Lowering Effect of Tempol
A. Signaling Studies in Cells and Tissues
Cellular signaling pathways activated by ROS have been reviewed (Griendling and Ushio-Fukai, 2000; Finkel, 2003; Griendling and FitzGerald, 2003; Touyz et al., 2003; Touyz, 2004; Cash et al., 2007).
1. Protein Kinase G and cGMP
Incubation of VSMCs with 30 mM glucose down-regulated the mRNA, protein, and activity of cGMP-dependent protein kinase G-1 (Liu et al., 2007b). This down-regulation was prevented by incubation with tempol (Liu et al., 2007b). Wang et al. (2003a,b) showed that tempol reversed the defective acetylcholine (ACh)-induced endothelium-dependent relaxations of renal afferent arterioles dissected from rabbits with oxidative stress caused by prolonged infusion of Ang II. This effect of tempol depended on cGMP. The authors proposed that tempol improved NO signaling via cGMP in models of oxidative stress.
2. Protein Kinase A and cAMP
Tempol did not alter isoproterenol-stimulated generation of cAMP in preglomerular microvessels (Jackson et al., 2004). Indeed activation of β1-adrenergic receptors in renal afferent arterioles from a rabbit model moderated oxidative stress. Only after blockade of cAMP was the contraction to norepinephrine (NE) enhanced by oxidative stress and normalized by coincubation with tempol (Wang et al., 2006b).
3. Mitogen-Activated Protein Kinases
Ang II is a potent activator of the MAPK cascade in cardiovascular tissue where it acts via a redox-sensitive mechanism. Tempol markedly suppressed Ang II-induced activation of vascular extracellular signal-regulated kinase (ERK) 1 and 2 and p38 (Zhang et al., 2007). This suppression was ascribed in part to an increase in NO bioactivity because it was prevented by NOS blockade (Zhang et al., 2007). Tempol prevented the phosphorylation of MAPKs, ERK1 and 2, c-Jun N-terminal kinase (JNK), and p38 in the aorta and heart of rats during infusions of Ang II or phenylephrine (PE) (Zhang et al., 2004a; Kimura et al., 2005a) and inhibited the phosphorylation of p38, MAPK, JNK, and ERKs in vascular tissue stimulated by Ang II or endothelin-1 (ET-1) (Touyz et al., 2004). Cerebral ischemia increased generation and phosphorylation of ERK1 and 2, which were prevented by tamoxifen or tempol (Wakade et al., 2008).
Prolonged administration of tempol has been found to be very effective in preventing MAPK activation in the tissues of several animal models of hypertension (Iglarz et al., 2004; Nishiyama and Abe, 2004). For example, tempol (3 mmol/l in drinking water for 6 weeks) prevented the increased activities of ERK1 and 2 and JNK in the renal cortex of rats with aldosterone- and salt-induced hypertension (Nishiyama et al., 2004a). Dahl salt-sensitive (DSS) rats fed salt had a major increase in the glomerular MAPK activity, including ERK1 and 2 and JNK which was prevented by oral tempol (3 mmol/l in drinking water for 4 weeks) (Nishiyama et al., 2004b). This effect was independent of BP reduction. The stimulation by a low-potassium diet of renal c-Jun phosphorylation and c-Src expression was prevented by 1 week of tempol administration (Babilonia et al., 2005).
Thus, tempol is very effective in preventing MAPK activation during oxidative stress both in vivo and in vitro.
4. Nuclear Factor κB
Tempol or pyrrolidine dithiocarbamate prevented activation of NF-κB in the aorta and kidney of rats with deoxycorticosterone acetate (DOCA)-salt induced hypertension (Beswick et al., 2001). Tempol also prevented activation of NF-κB and protein kinase C (PKC) in rats with oxidative stress caused by feeding buthionine sulfoximine (BSO) to deplete glutathione (Banday et al., 2007a).
5. Rho and Rho Kinase
ROS generated by xanthine plus xanthine oxidase in rat aortic rings led to incorporation of Rho into membranes (Jin et al., 2004). The associated phosphorylation of the myosin light chain phosphatase target subunit-1 and vascular contraction were blocked by the Rho kinase inhibitor Y-27632. Tempol blocked the ROS-induced Ca2+ sensitization of these rings by preventing activation of Rho and Rho kinase (Jin et al., 2004). This may be an important component of the effect of tempol to reduce contractility of VSMCs during oxidative stress.
6. Protein Kinase C
Pretreating blood vessels from diabetic rats with the PKC inhibitor bisindolylmaleimide I improved endothelium-dependent relaxant factor. (EDRF)/NO responses without moderating vascular (Coppey et al., 2003). The authors concluded that activation of PKC was downstream from oxidative stress. Indeed, tempol prevented PKC activation, generation (Coppey et al., 2003), downstream phosphorylation of target proteins (Banday et al., 2007a), and c-jun oncogene expression (Kuo et al., 1995) in proximal tubules from rats with glutathione depletion (Banday et al., 2007a) and in lung cells stimulated with the redox-cycling quinolone, paraquat (Kuo et al., 1995). However, the finding that tempol blocked increases in intracellular [Ca2+] and constriction of vasa recta pericytes after stimulation by the PKC agonist phorbol 12,13-dibutyrate demonstrated that tempol also can interrupt signaling downstream from PKC (Zhang et al., 2004c).
B. Antihypertensive Action in Animal Models
1. Overview of Antihypertensive Response to Tempol
Both acute and prolonged administration of tempol have been shown to reduce the BP in hypertensive models. However, two differences are apparent between these responses.
First, the acute response to intravenous tempol in hypertensive rodent models was very rapid in onset (maximal within 2 min of the intravenous bolus) and reversed fully within 15 min (Patel et al., 2006), whereas the response to tempol added to the drinking water has been a delayed reduction in BP over 24 h that took 2 or more weeks to develop fully (Welch et al., 2005b). Second, acute administration of tempol reduced the heart rate (HR) (Patel et al., 2006) and renal sympathetic nerve activity (RSNA) of rats (Xu et al., 2002, 2004). This contributed to the fall in BP in the SHR after intravenous tempol because blockade of ganglionic transmission reduced the antihypertensive response (Chen et al., 2007a). In contrast, Welch et al. (2005b) reported that prolonged subcutaneous infusion of tempol to SHR over 2 weeks did not alter the HR or plasma NE or renal catecholamine excretion. Thus, either the sympatholytic actions of intravenous tempol are a unique response to acute administration or compensatory mechanisms to override this effect develop during prolonged tempol administration.
Despite these differences, >85% of hypertensive models studied have shown a reduction in BP with tempol, whether given acutely or by prolonged administration. The hypertensive models to which tempol has been administered acutely and by prolonged administration are detailed in Tables 1 and 2, respectively.
TABLE 1. Blood pressure and heart rate in response to acute tempol administration.
Mean values are shown for systolic blood pressure (SBP) or mean arterial pressure (MAP) and percent blood pressure response or percent normalization of blood pressure and percent heart rate response to acute tempol administration. Rat models: Acute Ang II, short term infusion of angiotensin II; Chronic Ang II, prolonged (d) infusion of angiotensin II; Acute PE, short-term (minutes or hours) infusion of phenylephrine; Capsaicin-salt, rats given capsaicin to induce sensory denervation and fed a high-salt diet; DSS, Dahl salt-sensitive rats fed a high-salt diet; DOCA-salt, deoxycorticosterone acetate plus salt; HTG, rat transgenic for human renin gene; Inducible malignant HTN, rats with an inducible renin gene to cause malignant hypertension; Lead, rats fed lead; D5R(−/−), dopamine-5 receptor deficient; GRK4γA142V, G-coupled receptor kinase 4γ arginine for valine polymorphism at nucleotide 142.
| SBP or MAP |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Model | n | Tempol Maximum Dose or Dose Used to Compare Groups | Tempol Route of Delivery | Normotensive Control Group | Experimental Hypertensive Group without Tempol | Experimental Group with Tempol | BP Response | HR Response | Reference | |
| μmol/kg | mm Hg | % change | % normalization | % changed | ||||||
| Studies in hypertensive rats with intravenous tempol | ||||||||||
| Acute Ang II | 4 | 15 | i.v. over 5 min | 93† | 153† | 151† (N.S.) | −1 | 3 | N.D. | Zhang et al., 2004a |
| Acute Ang II [200 (ng · kg)/min] | 5 | 173 | i.v. bolus then 43 μmol/kg | 110† | 152† | 152† (N.S.) | 0 | 0 | N.D. | Kimura et al., 2004 |
| Acute Ang II [200 (ng ·g)/min from 1–24 h] | 4–6 | 173 | i.v. bolus then 43 μmol/kg | N.D. | 148† | 134* | −33 | N.D. | N.D. | Kimura et al., 2004 |
| Chronic Ang II [200 (ng · kg)/min for 2 wk] | 5 | 173 | i.v. bolus then 43 μmol kg over 15 min | 110† | 175† | 110†* | −37 | 100 | N.D. | Kimura et al., 2004 |
| Chronic Ang II [200 (ng ·kg)/min for 2 wk] | 4 | 175 | i.v. bolus then 0.5 (mg ·kg)/min | 110† | 165† | 115†* | −33 | 92 | N.D. | Kimura et al., 2005a |
| Acute PE | 4 | 15 | i.v. over 5 min | 95† | 148† | 153† (N.S.) | −3 | 0 | N.D. | Zhang et al., 2004a |
| Capsaicin-4% salt (WKY) | ? | 216 | i.v. bolus | N.D. | 149 | 131* | −12 | N.D. | N.D. | Song et al., 2004 |
| DSS 8% salt | 7 | 145 | i.v. bolus | 129† | 197† | 184† (N.S.) | −7 | 19 | N.D. | Zicha et al., 2001 |
| DSS 8% salt, young | 7 | 60 | i.v. bolus | 125 | 150 | 137* | −9 | 48 | N.D. | Zicha et al., 2001 |
| DSS 8% salt, old | 7 | 60 | i.v. bolus | 145 | 170 | 162* | −5 | 23 | N.D. | Zicha et al., 2001 |
| DOCA-salt | 5 | 300 | i.v. bolus | 74† | 140† | 80†* | −43 | 91 | −10* | Xu et al., 2004 |
| HTG rat | ? | 145 | i.v. bolus | 95† | 137† | 124†* | −9 | 31 | N.D. | Kunes et al., 2002 |
| HTG female | ? | 145 | i.v. bolus | 101† | 120† | 110†* | −8 | 53 | N.D. | Kunes et al., 2002 |
| Inducible malignant HTN rat | 5 | 300–400 | i.v. over 1 h | 123† | 184† | 151†* | −18 | 54 | N.D. | Patterson et al., 2005 |
| 2K,1C | 13 | 200 | i.v. over 1 h | 107† | 155† | 131†* | −15 | 50 | N.D. | Guron et al., 2006 |
| Lead | 6 | 90 | i.v. over 30 min | 138 | 168 | 138* | −18 | 100 | N.D. | Vaziri et al., 2003b |
| Lewis rats, Zn-deficient | 8 | 20 | i.v. bolus and infusion | 110 | 110 | 80* | −26 | 100 | N.D. | Kurihara et al., 2002 |
| SD + L-NNA for 2 wk | 7 | 300 | i.v. | 118† | 194† | 140†* | −28 | 72 | −16* | Thakali et al., 2006 |
| SHR | 6 | 270 | i.v. bolus | N.D. | 140† | 70†* | −50 | N.D. | −20* | Patel et al., 2006 |
| SHR | 6 | 173 | i.v. over 1 min | 108† | 166† | 123†* | −26 | 74 | −13* | Shokoji et al., 2003 |
| SHR | 6 | 72 | i.v. bolus | 96† | 145† | 104†* | −28 | 84 | N.D. | Schnackenberg et al., 1998 |
| SHR | 6 | 900 | i.v. over 30 min | 72† | 167† | 72†* | −57 | 100 | N.D. | Schnackenberg et al., 1998 |
| SHR | 10 | 174 | i.v. bolus | N.D. | 178† | 120†* | −33 | 100 | −12* | Chen et al., 2007a |
| SHR Zn-deficient diet | 7 | 100 | i.v. bolus | 148† | 162† | 130†* | −20 | 100 | N.D. | Sato et al., 2002 |
| Zinc-fed rats | 8 | 100 | i.v. | 107† | 128† | 93†* | −27 | 100 | N.D. | Yanagisawa et al., 2004 |
| Studies in normotensive rats with intravenous tempol | ||||||||||
| DOCA-sham | 5 | 300 | i.v. bolus | N.D. | 98† | 74†* | −24 | N.D. | +9 | Xu et al., 2004 |
| Lewis rat, male | ? | 145 | i.v. bolus | N.D. | 100† | 96†* | −4 | N.D. | N.D. | Kunes et al., 2002 |
| Lewis rat, female | ? | 145 | i.v. bolus | N.D. | 105† | 101† (N.S.) | −4 | N.D. | N.D. | Kunes et al., 2002 |
| 4% salt (WKY) | ? | 216 | i.v. bolus | N.D. | 102 | 88* | −14 | N.D. | N.D. | Song et al., 2004 |
| SD | 5 | 90 | i.v. over 1 h | N.D. | 104† | 76†* | −27 | N.D. | +8 | Campese et al., 2004 |
| SD | 4 | 15 | i.v. over 5 min | N.D. | 100† | 98† (N.S.) | −2 | N.D. | +1 | Zhang et al., 2004a |
| SD | 6 | 90 | i.v. over 30 min | N.D. | 138 | 133 (N.S.) | −4 | N.D. | N.D. | Vaziri et al., 2003b |
| SD | 8 | 216 | i.v. bolus | N.D. | 116† | 112† (N.S.) | −3 | N.D. | N.D. | Nishiyama et al., 2001 |
| WKY | 12 | 200 | i.v. over 1 h | N.D. | 116† | 107†* | −8 | N.D. | N.S. | Guron et al., 2006 |
| WKY | 6 | 173 | i.v. over 1 min | N.D. | 108 | 88* | −19 | N.D. | +7 | Shokoji et al., 2003 |
| WKY | 6 | 72 | i.v. bolus | N.D. | 118† | 96†* | −19 | N.D. | N.D. | Schnackenberg et al., 1998 |
| WKY | 6 | 900 | i.v. over 30 min | N.D. | 122† | 72†* | −41 | N.D. | N.D. | Schnackenberg et al., 1998 |
| WKY | 6 | 90 | i.v. over 30 min | N.D. | 122† | 107†* | −12 | N.D. | N.D. | Schnackenberg et al., 1998 |
| Studies in normotensive or hypertensive rats with intracerebroventricular tempol | ||||||||||
| DDS + 8% NaCl | 8 | 40 μmol | i.c.v. | 100 | 146 | 116* | −21 | 65 | −11* | Fujita et al., 2007 |
| DSR NS vs. DSS NS | 7 | 40 μmol | i.c.v. | 103 | 119 | 109* | −10 | 63 | −6 | Fujita et al., 2007 |
| SD | 6 | 50 or 100 | i.c.v. | 106† | N.A. | 75* | −29 | 100 | −15* | Lu et al., 2004 |
| SHR | 4 | 1.5 | i.c.v. over 1 min | 108† | 162† | 163† (N.S.) | −1 | 0 | +1 | Shokoji et al., 2003 |
| SHRSP | 5 | 3.8 | i.c.v. bolus RVLM | 105† | 170† | 133†* | −22 | 57 | −11* | Kishi et al., 2004 |
| WKY | 5 | 3.8 | i.c.v. bolus RVLM | N.D. | 112† | 105† (N.S.)* | −6 | N.D. | −1 | Kishi et al., 2004 |
| WKY | 4 | 1.5 | i.c.v. over 1 min | N.D. | 108† | 108† (N.S.)* | 0 | N.D. | +1 | Shokoji et al., 2003 |
| Studies in mice with intravenous tempol | ||||||||||
| D5R(−/−) mice (ROS) | 7 | 58 | i.v. bolus | 86 | 110 | 97* | −12 | 54 | N.D. | Wang et al., 2007 |
| GRK4γA142V mice (NO ROS) | 4 | 58 | i.v. bolus | 99 | 117 | 117 (N.S.) | 0 | 0 | N.D. | Wang et al., 2007 |
?, unknown; N.D., not done; L-NNA, L-nitroarginine; N.A., not applicable. DSR, Dahl salt-resistant rat; i.c.v., intracerebroventricular; NS, normal salt; wk, week(s).
Significant change or difference from without tempol.
Value is for MAP.
TABLE 2. Blood pressure response to prolonged tempol administration.
Mean values are shown for systolic blood pressure (SBP) or mean arterial pressure (MAP) and percent blood pressure response or percentage normalization of blood pressure. Tempol dose p.o. is concentration of tempol in the drinking water unless otherwise noted.
| SBP or MAP |
BP Response |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Model | Control Model | Tempol Route of Delivery and Duration | n | Tempol Maximum Dose or Dose Used to Compare Groups | Control Normotensive | Experimental Hypertensive Group without Tempol | Experimental Group with Tempol | Change | Normalization | Reference |
| mm Hg | % | |||||||||
| Studies in hypertensive rats with systemic tempol | ||||||||||
| ACTH (0.2 (mg · kg)/d s.c.) | Vehicle | p.o. from 4 d before to 8 d after ACTH | 10 | 1 mM | 119 | 134 | 4 d, 118* 8 d, 123 (N.S.) |
4 d, −12 8 d, −8 |
4 d, 106 8 d, 73 |
Zhang et al., 2003b |
| Aldosterone (0.75 μg/h s.c. + salt 1% for 6 wk) | Vehicle | p.o. for 6 wk | 8 | 3 mM | 118 | 165 | 125* | −24 | 85 | Nishiyama et al., 2004a |
| Aldosterone (0.75 μg/h s.c. for 6 wk) | Vehicle | p.o. for 6 wk | 7 | 1 mM | 123 | 170 | 149* | −12 | 45 | Iglarz et al., 2004 |
| Ang II [5 (ng · kg)/min i.v. for 15 d] | Vehicle | p.o. for 15 d | 6 | 1 mM | 119 | 151 | 119* | −21 | 100 | Ortiz et al., 2001a |
| Ang II [100 (ng · kg)/min s.c. ± 8% salt diet for 12 d] | Vehicle | p.o. for 12 d | 5 | 1 mM | 127 | 184 | 150* | −18 | 60 | Ogihara et al., 2002 |
| Ang II [200 (ng ·kg)/min s.c. for 2 wk] | Vehicle | s.c. minipump for 2 wk | 8–11 | 200 (nmol ·kg)/min | 104† | 146† | 116†* | −21 | 71 | Welch et al., 2005a |
| Ang II [300 (ng · kg)/min s.c. for 7 d] | Vehicle | p.o. for 7 d | 8 | 2 mM | 125 | 186 | 142* | −24 | 72 | Hattori et al., 2005 |
| BSO (30 mM for 2 wk] | SD | p.o. for 2 wk | 8 | 1 mM | 100† | 123† | 104†* | −15 | 82 | Banday et al., 2007a |
| BSO (30 mM) + HS for 2 wk | HS | p.o. for 12 d | 8 | 1 mM | 112† | 143† | 107†* | −25 | 116 | Banday et al., 2007c |
| Capsaicin-4% salt (WKY) | None | p.o. (gavage) for 3 wk | 5–6 | 1 (mmol · kg)/d | 113 | 150 | 150 (N.S.) | 0 | 0 | Song et al., 2004 |
| Cyclosporine [30 (mg · kg)/d s.c.) | Vehicle | p.o. for 3 wk | 7 | 3 mM | 119 | 145 | 115* | −21 | 115 | Nishiyama et al., 2003 |
| Dexamethasone [10 (μg · kg)/d s.c.] | Vehicle-infused | p.o. from 4 d before to 8 d after Dex | 10 | 1 mM | 122 | 136 | 4 d, 128* 8 d, 122* |
4 d, −6 8 d, −10 |
4 d, 57 8 d, 100 |
Zhang et al., 2004b |
| DOCA-salt | Sham | p.o. for 3 wk | 6 | 1 mM | 107† | 161† | 108†* | −33 | 100 | Ghosh et al., 2004 |
| DOCA-salt | SD/sham-salt | i.p. for 3 wk | 8 | 87 (μmol · kg)/d | 119† | 164† | 123†* | −25 | 91 | Adeagbo et al., 2003 |
| DOCA-salt | Sham | i.p. for 3 wk | 8 | 87 (μmol · kg)/d | 130 | 203 | 151* | −26 | 71 | Awe et al., 2003 |
| DOCA-salt | Sham | p.o. for 5 wk | 13 | 1 mM | 118 | 200 | 176* | −12 | 29 | Nakano et al., 2003 |
| DOCA-salt | Sham | p.o. for 4 wk | 10 | 1 mM | 113 | 199 | 142* | −29 | 66 | Beswick et al., 2001 |
| DSS/8% salt | DSS LS and DSR | p.o. for 10 wk | 9 | 3 mM | 120 | 220 | 191* | −13 | 29 | Guo et al., 2006 |
| DSS/8% salt | DSS LS | p.o. for 5 wk | 5 | 10 mM | 144 | 224 | 186* | −17 | 47 | Hisaki et al., 2005 |
| DSS/8% salt | DSS LS | p.o. for 4 wk | 8 | 3 mM | 107 | 184 | 128* | −30 | 73 | Kobori and Nishiyama, 2004 |
| DSS/8% salt | DSS/NS | p.o. for 3 wk | 34 | 1 mM | 148 | 233 | 199* | −15 | 40 | Bayorh et al., 2006 |
| DSS + salt | DSS LS | i.v. for 3 wk | 7 | 3 (mmol · kg)/d | 122† | 140† | 118†* | −16 | 102 | Meng et al., 2003 |
| DSS + 8% NaCl | DSS LS | p.o. for 4 wk | 8 | 3 mM | 113 | 185 | 128* | −31 | 79 | Nishiyama et al., 2004b |
| DSS + 8% NaCl | DSR | p.o. for 8–10 wk | 20 | 1 mM | 124 | 179 | 132* | −26 | 85 | Ozawa et al., 2004 |
| ET-1 [5 (pmol · kg)/min]-8% salt | Vehicle-treated, normal salt | p.o. for 12 d | 7–10 | 1 mM | 114† | 132† | 127† (N.S.) | −4 | 28 | Elmarakby et al., 2005 |
| ET-1 [5 (pmol · kg)/min]-8% salt | Tempol untreated | s.c. for 12 d | 7–10 | 170 (μmol ·kg)/d | 114† | 138† | 134† (N.S.) | −3 | 16 | Elmarakby et al., 2005 |
| ET-1 [5 (pmol ·kg)/min] i.v. | Vehicle-infused | i.v. for 9 d | 6 | 170 (μmol ·kg)/d | 125† | 141† | 127†* | −10 | 87 | Sedeek et al., 2003 |
| ET-B antagonist (A-192621) p.o. | Vehicle-treated | p.o. for 1 wk | 6 | 1 mM | 100† | 117† | 117† (N.S.) | 0 | 0 | Williams et al., 2004 |
| ET-B antagonist-10% salt p.o. | Vehicle-treated, 10% salt | p.o. for 1 wk | 6 | 1 mM | Day 3, 110† Day 7, 110† |
Day 3, 135† Day 7, 138† |
Day 3, 120†* Day 7, 138† (N.S.) |
Day 3, −11 Day 7, 0 |
Day 3, 60 Day 7, 0 |
Williams et al., 2004 |
| Fructose-fed | Fructose untreated | p.o. for 4 wk | 7 | 1 mM | 101† | 128† | 103†* | −19 | 93 | Onuma and Nakanishi, 2004 |
| Hyperthyroid (s.c. thyroxine for 6 wk) | Vehicle-infused | p.o. for 6 wk | 8 | 1 mM | 120† | 147† | 127†* | −14 | 74 | Moreno et al., 2005 |
| High salt 10% | Normal salt | p.o. for 1 wk | 6 | 1 mM | 98† | 112† | 100†* | −11 | 85 | Williams et al., 2004 |
| Inducible renin transgene-8% salt | Pre-salt loading | p.o. for 10 d | 6 | 2 mM | 137 | 171 | 148* | −13 | 63 | Howard et al., 2005 |
| Intermittent hypoxia (sleep apnea) | SD | p.o. for 2 wk | ? | 1 mM | 101 | 118 | 107* | −9 | 65 | Troncoso Brindeiro et al., 2007 |
| 1K, 1C | Sham UNX | p.o. for 2 wk | 6 | 2 mM | 95† | 159† | 139†* | −13 | 31 | Dobrian et al., 2001 |
| 1K, 1C (10 wk) | Sham | p.o. for 5 wk | 5 | 1 mM | 130 | 170 | 135* | −21 | 90 | Christensen et al., 2007b |
| 2K,1C | Sham | s.c. minipump for 13 d | 8 | 288 (μmol · kg)/d | 105† | 148† | 118†* | −20 | 70 | Welch et al., 2003 |
| Lead (100 ppm for 12 wk) | SD without lead in diet | i.p. for 2 wk | 6 | 15 (mmol · kg)/d | 122 | 173 | 143* | −17 | 59 | Vaziri et al., 2001 |
| Leptin | Leptin untreated | p.o. for 7 d | 8 | 2 mM | 126 | 152 | 128* | −16 | 92 | Beltowski et al., 2005 |
| Offspring of protein- malnourished mothers | Offspring of normal mothers | p.o. for 13 wk including 3 wk pre-HTN | 14–19 | 2 mM | 130 | 143 | 130* | −9 | 100 | Stewart et al., 2005 |
| Obese Zucker | Lean | p.o. for 15 d | 10 | 1 mM | 89† | 110† | 100†* | −9 | 48 | Banday et al., 2005 |
| Five-sixths nephrectomy (infarction) | Sham | IP for 10 d | 10 | 1.5 (mmol · kg)/d for 10 d | 118 | 145 | 122* | −8 | 85 | Hasdan et al., 2002 |
| Five-sixths nephrectomy | Sham | p.o. for 1 wk | 6 | 1 mM | 120 | 180 | 150* | −17 | 50 | Vaziri et al., 2003a |
| Five-sixths nephrectomy | Sham | IP for 10 d | 10 | 1.5 (mmol · kg)/d | 118 | 145 | 122* | −16 | 85 | Hasdan et al., 2002 |
| Ren-2 transgenic rat | SD | p.o. for 3 wk | 6 | 1 mM | 115† | 197† | 194† (N.S.) | −2 | 3 | Whaley-Connell et al., 2007 |
| SD + HS + BSO (30 mM for 12 d) | HS | p.o. for 12 d | 8 | 1 mM | 112† | 142† | 114†* | −20 | 93 | Banday et al., 2007d |
| SHR | WKY | p.o. or s.c. for 2 wk | 6–8 | 2 mM 200 (nmol · kg)/min by minipump | 104† 104† |
p.o., 147† s.c., 150† |
p.o., 128†* s.c., 126†* |
p.o., 13* s.c., −16* |
p.o., 45 s.c., 52 |
Welch et al., 2005b |
| SHR | WKY | p.o. from wk 6–11 | 6 | 1 mM | 143 | 171 | 137* | −20* | 105 | Nabha et al., 2005 |
| SHR | SD | p.o. for 5 – 15 d | 10 | 1 mM | 5 d, 118† 15 d, 124† |
5 d, 149† 15 d, 179† |
5 d, 143† (N.S.) 15 d, 165† (N.S.) |
5 d, −4 15 d, −8 |
20 25 |
de Richelieu et al., 2005 |
| SHR | WKY | p.o. from 0–15 wk | 6–12 | 170 (μmol ·kg)/d | 100† | 181† | 156†* | −14 | 30 | Fortepiani and Reckelhoff, 2005 |
| SHR | WKY | p.o. from 9–15 wk | 6–12 | 170 (μmol · kg)/d | 100† | 195† | 163†* | −16 | 34 | Fortepiani and Reckelhoff, 2005 |
| SHR | Untreated SHR | p.o. for 4 d at 13–14 wk | 10 | 1 mM | 108† (WKY) | 199† | 177†* | −11 | 24 | Feng et al., 2001 |
| SHR | Untreated SHR | p.o. for 7 wk from 5–12 wk | 7 | 1 mM | 108† (WKY) | 187† | 167†* | −11 | 25 | Feng et al., 2001 |
| SHR | WKY | p.o. for 2 wk | 8 | 1 mM | 118† | 162† | 134†* | −17 | 63 | Schnackenberg and Wilcox, 1999 |
| SHR | WKY | i.p. for 7 d | 7 | 1.5 (mmol · kg)/d | 97† | 133† | 120†* | −10 | 36 | Schnackenberg et al., 1998 |
| SHR aging (16 mo) | Untreated SHR | p.o. for 8 mo from 8–16 mo | 6 | 6 mM | 108† | 185† | 160†* | −14 | 34 | Fortepiani et al., 2003 |
| SHR aging (16 mo) | Untreated SHR | p.o. for 8 mo | 10 | 1 mM | 108† | 188† | 161†* | −14 | 33 | Payne et al., 2003 |
| SHR aging female (16 mo) | Untreated SHR female | p.o. for 8 mo from 8–16 mo | 6 | 6 mM | 108† | 195† | 195† (N.S.) | 0 | 0 | Fortepiani et al., 2003 |
| SHR female | WKY | p.o. from 0–15 wk | 6–12 | 170 (μmol · kg)/d | 101† | 172† | 127†* | −26 | 63 | Fortepiani and Reckelhoff, 2005 |
| SHR female | WKY | p.o. from 9–15 wk | 6–12 | 170 (μmol · kg)/d | 101† | 160† | 159† (N.S.) | −1 | 1 | Fortepiani and Reckelhoff, 2005 |
| SHRSP, Mg2+-deficient | Untreated SHRSP | p.o. for 7 wk | 6 | 1 mM | 108 | 240 | 195* | −19 | 30 | Touyz et al., 2002 |
| SHRSP, 4% salt | Untreated SHRSP | p.o. for 6 wk | 6 | 1 mM | 108 | 260 | 220* | −15 | 25 | Park et al., 2002 |
| UNX, aldosterone (0.75 μg/h), s.c. salt 8% for 6 wk | Vehicle | p.o. for 4 wk | 27 | 6 mM | 139 | 236 | 131* | −45 | 100 | Shibata et al., 2007 |
| UNX, aldosterone (75 pg/h), 1% NaCl for 3 wk | Vehicle | p.o. for 3 wk | ? | 2 mM | 118 | 186 | 158* | −8 | 41 | Hirono et al., 2007 |
| Studies in hypertensive rats with intracerebroventricular tempol | ||||||||||
| SHR | Untreated SHR | i.c.v. for 2 wk | 6 | 13.2 μmol/d | 163 | 209 | 210 (N.S.) | 0 | 0 | Kagiyama et al., 2000 |
| Studies in hypertensive mice with systemic tempol | ||||||||||
| Ang II (0.7 (mg · kg)/d), WT mice | Vehicle | s.c. for 2 wk | 6 | 28 (mmol · kg)/d | 95 | 154 | 128* | −17 | 44 | Dikalova et al., 2005 |
| Ang II (0.7 (mg · kg)/d), Nox-1-overexpressing mice | Vehicle | s.c. for 2 wk | 6 | 28 (mmol · kg)/d | 95 | 175 | 138* | −21 | 46 | Dikalova et al., 2005 |
| ET-B-deficient; 8% NaCl | WT; 8% NaCl | p.o. for 1 wk | 20 | 1 mM | 134 | 183 | 143* | −22 | 82 | Sullivan et al., 2006 |
| ET-B-deficient; 8% NaCl | WT; 8% NaCl | p.o. for 2 wk | 20 | 1 mM | 134 | 174 | 158* | −9 | 40 | Sullivan et al., 2006 |
HS, high salt; LS, low salt; DSR, Dahl salt-resistant rat; UNX, uninephrectomized; Dex, dexamethasone; NS, normal salt; HTN, hypertension; ?, unknown; i.c.v., intracerebroventricular; WT, wild type; ACTH, adrenocorticotropin; d, day(s); wk, week(s); mo, month(s).
Significant change with tempol.
Value is for MAP.
It is hard to compare responses to tempol among models with widely varying basal levels of hypertension. Because the absolute reduction in BP with antihypertensive agents increases with the basal levels of BP, one solution has been to assess the fractional (percent) changes in BP with tempol. However, clinicians require insight into the degree to which a new agent corrects established hypertension. These goals are better served by quantitating the fractional (percent) normalization of BP. Therefore, we have reported the effectiveness of tempol in Tables 1 and 2 both as percent reductions and percent normalizations of BP. We have used as “normal BP” that of a control group, for example, Wistar-Kyoto rats (WKY) in a study of SHR, when it has been provided by the investigator. For studies that have not reported data on a control model, we have estimated the normal level of BP from animals prepared under comparable conditions in other studies.
Of 26 studies in which tempol was given by acute intravenous injection or acute infusion to hypertensive rat models, 22 (85%) have recorded a fall in BP (Table 1, Studies in hypertensive rats with intravenous tempol). Of the four studies in which acute intravenous tempol failed to reduce the BP in a hypertensive rat model, three were in rats infused for only a few minutes with pressor doses of PE (Zhang et al., 2004a) or Ang II (Kimura et al., 2004; Zhang et al., 2004a). The fourth discordant study was in DSS rats fed a high-salt diet (Zicha et al., 2001). Two of these four negative studies used a dose of tempol of 15 μmol/kg (Table 1, Studies in hypertensive rats with intravenous tempol), which is below the effective dose for intravenous tempol in the anesthetized SHR, which is 72 to 90 μmol/kg (Patel et al., 2006). Parameters of ROS were not recorded in these four studies with negative results.
Isolated vessels incubated with Ang II took 10 to 20 min to develop a significant increase in ROS and a relaxation response to tempol (Wang et al., 2003b, 2004; Chen et al., 2007b). The two models in which hypertension was induced by prolonged infusion of Ang II into rats for 1 h to 2 weeks showed a 92% (Kimura et al., 2004) or 100% (Kimura et al., 2005a) normalization of BP with intravenous tempol. Thus, the failure of tempol to reduce the BP in studies in which PE (Zhang et al., 2004a) or Ang II (Kimura et al., 2004; Zhang et al., 2004a) was infused for only a few minutes may be explained by a failure of this protocol to induce vascular oxidative stress, but this hypothesis was not established.
Of two studies in hypertensive mice, intravenous tempol reduced the BP in D5R(−/−) mice but not in GRK4γA142V(−/−) mice (Wang et al., 2007) (Table 1, Studies in mice with intravenous tempol). Both of these models had modest hypertension yet only the D5R(−/−) mouse had evidence of increased ROS, and only this model had an acute antihypertensive response to tempol.
BP was reduced by acute intravenous administration of tempol in 9 of 13 studies (69%) of normotensive rats (Table 1, Studies in normotensive rats with intravenous tempol). Two studies that reported no fall in BP in normotensive rats included one that used a low dose of 15 μg/kg (Zhang et al., 2004a) that is below the effective threshold (Schnackenberg et al., 1998; Campese et al., 2004; Patel et al., 2006). Thus, when given in an effective dose, acute intravenous tempol reduced the BP in all hypertensive models with evidence of oxidative stress, but in only 8 of 12 studies in normotensive models.
Intravenous tempol reduced mean arterial pressure (MAP) by 28% in hypertensive SHR, which was significantly more than the 11% reduction in normotensive WKY. Likewise, tempol caused a significantly greater reduction in renal vascular resistance (RVR) in SHR (Schnackenberg et al., 1998). Clearly, the effects of intravenous tempol are greater in hypertensive than in normotensive models. No study has reported adverse effects from hypotension when tempol was given to hypertensive or normotensive rodents.
When recorded, the HR was reduced with intravenous tempol in six of seven hypertensive rat models (Table 1, Studies in hypertensive rats with intravenous tempol) including one study in which NOS was blocked (Thakali et al., 2006). Four studies in normotensive models reported a modest increase in HR with intravenous tempol (Table 1, Studies in normotensive rats with intravenous tempol).
Multiple studies have investigated the effect of prolonged tempol administration. Of 68 studies, 59 (87%) recorded a significant reduction in BP for at least one time point after administration (Table 2, Studies in hypertensive rats with systemic tempol). The majority (58 of 68) used oral tempol, three used subcutaneous infusions (Welch et al., 2003, 2005a; Dikalova et al., 2005), five used intraperitoneal injections (Schnackenberg et al., 1998; Vaziri et al., 2001; Hasdan et al., 2002; Adeagbo et al., 2003; Awe et al., 2003), and two used intravenous infusions (Meng et al., 2003; Sedeek et al., 2003). The BP during prolonged tempol seems to be dependent on the level of BP before tempol.
The antihypertensive effects of tempol were apparent across a wide range of models. Although all routes of tempol administration were effective, in all except one study in which tempol failed to reduce BP (Elmarakby et al., 2005), it was given orally. Of four studies in which prolonged administration of tempol was given to hypertensive mice, all recorded a fall in BP (Table 2, Studies in hypertensive mice with systemic tempol).
Of 63 studies in hypertensive rats reporting a measure of systemic, vascular, or renal ROS, 55 (87%) reported that tempol had reduced ROS, at least in some parameter of measurement (Table 3, Studies in hypertensive rat models).
TABLE 3.
Response of indices of oxidative stress to prolonged tempol administration
| Model | Control Model | Tempol Route of Delivery and Duration | n | Tempol Maximum Dose or Dose Used to Compare Groups | ROS Marker and Value in Hypertensive Group without Tempol | Control ROS Value in Normotensive Group | ROS Value in Hypertensive Group with Tempol | Change in ROS | Normalization of ROS | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| % | ||||||||||
| Studies in hypertensive rat models | ||||||||||
| ACTH [0.2 (mg ·kg)/d s.c.] | Vehicle | p.o. (4 d before to 8 d after ACTH) | 10 | 1 mM | Plasma 8-iso, 12.9 nM | 8.4 | 4 d pre, 16.3 8 d post, 13.2 |
+26/+28-iso d 4/8 (NS/NS) | −26/−2 | Zhang et al., 2003b |
| Aldosterone (0.75 μg/h s.c. + salt 1% for 6 wk) | Vehicle | p.o. for 6 wk | 8 | 3 mM | Renal cortex TBARS, 0.23 nmol/mg protein | 0.10 | 0.11 | −52* | 92 | Nishiyama et al., 2004a |
| Urine TBARS, 0.39 μmol/d | 0.10 | 0.13 | −67* | 90 | ||||||
| Aldosterone (0.75 μg/h s.c. for 6 wk) | Vehicle | p.o. for 6 wk | 7 | 1 mM | Plasma 8-iso, 16.8 ng/ml | 13.1 | 10.6 | −37* | 166 | Iglarz et al., 2004 |
| NADPH-generated in heart/aorta/kidney/mesenteric artery, 230/930/1500/670 cpm/mg tissue | 122/300/250/150 | 78/340/250/200 | −66*/−63*/−83*/70* | 141/94/100/90 | ||||||
| Ang II [5 (ng · kg)/min i.v. for 15 d] | Vehicle | p.o. for 15 d | 6 | 1 mM | Plasma/renal vein 8-iso, 193/353 pg/ml | 122/202 | 122/242 | −37*/−31* | 100/74 | Ortiz et al., 2001a |
| Plasma/renal vein TBARS, 1.7/1.9 nmol/ml | 0.8/0.7 | 1.2/1.1 | −33*/−36* | 56/67 | ||||||
| Ang II [100 (ng ·kg)/min s.c. ± 8% salt for 12 d] | Vehicle | p.o. for 12 d | 5 | 1 mM | Plasma cholesterol ester hydroperoxide levels, 0.27 μM | 0.13 | 0.13 | −52* | 100 | Ogihara et al., 2002 |
| Ang II [200 (ng ·kg)/min s.c. for 2 wk] | Vehicle | s.c. for 2 wk | 8–11 | 200 (nmol ·kg)/min | Kidney cortex NADPH oxidase activity, 3.6 nmol of /(min · mg protein) | 2.3 | 2.7 | −15* | 69 | Welch et al., 2005a |
| Ang II [300 (ng ·kg)/min s.c. for 7 d] | Vehicle | p.o. for 7 d | 8 | 2 mM | Aortic , 4.3 RLU | 0.7 | 1.3 | −70* | 83 | Hattori et al., 2005 |
| BSO (30 mM for 2 wk) | Vehicle | p.o. for 2 wk | 8 | 1 mM | Urine 8-Iso/Cr, 1.2 pg/mg | 0.80 | 0.82 | −33* | 98 | Banday et al., 2007a |
| BSO (30 + HS) | HS | p.o. for 12 d | 8 | 1 mM | Urine 8-Iso/Cr, 1.3 pg/mg | 0.84 | 0.86 | −34* | 96 | Banday et al., 2007c |
| Capsaicin-4% salt | 4% salt | p.o. (gavage) for 3 wk | 5–6 | 1 (mmol · kg)/d | Mesenteric artery , 1125 cpm/mg tissue | 730 | 950 | −16* | 44 | Song et al., 2004 |
| Cyclosporine [30 (mg · kg)/d s.c.] | Vehicle | p.o. for 3 wk | 7 | 3 mM | Kidney TBARS, 37 nmol/g | 24 | 19 | −49* | 138 | Nishiyama et al., 2003 |
| Aortic , 26 cpm | 17 | 16 | −38* | 111 | ||||||
| DETC [7.5 (mg · kg)/d into medullary interstitium; SD] | Tempol only, no DETC | Infused into medullary interstitium for 8 d before and during DETC | 8 | 58 (μmol · kg)/d | Urine 8-iso, 8.8 ng/d | 4.1 ng/d | 4.2 ng/d | −52* | 98 | Makino et al., 2003 |
| Dexamethasone [10 μg · kg)/d s.c.] | Vehicle | p.o. (4 d before to 8 d after Dex) | 10 | 1 mM | Plasma 8-iso, 12 nM | 8.8 | 4 d before, 10.4 8 d after, 10 |
−13/17, d 4/8 (NS/NS) | 50/63 | Zhang et al., 2004b |
| DOCA + salt 0.9% | Sham | p.o. for 3 wk | 6 | 1 mM | Aortic , 3166 (mU · mg)/min | 875 | 824 | −74* | 102 | Ghosh et al., 2004 |
| DOCA + salt | SD/sham high salt | i.p. for 3 wk | 8 | 87 (μmol · kg)/d | Plasma 8-iso, 0.77 ng/ml | 0.20 | 0.36 | −53* | 72 | Adeagbo et al., 2003 |
| DOCA + salt 1% | Sham | p.o. for 4 wk | 10 | 1 mM | aortic rings, 7153 cpm/mg tissue | 3055 | 2939 | −59* | 103 | Beswick et al., 2001 |
| DOCA + salt 1% | Sham | p.o. for 5 wk | 13 | 1 mM | Aortic , 1250 RLU/(min · mg) | 525 | 750 | −40* | 69 | Nakano et al., 2003 |
| DSS + 8% salt | DSR | p.o. for 10 wk | 9 | 3 mM | Plasma TBARS, 14 nmol/ml | 7 | 7 | −50* | 100 | Guo et al., 2006 |
| Cardiac NADPH oxidase, 361 cpm/mg protein | 245 | 185 | −49* | 152 | ||||||
| DSS + 8% salt | DSS LS | p.o. for 5 wk | 5 | 10 mM | 8-OHdG-positive cells, 347 cells/area | 159 | 259 | −25* | 47 | Hisaki et al., 2005 |
| DSS + 8% salt | DSS LS | p.o. for 4 wk | 8 | 3 mM | Urine TBARS, 0.66 μmol/d | 0.14 | 0.24 | −64* | 81 | Kobori and Nishiyama, 2004 |
| DSS + 8% salt | DSS LS | p.o. for 2 wk | 9 | 1 mM | Urine 8-iso, 14 ng/d | 7.4 | 8 | −43* | 91 | Hoagland et al., 2003 |
| DSS + 8% salt | DSS LS | p.o. for 4 wk | 8 | 3 mM | Kidney TBARS, 86 nmol/g | 41 | 48 | −46* | 88 | Nishiyama et al., 2004b |
| DSS + 8% salt + L-NAME | DSS LS | p.o. for 2 wk | 9 | 1 mM | Urine 8-iso, 15.2 ng/d | 7.4 | 7.5 | −51* | 99 | Hoagland et al., 2003 |
| DSS + 8% salt + HET- 0016 (20-HETE blocker) | DSS LS | p.o. for 2 wk | 9 | 1 mM | Urine 8-iso, 14.6 ng/d | 7.4 | 8.5 | −42* | 85 | Hoagland et al., 2003 |
| DSS + salt | DSS LS | i.v. for 3 wk | 7 | 3 (mmol · kg)/d | Renal cortical/medullary , 72/35 cpm/mg protein | 47/26 | 22/12 | −69*/−66* | 200/256 | Meng et al., 2003 |
| ET-1 [5 (pmol · kg)/min i.v. +8% salt] | Vehicle- infused, normal salt | p.o. for 12 d | 7–10 | 1 mM | Plasma 8-iso, 128 pg/ml | 52 | 72 | −44* | 74 | Elmarakby et al., 2005 |
| Aortic , 740 cpm/mg | 90 | 210 | −72* | 82 | ||||||
| ET-1 [5 (pmol ·kg)/min i.v. +8% salt] | Untreated | s.c. for 12 d | 7–10 | 170 (μmol ·kg)/d | Aortic , 740 cpm/mg | 90 | 240 | −68* | 77 | Elmarakby et al., 2005 |
| ET-1 [5 (pmol ·kg)/min i.v.] | Vehicle- infused | i.v. for 9 d | 6 | 170 (μmol ·kg)/d | Kidney TBARS, 462 ng/μg of protein | 48 | 287 | −38 (N.S.) | 42 (N.S.) | Sedeek et al., 2003 |
| Urine 8-iso, 11 ng/d | 7.5 | 8.9 | −19* | 60 | ||||||
| ET-B antagonist (A- 192621) p.o. | Vehicle- treated | p.o. for 1 wk | 6 | 1 mM | Plasma 8-iso, 25 pg/ml | 18 | 23 | −8 (N.S.) | 29 | Williams et al., 2004 |
| Urine H2O2, 4 nmol d | −1 | −1 | −75* | 100 | ||||||
| ET-B antagonist + 10% salt p.o. | Vehicle- treated | p.o. for 1 wk | 6 | 1 mM | Plasma 8-iso d 3/7, 75/66 pg/ml | 18 | 57/66 | −24/0 (NS/NS) | 32/0 | Williams et al., 2004 |
| ET-B deficient + 8% salt | Wild type; 8% NaCl | p.o. for 15 d | 20 | 1 mM | Urine TBARS, 1164 nmol/24 h | 1314 | 1169 | −11* | 11 | Sullivan et al., 2006 |
| High salt 10% | Normal salt | p.o. for 1 wk | 6 | 1 mM | Plasma 8-iso d 3/7, −64/50 pg/ml | 18 | 60/63 | −6/−26 (NS/NS) | 9/26 | Williams et al., 2004 |
| Urine H2O2, 3 nmol/d | −1 | 2 | −33* | 50 | ||||||
| Hyperthyroid (s.c. T4 for 6 wk) | Vehicle infused | p.o. for 6 wk | 8 | 1 mM | Plasma MDA, 10.2 μM | 6.8 | 7.5 | −26* | 79 | Moreno et al., 2005 |
| Urine 8-iso, 12.5 μg/d | 6.5 | 7.5 | −40* | 83 | ||||||
| 1K,1C | Sham uninephrec- tomized | p.o. for 2 wk 2 d | 6 | 2 mM | Aortic rings/ , 80 (RLU 15 min/mg) | 42 | 63 | −21* | 45 | Dobrian et al., 2001 |
| Renal nitrotyrosine, 59 ng/mg of protein | 14 | 32 | −46* | 60 | ||||||
| Plasma 8-iso, 240 pg/ml | 305 | 340 | −42* | 0 | ||||||
| 1K,1C (10 wk) | Sham | p.o. for 5 wk | 5 | 1 mM | DHE fluorescence in mesenteric arteries | 16 | 25 | −38* | 68 | Christensen et al., 2007b |
| 2K,1C | Sham | s.c. minipump for 13 d | 8 | 288 (μmol ·kg)/d | Urine 8-iso, 12.5 ng/d | 8 | 9 | −28* | 78 | Welch et al., 2003 |
| Urine MDA, 610 μmol/d | 400 | 330 | −46* | 133 | ||||||
| Leptin | Leptin untreated | p.o. for 7 d | 8 | 2 mM | Urine 8-iso, 325 ng/d | 190 | 225 | −31* | 43 | Beltowski et al., 2005 |
| Plasma 8-iso, 218 pg/ml | 130 | 138 | −37* | 91 | ||||||
| Obese Zucker | Lean | p.o. for 15 d | 10 | 1 mM | Renal MDA, 0.91 nmol/mg | 0.55 | 0.59 | −35* | 89 | Banday et al., 2005 |
| Offspring of protein malnourished mothers | Offspring of normal mothers | p.o. for 13 wk | 14–19 | 2 mM | Kidney nitrotyrosine, 1.42 (relative abundance) | 1.0 | 0.45 | −70* | 100 | Stewart et al., 2005 |
| Ren-2 transgenic rat | Control | p.o. for 3 wk | 6 | 1 mM | Cardiac MDA, 0.60 μm/mg of protein | 47 | 0.33 | −45* | 207 | Whaley- Connell et al., 2007 |
| Ren-2 transgenic rat | SD | p.o. for 3 wk | 6 | 1 mM | NADPH oxidase of mesenteric arteries, 18 | 11 | 13 | −28* | 71 | Whaley- Connell et al., 2007 |
| SD + HS (1% NaCl) + BSO (30 mM for 12 d] | HS | p.o. for 12 d | 8 | 1 mM | Urine 8-iso, 54 pg/mg Cr | 41 | 42 | −22* | 92 | Banday et al., 2007d |
| SHR | WKY | s.c. for 2 wk | 6–8 | 200 (nmol ·kg)/min | Urine 8-iso, 13.2 ng/d | N.A. | 9.6 | −27* | N.A. | Welch et al., 2005b |
| SHR | Untreated SHR | p.o. for 2 wk | 6–8 | 1 mM | Kidney cortex , 11,889 RLU | N.A. | 9315 | −22* | N.A. | Yanes et al., 2005 |
| Medulla, 6413 RLU | 5944 (N.S.) | −7 (N.S.) | ||||||||
| Plasma total anti- oxidant, 1.2 nM | 1.57 | −31* | ||||||||
| Urine 8-iso, 2.21 ng/mg of creatinine | 1.53 | −31* | ||||||||
| SHR | WKY | p.o. for 15 wk | 6–12 | 170 (μmol ·kg)/d | Kidney 8-iso, 5.2 ng/mg of tissue | 1.7 | 1.7 | −67* | 100 | Fortepiani and Reckelhoff, 2005 |
| SHR | WKY | p.o. for 6 wk | 6–12 | 170 (μmol ·kg)/d | Kidney 8-iso, 5.3 ng/mg of tissue | 1.7 | 4.8 | −9* | 14 | Fortepiani and Reckelhoff, 2005 |
| SHR | WKY | p.o. for 2 wk | 8 | 1 mM | Urine 8-iso, 9.8 ng/d | 6.8 | 6.0 | −39* | 127 | Schnackenberg and Wilcox, 1999 |
| SHR aging (16 mo) | Untreated SHR | p.o. for 8 mo | 6 | 6 mM | Urine 8-iso, 20 ng/d | N.A. | 12 | −40* | NA | Fortepiani et al., 2003 |
| SHR aging female (16 mo) | Untreated SHR female | p.o. for 8 mo | 6 | 6 mM | Urine 8-iso, 45 ng/d | N.A. | 37 | −18* | N.A. | Fortepiani et al., 2003 |
| SHR aging (16 mo) | Untreated SHR | p.o. for 8 mo | 10 | 1 mM | Urine 8-iso, 1.84 (ng ·mg Cr)/d | N.A. | 1.28 | −30* | N.A. | Payne et al., 2003 |
| SHR female | WKY | p.o. for 15 wk | 6–12 | 170 (μmol ·kg)/d | Kidney 8-iso, 3.7 ng/mg of tissue | 2.2 | 1.7 | −54* | 133 | Fortepiani and Reckelhoff, 2005 |
| SHR female | WKY | p.o. for 6 wk | 6–12 | 170 (μmol ·kg)/d | Kidney 8-iso, 3.6 ng/mg of tissue | 2.2 | 3.7 | −3 (N.S.) | 0 | Fortepiani and Reckelhoff, 2005 |
| SHR + L-NAME | Untreated SHR | p.o. for 2 wk | 6–8 | 1 mM | Kidney cortex , 10,423 RLU | N.A. | 9506 | −9* | N.A. | Yanes et al., 2005 |
| Medulla, 7422 RLU | 5248 | −29* | ||||||||
| Plasma total antioxidant, 1.25 nM | 1.46 | −17* | ||||||||
| Urine 8-iso, 1.89 ng/mg creatinine | 1.32 | −30* | ||||||||
| SHRsp low Mg2+diet | Untreated SHRsp | p.o. for 7 wk | 6 | 1 mM | Plasma TBARS, 2.7 μmol/ml | N.A. | 1.5 | −44* | N.A. | Touyz et al., 2002 |
| Vascular , 1.2 (nmol ·min)/mg tissue | 0.6 | −50* | ||||||||
| SHRsp 4% salt | Untreated SHRsp | p.o. for 6 wk | 6 | 1 mM | Vascular , 19 RLU | N.A. | 4 | −79* | N.A. | Park et al., 2002 |
| Plasma total antioxidants, 0.85 nM | 1.1 | −29* | ||||||||
| Streptozotocin (DM)/L-NAME | SD, tempol untreated | i.v. for 2 wk | 5 | 18 (μmol ·kg)/h | Urine 8-iso, 118 ng/d | 23 | 40 | −66* | 82 | Brands et al., |
| Studies in hypertensive mouse models | ||||||||||
| Ang II [0.7 (mg ·kg)/d], WT mice | Vehicle | s.c. for 2 wk | 28 (mmol ·kg)/d | Aortic , 125 pmol/mg of tissue | 50 | 85 | −32* | 53 | Dikalova et al., 2005 | |
| Ang II infused [0.7 (mg · kg)/d] Nox-1- overexpressing mice | Vehicle | s.c. for 2 wk | 28 (mmol ·kg)/d | Aortic , 250 pmol/mg of tissue | 75 | 160 | −36* | 51 | Dikalova et al., 2005 | |
Mean values are shown.
8-iso, 8-isoprostane PGF2α; TBARS, thiobarbituric acid reactive agent; RLU, relative light unit; HS, high salt; Dex, dexamethasone; DSR, Dahl salt-resistant rat; LS, low salt; 8OHdG, 8-hydroxy-2′-deoxyguanosine; HET-0016, N-hydroxy-N′-(4-butyl-2-methylphenyl)-formamidine; N.A., not applicable; adrenocorticotropin; d, day(s); wk, week(s); mo, month(s).
Significant change with tempol.
Of the 45 studies in which measurements were made of the BP and some parameter of ROS, 34 (76%) reported a reduction in both, 7 (16%) reported a reduction in BP but not in ROS (Hasdan et al., 2002; Fortepiani et al., 2003; Sedeek et al., 2003; Zhang et al., 2003b; Williams et al., 2004; Dikalova et al., 2005; Sullivan et al., 2006), 3 (7%) reported unchanged BP despite a reduction in ROS (Song et al., 2004; Elmarakby et al., 2005; Whaley-Connell et al., 2007), and 1 (2%) reported no change in BP or ROS (Song et al., 2004). Thus, BP and ROS were directionally concordant in 35 of 45 studies (78%).
Tempol has been an effective antihypertensive agent in Ang II-dependent models (e.g., Ang II-infused rats), renin-dependent models [e.g., two-kidney, one-clip (2K,1C) Goldblatt hypertensive rats] and salt- and volume-dependent, low-renin models (e.g., DOCA-salt rats). Clearly, there is no absolute requirement for an activated systemic renin-angiotensin-aldosterone system (RAAS) or volume expansion for a model to be responsive to tempol. The antihypertensive action of tempol in DSS rats was additive with the mineralocorticoid receptor antagonist eplerenone, which suggests that tempol and eplerenone reduce BP by largely independent means in this model (Bayorh et al., 2006).
Tempol has been as effective in prevention as in reversal of established hypertension. This fact was illustrated in two studies from Zheng et al. (2003b, 2004b), who reported that tempol was equally effective in preventing or normalizing the elevation in BP in rats whether given 4 days before or 8 days after prolonged infusions of adrenocorticotropin or dexamethasone (DEXA). However, tempol generally has been most effective when administered before the onset of hypertension.
Of the seven reports in which prolonged administration of tempol failed to reduce BP, two were in models that were barely hypertensive (Williams et al., 2004; Elmarakby et al., 2005) and one showed reductions in MAP of 8% (de Richelieu et al., 2005), but one study in rats transgenic for the renin-2 (ren-2) gene (Whaley-Connell et al., 2007) and two in the SHR (Fortepiani et al., 2003; de Richelieu et al., 2005) showed no changes in BP despite considerable baseline hypertension. These reports do not indicate a specific lack of effect of tempol for reducing BP in the Ren-2 or SHR models. Thus, another study at a somewhat earlier stage of Ren-2 hypertension showed a significant fall in BP, and 13 other studies in SHR (Table 2, Studies in hypertensive rats with systemic tempol) reported significant falls in BP with prolonged tempol administration (Howard et al., 2005). The response of these models to tempol is described in greater detail in sections II.B.2.a and II.B.2.d.
2. Action in Animal Models of Hypertension
a. Spontaneously Hypertensive Rat
The SHR has been particularly well studied. In five studies, tempol was administered acutely by intravenous injection to SHR in doses of 72 to 900 μmol/kg and reduced BP in all five (Schnackenberg et al., 1998; Sato et al., 2002; Shokoji et al., 2003; Patel et al., 2006; Chen et al., 2007a) with a 26% (Shokoji et al., 2004) to 100% (Schnackenberg et al., 1998) normalization of hypertension. Tempol has been added to the drinking water of SHR in nine studies (Schnackenberg et al., 1998; Schnackenberg and Wilcox, 1999; Feng et al., 2001; Fortepiani et al., 2003; Payne et al., 2003; de Richelieu et al., 2005; Fortepiani and Reckelhoff, 2005; Nabha et al., 2005; Welch et al., 2005b) in doses from 1 (Schnackenberg and Wilcox, 1999; Feng et al., 2001; Payne et al., 2003; de Richelieu et al., 2005; Nabha et al., 2005) to 6 (Fortepiani et al., 2003) mmol/l over 5 days (de Richelieu et al., 2005) to 8 months (Fortepiani et al., 2003). It reduced the BP in at least one group of SHR in all except one study (de Richelieu et al., 2005) in which it produced a 20 to 25% normalization of MAP over a short period of 5 days that was not statistically significant. The mean normalization of BP in the studies in SHR was 43%. Oral tempol prevented the age-dependent rise in BP in the SHR (Nabha et al., 2005). Tempol was also effective when given intraperitoneally to SHR at 1.5 mmol · kg−1 · day−1 (Schnackenberg and Wilcox, 1999) or by subcutaneous infusion via an osmotic minipump at 200 nmol · kg−1 · min−1 (Welch et al., 2005b).
Although a fall in BP during prolonged administration of tempol to male SHR has been a remarkably consistent finding, an exception was the absence of a significant fall in BP of 10- to 12-week-old SHR after 5 to 15 days of oral tempol (1 mmol/l in drinking water) (de Richelieu et al., 2005). BP was directly measured only after surgery and during mechanical ventilation and muscle paralysis, which might have obscured an earlier antihypertensive effect of tempol in this study.
A surprising finding has been the variable BP response to tempol in female SHR (Sartori-Valinotti et al., 2007). Fortepiani and coworkers reported that whereas male SHR had an antihypertensive response to tempol (6 mmol/l in drinking water) (Fortepiani et al., 2003) as did female SHR administered tempol for the first 15 weeks of their life (Fortepiani and Reckelhoff, 2005), no response was observed in postmenopausal female SHR (Fortepiani et al., 2003) or premenopausal female SHR when dosed from 9 to 15 weeks of age (Fortepiani and Reckelhoff, 2005). These data demonstrate a complex interaction between gender or sex hormones and age in the response to tempol. Remarkably, tempol was more effective in lowering the BP of young female than young male SHR and became less effective after menopause. The finding that aged, postmenopausal female SHR, which lack estrogen, would lack an antihypertensive response to tempol was unexpected because other observations by this group attested to increased ROS generation in postmenopausal rats (Fortepiani et al., 2003). Disparate responses between male and female SHR have also been noted by these authors with an anti-oxidant regimen of vitamins C and E. However, in the vitamin study, the postmenopausal females had an antihypertensive response to the antioxidants, whereas the males were resistant (Fortepiani and Reckelhoff, 2005). No clear explanation for the opposite effects in these studies of age and gender on the antihypertensive response to tempol or vitamins is apparent presently (Sartori-Valinotti et al., 2007). Sullivan et al. (2006) also reported sex differences in the response to prolonged oral tempol administration to salt-fed endothelin type B receptor-deficient rats (Sullivan et al., 2006). Whereas oral tempol caused almost complete reversal of hypertension initially in both males and females, these effects waned over 2 weeks at which time the BP was higher in females. This higher BP was accompanied by elevated plasma levels of ET-1. The authors concluded that ET-1 may have caused the elevated BP in females given tempol. These results suggest that mechanisms compensating for the effects of tempol that are mediated by ET-1 may be more important in females. However, this explanation is unsatisfactory because tempol reduced the BP of rats made hypertensive by infusion of ET-1 (Sedeek et al., 2003).
b. Renovascular Effects
There is evidence of oxidative stress in renovascular disease. Patients with renal artery stenosis and renovascular hypertension had increased plasma levels of lipid peroxidation products that were corrected by a successful intervention to correct the renal artery stenosis (Higashi et al., 2002).
Intravenous tempol (200 μmol · kg−1 · h−1) produced a 50% normalization of the hypertension that developed 1 month after clipping of one renal artery (2K,1C model) in the rat (Guron et al., 2006). Tempol given subcutaneously. by minipump over 13 days at 288 μmol · kg−1 · day−1 produced a 70% normalization of MAP in this model (Welch et al., 2003). Oral tempol given to the less renin-dependent 1K,1C rat model at 1 (Christensen et al., 2007b) or 2 (Dobrian et al., 2001) mmol/l in the drinking water for 2 (Dobrian et al., 2001) to 5 (Christensen et al., 2007b) weeks produced a 31% (Dobrian et al., 2001) and 90% (Christensen et al., 2007b) normalization of MAP.
Rats at the early (2–4 weeks) phase of 2K,1C hypertension, which is strongly Ang II-dependent, had increased excretion of 8-iso-PGF2α and MDA and reduced glomerular filtration rate (GFR) and kidney weight downstream from the renal artery clip (clipped kidney). These were accompanied by reduced outer cortical pO2 and reduced renal tubular Na+ transport (TNa) per oxygen used (QO2) by the clipped kidney (Welch et al., 2003). All of these parameters were prevented by 2 weeks of tempol infusion (200 nmol · kg−1 · min−1 s.c.) but not by 2 weeks of administration of an angiotensin receptor blocker (ARB) despite a similar moderation of hypertension (Welch et al., 2003). This result was remarkable because the 2K,1C rat is the quintessential model of Ang II-induced hypertension. These findings point to potential advantages of tempol over an ARB or an ACEI in renovascular disease that merit further study.
c. Angiotensin II-Infused and Angiotensin II-Dependent Hypertension
Incubation of many vascular tissues with Ang II increased generation (reviewed in Wilcox, 2005). The addition of tempol to blood vessels in which had been stimulated by prolonged incubation with Ang II generally prevented the increase in and reduced the contraction (Cai et al., 2003; Wilcox, 2005). However, one study of rat aortic rings and mesenteric resistance vessels incubated acutely with Ang II, ET-1, PE, and KCl demonstrated that coincubation with 10−4 M tempol reduced the sensitivity and responsiveness to Ang II selectively in an endothelium-dependent manner. This reduction was associated with a quenching of vascular and an enhancement of NO signaling by Ang II (Shastri et al., 2002). A selective effect of tempol on Ang II responses was also seen in a mild model of oxidative stress. Thus, Wang et al. (2003b) studied the contractility of perfused renal afferent arterioles isolated from rabbits with oxidative stress caused by a 2-week infusion of Ang II at two different rates. There was a selective enhancement of contractions to Ang II in vessels from rabbits infused with Ang II at the lower, nonpressor rate that was prevented by tempol but a more general enhancement of contraction to Ang II, ET-1, and U-46,619 in those infused with Ang II at a higher pressor rate that were all prevented by tempol.
Ang II treatment of porcine isolated coronary arterioles elicited Ang type 1 receptor (AT1-R)-dependent contractions at low concentrations and Ang type 2 receptor (AT2-R)- and NOS-dependent dilations at higher concentrations that were apparent after AT1-R blockade (Zhang et al., 2003a). Tempol moderated the AT1-R- dependent contraction, consistent with AT1-R mediating ROS production (Chabrashvili et al., 2003). Incubation of mesenteric or renal afferent arterioles from rabbits with oxidative stress with Ang II further impaired their EDRF/NO responses, which were restored by tempol (Wang et al., 2004, 2006a). Moreover, the relaxation responses to Ang II of aortas from diabetic rats in the presence of AT1-R blockade that were mediated by AT2-Rs were enhanced by tempol (Arun et al., 2004). However, elderly rats were shown to have enhanced expression of AT2-Rs, which mediated a paradoxical endothelium-dependent contractile response that was prevented by tempol (Pinaud et al., 2007). Thus, tempol normally resets the balance of vasoconstriction: vasodilation induced by Ang II toward a moderation of vasoconstriction.
Tempol (3 μmol · kg−1 · min−1) infused into the renal arteries of dogs pretreated with L-nitroarginine to block NOS attenuated reductions in renal blood flow (RBF), GFR, and sodium and fluid excretion in response to intra-arterial infusion of Ang II (Majid et al., 2005). Thus, tempol has an NO-independent component of action to blunt renal responses to Ang II.
The infusion of Ang II into rats (Chabrashvili et al., 2003; Welch et al., 2005a) or mice (Kawada et al., 2002; Dikalova et al., 2005; Welch et al., 2006) at a slow pressor rate increased the expression of NADPH oxidase components, increased generation in the blood vessels and kidneys, and increased the excretion of 8-iso-PGF2α and MDA. These effects and the rise in BP were prevented by coinfusion of tempol (Kawada et al., 2002; Kimura et al., 2005a; Welch et al., 2005a). In a discordant study, tempol failed to reduce the BP of rats during a 2-week infusion of Ang II despite a reduction in aortic unless the Ang II was given with enalapril to block angiotensin-converting enzyme (Elmarakby et al., 2007). Mice with vascular oxidative stress due to overexpression of neutrophil oxidase-1 (Nox-1) in VSMCs had an exaggerated increase in BP, vascular hypertrophy, and ROS during infusion of Ang II that were moderated by coinfusion of tempol (Dikalova et al., 2005).
Tempol given by acute intravenous injection at 15 (Zhang et al., 2004a) or 173 μmol/kg followed by an infusion at 43 μmol/kg (Kimura et al., 2004) did not reduce the BP of rats during a 5-min intravenous infusion of Ang II but, when given at a later stage at 1 to 24 h of Ang II infusion, tempol reduced the BP by up to 33% (Kimura et al., 2004). Tempol given intravenously at 43 (Kimura et al., 2004) or 170 μmol/kg or infused at 3 μmol · kg−1 · min−1 (Kimura et al., 2005a) produced 100 and 92% normalization of BP of rats infused with Ang II for 2 weeks at a slow pressor rate of 200 ng · kg−1 · min−1. When tempol was given in the drinking water to Ang II-infused rats (Ortiz et al., 2001a; Ogihara et al., 2002; Hattori et al., 2005) at 1 to 2 mmol/l or by subcutaneous infusion at 200 nmol · kg−1 · min−1 (Welch et al., 2005a) or 28 μmol · kg−1 · day−1 (Dikalova et al., 2005) over 7 (Hattori et al., 2005) to 15 (Ortiz et al., 2001a) days, it produced a 44 (Dikalova et al., 2005) to 100% (Ortiz et al., 2001a) normalization of the BP. These data demonstrate that tempol does not act as a direct antagonist of circulating Ang II but is highly effective in moderating the sustained increase in BP during prolonged Ang II infusion in rats and mice.
Transgenic rats overexpressing the ren-2 gene develop extreme hypertension that is lethal unless they receive an ARB or an ACEI. In one study, the hypertension was not significantly moderated by 3 weeks of oral tempol (1 mmol/l) administration, despite normalization of cardiac levels of NADPH oxidase and MDA and p22phox expression (Whaley-Connell et al., 2007). Administration of an ARB or tempol to this model prevented the activation of insulin-stimulated protein kinase B, which is required for phosphorylation and activation of phosphoinositol 3-kinase, and prevented coronary artery adventitial fibrosis (Whaley-Connell et al., 2007). This outcome is remarkable because in a second study in Ren-2 rats at a somewhat earlier stage with less severe hypertension, tempol (2 mmol/l of water) produced a robust 63% normalization of BP over 10 days (Howard et al., 2005). Moreover, in prehypertensive Ren-2 transgenic rats, intrarenal arterial infusions of tempol increased the RBF, GFR, and sodium excretion more than in control rats (Kopkan et al., 2007). Presumably the very high levels of renin and Ang II in the later stages of this severely hypertensive model can sustain hypertension even after inhibition of excessive ROS.
These animal studies could have clinical relevance because human brachial (Hussain et al., 2006) or coronary arteries (Püntmann et al., 2005) from subjects with cardiovascular disease had increased generation in response to Ang II. Coincubation with tempol prevented the increase in and reduced the contractions to Ang II (Püntmann et al., 2005; Hussain et al., 2006).
d. Deoxycorticosterone Acetate- or Aldosterone-Salt Hypertension
The administration of a mineralocorticosteroid such as DOCA with a high salt intake to unine-phrectomized rats produces severe hypertension with suppression of circulating renin. Therefore, it is considered a model of human low-renin hypertension.
A high salt intake in the rat increased lipid peroxidation and NADPH oxidase activity and reduced the expression of Cu/Zn- and Mn-SOD in the kidneys (Kitiyakara et al., 2003). Therefore, an increase in dietary salt itself can cause oxidative stress. Tempol has been very effective in preventing or moderating hypertension in uninephrectomized rats given a high-salt diet and DOCA (Beswick et al., 2001; Adeagbo et al., 2003; Awe et al., 2003; Nakano et al., 2003; Ghosh et al., 2004) or aldosterone (Nishiyama et al., 2004a; Hirono et al., 2007; Shibata et al., 2007). Tempol given by intravenous infusion (300 μmol/kg) to DOCA-salt hypertensive rats produced a 91% normalization of BP (Xu et al., 2004). Tempol given in the water at 1 (Beswick et al., 2001; Nakano et al., 2003; Ghosh et al., 2004; Iglarz et al., 2004) to 6 (Shibata et al., 2007) mmol/l or infused subcutaneously at 87 μmol · kg−1 · day−1 (Adeagbo et al., 2003; Awe et al., 2003) produced a 29 (Nakano et al., 2003) to 106% (Shibata et al., 2007) normalization of BP, which averaged 73% among these studies. Oral tempol (3 mmol/l for 6 weeks) was as effective as the mineralocorticosteroid antagonist eplerenone in preventing hypertension and proteinuria in rats given 1% NaCl to drink and infused with aldosterone (Nishiyama et al., 2004a). Tempol also reduced the BP of rats infused with aldosterone for 6 weeks without salt loading but did not reverse the remodeling of the resistance vessels in this model although it did prevent cardiac, renal, and aortic fibrosis and the associated oxidative stress (Iglarz et al., 2004). Aldosterone infusion for 3 weeks into uninephrectomized rats given saline to drink reduced plasma Ang II concentrations predictably, yet aortic tissue Ang II concentrations were increased (Hirono et al., 2007). Tempol and an ARB were equally effective in this model in moderating the hypertension and reducing the vascular expression of inflammatory mediators (Hirono et al., 2007). Tempol (3 mmol/l in water) prevented MAPK activation and glomerular sclerosis in the rat DOCA-salt model (Nishiyama and Abe, 2004). These results indicate that mineralocorticosteroid-salt models of low-renin hypertension in the rat are associated with rather severe oxidative stress, perhaps related to activation of a local tissue RAAS. The increased ROS signaling via MAPK and the hypertension can be largely prevented by the administration of tempol.
e. Dahl Salt-Sensitive Rat
As recently reviewed (Manning et al., 2005), the DSS rat is considered a model of salt sensitivity and nephrosclerosis. After 3 to 5 weeks of salt feeding, these rats developed severe oxidative stress, hypertension, and renal damage associated with reduced renal SOD activity. These defects were ameliorated by administration of tempol or vitamins E plus C (Manning et al., 2003, 2005). An intravenous bolus of tempol given to DSS rats (Zicha et al., 2001; Dobesová et al., 2002) at 60 (Dobesová et al., 2002) or 145 (Zicha et al., 2001) μmol/kg produced a 48 and 19% normalization of the BP. The fall in BP with intravenous tempol (142 μmol/kg) was greater in young than in elderly DSS rats (Dobesová et al., 2002). Prolonged tempol administration for 3 (Bayorh et al., 2006) to 10 weeks (Ozawa et al., 2004; Guo et al., 2006) in seven studies (Meng et al., 2003; Kobori and Nishiyama, 2004; Nishiyama et al., 2004b; Ozawa et al., 2004; Hisaki et al, 2005; Bayorh et al., 2006; Guo et al., 2006) produced a 29 (Guo et al., 2006) to 102% (Meng et al., 2003) normalization of BP, which averaged 65% among these studies. Thus, tempol is an effective antihypertensive agent in this highly salt-sensitive rat model of hypertension.
f. Endothelin Models
An infusion of ET-1 into rats increased their lipid peroxidation, RVR, and BP, all of which were reduced by coinfusion of tempol (110 μmol · kg−1 · 24 h−1) (Sedeek et al., 2003). However, in another study of ET-1-infused rats, there was no significant reduction in MAP with a similar rate of tempol infusion or with the addition of tempol to the drinking water (1 mmol/l) for 12 days (Elmarakby et al., 2005), perhaps because ET-1 produced only a modest increase in MAP of 15% in this protocol.
The administration of an endothelin type B receptor (ET-B) antagonist (A-192621) to normal rats for 1 week raised their BP by 17 to 25%. The coadministration of tempol for 1 week produced an initial 60% normalization of the BP, but this effect was lost after 1 week (Williams et al., 2004). ET-B-deficient rats given salt developed a more robust increase in BP of 37%, which was 40% normalized initially by oral tempol (Sullivan et al., 2006). However, over 15 days of tempol administration, the antihypertensive response again waned, especially in female rats, which had an increase in plasma ET-1. These results suggest that the modest and inconsistent effects of tempol to reduce the BP of rats infused with ET-1 or in ET-B-deficient rats may relate to a combination of the modest levels of hypertension, because tempol is not effective in reducing the BP of normotensive models, and sex-dependent compensatory changes in ET-1 generation. This result is surprising because tempol was quite effective in preventing increases in ET-1 generation both in vitro (An et al., 2007) and in vivo (Ortiz et al., 2001a; Fujii et al., 2005; Bell et al., 2007; Troncoso Brindeiro et al., 2007) and in moderating vasoconstriction of isolated blood vessels to ET-1 in several models of hypertension and oxidative stress (Wang et al., 2004, 2006c).
g. Lead- and Zinc-Induced Hypertension
Prolonged exposure of rats to lead in vivo or of ECs to lead in vitro generated •OH and . These increases in ROS were prevented by tempol (Vaziri and Ding, 2001; Vaziri et al., 2003b). Tempol given intravenously or by subcutaneous injection moderated the oxidative stress and the hypertension of rats given a diet with added lead (Vaziri et al., 2001). Rats fed a diet with added zinc for 5 weeks also developed hypertension that was reversed by acute intravenous tempol (Yanagisawa et al., 2004). SHR given a Zn-free diet also had an increased MAP perhaps because of defective function of Cu/Zn-SOD. The component of hypertension that was related to Zn deficiency was abolished by intravenous tempol (100 μmol/kg) (Sato et al., 2002).
h. Nitric-Oxide Synthase Inhibitor Hypertension
Oral administration of L-nitroarginine methyl ester (L-NAME) to block NOS increased the MAP of conscious rats substantially (Elmedal et al., 2004; Thakali et al., 2006). The hypertension was 72% normalized by acute intravenous tempol but was not modified by prolonged administration of tempol in the water (1.3 mmol · kg−1 · day−1) (Elmedal et al., 2004). The hypertension of rats given L-NAME for 2 months was not prevented by coad-ministration of tempol (142 μmol · kg−1 · day−1) plus vitamin C despite correction of cardiac indices of ROS (Bell et al., 2007). Thus, the hypertension accompanying acute, but not prolonged, NOS inhibition is responsive to tempol. This finding is consistent with the concept that a component of the acute antihypertensive response to tempol entails an improvement in NO bioactivity (Chen et al., 2007a).
i. Reduced Renal Mass Models
Removal of one kidney and two-thirds of the other produces a five-sixths nephrectomy rat model of progressive chronic kidney disease (CKD) related to a reduced renal mass (RRM). The BP and plasma renin activity (PRA) rose steeply in rats in which the tissue of the remaining kidney was infarcted by ligating the upper and lower pole renal arteries to produce a renal ischemia model (Hasdan et al., 2002; Vaziri et al., 2003a). However, there was little change in BP or PRA of rats in which the tissue of the remaining kidney was surgically resected (Griffin et al., 1994, 2004; Ibrahim and Hostetter, 1998; Griffin et al., 2004) unless these rats were fed a high-salt diet in which case they had a steady rise in BP (Ylitalo et al., 1976; Bidani et al., 1987; Li et al., 2007). Oral tempol (1 mmol/l to the drinking water) (Hasdan et al., 2002; Vaziri et al., 2003a; Li et al., 2007) or tempol given by intraperitoneal injection at 1.0 to 1.5 mmol ·kg−1 · day−1 for 10 days (Hasdan et al., 2002), 5 weeks (Vaziri et al., 2003a), or 12 weeks (Li et al., 2007) produced a 50 (Vaziri et al., 2003a) to 85% (Hasdan et al., 2002; Li et al., 2007) normalization of hypertension in these two models of CKD. Thus, the hypertension in the RRM rat model is highly responsive to tempol whether or not it is accompanied by a stimulated circulating RAAS.
j. Catecholaminergic and Dopaminergic Hypertension
Tempol did not affect contractions to NE in human brachial artery segments (Püntmann et al., 2005) or renal afferent arterioles from rabbits with Ang II-induced oxidative stress (Wang et al., 2003b, 2006b) or to PE in the SHR mesenteric vascular bed (Girouard and de Champlain, 2004). The failure of NE to enhance vascular in vessels from Ang II-infused rabbits was due to inhibition of generation by β1-adrenoceptor signaling via protein kinase A (Wang et al., 2006b).
The role of abnormal dopamine receptor (DR) signaling in hypertension has been studied extensively by Josè and colleagues (Albrecht et al., 1996; O’Connell et al., 1997; Felder and Jose, 2006; Wang et al., 2007) and Lokhandwala and colleagues (Banday et al., 2005, 2005a,b,c; Fardoun et al., 2006; Marwaha and Lokhandwala, 2006). The effect of tempol on the renal tubular actions of dopamine are reviewed in section II.E.6, where it is shown that the ability of dopamine-1-like receptors to decrease tubular NaCl transport is impaired in several hypertensive models because of the uncoupling of the D1R from the GRK type 4γ receptor (Wang et al., 2007), which participates in D1R desensitization (Felder and Jose, 2006). GRK4γ A142V transgenic mice have hypertension but do not have oxidative stress. These mice had no fall in BP with acute intravenous tempol (Wang et al., 2007). D1R(−/−) (Wang et al., 2007) and D5R(−/−) mice (Wang et al., 2007) also had hypertension, but this model had extensive evidence of oxidative stress and a significant reduction of hypertension with acute intravenous tempol.
Lokhandwada and colleagues reported that rats with oxidative stress from the administration of BSO for 2 weeks had hypertension (Banday et al., 2007a) that was exacerbated by a high-salt diet (Banday et al., 2007c). Administration of tempol for 2 weeks produced an 85% normalization of hypertension and restored normal signaling via the D1R to inhibit renal Na+/K+-ATPase (Banday et al., 2007c), G-protein coupling, NF-κB translocation, PKC activation, GRK-2 sequestration, and D1 receptor phosphorylation (Banday et al., 2007a). Other studies by this group demonstrated potent effects of tempol to restore dopamine D1R signaling in the kidneys of old Fisher 344 rats (Fardoun et al., 2006), in insulinopenic diabetic rats given streptozotocin (STZ) (Marwaha and Lokhandwala, 2006), and in obese Zucker rats (Banday et al., 2005, 2007b). Tempol caused a 50% normalization of the elevated BP in the Zucker model (Banday et al., 2005).
Collectively, these results point to an important role for impaired renal D1R signaling in the kidneys of animal models of oxidative stress, hypertension, or type I or II DM. These effects are responsive to tempol administration.
k. Hypoxia
Rats subjected to intermittent hypoxia for 14 days to mimic sleep apnea developed oxidative stress and hypertension, which were prevented by coadministration of tempol (1 mmol/l in water) (Troncoso Brindeiro et al., 2007). However, prolonged administration of tempol to neonatal rats chronically exposed to hypoxia led to stunted growth and impaired cellular proliferation in the airspaces although pulmonary vascular remodeling was prevented (Jankov et al., 2008). This result raises a note of caution for the use of tempol in chronic hypoxia.
l. Blood Pressure Programming
The perinatal milieu can program subsequent levels of BP in the adult (Racasan et al., 2005). Tempol (1 mmol/l in the drinking water) or a mixture of vitamins C and E or the NO donor compound molsidomine given to the dam for the last 2 weeks of gestation and to the offspring for the first 4 weeks after birth each prevented age-related increases in BP and proteinuria in SHR (Racasan et al., 2005). The authors attributed these effects of tempol to prevention of -induced activation of inducible NOS, because the inducible nitric-oxide synthase (iNOS) inhibitor, L-N6-(1-iminoethyl) lysine reduced the BP of SHR offspring (Racasan et al., 2002, 2005).
Feeding rats a low-protein diet during pregnancy caused oxidative stress, nitrotyrosine deposition, immune cell infiltration of the kidneys, and subsequent hypertension in the offspring (Stewart et al., 2005). Administration of tempol (2 mmol/l of water) or the anti-inflammatory agent mycophenolate mofetil to the pups for 3 weeks prevented these changes (Stewart et al., 2005).
m. Oxidant Protocols
The addition of BSO (30 mM) to the drinking water of rats for 2 weeks reduced their cellular levels of glutathione, induced lipid peroxidation, and raised the BP (Banday et al., 2007a). These changes were prevented by oral tempol (1 mmol/l) (Banday et al., 2007a). When combined with a high-salt diet, oral tempol was fully effective in preventing the rise in BP (Banday et al., 2007c,d).
n. Other Hypertensive Models
Rats given cyclosporin A developed vascular and renal oxidative stress and increased renal concentrations of Ang II and hypertension, which were reversed by oral tempol (3 mmol/l in the water) (Nishiyama et al., 2003). Tempol (1 mmol/l in the water for 6 weeks) was fully effective in preventing the rise in BP, oxidative stress, and vascular remodeling with dietary magnesium deficiency in the stroke-prone spontaneously hypertensive rat (SHRSP) (Touyz et al., 2002).
3. Mechanism of Antihypertensive Response to Acute Administration of Tempol
The fall in BP accompanying an acute intravenous dose of tempol (216 μmol/kg i.v.) in Ang II-infused rats (Nishiyama et al., 2001) or miniature swine (Hahn et al., 1999) has been ascribed to a fall in total peripheral resistance with a maintained or increased cardiac output.
The short-lived fall in BP after bolus intravenous dosing of tempol has been related to a rapid conversion of the plasma concentrations of the nitroxide radical to the hydroxylamine, as assessed by EPR (Hahn et al., 1999). Early studies in mice (Hahn et al., 1998, 2000) and miniature pigs (Hahn et al., 1999) demonstrated that acute administration of tempol reduced the BP and the HR, whereas acute intraperitoneal administration of the reduced form, tempol-H, had no immediate effect on BP. However, there was a modest reduction in BP after a delay of 5 to 10 min at which time whole-body EPR studies demonstrated that some tempol-H had been oxidized to the nitroxide form (Hahn et al., 2000). These findings relate the acute antihypertensive response to tempol to the nitroxide radical and probably to its facility for reducing tissue levels of . Indeed, Hahn et al. (1999) demonstrated that the catalytic rate constants for superoxide dismutation by a series of nitroxides predicted their effects on systemic NO. Patel et al. (2006) demonstrated that the effectiveness of six-member ring nitroxides to reduce the BP of anesthetized SHR was predicted by their in vitro SOD-mimetic activity. In contrast, five-member ring nitroxides such as 3-CP did not lower BP in SHR (Patel et al., 2006), miniature swine (Hahn et al., 1999), or mice (Hahn et al., 1998) despite in vitro SOD-mimetic activity (Patel et al., 2006). This finding is consistent with the hypothesis that the acute antihypertensive response is due to rapid metabolism of , which is facilitated by a boat-and-chair conformational change that occurs with six- but not five-member ring nitroxides. This conformational change greatly accelerates the dismutation reaction that is apparently required for effective antihypertensive action in vivo. Thus, the rapid reversal of hypertension after intravenous injections of tempol may relate to the reduction of the nitroxide to the hydroxylamine, which does not lower BP itself. In addition, tempol is highly permeable and will leave the plasma compartment as it partitions into cells (Patel et al., 2006).
An acute infusion of Ang II over 30 min into conscious rats increased the MAP and the activation of MAPKs in the aorta and heart (Zhang et al., 2004a). Whereas tempol prevented MAPK activation and lipid peroxidation, it did not prevent the early rise in BP during the first 5 min of an Ang II infusion in this model (Zhang et al., 2004a). Likewise, tempol did not relax blood vessels that had been contracted with Ang II, U-46,619, or ET-1 for 5 min, yet relaxed them after they had been exposed to these agonists for 10 to 30 min or more (Wang et al., 2003b, 2004; Chen et al., 2007b). Three conclusions follow from these findings. First, the acute antihypertensive effect of tempol can be dissociated from its effects on MAPK signaling. Second, the vascular effects of tempol are not due to interruption of agonist-receptor interactions. Third, a period of agonist stimulation is required to generate vascular and create the condition for a BP-lowering effect of tempol.
Xu, Fink, and colleagues reported that the acute reduction in BP with intravenous tempol in normotensive (Xu et al., 2001) and hypertensive rats (Xu et al., 2002, 2004) was accompanied by inhibition of the renal sympathetic nerves that was independent of NOS (Xu et al., 2002). These studies are described further in section II.D.2.
Chen et al. (2007a) further probed the mechanism of the acute hypotensive response to graded doses of intravenous tempol in the anesthetized SHR (Chen et al., 2007a). The response was unaffected by blockade of catalase with 3-aminotriazole, by infusion of pegalated catalase, by glutathione depletion with BSO, by blockade of BK channels with iberiotoxin, or by inhibition of hemoxygenase with tin mesoporphyrin. Thus, the acute hypotensive response to tempol does not depend on the generation of H2O2 or the activation of BK channels or the generation of carbon monoxide or biliverdin by hemoxygenase. However, the hypotensive response was blunted by activation of KATP channels with cromakalim during maintenance of BP with infused NE or by blockade of these channels by glibenclamide. This finding implicated KATP channels in the hypotensive response. Moreover, the hypotensive response was reduced by blockade of NOS with L-NAME or by blockade of ganglionic transmission with hexamethonium (Chen et al., 2007a). Because L-NAME and hexamethonium were additive but glibenclamide and hexamethonium were less than additive, the authors concluded that the acute antihypertensive response to tempol depended on the independent effect of potentiation of NO and inhibition of the peripheral SNS and that the latter involved the activation of KATP channels.
4. Mechanism of Antihypertensive Response to Prolonged Tempol
a. Studies of Dose, Duration, and Route of Administration
Tempol infused subcutaneously over 2 weeks into conscious SHR in doses of 50, 100, and 200 nmol · kg−1 · min−1 reduced the MAP and the excretion of 8-iso-PGF2α at the higher rates of infusion (Welch et al., 2005b). When tempol was given to rats in the drinking water within a range of 1 to 6 mmol/l, there was no clear indication that the fall in BP increased with the dose of tempol or with the duration of tempol administration (Table 2, Studies in hypertensive rats with systemic tempol). These oral doses are approximately equivalent to 100 to 600 nmol · kg−1 · min−1, which is within the effective dose range for infused tempol. Indeed, intraperitoneal and oral dosing of tempol yielded apparently similar reductions in BP in a single study. A 1.5 mmol · kg−1 · day−1 intraperitoneal dose of tempol given to SHR produced a 36% normalization of MAP, which was comparable with the response to an equivalent oral dose of tempol (Schnackenberg et al., 1998). This result suggests that rats respond similarly to infused and to oral tempol and that the bioactivity of tempol is probably quite high, but these theories remain to be tested formally. Feng et al. (2001) reported an equivalent reduction in BP of SHR given tempol for 4 days or 7 weeks. Welch et al. (2005b) reported that the MAP of conscious SHR infused subcutaneously with tempol (200 nmol · kg−1 · min−1) was reduced within the first 12 h and fell further over the subsequent 12 days. In contrast, Sullivan et al. (2006) reported that oral tempol (1 mmol/l) given to salt-loaded ET-B-deficient rats entirely prevented the rise in BP during the first week of a high-salt diet but was no longer effective after 15 days at which time the ET-B-deficient rats had enhanced excretion of 8-iso-PGF2α. These authors identified a delayed increase in plasma ET-1 as a potential compensatory mechanism that may have overridden the antioxidant and antihypertensive actions of tempol. However, tachyphylaxis to tempol has not been apparent in other models.
Some of the reports in which oral tempol failed to reduce the BP in hypertensive models may relate to loss of pharmacological activity of tempol in the drinking water. We have noted discoloration of tempol solutions exposed to light after approximately 2 days. We recommend protecting tempol from light in foil-wrapped drinking bottles and providing fresh tempol solutions daily to prevent this apparent degradation.
b. Relationships to Antioxidant Action
Tempol reduced the levels of markers of oxidative stress, such as lipid peroxidation products, in the kidney cortex, kidney medulla, renal blood vessels, plasma, and urine of many hypertensive rat and mouse models (Oberley et al., 1993; Ortiz et al., 2001a; Nishiyama et al., 2003; Welch et al., 2003, 2005b). Prolonged infusions of Ang II at a slow pressor rate enhanced parameters of oxidative stress in the kidney and enhanced the renal cortical NADPH oxidase activity (Chabrashvili et al., 2003; Wang et al., 2003b; Welch et al., 2005a), both of which were prevented by coinfusion of tempol (Welch et al., 2005a). Thus, prolonged administration of tempol can reduce oxidative stress and can reset the endogenous redox machinery toward an antioxidant profile in the kidneys, which could be important for its prolonged hypotensive action.
Mitchell and coworkers demonstrated that tempol added to hamster lung fibroblasts interacts with heme proteins to exert a catalase-like metabolism of H2O2 (Krishna et al., 1996b). In contrast, Chen et al. (2007a) reported that the addition of tempol to the aorta of rats with oxidative stress caused an abrupt increase in H2O2 and showed further that H2O2 was required for the early, transient vasodilator response. H2O2 can produce vasodilation (Chen et al., 2007b), vasoconstriction (Schnackenberg et al., 2000) via activation of thromboxane-prostanoid receptors (TP-Rs) (Gao and Lee, 2001), or a biphasic response (Gao et al., 2003), depending on the vascular bed, the concentration, and the experimental conditions.
Pollock, Makino, and coworkers have demonstrated an accumulation of H2O2 in the urine and kidneys of rats given tempol over a prolonged period (Makino et al., 2003; Elmarakby et al., 2005). However, pretreatment with intravenous PEG-catalase did not blunt the acute hypotensive response to intravenous tempol in the SHR (Chen et al., 2007b). Moreover, PEG-catalase was actually required to permit a hypotensive response to infusion of tempol into the renal medulla of the rat, suggesting that tempol-induced generation of H2O2 at this site prevented a fall in BP. Therefore, the present evidence suggests that the accumulation of H2O2 in the blood vessels after acute tempol administration may contribute to a transient vasodilation but probably is not required for the sustained antihypertensive response to tempol. However, tempol-induced increases in H2O2 in the renal medulla may enhance NaCl reabsorption and maintain hypertension.
Many studies have shown rather directly that tempol decreased tissue levels of ROS using assays that include lipid peroxidation, protein or DNA oxidation, dihydroethidium (DHE) fluorescence, or lucigenin-enhanced chemiluminescence (Beswick et al., 2001; Dobrian et al., 2001; Park et al., 2002; Touyz et al., 2002; Meng et al., 2003; Nakano et al., 2003; Nishiyama et al., 2003; Ghosh et al., 2004; Iglarz et al., 2004; Elmarakby et al., 2005; Hattori et al., 2005; Yanes et al., 2005). Figure 3 depicts values from individual studies in which tempol has been given by prolonged administration to hypertensive rat models. Significant correlations are apparent between the changes in BP and plasma indices of ROS (Fig. 3A) and especially in changes in renal excretion of lipid peroxidation products (Fig. 3B). Figure 4 depicts values from individual rat studies of the degree of normalization of BP during prolonged tempol administration and the normalization of vascular (Fig. 4A) or of kidney tissue indices of ROS (Fig. 4B). The close correlations are remarkable, given the variability in the measurement of ROS in vivo. These relationships are quite compatible with the hypothesis that prolonged tempol administration reduced hypertensive levels of BP by reducing systemic, vascular, and/or renal oxidative stress. The observation that prolonged tempol administration did not lower BP in normotensive control animals with normal parameters of oxidative stress provides further support for this hypothesis.
Fig. 3.

Individual mean study values from hypertensive rat models showing the relationships between changes in plasma indices of ROS (A) or change in renal excretion of lipid peroxidation products (B) and changes in BP with prolonged tempol administration. The data were derived from the following animal models and studies: spontaneously hypertensive rats (SHR) (Fortepiani et al., 2003; Payne et al., 2003; Welch et al., 2005b; Yanes et al., 2005); Dahl salt-sensitive rats and fed salt (DSS + salt) (Hoagland et al., 2003; Kobori and Nishiyama, 2004; Guo et al., 2006); rats administrated deoxycorticosterone acetate, uninephrectomized, and fed salt (DOCA + salt) (Adeagbo et al., 2003); rats infused with dexamethasone or adrenocorticotropic hormone (DEXA/adrenocorticotropin) (Zhang et al., 2003b, 2004b); rats infused with angiotensin II (Ang II) (Ortiz et al., 2001a; Ogihara et al., 2002); rats infused with aldosterone and fed a diet with or without extra salt (Aldo ± salt) (Iglarz et al., 2004; Nishiyama et al., 2004a); two kidney, one clip Goldblatt rat model of renovascular hypertension (2K,1C) (Welch et al., 2003); one kidney, one clip Goldblatt rat model of renovascular hypertension (1K,1C)(Dobrian et al., 2001); rats infused with endothelin-1 (ET models) (Sedeek et al., 2003; Elmarakby et al., 2005; Sullivan et al., 2006); rats administered buthionine sulfoximine (BSO) (Banday et al., 2007a,c); and other hypertensive models (Makino et al., 2003; Song et al., 2004; Beltowski et al., 2005; Moreno et al., 2005).
Fig. 4.

Individual mean study values from hypertensive rat models showing the relationships between normalization of vascular superoxide (A) or normalization of kidney indices of ROS (B) and normalization of BP with prolonged tempol administration. The data were derived from the following animal models and studies: spontaneously hypertensive rats (SHR) (Fortepiani and Reckelhoff, 2005); Dahl salt-sensitive rats and fed salt rat (DSS + salt) (Meng et al., 2003; Nishiyama et al., 2004b); rats administrated deoxycorticosterone acetate, uninephrectomized, and fed salt (DOCA + salt): (Beswick et al., 2001; Nakano et al., 2003; Ghosh et al., 2004); rats infused with angiotensin II (Ang II) (Ortiz et al., 2001a; Hattori et al., 2005; Welch et al., 2005a); rats infused with aldosterone and fed salt (Aldo + salt) (Iglarz et al., 2004; Nishiyama et al., 2004a); rats transgenic for the human renin-2 gene (Ren-2) (Whaley-Connell et al., 2007); one kidney, one clip Goldblatt rat model of renovascular hypertension (IK,1C) (Dobrian et al., 2001; Christensen et al., 2007b); rats infused with endothelin-1 (ET models): (Sedeek et al., 2003; Elmarakby et al., 2005); and other hypertensive models: (Nishiyama et al., 2003; Banday et al., 2005; Stewart et al., 2005).
On the other hand, some reports have dissociated the ability of tempol to reduce BP from its ability to reduce oxidative stress. Zhang et al. (2003a,b) reported that the administration of 1 mmol/l tempol in the drinking water of Sprague-Dawley (SD) rats made hypertensive by the administration of DEXA or adrenocorticotropin reduced their BP without an accompanying decrease in plasma levels of 8-iso-PGF2α. It is notable that the systolic blood pressure was increased by only 15 mm Hg in these models. Hasdan et al. (2002) reported that 10 days of intraperitoneal administration of tempol (1.5 mmol · kg−1 · day−1) to rats subjected to five-sixths nephrectomy prevented the early increase in BP but did not reduce the plasma levels of advanced oxidation protein products. However, mesenteric arterioles dissected from these rats had an impaired relaxant response to ACh that was improved by tempol. This finding suggests that tempol administration had corrected oxidative stress within the small blood vessels. Pollock and coworkers noted that oral tempol (1 mmol/l) decreased the hypertension of rats fed a high-salt diet despite no change in the excretion of 8-iso-PGF2α (Williams et al., 2004; Elmarakby et al., 2005), whereas the same dose of tempol did not consistently decrease the hypertension of rats infused with ET-1 or with the ET-B receptor antagonist A-192621 despite decreasing plasma levels of 8-iso-PGF2α and vascular . These authors proposed that an increase in the renal production of H2O2 had compromised the reduction in BP with tempol. However, a prior study by Sedeek et al. (2003) reported that intravenous infusion of tempol (173 μmol · kg−1 · day−1) into rats infused with ET-1 caused a robust 87% normalization of BP. In this study, the fall in BP was coupled with significant reductions in the malonaldehyde content of the kidney and in the renal excretion of 8-iso-PGF2α. These conflicting results of tempol administration in rat models of ET-1-induced hypertension may relate to the modest increase in BP with ET-1. Thus, Pollock and coworkers noted a 28% normalization of BP and 44% normalization of the excretion of 8-iso-PGF2α with tempol in rats infused with ET-1, but the change in BP did not represent a significant change.
Recent studies in the Ren-2 rat model of malignant hypertension showed that 21 days of oral tempol (1 mmol/l) reduced parameters of ROS and the remodeling in the aorta (Wei et al., 2007) and heart (Whaley-Connell et al., 2007) without significantly reducing the BP. However, these studies used the tail-cuff method to assess BP, which provides a stressed measure that may not reflect the BP measured telemetrically in unrestrained rats (Sasser et al., 2002).
Tempol has been found to be ineffective as an antihypertensive agent in animal models that are not associated with heightened oxidative stress. The antihypertensive response to an intravenous infusion of tempol in SD rats infused with pressor doses of Ang II was negligible during the first 10 min but became increasingly effective as the duration of the Ang II infusion increased up to a maximum at 12 h at which time tempol prevented 96% of the increase in BP (Kimura et al., 2004). It has been reported that it takes 5 to 20 min of incubation with Ang II for isolated blood vessels to develop oxidative stress (Wang et al., 2003b). These studies show that tempol does not block the immediate pressor effects of Ang II, but diminishes, or even prevents, the effects that develop during a prolonged Ang II infusion that are accompanied by increased ROS.
Thus, the balance of evidence favors the hypothesis that prolonged administration of tempol reduces BP in hypertensive models as a consequence of its antioxidant actions, although some discordant results are apparent.
c. Interaction with Endogenous Oxidant/Antioxidant Pathways
Tempol can down-regulate the expression of the p22phox subunit of NADPH oxidase and thereby reduce the activity of the enzyme in target tissues. Slow pressor infusions of Ang II increased p22phox expression and NADPH oxidase activity in the kidney (Chabrashvili et al., 2003; Welch et al., 2005a; Modlinger et al., 2006) and the renal afferent arteriole (Wang et al., 2003b). Coinfusion of tempol with Ang II prevented the up-regulation of p22phox in these models (Nishiyama et al., 2004a; Welch et al., 2005a). Hattori et al. (2005) reported that tempol prevented an increase in the expression in the aorta, cardiac, and adipose tissue of the NADPH oxidase components p47phox, Nox-2, p22phox, and Rac1 in rats infused with a slow pressor dose of Ang II for 7 days. Prolonged tempol administration to DSS rats normalized the NADPH oxidase activity and p22phox and gp91phox expression in the left ventricle (Guo et al., 2006).
Tempol also can promote endogenous antioxidant defense systems. For example, tempol administered to DOCA-salt rats challenged with the pro-oxidant tert-butyl hydroperoxide prevented the down-regulation of Cu/Zn-SOD in the kidneys and mesenteric vessels (Awe et al., 2003). A slow pressor infusion of Ang II reduced the expression of EC SOD in the kidneys, and reduced the SOD activity of the plasma, aorta, and kidneys (Welch et al., 2006), which was preserved by the administration of tempol (Welch et al., 2005a).
These effects of tempol administration to reduce endogenous levels could be important in providing sustained reductions in oxidative stress in the tissues. They may thereby contribute to the reduction in oxidative stress that is correlated with the reduction in BP (Figs. 3 and 4) and to the rather uniform reductions of BP throughout the day seen in SHR given tempol in the drinking water (Welch et al., 2005b), because these effects on endogenous generation and metabolism may outlive the direct redox effects of circulating tempol. On the other hand, tempol prevented the vascular expression of the inducible (type 1) isoform of heme oxygenase, which is an important endogenous antioxidant and vasodilator pathway (Lee et al., 2005).
d. Role of Nitric-Oxide Synthase
Several studies have addressed the hypothesis that the antihypertensive effects of tempol entail restoration of an action of NO whose bioactivity in the blood vessels and kidneys is often curtailed in hypertension (Wilcox, 2005). Tempol could enhance the effects of NO by preventing its bioinactivation by (Rubanyi and Vanhoutte, 1986; Zhang et al., 2005), by enhancing the stimulus to endothelial NO generation by stimulating blood flow and endothelial shear forces, by interrupting the incorporation of NO into glutathione to form S-nitrosoglutathione (Schrammel et al., 2003), by increasing the activity of the redoxsensitive dimethylarginine dimethylaminohydrolase, which metabolically inactivates the endogenous NOS inhibitor asymmetric dimethylarginine (Palm et al., 2007), or by recoupling NOS by improving the availability of the reduced form of tetrahydrobiopterin (BH4) (Cai and Harrison, 2000). Indeed, vascular eNOS was uncoupled from NO formation by oxidation of BH4 in the blood vessels from DOCA-salt rats (Zheng et al., 2003). Treatment of these blood vessels with apocynin or tempol reduced , increased BH4, and restored NO activity and EDRF responses (Zheng et al., 2003).
Schnackenberg et al. (1998) first reported that acute intravenous infusions of tempol into anesthetized SHR caused substantial reductions in MAP that were blocked during inhibition of NOS by L-NAME. This effect was not due to the increase in BP with L-NAME because SHR infused with a pressor dose of NE retained a full hypotensive response to tempol. They concluded that the hypotensive response to short-term administration of tempol to the SHR depended on NOS. Nishiyama et al., (2001) showed further that L-NAME almost abolished the falls in BP and in systemic and renal vascular resistances produced by tempol infused intravenous into Ang II-infused hypertensive rats. Indeed, tempol increased NO activity measured electrochemically in vivo in rats infused with Ang II (López et al., 2003). Prolonged administration of tempol to aging SHR reduced their BP and the PE- induced “active stress” and increased the ACh-induced relaxations of aortic strips isolated from these rats (Payne et al., 2003). These vascular effects of tempol were prevented by inhibitors of NOS or cGMP, implying that tempol had restored vascular NO signaling. L-NAME given to aged SHR prevented the fall in BP produced by a prolonged 2-week administration of oral tempol (Yanes et al., 2005). Tempol (150 μmol/kg i.v.) given acutely to hypertensive DSS rats with oxidative stress restored a pressor response to NOS inhibition with L-NAME, suggesting that tempol had restored vasoactive NO generation (Zicha et al., 2001). Thus, both the acute and the prolonged lowering of the BP by tempol has been related to enhancing the production or action of endogenous NO. Interestingly, the bradycardia that accompanies acute tempol administration has been little affected by NOS blockade (Table 1).
On the other hand, several studies have shown that tempol can reduce the BP of rats made hypertensive by prolonged blockade of NOS. Two weeks of NOS inhibition in normal rats by L-nitroarginine (0.5 g/l in the drinking water) increased their MAP by approximately 75 mm Hg (Thakali et al., 2006). The acute intravenous administration of tempol to rats in this study caused dose-dependent reductions in MAP of up to 54 mm Hg. Clearly, tempol given acutely can reduce the BP independent of NOS. However, the effect of prolonged NOS blockade to modify the hypotensive response to tempol has been inconsistent. The administration of L-NAME to rats over 7 days increased the MAP by approximately 70 mm Hg and increased the reactivity of the aorta to PE (Preti et al., 2005), neither of which was modified by oral administration of tempol (1.2 mmol · kg−1 · day−1) (Preti et al., 2005). NOS blockade did not affect the lowering of BP by acute intravenous tempol in one study (Xu et al., 2002) but blunted (Shokoji et al., 2003; Thakali et al., 2006) or blocked (Schnackenberg et al., 1998; Nishiyama et al., 2001) the response in other studies. Prolonged L-NAME administration to rats for 2 months caused hypertension and cardiac oxidative stress. Coadministration of tempol (150 μmol · kg−1 · day−1) and vitamin C prevented the oxidative stress but not the hypertension (Bell et al., 2007). NOS blockade blunted (Majid and Navar, 2001; Hoagland et al., 2003; Majid et al., 2005; Sainz et al., 2005) or blocked (Yanes et al., 2005) the hypotensive or renal responses to prolonged oral tempol in several studies. The reason for these discordant results requires further study. However, the reports that tempol retains efficacy in reducing the BP in some studies in which NOS has been blocked demonstrate the presence of NOS-independent pathways of BP reduction by tempol.
C. Vascular Actions of Tempol
1. Endothelium-Dependent Relaxant Factor/Nitric Oxide
The reaction of with NO not only biodegrades NO (Rubanyi and Vanhoutte, 1986) but generates ONOO−, which is a potent oxidant and nitrosating species that can modify protein structure and function. For example, ONOO− inactivated vascular prostacyclin (PGI2) synthase (Zou et al., 1997). Thus, a reduction of in ECs by tempol should increase vascular NO and PGI2 signaling, both of which should enhance EDRF responses. However, an increase in bioactive NO is not necessarily accompanied by an increase in the excretion of NO metabolites [nitrite (NO2) and nitrate (NO3)] (Welch et al., 2005b). Moreover, tempol increased vascular NO bioactivity (Zhang et al., 2005) but reduced renal cortical nNOS protein expression in the 1K,1C rat model of Goldblatt hypertension (Dobrian et al., 2001; Dutta et al., 2006).
Several studies have shown that tempol added to vessels from hypertensive models can enhance NO generation (Park et al., 2002; Lu and Kassab, 2004; Arrick et al., 2007). NO activity measured with a catheter-type NO sensor placed in the aorta of rabbits was reduced during prolonged infusion of Ang II but was restored by tempol (Imanishi et al., 2006). Treatment of bovine aortic or atrial ECs with SOD, tempol, or two other nitroxides, 3-CP or 3-ethoxycarbonyl-peroxyl (Zöllner et al., 1997), or the addition of tempol to the bath of rat perfused vasa recta capillary blood vessels increased NO release or activity (Zhang et al., 2005).
The EDRF/NO response was diminished or absent in blood vessels from many hypertensive or diabetic models of oxidative stress (Didion et al., 2006; Viswanad et al., 2006; Blanco-Rivero et al., 2007) or from mice with deletion of the gene for EC SOD (Kitayama et al., 2006). The EDRF/NO responses in vessels from these models of oxidative stress were enhanced by tempol (Haj-Yehia et al., 1999). Tempol restored NO-dependent vasodilation of blood vessels perfused at high pressure, which enhanced vascular generation (Christensen et al., 2007a). Tempol added to the bath of aortas from rats with enhanced ROS due to a high-salt diet restored NO bioactivity and relaxation responses to methacholine (cholinergic agonist) or histamine (Zhu et al., 2004) without moderating the increased EC [Ca2+] induced by these agonists (Zhu et al., 2006). This finding indicates that tempol preserved bioactive NO, whether generated in response to endothelial shear stress or agonist stimulation rather than raising EC calcium concentration to stimulate eNOS activity.
Multiple studies have demonstrated that tempol can increase the vascular relaxation response to ACh. This increase could contribute to the blunting by tempol of the constrictor responses to agonists in vessels from animals with hypertension or oxidative stress (Romanko and Stepp, 2005). Thus, incubation with tempol improved EDRF/NO responses and reduced contractile responses to agonists in mouse mesenteric arterioles (Wang et al., 2006a), in rabbit renal afferent arterioles from animals with oxidative stress due to Ang II infusion (Wang et al., 2003b, 2004), and in subcutaneous resistance arteries taken from patients with oxidative stress due to cardiovascular disease or hypertension (Hussain et al., 2006). Incubation with tempol of carotid arteries from DOCA-salt hypertensive rats increased the ratio of BH4/dihydrobiopterin, thereby improving NO generation by recoupling NOS (Zheng et al., 2003). Tempol can do more than just prevent oxidation of BH4. Thus, the administration of tempol to rats with an uncoupled eNOS from STZ-induced DM enhanced the expression of GTP cyclohydrolase 1, which is required for biopterin synthesis (Xu et al., 2007). Tempol protected blood vessels from impaired EDRF/NO responses produced by the oxidants homocysteine (Hucks et al., 2004) or C-reactive protein (Qamirani et al., 2005). Because the plasma levels of these compounds are increased in many patients with cardiovascular or inflammatory diseases (Qamirani et al., 2005), moderation of their vascular effects by tempol could be beneficial in these conditions.
Vaziri et al. (2001) reported that rats with lead-induced hypertension and oxidative stress had reduced excretion of nitrate and nitrite, despite an up-regulation of eNOS and iNOS in the kidneys. They attributed the up-regulation of NOS to reduced bioactive NO because it was restored by 2 weeks of tempol administration (Vaziri and Ding, 2001; Vaziri et al., 2001).
Thus, tempol may enhance EDRF responses in models of oxidative stress and inflammation by reducing metabolism of NO to ONOO−, preventing inactivation of PGI2 synthase, by enhancing NOS expression, and by enhancing NOS activity by preventing the uncoupling of the enzyme during reduced availability of its cofactor, BH4. Because blockade of NOS increases BP and RVR substantially (Gilani et al., 2007), an improvement in small vessel EDRF/NO by tempol could be an important component of its hypotensive action.
Studies have disclosed additional NO-independent mechanisms of endothelium-dependent relaxation with tempol. Thus, nitronyl nitroxides that trap NO blocked the coronary vasodilation response to NO donor compounds but not to tempol. This identified an NO-independent pathway (Konorev et al., 1995). Acute intravenous and 8- to 10-week prolonged oral administration of tempol improved both the defective NOS-dependent and NOS-independent ACh-induced vasodilation of renal afferent arterioles in hydronephrotic kidneys from DSS rats (Ozawa et al., 2004).
Rabbits developed nitrate tolerance, endothelial dysfunction, and a reduction in plasma NO activity after 7 days of treatment with nitroglycerin patches. Nitroglycerin responses were restored by tempol (10 mmol/l), whereas an ACEI or ARB was less effective (Imanishi et al., 2007). Nitrate tolerance developed after 90 min of incubation of aortic rings with nitroglycerin (Ghatta et al., 2007). This was prevented by coincubation with tempol or H2O2 but was exacerbated by catalase or ebselen. Because tempol released H2O2 from nitroglycerin-tolerant rings, the nitrate tolerance was ascribed to decreased endogenous formation of H2O2, which was restored by tempol (Ghatta et al., 2007).
2. Endothelium-Dependent Hyperpolarizing Factor/Hydrogen Peroxide
EDHF is released by Ca2+-mobilizing endothelial agonists or shear stress but is distinct from NO or PGI2. It causes hyperpolarization and vasodilation of adjacent VSMCs (Miura et al., 2003). Studies have shown that the EDHF response can depend on H2O2 (Matoba et al., 2000; Yada et al., 2008), epoxyeicosatrienoic acid (Wang et al., 2003a), endocannabinoids (Randall and Kendall, 1997), a local rise in extracellular [K+] (Edwards et al., 2001), or electromechanical coupling via gap junctions (Figueroa et al., 2006), depending on the species, conditions, and type of blood vessel.
Sainz et al. (2005) attributed the antihypertensive effects of tempol in rats with L-NAME-induced hypertension to increased EDHF activity. Likewise, the reduction in PE-induced contractions of mesenteric arteries from cholesterol-fed mice with oxidative stress by tempol also was attributed to the release of an EDHF (Kutala et al., 2006). Tempol restored the blunted EDHF-dependent vasodilation in mesenteric vessels from DOCA-salt hypertensive rats (Adeagbo et al., 2003).
Tempol may enhance EDHF by increasing the generation of H2O2 that Ghatta et al. (2007) and Chen et al. (2007a) demonstrated directly in rat aortic rings from Amplex red and luminol fluorometry, respectively. Tempol improved EDHF responses in blood vessels from several models of hypertension or oxidative stress including the coronary (Morikawa et al., 2003) and the mesenteric (Yada et al., 2008) arteries from Cu/Zn-SOD(−/−) mice in which endogenous H2O2 was severely compromised. Indeed, enhancement of EDHF responses by tempol was related to H2O2 formation because it was blocked by catalase (Yada et al., 2008).
However, it is unclear whether tempol generates functionally significant quantities of H2O2 in vivo (Kopkan et al., 2006). Moreover, some studies have dissociated H2O2-dependent relaxations to tempol from EDHF responses. Chen et al. (2007b) reported that the addition of tempol to the perfusate of rat isolated mesenteric resistance vessels precontracted with U-46,619 caused a transient dilation that was accompanied by increased H2O2. The relaxation was prevented by catalase but not by endothelium removal or by a high bath [K+]. This result related the vasodilator response to tempol to H2O2 but dissociated it from EDHF. The moderation by tempol of stretch-induced tone in aortic rings from DOCA-salt rats was prevented by catalase or SOD independent of the endothelium or of NOS. This result again identified an H2O2-dependent but endothelium-independent pathway for VSMC relaxation by tempol (Itoh et al., 2003; Ghosh et al., 2004). Presently, it is not clear how tempol generates H2O2 in VSMCs and how H2O2 elicits relaxation independent of K+ channels.
There are several pathways of interaction between NO and H2O2 that are potentiated by tempol. For example, H2O2 enhanced phosphoinositol-dependent phosphorylation of eNOS at Ser-1177 thereby increasing NOS activity (Douthwaite et al., 1999; Thomas et al., 2002) and up-regulated eNOS expression by transcriptional and post-transcriptional mechanisms (Drummond et al., 2000). Tempol prevented the reduction in calcium-stimulated NO generation by H2O2 in ECs (Douthwaite et al., 1999). Both NO and H2O2 generated in mitochondria can mediate flow-dependent dilation in blood vessels (Liu et al., 2003; Gutterman, 2005). Small mesenteric arteries from Ang II-infused rats retained a vasodilator response to ACh that was mediated both via NO generated by a coupled eNOS and via H2O2 generated by an uncoupled eNOS, because relaxation responses were blunted by NOS blockade and by catalase (Kang et al., 2007). The vasodilation of rabbit mesenteric arterioles to an NO donor was enhanced by tempol and was prevented by catalase, indicating a role for H2O2 to increase NO signaling. The effect of tempol to enhance the response to the NO donor was attributed to a reduction in the generation of •OH from H2O2 by tempol (Douthwaite et al., 1999).
These vasodilator actions of tempol that are mediated by H2O2 in vitro must be contrasted with the prohypertensive effects of H2O2 produced by infusion of tempol into the renal medulla of rats with oxidative stress (Makino et al., 2003) and with the absence of an effect of PEG-catalase on the antihypertensive response to acute administration of tempol to the SHR (Chen et al., 2007a). Presently, it is unclear whether the effects of tempol on vascular H2O2 detected ex vivo are relevant to in vivo responses.
3. Endothelium-Dependent Contracting Factor
Blood vessels from some models of hypertension, when studied under spontaneous tone, display a paradoxical constrictor response to ACh that is abolished by endothelium removal. This EDCF response occurs in human coronary arteries at sites of atherosclerosis (Lavi et al., 2008) and may contribute to coronary spasm and myocardial ischemia.
Aortic rings (Jerez et al., 2005) or renal afferent arterioles (Wang et al., 2004) from rabbits infused with Ang II have an enhanced contraction to Ang II that is mediated in part by the endothelium. These endothelium-dependent responses were diminished by incubation with indomethacin to block cyclooxygenase, by SQ-29,548 to block TP-Rs, or by tempol to reduce ROS. These findings suggest that tempol prevented the endothelial generation of vasoconstrictor prostanoids that activated TP-Rs on VSMCs of blood vessels from animals with oxidative stress.
There are several other examples of tempol moderating contractile responses that were mediated by an EDCF. ACh or a low ambient pO2 contracted blood vessels from DSS rats even when the rats were maintained on a low-salt diet. These contractions were reversed by bath addition of tempol (10−4 M) (Drenjancevic-Peric and Lombard, 2005). AT2-Rs mediated a paradoxical endothelium-dependent contractile response in mesenteric resistance vessels from aged rats that was prevented by incubation with tempol (Tatchum-Talom and Martin, 2004). Stretching of the aorta isolated from DOCA-salt rats increased the generation via an endothelium-dependent mechanism. This generation was prevented by 3 weeks of oral tempol administration (Ghosh et al., 2004).
Thus, tempol not only promotes responses mediated by EDRF/NO and EDHF but also prevents responses mediated by EDCF. The outcome should be vasodilation and a fall in BP, but it is not possible to study directly the role of the endothelium in hypertension. Indeed, caution is warranted because there are examples in which pharmacological treatment of a hypertensive model has improved the endothelial function of isolated blood vessels without a corresponding fall in BP (Tesfamariam and Ogletree, 1995).
4. Endothelin-1
Ang II (Moreau et al., 1997; An et al., 2006., 2007), 8-iso-PGF2α (Yura et al., 1999), H2O2 (Ruef et al., 2001), and a high-salt diet (Pollock and Pollock, 2001; Sasser et al., 2002) all increased ET-1 generation or release from VSMCs. ET-1 synthesis in rat cardiac fibroblasts was stimulated by ROS via an ERK pathway (Cheng et al., 2003). ET-1 generation in the rat aorta stimulated the generation of via cooperative effects of endothelin type A receptors (ET-A-Rs) and endothelin type B receptors that engaged NADPH oxidase and an uncoupled NOS (Loomis et al., 2005). Thus, ET-1 can stimulate generation in VSMCs and can itself stimulate ET-1 release. Other studies demonstrate that this feed-forward mechanism could sustain ROS production in vascular tissue and could be interrupted by tempol (Pollock, 2005).
There are several examples of tempol reducing ET-1 generation. Bath addition of tempol reduced ET-1-induced contractions of renal afferent arterioles from rabbits with oxidative stress (Wang et al., 2003b). Tempol (10−5 M), 4,5-dihydroxy-1,3-benzene disulfonic acid (tiron), diphenyleneiodonium, apocynin, and SOD all prevented the effect of Ang II to increase preproendothelin-1 mRNA and ET-1 release from vascular adventitial fibroblasts (An et al., 2007). A 2-month administration of L-NAME to rats increased cardiac ROS and mRNA for preproendothelin-1 (Bell et al., 2007). The coadministration of tempol (200 μmol · kg−1 · day−1) and vitamin C prevented the ET-1 response without modifying the hypertension (Bell et al., 2007). Intravenous tempol caused dose-dependent (55–600 μmol/kg) reductions in renal venous ET-1 release after ischemia-reperfusion injury in the rat (Fujii et al., 2005). A 2-week infusion of Ang II into SD rats increased renal venous concentrations of 8-iso-PGF2α, MDA, and ET-1. These were all prevented by oral tempol (1 mmol/l) (Ortiz et al., 2001a). Rats subjected to intermittent hypoxia for 2 weeks developed oxidative stress, hypertension, and increased plasma ET-1 that were normalized by oral tempol (1 mmol/l) (Troncoso Brindeiro et al., 2007).
Tempol also blunts responses to ET-1. Thus, ET-1 increased and intracellular [Ca2+] in VSMCs of the shark. These effects were blocked by inhibition of ROS by tempol, by inhibition of NADPH oxidase by apocynin, or by inhibition of cyclic adenine diphosphate ribose cyclase with nicotinamide and Zn (Fellner and Parker, 2005). These authors concluded that tempol blocked the adenine diphosphate ribose cyclase-induced Ca2+ response initiated by ET-1. Tempol moderated the enhanced ET-1 contractions in blood vessels from several animal models of hypertension (Li et al., 2003; Wang et al., 2004, 2006a).
The ET-A-Rs of mesenteric resistance arteries from the EC-SOD(−/−) mouse were up-regulated and mediated enhanced generation and contraction to ET-1 that were normalized by PEG-SOD or tempol (Wang et al., 2006c).
5. Potassium Channels
As recently reviewed by Liu, Gutterman, Harder, and coworkers, and H2O2 both enhance BK channel activity in rat and cat cerebral arterioles (Liu and Gutterman, 2002; Gebremedhin et al., 2008). Tempol activated K+ currents in cells transfected with the gene for the BK channel (Xu et al., 2005) and activated BK channels directly in VSMCs from mesenteric arteries of rats (Xu et al., 2006). Superfusion with tempol (1–3 mmol/l for 10 min) during patch-clamp studies increased by approximately 4-fold the peak outward current (Io) through BK channels in VSMCs from control and DOCA-salt hypertensive rats. The authors concluded that tempol activated BK channels directly on VSMCs. However, some inconsistencies were apparent. Thus, tempol did not change the mean open time or the single channel conductance. The Io was not increased in VSMCs from DOCA-salt rats, which have increased BK channel expression. The expression of the β1 subunit of the BK channel is down-regulated in VSMCs from SHR and Ang II-infused rats (Amberg and Santana, 2003; Amberg et al., 2003), which are models that exhibit a strong antihypertensive response to tempol (Thakali et al., 2006). Pretreatment of SHR with iberiotoxin in a dose 3-fold higher than that required to block BK channels did not affect basal BP or the antihypertensive response to an acute intravenous dose of tempol (Chen et al., 2007a). Indeed, ROS generation by rat hypoxic cerebral VSMCs activated BK channels and this effect was actually blocked by tempol (Gebremedhin et al., 2008). Thus, the role of BK channel activation in the BP-lowering action of tempol requires further study.
Other studies have implicated tempol in the regulation of the KATP channel (Kir6.1) (Hanna et al., 2005). Superoxide enhanced KATP channel activity in guinea pig cardiac myocytes yet decreased the KATP channel opening probability in cerebral vessels (Liu and Gutterman, 2002; Gebremedhin et al., 2008). Other studies have shown that tempol can enhance relaxation responses of VSMCs via KATP channel activation. Thus, Ang II added to the aorta of rats with STZ-induced DM during blockade of AT1-Rs caused relaxation via the AT2-Rs that were augmented by 100 μM tempol. This effect of tempol was due to activation of KATP channels because it was prevented by the KATP channel antagonist glipizide (Arun et al., 2004). The KATP channel activator, cromakalim, reduced the BP of the SHR and, when given with NE to maintain the BP, cromakalim prevented approximately 40% of the reduction in BP and HR with intravenous tempol. Likewise, blockade of the KATP channel with glipizide prevented approximately 40% of the hypotensive response to tempol (Chen et al., 2007a). These studies related the BP-lowering action of acute, intravenous doses of tempol in the SHR to activation of KATP channels. Tempol may have activated KATP channels on sympathetic neurons in this preparation because the blunting of the hypotensive action of intravenous tempol by glibenclamide was diminished in SHR pretreated with the ganglion blocking drug, hexamethonium (Chen et al., 2007a). Activation of KATP channels also can generate ROS that were blocked by tempol (Hanna et al., 2005).
Thus, although tempol can activate BK channels on VSMCs, these channels do not seem to mediate the acute hypotensive response to intravenous tempol in the SHR. In contrast, the activation of KATP channels by tempol is implicated in the acute hypotensive response in this preparation. This effect of tempol to activate KATP channels may be explained by a reduction of vascular or perhaps an increase in vascular H2O2 by tempol that removes an inhibitory influence on KATP channel activity. However, this hypothesis requires further study because tempol both activates and inhibits KATP channels in different preparations.
6. Contractility
Tempol can moderate vascular contractions due to . Thus, the addition of the oxidant drug tert-butyl hydroperoxide to the perfusate of a rat kidney or mesenteric artery increased ROS and led to vasoconstriction, which were moderated by tempol (Awe et al., 2003) or the redox-cycling spin trap, nitroblue tetrazolium but not by catalase (Ghosh et al., 2002). Because nitroblue tetrazolium reduced vascular without the formation of H2O2 (Chen et al., 2007b), these observations imply that tempol can reduce vascular contractility by reducing .
There are several examples in which the addition of tempol to the bath of blood vessels from hypertensive models reduced their sensitivity and responsiveness to agonists (Tatchum-Talom and Martin, 2004). These findings have been variously ascribed to an enhancement of the effect of NO (Shastri et al., 2002), to release of an EDHF (Kutala et al., 2006), or to prevention of the generation of an EDCF (Wang et al., 2004). The enhanced contractile responses of mesenteric vascular beds isolated from 40-week-old (aged) SD rats to NE and 5-hydroxytryptamine were normalized by 3 weeks of oral tempol (1 mmol/l) (Tatchum-Talom and Martin, 2004). Moreover, the addition of tempol to the bath of blood vessels from rats or mice with oxidative stress moderated the contractions to Ang II (Shastri et al., 2002; Wang et al., 2003b, 2004b, 2006a; Hussain et al., 2006), ET-1 (Wang et al., 2003b, 2004, 2006a), U-46,619 (Schnackenberg et al., 2000; Wang et al., 2003b, 2004), arginine vasopressin (Faraci et al., 2006), serotonin (Tatchum-Talom and Martin, 2004), or PE (Wang et al., 2006b) but generally did not moderate the contractions to NE (Wang et al., 2003b, 2004, 2006a,Wang et al., b). The discordant effects of NE were ascribed to its activation of β1 adrenoceptors on VSMCs that prevented vascular generation and thereby prevented a response to tempol (Wang et al., 2006b).
7. Cyclooxygenase, Vasoconstrictor Prostaglandins, and Thromboxanes
The addition of nitroxides to cells cultured with pro-oxidants increased prostaglandin (PG) synthesis, perhaps by increasing production of peroxide that is required for cyclooxygenase activity (Taylor et al., 1983; Smith and Marnett, 1991; Smith et al., 1996). Superoxide activated PKC and increased the expression of cyclooxygenase-2 (COX-2) (Cosentino et al., 2003; Kiritoshi et al., 2003; Li et al., 2005a) via the formation of peroxynitrate (Eligini et al., 2001; Chen et al., 2006). Administration of tempol to rats with STZ-induced DM moderated the increased expression of COX-2 in their kidneys (Li et al., 2005a; Chen et al., 2006).
Cyclopentane isoprostanes are generated nonenzymically by interaction of with arachidonate. Tempol reduced the excretion of 8-iso-PGF2α in the SHR (Schnackenberg and Wilcox, 1999) and many rat models of oxidative stress (Ortiz et al., 2001a; Welch et al., 2003, 2005a). Tempol inhibited the production of 8-iso-PGF2α in RAW264.7 macrophage cells, in which it prevented the activation of NF-κB, iNOS, and the generation of NO from iNOS (Musiek et al., 2005). Isoprostones are agonists at TP-Rs (Wang et al., 2004). Prolonged infusions of Ang II into rats or mice increased renal excretion of thromboxane B2 (Luft et al., 1989) and 8-iso-PGF2α (Kawada et al., 2002). Similar infusions of Ang II into rabbits increased the mRNA for COX-2 but not COX-1 in renal afferent arterioles, which had enhanced contractions to the TP-R agonist U-46,619 that were prevented by bath addition of 10−4 M tempol (Wang et al., 2004). Thus, tempol can moderate vascular contractility in oxidative stress by reducing TP-R signaling.
Lipoxygenase-1 requires ROS for activity. It was inhibited by tempol (Jang et al., 2007).
Rat cerebral VSMCs exposed to hypoxia developed oxidative stress, a reduction in 20-hydroxyeicosatetraenoic acid (20-HETE) formation and increased BK channel currents. Both tempol and 20-HETE blocked the activation of BK channels (Gebremedhin et al., 2008). It is possible that tempol may reduce the formation of 20-HETE because blocking 20-HETE generation prevented the antihypertensive response to tempol in the DSS rat (Hoagland et al., 2003).
COX-2 is up-regulated in the blood vessels (Wang et al., 2004), glomeruli (Jaimes et al., 2005), and kidney cortex of several models of hypertension and vascular disease, including the RRM model of CKD (Wang et al., 1998), the 2K,1C model of renal artery stenosis (Mann et al., 2001), models of DM (Komers et al., 2001; Chen et al., 2006), SHRSP (Suganami et al., 2003), DSS rats (Jaimes et al., 2008), and rats with slow pressor infusions of Ang II (Wang et al., 2003b, 2004; Jaimes et al., 2005). COX-1 products maintain hypertension in early 2K,1C hypertensive rats, but this enzyme is expressed constitutively (Welch et al., 2007).
Thromboxane A2, prostaglandin endoperoxide, and isoprostanes are vasoconstrictor PGs that activate TP-Rs. The TP-R has been implicated in the vasoconstriction and the hypertension of Ang II-infused and Goldblatt hypertensive models of hypertension and oxidative stress (Lin et al., 1991; Wilcox and Lin, 1993; Welch et al., 2007). COX requires peroxide for full activity. Thus, H2O2 may contribute to the increased generation of COX-1 and -2 products activating TP-Rs in models of oxidative stress. ROS also can increase the expression of COX-2 (Smith and Marnett, 1991).
Several studies have shown that up-regulation of COX-2 during high salt intake is dependent on ROS and can be prevented by tempol. DSS rats fed salt had oxidative stress, up-regulation of renal cortical COX-2, and increased PGE2 excretion that were prevented by the administration of candesartan or tempol (Jaimes et al., 2008). The microsomal fraction of aortas from DOCA-salt hypertensive rats produced excessive ROS when stimulated by arachidonate (Adeagbo et al., 2003). This effect was mediated by COX-2 products and was prevented by 3 weeks of tempol administration (90 μmol/kg i.p.) (Adeagbo et al., 2003). Exposure of mouse CD cells to a hypertonic NaCl solution increased the phosphorylation of ERK1/2 and p38 within 20 min and, after 16 h, increased COX-2 expression by 6-fold. These increases were accompanied by increased PGE2 release (Yang et al., 2005). Coincubation with 2 mM TEMPO reduced the levels of COX-2 by 80%. This is an interesting model because these effects of TEMPO to reduce COX-2 were shown to depend on a reduction in ROS generated from mitochondria, rather than from NADPH oxidase.
Tempol not only reduces PG generation but also reduces the response to TP-R agonists. Thus, tempol moderated the contractions of rabbit renal afferent arterioles to prolonged stimulation with a stable TP-R agonist, U-46,619 (Schnackenberg et al., 2000), and moderated EDCF responses in renal afferent arterioles from rabbits with oxidative stress. These EDCF responses were mediated by COX-2 products activating TP-Rs on VSMCs (Wang et al., 2004). Recent studies in mesenteric resistance vessels from EC-SOD(−/−) mice showed enhanced contractions to ET-1 mediated by COX-1-derived vasoconstrictor PGs activating TP-Rs that were normalized by bath addition of tempol (Wang et al., 2006c).
COX-derived PGs mediate constrictor responses in blood vessels and the kidneys of many models of hypertension and oxidative stress in contrast with the vasodilator responses that are characteristic of normal animals. Prostacyclin synthase can be nitrosated and inactivated by low concentrations of peroxynitrite (Zou et al., 1997). The recycling of the TP-R from the membrane can be interrupted by H2O2 (Valentin et al., 2004). These are mechanisms that could contribute to a resetting of PG action by ROS. Moreover, studies in the TP-R knockout mouse by Kawada et al. (2004) have shown that this receptor is required for the generation of oxidative stress and thereby a response to tempol in Ang II-infused mice. Thus, ROS generated in hypertensive models can enhance activation of TP-Rs and thereby enhance vasoconstriction and further ROS generation. This process can be interrupted by tempol, which may contribute to its moderation of vasoconstriction and ROS production in blood vessels from hypertensive animal models.
8. Comparison with Other Antioxidants
It is beyond the scope of this review to detail the activity of other antioxidants. However, a brief description of studies that have compared antioxidants with tempol is included below.
Nitroxides inhibited lipid peroxidation and protein carbamylation better than the commercial antioxidant chemicals butylated hydroxytoluene and butylated hydroxyanisole or the natural phenolic antioxidants α-hydroxytyrosol, tyrosol, caffeic acid, and α-tocopherol (Damiani et al., 2003).
Cu/Zu-SOD and catalase were not taken up into alveolar cells in culture, even over 24 h of incubation, unless they were covalently linked to PEG, which provided cellular entry and defense against oxidant damage (Walther et al., 1991). The half-time for this uptake was approximately 4 h. This relatively slow time course of cellular uptake may explain that whereas intravenous tempol reduced MAP maximally in the SHR within 3 min, PEG-SOD had no immediate effect on BP but reduced MAP over 110 min (Patel et al., 2006) and reduced oxidative stress over 1 week (Mügge et al., 1991). SOD that was encapsulated in liposomes (Laursen et al., 1997) or bonded to heparin (Nakazono et al., 1991) was also effective in lowering BP or parameters of oxidative stress when given over several days but was no more effective than native SOD in reducing BP when given acutely by intravenous injection (Patel et al., 2006).
Mn(III)tetrakis[1-methyl-4-pyridyl] porphyrin (MnT-MPyP) was the most effective agent studied for restoring nitrergic neurotransmission in the bovine retractor penis muscle during oxidative stress (Mok et al., 1998). After incubation of aortic rings with diethyldithiocarbamate (DETC) to block SOD and xanthine plus xanthine oxidase to generate , the most effective agents in restoring EDRF/NO responses were Cu(II)-[diisopropylsalicylate]2, MnTMPyP, tempol, and 4,5-dihydroxy-1,3-benzene-disulfonic acid (MacKenzie and Martin, 1998). MacKenzie and Martin (1998) concluded that metal-based antioxidants were more effective than spin traps. This conclusion may reflect the specific experimental conditions in which ROS were generated by chelation of metals with DETC (MacKenzie and Martin, 1998) and should not be generalized. Indeed, in a study of rat aorta, MnTMPyP caused graded enhancement of PE-induced contractions by destruction of NO via a paradoxical increase in generation. This effect was blocked by SOD (MacKenzie et al., 1999).
Tiron has been used widely to scavenge . However, studies in blood vessels and in solutions demonstrated that it chelated Ca2+ at concentrations well below those at which it scavenged and that this effect on Ca2+ was responsible for its vasorelaxant properties (Ghosh et al., 2002). Moreover, its vasodilator action in the rat superior mesenteric vascular bed was not perturbed by coadministration of tempol (100 μmol/l), which led to the conclusion that its biological effects may not be due to scavenging of (Ghosh et al., 2002).
MnTMPyP (Day and Crapo, 1996; Mollace et al., 2003) and EUK-134 (Baudry et al., 1993; Sharpe et al., 2002) are SOD mimetics with catalase-like activity. They had efficacy similar to that of tempol in protection against oxidative stress induced by the redox-cycling quinolone paraquat (Samai et al., 2007).
Kruglov et al. (2008) compared the efficacy of antioxidants in preventing the generation of in permeabilized mitochondrial membranes. Tempo was almost as effective as SOD and 8-fold more effective than a triphenylphosphonium-linked TEMPO compound termed mitoTEMPO that was designed to partition into mitochondria. 2,2,5,7,8-Pentamethyl-6-chromanol and 2,6-di-tert-butyl-4-methylphenol, two phenolic antioxidants, and α-tocopherol (vitamin E) were almost ineffective. Luo et al. (2007) reported preliminary results from a comparative study of the effectiveness and sensitivity of 11 drugs in extinguishing (detected by lucigenin-enhanced chemiluminescence) generated by Ang II stimulation of SHR preglomerular VSMCs (Luo et al., 2007). The catalytic antioxidants, SOD, PEG-SOD, and tempol, were the most effective followed by N-acetylcysteine (NAC), Mn(III)tetrakis(4-benzoic acid)porphyrin, epicachin, nitroblue tetrazolium, and ebselen. Vitamins C or E or trilox (soluble form of vitamin E) were almost ineffective. Nitroblue tetrazolium and N-acetylcysteine elicited a paradoxical increase in at low concentrations. The authors concluded that cell-permeable catalytic antioxidants such as PEG-SOD or tempol are the ideal agents for cellular dismutation of .
D. Sympatholytic Actions
Tempol can interrupt the actions of the SNS at several sites. These effects are more prominent in response to acute than prolonged administration of tempol.
1. Afferent Actions
Intraperitoneal administration of tempol (300–1200 μmol/kg) to mice reduced nociceptive responses to intraplantar injections of phenol (Hacimuftuoglu et al., 2006). This result probably involved a spinal action because intrathecal injections of tempol were also highly effective. Studies in the rat by Campese and Krol (2002) disclosed an important role for renal nociceptive responses in causing hypertension. Stimulation of rat renal afferent nerves by an intrarenal injection of phenol increased NADPH oxidase activity in the hypothalamus and brainstem and increased the RSNA and the BP (Ye et al., 2006). All of these effects were abolished in rats given intracerebroventricular injections of tempol or PEG-SOD.
2. Peripheral Sympathetic Nervous System
ROS can stimulate the peripheral SNS and the release of NE (Yoshino et al., 2002). Renal ischemia for 45 min in the rat, followed by reperfusion, increased renal venous NE spillover, which was reduced by pretreatment with tempol (55–550 μmol/kg i.v.) (Fujii et al., 2005). Xu and coworkers first demonstrated that the acute fall in BP with intravenous tempol was accompanied by a sympatholytic action in normotensive (Xu et al., 2001) and DOCA-salt hypertensive rats (Xu et al., 2002, 2004). They noted a robust, dose-dependent, and immediate reduction in the BP after tempol in DOCA-salt rats despite the absence of any change in DHE-induced fluorescence in the aorta or vena cava dissected from these rats, indicating a maintained level of vascular (Xu et al., 2004). Because the administration of apocynin to inhibit the p47phox component of NADPH oxidase or SOD or PEG-SOD all failed to reduce the BP acutely in this model, they concluded that the BP-lowering effect of acute intravenous tempol was independent of vascular SOD-mimetic effects. The fall in BP with tempol was blocked by inhibition of the SNS but not by inhibition of NOS; thus, they concluded that the response to tempol was mediated by direct inhibition of sympathetic nerve discharge independent of NOS. However, the failure of SOD or PEG-SOD to exert an abrupt hypotensive action in this study (Xu et al., 2004) may relate to the initial retention of these large molecular weight substances within the vascular system, thus limiting their diffusion to sites around the sympathetic nerves. Consistent with this concept, Patel et al. (2006) showed that SOD and liposome-encapsulated SOD do indeed reduce the BP of anesthetized SHR to a level comparable to that produced by intravenous tempol, but whereas the effects of tempol were maximal within 1 to 3 min, the hypotensive effects of SOD and even liposomal-encapsulated SOD were delayed more than 90 min, perhaps reflecting the time for these agents to escape from the bloodstream. The failure to detect a reduction in in the blood vessels of rats shortly after tempol administration is surprising because tempol has an almost instantaneous effect to reduce in isolated blood vessels and cultured VSMCs (Schnackenberg et al., 2000; Chen et al., 2007b). Finally, recent studies have concluded that apocynin inhibits phagocytic (Nox-2-dependent) but not vascular (Nox-1-dependent) NADPH oxidases (Stolk et al., 1994; Vejrazka et al., 2005; Ximenes et al., 2007; Heumuller et al., 2008; Touyz, 2008) and so would not be anticipated to mimic the vascular effects of tempol.
Subsequent studies have confirmed that tempol given acutely inhibits the SNS. Direct application of tempol to renal sympathetic nerves reduced their activity (Shokoji et al., 2003). This was a manifestation of SOD-mimetic activity because inhibition of SOD activity by local application of DETC to renal nerves increased their spontaneous traffic, which was reversed by local application of tempol (Shokoji et al., 2004). Because blockade of voltage-gated potassium channels by local application of 4-aminopyridine prevented the increase in renal nerve activity induced by DETC, the authors suggested that tempol activated these channels, but this effect was not studied directly (Shokoji et al., 2004).
3. Baroreflex Inhibition
Inhibition of sympathetic nerves probably underlies the paradoxical slowing of the HR with acute intravenous administration of tempol despite a sharp fall in the BP that should engage a baroreflex activation of the SNS. The observation by Shokoji et al. (2003) that the acute reduction in HR with tempol was less prominent in normotensive WKY than in hypertensive SHR suggests further that this sympatholytic action of tempol is enhanced under conditions of hypertension and that the degree of oxidative stress may set the level of SNS activity. A deficiency of NO within the rostroventrolateral medulla (RVLM) has been implicated in baroreceptor dysfunction (Mayorov, 2005). However, although blockade of nNOS in the RVLM of conscious rabbits reduced sympathetic baroreflex transmission, this was unaffected by local microinjection of tempol (Mayorov, 2005). Thus, the baroreflex inhibition that accompanies intravenous tempol probably relates to its established actions to reduce the peripheral SNS discharge or the central sympathetic drive rather than to resetting of the baroreflex itself.
4. Central Actions
Although some studies have established that tempol can reduce the activity of the SNS by direct effects on postganglionic sympathetic nerves, others have documented additional central actions to reduce the SNS activity (Tables 1 and 2, Studies in normotensive or hypertensive rats with intracerebroventricular tempol). Thus, central infusions of Ang II increased the MAP only in male mice that had evidence of greater ROS in the brain (Xue et al., 2007). This effect of Ang II was prevented by the central administration of tempol (Xue et al., 2007). Infusions of tempol (20 or 40 μmol) into the lateral ventricle of DSS or Dahl salt-resistant rats reduced the BP, SNS activity, and HR (Fujita et al., 2007). Salt-sensitive rats fed salt had enhanced hypothalamic NADPH oxidase activity, enhanced hypothalamic mRNA expression of p22phox, p47phox, and Nox-2, and an enhanced response to central tempol (Fujita et al., 2007). The central administration of tempol at doses greater than 5 μmol/kg caused dose-dependent reductions in BP, HR, and SNS discharge in baroreceptor-denervated, anesthetized rats (Lu et al., 2004). Campese et al. (2004) infused approximately 4 μmol of tempol over 1 h into the lateral cerebral ventricle of SD rats. This infusion reduced MAP, HR, RSNA, and hypothalamic NE secretion, thereby demonstrating a central action of tempol to inhibit the SNS in normotensive rats. The authors contrasted this finding with the effects of intravenous infusions of a high dose of 20 μ mol of tempol over 1 h, which also decreased MAP but increased HR, hypothalamic NE secretion, and RSNA. Although sinoaortic denervation and cervical vagotomy blunted the effects of intravenous tempol to reduce hypothalamic NE secretion, the reduction in BP after intracerebroventricular. administration of tempol remained largely intact and was accompanied by an increase in HR. The authors concluded that tempol had contrasting actions: central effects to reduce SNS discharge and BP and peripheral effects to reduce the BP but reflexly activate the SNS. It is unclear why acute intravenous administration of tempol in this study increased SNS discharge and HR in contrast to the previously discussed examples in which the acute intravenous administration of tempol elicited the opposite effects.
The RVLM and the paraventricular nucleus (PVN) are important brainstem sites for regulation of the SNS. Both are responsive to local tempol microinjection. Microinjection of tempol into the RVLM attenuated pressor responses to local Ang II and attenuated the accompanying phosphorylation of ERK1 and 2 but not the phosphorylation of stress-activated protein kinase/Jun N-terminal kinase (Chan et al., 2005). Infusions of tempol (10–100 pmol) over 1 min into the RVLM of SHRSP caused graded decreases in MAP and HR (Kishi et al., 2004). Consistent with this result was the observation by Kimura et al. (2005b) that overexpression of Mn-SOD in the RVLM of SHRSP decreased MAP and SNS activity and that central infusions of tempol at 0.5 μmol/h for 1 week attenuated the hypertension in rats with cerebral oxidative stress induced by overexpression of iNOS in the RVLM. Microinjection of tempol (200 nmol) or tiron (10 nmol) into the PVN of anesthetized rats blocked the reflex increase in RSNA after epicardial bradykinin injection and blocked the increase in RSNA and MAP accompanying injection of Ang II into the PVN (Han et al., 2005).
Ang II acting on AT1 receptors has important effects within the brain to activate the SNS and raise BP, which are targets for tempol. Zimmerman et al. (2004) reported that injection of an adenovirus expressing Cu/Zn-SOD into the subfornical organ of the hindbrain of rats blunted the rise in MAP produced by a 2-week infusion of Ang II at a slow pressor rate. Ang II increased the rate of firing of neuronal cells cultured from the hypothalamus and brainstem by inhibiting the delayed rectifier potassium current (IKV) (Sun et al., 2005). There were accompanying increases in neuronal cell ROS and NADPH oxidase activity that were blocked by gp91ds-tat, which inhibits NADPH oxidase/Nox-2, or by tempol (Sun et al., 2005). Because Ang II activates NADPH oxidase (Chabrashvili et al., 2003; Wang et al., 2004), it is likely that increased formation by Ang II caused the increase in SNS activity. Indeed, an intracerebroventricular injection of a relatively large dose of tempol (75 μ mol/kg) prevented the increase in SNS discharge and BP after intracerebroventricular Ang II (Lu et al., 2004). These central effects of tempol reduced brain levels of markers of ROS and were specific for Ang II because intracerebroventricular tempol did not prevent the increased SNS discharge after acute heat stress (Lu et al., 2004). Moreover, the microinjection of tempol (20 nmol) into the rostroventral medulla of the rabbit reduced the hypertensive response to microinjection of Ang II but not glutamate (Mayorov et al., 2004) and reduced the hypertension and tachycardia with air-jet stress (De Matteo et al., 2006), whereas 3-CP, a nitroxide with little SOD-mimetic activity in vivo (Adler et al., 2003; Patel et al., 2006), was not effective.
Adrenomedullin is an endogenous antioxidant peptide. Fujita et al. (2005) reported that adrenomedullin knockout mice develop an exaggerated increase in BP and RSNA when fed a high-salt diet and infused intracerebroventricularly with hypertonic saline. These effects were prevented by intracerebroventricular tempol, which also prevented the NaCl-induced increase in brain . This result suggests that the effect of tempol to correct salt sensitivity could entail a central action (Meng et al., 2003; Kopkan and Majid, 2005; Welch et al., 2005b; Banday et al., 2007d).
In contrast to these studies that have documented the BP-lowering effects of centrally administered tempol in the rat, Patel et al. (2006) reported that whereas acute intravenous injection of tempol into anesthetized SHR elicited graded reduction in BP and HR, intracerebroventricular injections of 0.85 to 13.5 μmol/kg (up to 5% of the effective intravenous dose) had no antihypertensive effect. As a positive control, these authors showed that intracerebroventricular injections of Ang II raised the BP in this model. Likewise, Shokoji et al. (2003) reported that intracerebroventricular doses of tempol up to approximately 1.7 μmol over 1 min did not alter MAP or RSNA of anesthetized SHR or WKY. Kagiyama et al. (2000) infused tempol intracerebroventricularly at 0.55 μmol/h into 12-week-old SHR and reported no reduction in MAP over 2 weeks.
Thus, although SOD and tempol reduced the BP by central actions in many studies, these effects have been inconsistent. This inconsistency may relate to insufficient passage of tempol from the lateral ventricle to brain sites that activate the SNS in the rostroventral medulla and RVLM wherein local application of tempol has been more effective. However, it remains unclear to what extent central effects of tempol contribute to its antihypertensive action. Because prolonged infusion of tempol lowers the BP in conscious SHR without changes in catecholamines or HR (Welch et al., 2005b), it is likely that central effects on the SNS are not a prominent part of the antihypertensive response to prolonged tempol, at least in the SHR model.
E. Renal Actions
Prolonged administration of tempol has multiple effects on the kidney, which could contribute to its anti-hypertensive action.
1. Renal Hemodynamics and Autoregulation
Tempol can increase the RBF in models of oxidative stress by enhancing the renal actions of NO (Majid and Kopkan, 2007). Tempol increased tissue levels of NO in the renal medulla of rats infused with Ang II, as detected with an NO-sensitive electrode (Badzyńska et al., 2004) or by 3-amino-4-aminomethyl-2′,7′-difluorescein fluorescence studies of isolated, perfused vasa recta capillaries (Zhang et al., 2005). Zinc deficiency increased RVR in rats, perhaps because it limited the activity of Cu/Zn-SOD. Tempol led to a steep reduction in RVR in this model (Kurihara et al., 2002). However, the effect of tempol to improve renal EDRF/NO responses has been dissociated from a reduction in RVR in a model of experimental atherosclerotic renovascular disease in swine (Chade et al., 2004).
Tempol (216 μmol/kg i.v.) infused into rats during an Ang II infusion reduced the MAP, but did not change the RBF, implying that it had reduced the RVR (Nishiyama et al., 2001). This result may be more than a manifestation of an autoregulatory response to a fall in BP because, in other studies, a direct intrarenal arterial infusion of tempol into Ang II-infused rats increased the cortical, medullary, and total RBF and increased the GFR, urine flow, and Na+ excretion without changing the BP (Kopkan et al., 2006).
Tempol also can increase RBF by NO-independent means. Oral tempol normalized the increased RVR and the exaggerated increase in RVR produced by an infusion of Ang II into SHR kidneys despite the administration of L-NAME (de Richelieu et al., 2005). Indeed, the acute effect of tempol to moderate renal vasoconstriction with Ang II was enhanced after NOS inhibition perhaps because of NOS-inhibition enhanced ROS generation (Just et al., 2007). The GFR and the RBF in models of DM were increased by tempol despite blockade of NOS (Brands et al., 2004).
Tempol has produced prominent renal vasodilation in Ang II-dependent models of hypertension. Guron et al. (2006) reported a sharp reduction in RVR by intravenous tempol (200 μmol · kg−1 · h−1) in the clipped and contralateral kidneys of rats with early (3-week) 2K,1C Goldblatt hypertension. Tempol increased the GFR and the RBF of the clipped kidney, despite a fall in MAP (Guron et al., 2006). A 2-week administration of tempol (200 nmol · kg−1 · min−1 s.c.) or an ARB to early 2K,1C rats moderated hypertension. However, only tempol increased the GFR and reduced the RVR of the clipped kidney (Welch et al., 2003).
Tempol has increased blood flow to the renal medulla more than to the renal cortex in several models. Interstitial infusion of tempol (30 μmol · kg−1 · min−1) into the renal medulla increased medullary blood flow and sodium excretion by NOS-independent means (Zou et al., 2001; Chen et al., 2003). Oral tempol (1 mmol/l in the water for 4 weeks) reduced the BP and increased the medullary but not the cortical or total RBF of fructose-fed rats (Onuma and Nakanishi, 2004). Oral tempol (1 mmol/l over 4 days or 7 weeks) reduced the MAP of SHR by 20 mm Hg yet increased the medullary blood flow by 35 to 50% without changing cortical blood flow or total RBF (Feng et al., 2001). Thus, tempol produces pronounced vasodilation of medullary blood vessels in the rat (Feng et al., 2001; Onuma and Nakanishi, 2004). An increase in medullary blood flow in hypertensive models can contribute to natriuresis and a reduction in BP (Cowley et al., 2003).
An intrarenal infusion of tempol increased RBF in denervated kidneys from salt-depleted dogs (Dutta et al., 2006). This result indicates that although tempol can reduce RSNA (Xu et al., 2002, 2004), a reduction in RSNA is not required for tempol to reduce RVR.
Tempol given over 2 weeks to SHR, DSS, or Ang II-infused rats increased the GFR (Schnackenberg and Wilcox, 1999; Hoagland et al., 2003; Just et al., 2007). Presently, there are no studies of the effects of tempol on glomerular capillary pressure to assess the hemodynamic mechanism of this effect.
Impaired renal autoregulation and glomerular hypertension in CKD predisposes to progressive kidney damage (Kotchen et al., 2000; Bidani and Griffin, 2004). Renal damage is accompanied by an increase in the circulating levels and glomerular expression of transforming growth factor β (Sharma et al., 2005). Infusion of transforming growth factor β into rats increased ROS generation in renal blood vessels and prevented afferent arteriolar constrictor responses to increased renal perfusion pressure (autoregulation). Tempol or apocynin enhanced autoregulation in this model (Sharma et al., 2005). This finding is interesting because tempol impaired renal afferent arteriolar contractions to agonist drugs in several models of oxidative stress (Wang et al., 2003b, 2004) and impaired vasoconstriction during activation of the tubuloglomerular feedback (TGF) response (Welch and Wilcox, 2001) yet enhanced myogenic contractions to increased stretch in this model. On the other hand, oral tempol (1 mmol/l) given to SHR over 4 days to 7 weeks did not alter autoregulation (Feng et al., 2001) but blocked the enhanced autoregulation of RBF produced by Ang II in isolated perfused kidneys (Guan et al., 2003). Reports from studies in which tempol preserved or improved RBF, despite a fall in BP, are consistent with the conclusion that tempol preserved or enhanced renal autoregulation (Kawada et al., 2002; Welch et al., 2005b).
2. Afferent Arteriole and Tubuloglomerular Feedback Response
Schnackenberg et al. (2000) reported that isolated, perfused renal afferent arterioles dissected from normal rabbits developed strong contractions when incubated for 20 to 30 min with the TP-R-mimetic U-46,619. These contractions were moderated by the addition of 10−3 M tempol to the bath. The addition of tempol to the bath had no effect on the immediate contractions to Ang II or U-46,619 but moderated the contractions of arterioles incubated with these agonists for 20 to 30 min (Wang et al., 2003b). Chen et al. (2007b) demonstrated that it takes some minutes for Ang II or U-46,619 to increase in rat mesenteric resistance vessels ex vivo or mouse cremasteric vessels in vivo, which may explain why normal vessels have to be incubated with Ang II or U-46,619 for some time before tempol becomes effective in moderating contractile responses. In contrast to these effects in vessels from normal animals, Wang et al. (2003b, 2004) have shown that tempol moderated the immediate contractions produced by Ang II, U-46,619, and ET-1 in afferent arterioles isolated from rabbits infused with slow pressor doses of Ang II for 2 weeks. The Ang II infusion had up-regulated the expression of p22phox in the afferent arterioles (Wang et al., 2004) and the kidneys (Chabrashvili et al., 2003) and increased the renal cortical NADPH oxidase activity (Wang et al., 2003b). Apparently, the slow pressor infusion of Ang II had induced the machinery for generation in the renal afferent arteriole, thereby creating the conditions for an abrupt increase in ROS when agonists were added to these arterioles, which now became responsive to the moderating effects of tempol.
The replacement of an EDRF/NO response in normal mesenteric and renal afferent arterioles (Wang et al., 2003a, 2006a) by an EDCF response in vessels from Ang II-infused rodents (Wang et al., 2003b, 2006a) enhanced their contractility to Ang II, ET-1, and TP-R activation. These effects were moderated by bath addition of tempol. Likewise, Ozawa et al. (2004) reported impaired EDRF/NO and EDHF responses of renal afferent arterioles from DSS rats fed a high-salt diet that were restored by oral tempol over 10 weeks or after acute bath addition of tempol to the vessel.
Guyton’s model of body fluid and BP homeostasis predicts that the level of BP is sensed in the kidneys wherein appropriate changes in salt and fluid excretion stabilize the pressure despite perturbations caused by vasoconstriction or salt intake (Guyton et al., 1995). The pressure sensed within the kidney must represent the integrated effects of the perfusion pressure and the preglomerular tone that regulates the transmission of this pressure into the kidney. Thus, a reduction in renal afferent arteriolar vasoconstriction by tempol in hypertensive models should permit better transmission of pressure into the kidneys and thereby restore a normotensive set point for the regulation of BP, which should lead to a lowering of BP. However, set against this result, is the finding that tempol can restore renal autoregulation in some models of hypertension (Sharma et al., 2005). This ability to enhance afferent arteriolar vasoconstriction during increased perfusion pressure should limit the transmission of the arterial pressure into the kidneys. A study of the effects of tempol on glomerular capillary pressure at different levels of perfusion pressure would be helpful in resolving these apparent contradictions.
NaCl delivery and reabsorption at the macula densa segment elicits an increase in renal afferent arteriolar tone mediated by the TGF response. The same signal also inhibits renin secretion. nNOS is heavily expressed in the macula densa cells. Generation of NO by nNOS in macula densa cells blunted TGF responses during NaCl reabsorption (Wilcox et al., 1992). Tempol (10−4 M) dampened TGF responses when perfused through the loop of Henle of normotensive Sprague-Dawley rats (Wilcox and Welch, 2000) and especially SHR (Welch and Wilcox, 2001) in which NADPH oxidase components were overexpressed in macula densa cells (Chabrashvili et al., 2002). Welch and Wilcox (2001) demonstrated further that the local microperfusion of tempol into the interstitium of the juxtaglomerular apparatus of the SHR blunted the TGF responses in adjacent nephrons. This effect was attributed to a restoration by tempol of NO signaling in the juxtaglomerular apparatus because a local intersitial infusion of tempol to this region restored the enhanced TGF response to microperfusion of the neuronal NOS inhibitor, 7-nitroindazole, into the macula densa segment, implying that tempol had restored the blunting of the TGF response by NO derived from nNOS. Further experiments were conducted in SHR given the ARB, candesartan, or equally antihypertensive therapy with hydralazine, hydrochlorothiazide, and reserpine for 2 weeks. Only candesartan prevented a TGF response to tempol microperfused into the interstitium and restored a TGF response to 7-nitroindazole microperfused into the macula densa segment (Welch and Wilcox, 2001). The authors concluded that tempol had reversed oxidative stress and restored local NO signaling in the juxtaglomerular apparatus of the SHR and that the oxidative stress was caused by prolonged AT1-receptor activation. This ability of tempol to blunt TGF responses was confirmed by Ichihara et al. (2001) in the juxtamedullary nephron preparation.
Microperfusion of tempol (10−4 M) via the tubular lumen of the macula densa segment or the addition of tempol to the bath of a perfused juxtaglomerular apparatus dissected from rabbit kidneys blunted the TGF responses (Ren et al., 2002). These effects of tempol were ascribed to actions within the macula densa cells rather than the afferent arterioles because perfusion of tempol via the lumen of the afferent arteriole was not effective. Furthermore, they were ascribed to an action of tempol to reduce within the macula densa cells because the addition of the impermeable SOD to the bath was not effective. Blockade of neuronal NOS in macula densa cells prevented the blunting of TGF by bath addition of tempol, thereby relating the effect of tempol to nNOS in the macula densa. The authors concluded that tempol preserved the effect of NO derived from nNOS to inhibit the luminal solute entry into macula densa cells via the Na+/K+/2Cl− transporter. NO generated within the macula densa inhibited solute transport by inactivating the Na+/K+/2Cl− transporter. Solute reabsorption by this pathway is the signal for activation of the TGF response (Ren et al., 2002). These conclusions were supported by the direct observation that tempol (10−4 M) blocked the increase in , as detected by DHE fluorescence in the macula densa segment during luminal perfusion of NaCl (Liu et al., 2007a). This finding led to an intriguing hypothesis that NaCl reabsorption by macula densa cells enhanced generation, which impaired NO bioactivity, thereby facilitating further NaCl reabsorption via the Na+/K+/2Cl− luminal transporter. Such a response would generate a strong signal to activate the TGF response that could be interrupted by tempol acting within macula densa cells. Later studies showed that tempol also can blunt TGF by moderating afferent arteriolar contractions in response to macula densa activation (Liu et al., 2004). These dual effects of tempol to moderate the TGF response via reducing the signal in the macula densa and reducing the response in the afferent arteriole could contribute to renal vasodilation and to a fall in BP.
3. Glomerulus and Podocyte
Glomerular podocytes are prominent sites for the expression of NADPH oxidase components whose expression was enhanced in the SHR (Chabrashvili et al., 2002) and a model of type I DM (Asaba et al., 2005, 2007; Tojo et al., 2007). Blockade of mineralocorticosteroid receptors with eplerenone or the administration of tempol to uninephrectomized rats infused with aldosterone and fed an 8% NaCl diet blocked the development of hypertension, proteinuria, oxidative stress, podocyte damage, and up-regulation of the aldosterone effector kinase-1 in glomerular podocytes (Shibata et al., 2007). Tempol was as effective as a mineralocorticosteroid antagonist in interrupting aldosterone signaling in podocytes. The effects of tempol on the glomerular podocytes and mesangial cells (Kwan et al., 2005) in models of oxidative stress might contribute to a reduction in proteinuria or glomerular damage.
4. Salt and Fluid Reabsorption and Excretion and Salt Sensitivity
Tempol increased Na+ excretion in rats with oxidative stress due to infusion of Ang II (López et al., 2003). Some studies have related changes in Na+ reabsorption with tempol to facilitation of NO-dependent actions. Thus, tempol blocked the effect of an intraarterial infusion of xanthine plus xanthine oxidase to increase Na+/K+-ATPase activity in the renal medulla. This effect was dependent on NOS and cGMP and was specific because tempol did not affect cortical Na+/K+-ATPase or H+/K+-ATPase (Beltowski et al., 2004).
Studies by Majid and Navar (2001) have demonstrated the NOS-independent effects of tempol. Thus, blockade of NOS actually enhanced the effect of tempol to reduce the Na+ reabsorption in the kidneys of anesthetized dogs. Tempol (3 μmol · kg−1 · min−1) infused intrarenally into anesthetized dogs did not change renal hemodynamics or fluid excretion, but after blockade of NOS tempol increased urine flow and Na+ excretion (Majid et al., 2004) and moderated the fall in Na+ excretion during Ang II infusion (Majid et al., 2005). The authors proposed that NOS blockade enhanced renal ROS generation and thereby enhanced the renal response to tempol.
On the other hand, Sainz et al. (2005) failed to detect an effect of tempol on diuresis, natriuresis, kaliuresis, proteinuria, or creatinine clearance in rats made hypertensive with L-NAME. However, a preserved rate of Na+ excretion, despite lower BP in the rats given tempol, led these authors to conclude that tempol had reset the pressure natriuresis, which contributed to the fall in BP.
Garvin, Ortiz, Stoos, and coworkers found that NO blocked luminal Na+ uptake into isolated, perfused thick ascending limb (TAL) segments of the loop of Henle (García et al., 1999; Garvin and Hong, 1999; Ortiz and Garvin, 2001; Ortiz et al., 2001b; Herrera et al., 2006) and the CD (Stoos et al., 1992, 1994, 1995). In contrast, NO can enhance Na+ and fluid reabsorption in the proximal tubule of the rat and mouse (Wang, 1997, 2000, 2002; Wang et al., 2000; Wu and Johns, 2004). However, the effects of NO on proximal reabsorption are controversial (Wilcox, 2000).
The effects of tempol on tubular reabsorption have been studied at several nephron sites. Although the effects of tempol on the proximal tubule have not been studied directly, Wu and Johns (2004) reported that luminal perfusion of SOD into the proximal tubule of the SHR increased fluid reabsorption. In contrast, Banday, Lokhandwala, Josè, and coworkers have shown that tempol restored proximal tubule dopamine D1 receptor signaling in hypertensive models. This effect was predicted to reduce proximal reabsorption via a cAMP-dependent mechanism (see section II.E.6) (Bek et al., 2001; Asghar and Lokhandwala, 2004; Banday et al., 2005; Fardoun et al., 2006; Felder and Jose, 2006; Marwaha and Lokhandwala, 2006; Yang et al., 2006; Banday et al., 2007a,b). Direct studies of proximal tubular fluid reabsorption are required to settle this controversy.
The addition of tempol (50 μmol/l) to isolated, perfused TAL segments increased the release of NO in response to L-arginine and inhibited Cl− reabsorption (Ortiz and Garvin, 2002a). This result was ascribed to a reduction in rather than to an increase in H2O2, because H2O2 did not affect Cl− reabsorption from this segment (Ortiz and Garvin, 2002b). Ortiz and Garvin (2002a) demonstrated a negative interaction between NO and on tubular reabsorption from isolated TAL segments. An increase in tubular fluid reabsorption via the Na+/K+/2Cl− luminal transporter in the perfused TAL segments increased tubular , as assessed by DHE fluorescence (Hong and Garvin, 2007). The increase in was prevented by luminal perfusion of tempol. Tempol inhibited Cl− reabsorption in the isolated perfused TAL segments of SD rats by promoting the inhibitory action of NO on luminal Na+ entry (Ortiz and Garvin, 2002a). This effect of tempol was mediated by the combined effects of blocking the luminal Na+/K+/2Cl− cotransporter (Ortiz and Garvin, 2002b; Juncos and Garvin, 2005), blocking the Na+/H+ exchange (Juncos et al., 2006), and blocking Na+/K+-ATPase (Varela et al., 2004).
Juncos et al. (2006) studied the effects of ROS on Na+/H+ exchange in isolated perfused TAL segments dissected from rat kidneys (Juncos et al., 2006). Generation of by xanthine plus xanthine oxidase doubled luminal Na+/H+ exchange but reduced basolateral Na+/H+ exchange. Tempol prevented both effects. The authors proposed that a primary effect of to reduce basolateral Na+/H+ exchange increased intracellular [H+] and thereby stimulated luminal Na+/H+ exchange and NaHCO3 absorption. Tempol inhibited this process and thereby inhibited Na+ and HCO3− reabsorption.
An increase in luminal flow or an increase in tubular Na+ absorption increased in perfused TAL segments (Garvin and Hong, 2008). These effects were prevented by luminal tempol (Garvin and Hong, 2008). These authors demonstrated an important regulatory role for ROS in the TAL. An increase in tubular fluid flow enhanced tubular reabsorption, which increased and facilitated luminal NaCl uptake and Na+/K+ ATPase activity. This process was interrupted by tempol, which therefore should prevent Na+ reabsorption during high flow states. Recently, Garvin and Hong (2008) have controlled for the effects of Na+ reabsorption to increase ROS by using a Na+-free perfusate and controlled for the effects of flow by obstructing the tubule. They demonstrated that stretch per se enhanced production in TAL segments. This was prevented by luminal tempol. The authors proposed that an increase in tubular stretch accompanying an increase in tubular fluid filtration and nephron flow that can occur with hypertension, salt loading, or DM may contribute to generation in the TAL. This generation would be anticipated to enhance tubular NaCl reabsorption and could thereby contribute to salt sensitivity and an increase in BP. Therefore, tempol can interrupt an increase in generation in the nephron whether caused by increased tubular transport, luminal flow, or stretch. This effect could be an important component of the action of tempol to prevent salt sensitivity or hypertension.
Tempol has been found to reduce the open probability of the epithelial sodium channel (ENaC) in aldosterone-stimulated distal nephron cells (Yu et al., 2007). Reduced activity of ENaC is anticipated to reduce the luminal membrane potential and to reduce tubular K+ secretion. However, this hypothesis is not consistent with the finding that 7 days of tempol administration to rats fed a low-K+ diet prevented renal generation and increased renal K+ excretion (Babilonia et al., 2005). This kaliuretic response to tempol may be related to the observation that tempol prevented the phosphorylation and inactivation of the ROMK (Kir 1.1) channel in the CDs of rats with oxidative stress (Babilonia et al., 2006). Activation of luminal ROMK channels in the CDs by tempol would be anticipated to enhance tubular K+ secretion. Further work is required to establish more clearly the effects of tempol on renal potassium excretion and K+ transport in the collecting ducts.
Thus, a prominent effect of tempol is to reduce luminal Na+ entry and thereby reduce Na+ reabsorption in the TAL and CDs. However, the anticipated increases in net Na+ and fluid excretion are not prominent with tempol. This might relate to opposite effects of tempol on reabsorption from the proximal tubule but has yet to be studied directly.
Guyton’s theory predicts that the BP will rise with salt intake (“salt-sensitivity”) if tubular NaCl reabsorption is not reduced appropriately by the high salt intake at a nephron segment at or beyond the macula densa (Guyton et al., 1995; Guyton and Coleman, 1999). Ongoing NaCl reabsorption in the distal nephron would impair the efficient elimination of the salt load and lead to increased blood volume, venous return, and cardiac output. After time, whole-body autoregulation would dictate a rise in peripheral resistance that would sustain the rise in BP during the high salt intake (Hinojosa-Laborde et al., 1992). Salt-sensitive hypertension develops if there is a failure to adjust nephron reabsorption or peripheral resistance appropriately to changes in dietary salt. A high-salt diet increased oxidative stress in the kidneys of the rat (Kitiyakara et al., 2003). This increase was accompanied by up-regulation of p47phox and Nox-1, down-regulation of Cu/Zn-SOD, and increased activity of NADPH oxidase in the kidney cortex (Kitiyakara et al., 2003). ROS have been implicated in causing salt-sensitive hypertension (Manning et al., 2003; Welch et al., 2005b). The ability of tempol to reduce luminal entry of Na+ into the TAL and to reduce ENaC activity in the CDs should combat salt sensitivity of BP. These effects have been studied in several models.
The administration of BSO to rats to deplete glutathione caused marked oxidative stress and salt-sensitive hypertension. Oral administration of tempol (1 mmol/l for 12 days) prevented the hypertension, oxidative stress, and endothelial dysfunction in this model (Banday et al., 2007d).
Tempol (50 μM) or apocynin restored both defective endothelial signaling and NO activity in mesenteric resistance vessels from rats fed a high-salt diet for 3 days. The high salt intake had provoked an increase in vascular , as assessed from DHE fluorescence (Zhu et al., 2007).
Welch et al. (2005b) reported that a high salt intake led to a greater fall in MAP during administration of tempol to SHR (Welch et al., 2005b). This was not due to a natriuretic action of tempol because the cumulative balance for Na+ and the body weight of the SHR were not perturbed. These observations imply that tempol had corrected the salt sensitivity in this model. Meng et al. (2003) reported that tempol prevented the salt-induced increase in BP in DSS rats, whereas Kopkan and Majid (2005) reported that tempol prevented the salt-induced increase in BP of rats given L-NAME. Thus, tempol corrects salt sensitivity independent of NOS, consistent with its natriuretic actions in the dog that also are independent of NOS (Majid et al., 2004, 2005; Majid and Kopkan, 2007). The observation in these studies that a high salt intake potentiated the reduction in BP with tempol contrasts with the response to all other antihypertensive agents whose effects are reduced during increases in dietary salt (Cappuccio, 2008).
5. Renin-Angiotensin-Aldosterone System
Navar, Nishiyama, and coworkers reported that 4 weeks of oral administration of tempol to DSS rats fed a high-salt diet prevented an increase in intrarenal angiotensinogen, whereas an equally antihypertensive dose of hydralazine was not effective (Kobori et al., 2003). Because intrarenal angiotensinogen correlated with the levels of Ang II in the renal tissues (Kobori et al., 2006), they concluded that tempol prevented Ang II generation in the kidneys in this salt-sensitive model. Indeed, direct measurement by Bayorh et al. (2006) have confirmed that 3 weeks of oral tempol administration to hypertensive DSS rats reduced the tissue levels of Ang II in the kidneys, but not in the heart. An effect of tempol to reduce renal tissue levels of Ang II could contribute to an NO-independent reduction in Na+ reabsorption (Majid and Nishiyama, 2002). However, Welch et al. (2005b) reported that prolonged oral administration of tempol to SHR increased the PRA. The functional significance of an increase in circulating renin-angiotensin-aldosterone components is not clear because tempol prevented many of the effects of an activated renin-angiotensin system, including the ability of Ang II to raise BP (Ortiz et al., 2001a; Kawada et al., 2002; Dikalova et al., 2005; Hattori et al., 2005; Welch et al., 2005a) and RVR (Nishiyama et al., 2001; Kawada et al., 2002; Welch et al., 2005a) and to constrict renal afferent arterioles (Wang et al., 2003b, 2004).
6. Dopamine Receptor Signaling
D1-like receptors include the dopamine type 1 and 5 receptors whose activation moderated ROS and reduced the NaCl and fluid reabsorption in the proximal tubule, reduced the RSNA, and reduced the renal expression of AT1-Rs. These effects could have contributed to a fall in BP with dopamine infusion (Hollon et al., 2002; Zeng et al., 2005; Felder and Jose, 2006; Yang et al., 2006).
Tempol prevented the down-regulation and hyper-phosphorylation of dopamine D1 receptors in the proximal tubules of rats with oxidative stress (Fardoun et al., 2006). Banday, Lokhandwala, and coworkers evaluated the effects of oral tempol (1 mmol/l for 2 weeks) in obese Zucker rats that had hypertension, hyperglycemia, and hyperinsulinemia, increased renal oxidative stress, and increased PKC activity in the proximal tubules (Banday et al., 2005), which inactivated the D1 receptor (Banday et al., 2007b). Tempol improved each of these defects, thereby restoring D1 receptor signaling and a natriuretic response to a D1 receptor agonist. These authors also reported that prolonged tempol administration to diabetic rats (Marwaha and Lokhandwala, 2006) or elderly Fischer 344 rats (Asghar and Lokhandwala, 2004, 2006) corrected renal lipid peroxidation and moderated hyperglycemia (Banday et al., 2007b). They proposed that tempol both normalized MAPK in renal proximal tubules and prevented D1-R inactivation, thereby restoring D1-R G-protein coupling and signaling via adenylate cyclase. These restorative effects of tempol on D1-R signaling in the proximal tubule are predicted to reduce proximal reabsorption of NaCl and fluid and moderate hypertension (Bek et al., 2001).
DR signaling has also been implicated in moderating oxidative stress. Activation of vascular D1-like receptors inhibited oxidative stress in VSMCs provoked by platelet-derived growth factor (Yasunari et al., 2000). The D5R knockout mouse had enhanced NADPH oxidase activity in proximal tubules and hypertension, both of which were reversed by tempol (Yang et al., 2006).
Thus, signaling via the D1- and D5-R in renal proximal tubules is reduced by oxidative stress and can itself reduce the generation of . Correction of D1R and D5-R signaling in the proximal tubule by tempol may contribute to an antioxidant and natriuretic action.
7. Adenosine
Adenosine generated within the kidneys during Ang II infusion enhanced renal vasoconstriction and tubular NaCl reabsorption via activation of adenosine type 1 receptors (A1-Rs) (Welch, 2002). Activation of A1-Rs constricted the renal afferent arteriole, enhanced proximal tubule Na+ and fluid reabsorption, activated the TGF response, and inhibited renin secretion (Welch, 2002). Adenosine released within the kidneys may contribute to the renal effects of ROS. Thus, generation in renal tissue homogenates increased the maximal velocity of the adenosine-generating enzyme, 5′-nucleotidase, and doubled the release of adenosine (Chen et al., 2001). Moreover, blockade of SOD with DETC caused oxidative stress and renal vasoconstriction. These effects were mediated by adenosine and prevented by tempol (Chen et al., 2001). Interestingly, the increase in tissue concentrations of adenosine in kidneys of rats given DETC or subjected to ischemia and reperfusion were blocked by infusion of tempol (30 μmol · kg−1 · min−1) (Chen et al., 2001). Long et al. (2007) reported that rats infused with Ang II for 2 weeks had an increased level of renal cortical interstitial adenosine that was corrected by coinfusion of tempol. Thus, tempol blocks renal adenosine generation during oxidative stress. Because A1-Rs enhance proximal tubular Na+ and fluid reabsorption and enhance renal vasoconstriction, a reduction in renal adenosine by tempol could contribute to natriuresis, vasodilation, and a fall in BP.
8. Renal and Systemic Oxygenation and Hypoxia-Inducible Factor
Welch and coworkers reported that a 2-week administration of tempol to rats corrected the reduced renal cortical pO2 and the reduced tubular Na+ transport per O2 used (TNa/QO2) in the kidneys of rats with oxidative stress caused by a slow pressor infusion of Ang II (Welch et al., 2005a) or in the clipped kidneys of the early 2K,1C rat model of Goldblatt hypertension (Welch et al., 2003). The administration of tempol also increased the pO2 of the renal cortex and the renal medulla of the SHR but not the WKY kidney when studied noninvasively by blood oxygen level-dependent MRI (Li et al., 2005b). The administration of bradykinin to the rat kidney increased NO generation and reduced O2 usage. This effect of bradykinin was blunted in the SHR model of oxidative stress but was restored to levels of WKY kidneys by tempol (Adler and Huang, 2002).
The mechanisms whereby tempol improves renal oxygenation have not yet been established. They entail a direct renal action because tempol suppressed agonist-stimulated O2 consumption in rat renal cortical homogenates and prevented increased O2 consumption in the renal tissues from elderly Fisher 344 rats (Adler et al., 2004). Because the proximal tubule is the major site for Na+ reabsorption and O2 usage and the changes in TNa/QO2 in the kidneys with tempol are profound, it is likely that tempol affects the energetics of proximal transport. The improvement in renal oxygenation with tempol could represent an increase in NO bioavailability because NOS blockade reduced renal cortical pO2 and the TNa/QO2 (Adler et al., 2001, 2004; Adler and Huang, 2002). Because NO competes with O2 in the mitochondrial respiratory chain, an increase in bioactive NO with tempol should reduce mitochondrial O2 usage (Wolin et al., 1999; Nisoli et al., 2007). Adler and Huang (2002) reported that bradykinin or enalaprilat administered to anesthetized rats stimulated endogenous NO and decreased renal O2 consumption. The responses to these agents were impaired in the kidneys of SHR but were restored by inhibition of NADPH oxidase with apocynin or AT1-Rs with an ARB (Adler and Huang, 2004). They suggested that activation of AT1-R in the kidneys enhanced renal cortical ROS production via NADPH oxidase, limited bioactive NO, and thereby impaired renal O2 usage. These results are consistent with the finding that the oral administration of tempol prevented the fall in renal cortical pO2 and in TNa/QO2 in the kidneys of rats infused with Ang II (Welch et al., 2005a).
The protein expression for HIF-1α in renal medullary interstitial cells was reduced by but not by •OH and was increased by tempol, PEG-SOD, or blockade of NADPH oxidase (Yang et al., 2003). Tempol increased the expression of HIF-1α, HIF-2α, and hemeoxygenase-1 in the outer medulla of diabetic kidneys (Rosenberger et al., 2008).
Thus, prominent effects of tempol in the kidney are to improve the efficiency of O2 usage and increase tissue pO2. Because prolonged hypoxia in rats increases their BP (Mazzali et al., 2003), an improvement in kidney pO2 with tempol may contribute to combating hypertension or progressive kidney disease, but this action is speculative. Despite an improvement in renal oxygenation, tempol increased HIF expression, presumably because of its ability to reduce renal generation.
III. Toxicity of Tempol
The intraperitoneal doses of tempol or tempol-H causing death of 50 to 70% of mice (LD50–70) were found to be 1.6 (Hahn et al., 1992a) and 2.0 mmol/kg, respectively (Hahn et al., 2000). A dose of 1.85 mmol/kg tempol-H given intraperitoneally was well tolerated (Hahn et al., 2000). The maximum tolerated doses of nitroxides after acute intravenous injection into the tail vein of mice were 0.25 to 1.5 mmol/kg (Matsumoto et al., 2004), which were similar to the maximum tolerated doses after intraperitoneal administration (Hahn et al., 1998). Of five nitroxides tested, the least toxic was tempol (Matsumoto et al., 2004). The toxicity of tempol-H was similar to that of tempol. These toxic or lethal doses of tempol are approximately 30-fold higher than the effective doses for reduction in BP.
The toxic manifestations of high doses of tempol entailed restlessness and seizures 10 to 20 min after intraperitoneal injections (Gallez et al., 1992; Hahn et al., 1992a). Nitroxides added to slices of guinea pig hippocampus in the concentration range of 1 to 5 mM increased the neural excitability, but tempol was less toxic than tempo or tempamine (Hahn et al., 1995).
Tempol has been reported either not to change (An and Hsie, 1993) or to decrease the frequency of gene mutations in Chinese hamster ovary cells stimulated by bleomycin (An and Hsie, 1992). Tempol and other nitroxides in concentrations up to 1 mM had no adverse effects on the growth and viability of these cells (Ankel et al., 1987). Even at concentrations of 50 mM, tempol did not induce chromosomal aberrations in cultured human peripheral blood lymphocytes (Johnstone et al., 1995). In another study, concentrations of tempol up to 100 μmol/l protected lymphocytes from metal-induced toxicity but, in the absence of metals, tempol was toxic at more than 100 to 1000 μmol/l (Lewinska et al., 2008). The nitroxides 2,2,5,5-tetramethyl–1-pyrrolidinyl-oxy-3-carboxylic acid and 2,2,6,6-tetramethyl–1-oxido-4-piperidinyl–1-succinic acid and their hydroxylamine and amine derivatives did not induce sister chromatid exchanges or mutations in Chinese hamster ovary cells (Afzal et al., 1984). The LD50 doses of these nitroxides in rats exceeded 15 mmol/kg, suggesting that they had a very low toxicity (Afzal et al., 1984).
High concentrations of nitroxides decreased the osmotic fragility of erythrocytes and caused dysmorphic changes that might predispose to hemolysis (Bieri et al., 1974). A study of 58 nitroxides in V79 cells detected no cytotoxicity at doses of 100 μmol/l (Krishna et al., 1998). The acute toxicity of nitroxides after intravenous injection in mice followed the order amino-TEMPO > tempone > tempol > carboxy-TEMPO = carbamoyl-PROXYL > carboxy-TEMPO. When given in vivo to mice, tempone is metabolized rapidly to the less toxic tempol (Kroll et al., 1999). These studies suggest that nitroxides are generally free from toxic effects except at exceptionally high concentrations.
IV. Conclusions Concerning Blood Pressure-Lowering Actions of Tempol
The available data are compatible with the hypothesis that the immediate reduction in BP with intravenous tempol is due predominantly to vasodilation that can be ascribed in part to potentiation of vascular NO by a reduction of its interaction with . However, a component of the early fall in BP is independent of NOS and is accompanied by a fall in HR. This response represents an inhibition of the afferent, peripheral, and central activation of the SNS. The reduction in peripheral SNS activity may represent local action of tempol on the neurons to activate BK or KATP channel conductances, thereby leading to hyperpolarization that decreases neural discharge, but this hypothesis requires further study. The reduction in central sympathetic drive with tempol probably entails a reduction in in the RVLM and PVN, which are brainstem nuclei that are very responsive to microinjection of tempol and that coordinate the central sympathetic drive.
The reason for the transient ability of an acute intravenous bolus of tempol to reduce BP, sympathetic tone, and HR may relate to the rapid reduction of tempol to the hydroxylamine that does not directly reduce BP. Tempol can cause a transient increase in vascular H2O2 and can activate BK and KATP channels on blood vessels and neurons that lead to hyperpolarization and thereby to vasorelaxation or reduced neural discharge.
Besides actions that promote vasodilation in models of oxidative stress, tempol also diminishes vasoconstriction by several mechanisms. Tempol diminishes the activation of AT1-Rs by Ang II, diminishes COX activity, diminishes vasoconstriction by PGs acting on TP-Rs, and diminishes the effects of ET-1 acting on ET-A-Rs and of catecholamines acting on α-adrenoceptors. Tempol prevents ET-1 release. Tempol corrects endothelial function by restoring EDRF/NO and EDHF responses and preventing EDCF responses in blood vessels from hypertensive models during agonist stimulation of the endothelium.
The constellation of acute vascular actions of tempol in hypertensive models demonstrates that it can function to potentiate NO and EDRF, as an EDHF mimetic, as an EDCF antagonist, as a potassium channel opener, and as a sympatholytic agent. This is a unique profile.
Although a bolus intravenous dose of tempol produces an abrupt fall in BP with a recovery over 10 to 15 min, an oral dose produces a gradual decline in BP that is maximal at 18 to 24 h. This probably represents the restoration of bioactive tempol nitroxide from the reduced hydroxylamine that occurs gradually in the circulation.
The effects of tempol that develop over days or weeks of administration to hypertensive models seem to be largely independent of the SNS, as indicated by the absence of any change in HR, plasma NE, or renal catecholamine excretion. The close correlation that is apparent in data from multiple studies of hypertensive rats between the reductions in BP and the reductions in systemic, renal, or vascular ROS with tempol supports the proposal that the antihypertensive response to prolonged tempol administration depends on a reduction in tissue and oxidative stress.
Proposed mechanisms for the sustained fall in BP during prolonged tempol administration include resetting of the renal pressure natriuresis mechanism, correction of salt sensitivity, an increase in the rates of Na+ and fluid excretion by NOS-dependent and -independent means, a reduction in renal adenosine release and intrarenal angiotensin II, prevention of phosphorylation and inactivation of DRs in renal proximal tubules, a reduction in NaCl reabsorption from the TAL of the loop of Henle and CDs, a reduction in the reactivity of the renal afferent arteriole to constrictor agonists and blunting of TGF responses leading to reduced RVR and better transmission of pressure into the kidneys, and improved renal usage of O2 and increased renal oxygen tension yet increased levels of HIF-1α. Presently, it is not clear which of these mechanisms is of predominant importance, but this probably varies among models.
Animal studies show that tempol is free of serious toxic effects at doses that reduce the BP. Despite these apparently beneficial effects in a wide range of animal models, tempol has yet to be developed as a drug for human hypertension.
Acknowledgments
The work from the authors’ laboratory described in this review was supported by the National Institute of Diabetes and Digestive and Kidney Diseases [Grants DK36079, DK49870, DK59274] and the National Heart, Lung, and Blood Institute [Grant HL68686] and by the George E. Schreiner Chair of Nephrology.
C.W. holds patents for the use of tempol to treat hypertension, SOD deficiency, iron toxicity, and skin ulceration and is a member of the scientific advisory board of Mitos Inc., which is developing topical tempol to prevent radiation-induced alopecia.
We thank Emily Wing Kam Chan for preparing and editing the manuscript.
Footnotes
Abbreviations: 1K,1C, one-kidney, one-clip; 20-HETE, 20-hydroxyeicosatetraenoic acid; 2K,1C, two-kidney, one-clip; 3-CP, 3-carbamoyl-PROXYL; 8-iso-PGF2α, 8-isoprostane prostaglandin F2α; A-192621, (±)-trans,trans-2-(4-n-propoxyphenyl)-4-(1,3-benzodioxol-5-yl)-1-[(2,6-dienthylphenyl) aminocarbonylmethyl]pyrrolidine-3-carboxylic acid; A1-R, adenosine type 1 receptor; ACEI, angiotensin-converting enzyme inhibitor; ACh, acetylcholine; Ang II, angiotensin II; ARB, angiotensin receptor blocker; AT1-R, angiotensin type 1 receptor; AT2-R, angiotensin type 2 receptor; BH4, tetrahydrobiopterin; BK, large-conductance, Ca2+-activated potassium; BP, blood pressure; BSO, buthionine sulfoximine; CAT-1, 4-trimethylammonium-2,2,6,6-tetramethylpiperidine-1-oxyl iodide; CD, collecting duct; CKD, chronic kidney disease; COX, cyclooxygenase; Cu/Zn-SOD, copper-zinc superoxide dismutase; D1, dopamine-1; D1R, dopamine-1 receptor; DEXA, dexamethasone; DHE, dihydroethidium; DM, diabetes mellitus; DOCA, deoxycorticosterone acetate; DR, dopamine receptor; DSS, Dahl salt-sensitive rat; EC, endothelial cell; EDCF, endothelium-dependent contracting factor; EDHF, endothelium dependent hyperpolarizing factor; EDRF, endothelium dependent relaxant factor; ENaC, epithelial sodium channel; eNOS, endothelial nitric-oxide synthase; EPR, electron paramagnetic resonance; ERK, extracellular signal regulated kinase; ET-1, endothelin-1; ET-A-R, endothelin type A receptor; ET-B, endothelin type B; EUK-134, manganese 3-methoxy-N,N′-bis(salicylidene)ethylenediamine chloride; GFR, glomerular filtration rate; gp91ds-tat, [H]RKKRRQRRR-CSTRIRRQL[NH3]; GRK, G-protein-coupled receptor kinase; H2O2, hydrogen peroxide; HIF, hypoxia inducible factor; HR, heart rate; iNOS, inducible nitric oxide synthase; JNK, c-Jun N-terminal kinase; KATP, ATP-dependent potassium; L-NAME, L-nitroarginine methyl ester; MAP, mean arterial pressure; MAPK, mitogen-activated protein kinase; MDA, malondialdehyde; Mn-SOD, manganese superoxide dismutase; MnTMPyP, Mn(III)tetrakis[1-methyl-4-pyridyl] porphyrin; MRI, magnetic resonance imaging; NE, norepinephrine; NF-κB, nuclear factor κB; nNOS, neuronal nitric-oxide synthase; NO, nitric oxide; NOS, nitric-oxide synthase; Nox-1, neutrophil oxidase-1; , superoxide anion; •OH, hydroxyl radical; ONOO−, peroxynitrite; paraquat, 1,1′-dimethyl-4,4′-bipyridinium dichloride; PE, phenylephrine; PEG, polyethylene glycol; PG, prostaglandin; PGI2, prostacyclin; PKC, protein kinase C; pO2, partial pressure of oxygen; PRA, plasma renin activity; PVN, paraventricular nucleus; RAAS, renin-angiotensin-aldosterone system; RBF, renal blood flow; Ren-2, renin-2; ROS, reactive oxygen species; RRM, reduced renal mass; RSNA, renal sympathetic nerve activity; RVLM, rostroventrolateral medulla; RVR, renal vascular resistance; SD, Sprague-Dawley; SHR, spontaneously hypertensive rat(s); SHRSP, stroke-prone spontaneously hypertensive rat; SNS, sympathetic nervous system; SOD, superoxide dismutase; SQ-29,548, 7-(3-((2-((phenylamino)carbonyl)hydrazino)methyl)-7-oxabicyclo(2.2.1)hept-2-yl)-5-heptenoic acid; STZ, streptozotocin; TAL, thick ascending limb; tempamine, 4-amino-2,2,6,6,-tetromethylpiperiodine-N-oxyl; TEMPO, 2,2,6,6,-tetramethylpiperidine-1-oxyl; tempol, 4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl; tempol-H, tempol hydroxylamine; tempone, 4-oxo-2,2,6,6-tetramethylpiperidine-N-oxyl; TGF, tubuloglomerular feedback; tiron, 4,5-dihydroxy-1,3-benzene disulfonic acid; TP-R, thromboxane-prostanoid receptor; U46,619, 9,11-dideoxy-9,11-methanoepoxy-prostaglandin F2; VSMC, vascular smooth muscle cell; WKY, Wistar-Kyoto rat(s); Y-27632, (+)-(R)-trans-4-(1-aminoethyl-N-4-pyridil)cyclohexanecarboxamide dihydrochloride.
References
- Adeagbo AS, Joshua IG, Falkner C, Matheson PJ. Tempol, an antioxidant, restores endothelium-derived hyperpolarizing factor-mediated vasodilation during hypertension. Eur J Pharmacol. 2003;481:91–100. doi: 10.1016/j.ejphar.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Adler A, Messina E, Sherman B, Wang Z, Huang H, Linke A, Hintze TH. NAD(P)H oxidase-generated superoxide anion accounts for reduced control of myocardial O2 consumption by NO in old Fischer 344 rats. Am J Physiol Heart Circ Physiol. 2003;285:H1015–H1022. doi: 10.1152/ajpheart.01047.2002. [DOI] [PubMed] [Google Scholar]
- Adler S, Huang H. Impaired regulation of renal oxygen consumption in spontaneously hypertensive rats. J Am Soc Nephrol. 2002;13:1788–1794. doi: 10.1097/01.asn.0000019781.90630.0f. [DOI] [PubMed] [Google Scholar]
- Adler S, Huang H. Oxidant stress in kidneys of spontaneously hypertensive rats involves both oxidase overexpression and loss of extracellular superoxide dismutase. Am J Physiol Renal Physiol. 2004;287:F907–F913. doi: 10.1152/ajprenal.00060.2004. [DOI] [PubMed] [Google Scholar]
- Adler S, Huang H, Loke KE, Xu X, Tada H, Laumas A, Hintze TH. Endothelial nitric oxide synthase plays an essential role in regulation of renal oxygen consumption by NO. Am J Physiol Renal Physiol. 2001;280:F838–F843. doi: 10.1152/ajprenal.2001.280.5.F838. [DOI] [PubMed] [Google Scholar]
- Adler S, Huang H, Wolin MS, Kaminski PM. Oxidant stress leads to impaired regulation of renal cortical oxygen consumption by nitric oxide in the aging kidney. J Am Soc Nephrol. 2004;15:52–60. doi: 10.1097/01.asn.0000101032.21097.c5. [DOI] [PubMed] [Google Scholar]
- Afzal V, Brasch RC, Nitecki DE, Wolff S. Nitroxyl spin label contrast enhancers for magnetic resonance imaging: studies of acute toxicity and mutagenesis. Invest Radiol. 1984;19:549–552. doi: 10.1097/00004424-198411000-00014. [DOI] [PubMed] [Google Scholar]
- Alayash AI. Hemoglobin-based blood substitutes: oxygen carriers, pressor agents, or oxidants? Nat Biotechnol. 1999;17:545–549. doi: 10.1038/9849. [DOI] [PubMed] [Google Scholar]
- Albrecht FE, Drago J, Felder RA, Printz MP, Eisner GM, Robillard JE, Sibley DR, Westphal HJ, Jose PA. Role of the D1A dopamine receptor in the pathogenesis of genetic hypertension. J Clin Invest. 1996;97:2283–2288. doi: 10.1172/JCI118670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpert E, Altman H, Totary H, Gruzman A, Barnea D, Barash V, Sasson S. 4-Hydroxy tempol-induced impairment of mitochondrial function and augmentation of glucose transport in vascular endothelial and smooth muscle cells. Biochem Pharmacol. 2004;67:1985–1995. doi: 10.1016/j.bcp.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Amberg GC, Bonev AD, Rossow CF, Nelson MT, Santana LF. Modulation of the molecular composition of large conductance, Ca2+ activated K+ channels in vascular smooth muscle during hypertension. J Clin Invest. 2003;112:717–724. doi: 10.1172/JCI18684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberg GC, Santana LF. Downregulation of the BK channel beta1 subunit in genetic hypertension. Circ Res. 2003;93:965–971. doi: 10.1161/01.RES.0000100068.43006.36. [DOI] [PubMed] [Google Scholar]
- An J, Hsie AW. Effects of an inhibitor and a mimic of superoxide dismutase on bleomycin mutagenesis in Chinese hamster ovary cells. Mutat Res. 1992;270:167–175. doi: 10.1016/0027-5107(92)90127-n. [DOI] [PubMed] [Google Scholar]
- An J, Hsie AW. Polymerase chain reaction-based deletion screening of bleomycin induced 6-thioguanine-resistant mutants in Chinese hamster ovary cells: the effects of an inhibitor and a mimic of superoxide dismutase. Mutat Res. 1993;289:215–222. doi: 10.1016/0027-5107(93)90072-n. [DOI] [PubMed] [Google Scholar]
- An SJ, Boyd R, Wang Y, Qiu X, Wang HD. Endothelin-1 expression in vascular adventitial fibroblasts. Am J Physiol Heart Circ Physiol. 2006;290:H700–H708. doi: 10.1152/ajpheart.00326.2005. [DOI] [PubMed] [Google Scholar]
- An SJ, Boyd R, Zhu M, Chapman A, Pimentel DR, Wang HD. NADPH oxidase mediates angiotensin II-induced endothelin-1 expression in vascular adventitial fibroblasts. Cardiovasc Res. 2007;75:702–709. doi: 10.1016/j.cardiores.2007.02.015. [DOI] [PubMed] [Google Scholar]
- Anastassopoulou JD, Rakintzis NT. Reaction of OH radicals with 2,2,6,6-tetramethyl-4-piperidinol-oxyl (TEMPOL) in a aqueous solution. Z Phys Chem Neu Folge. 1984;141:S53–S58. [Google Scholar]
- Ankel EG, Lai CS, Hopwood LE, Zivkovic Z. Cytotoxicity of commonly used nitroxide radical spin probes. Life Sci. 1987;40:495–498. doi: 10.1016/0024-3205(87)90116-0. [DOI] [PubMed] [Google Scholar]
- Anzai K, Ueno M, Yoshida A, Furuse M, Aung W, Nakanishi I, Moritake T, Takeshita K, Ikota N. Comparison of stable nitroxide, 3-substituted 2,2,5,5-tetramethylpyrrolidine-N-oxyls, with respect to protection from radiation, prevention of DNA damage, and distribution in mice. Free Radic Biol Med. 2006;40:1170–1178. doi: 10.1016/j.freeradbiomed.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Arrick DM, Sharpe GM, Sun H, Mayhan WG. nNOS-dependent reactivity of cerebral arterioles in type 1 diabetes. Brain Res. 2007;1184:365–371. doi: 10.1016/j.brainres.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arun KH, Kaul CL, Poduri R. Tempol augments angiotensin II-induced AT2 receptor-mediated relaxation in diabetic rat thoracic aorta. J Hypertens. 2004;22:2143–2152. doi: 10.1097/00004872-200411000-00017. [DOI] [PubMed] [Google Scholar]
- Asaba K, Tojo A, Onozato ML, Goto A, Fujita T. Double-edged action of SOD mimetic in diabetic nephropathy. J Cardiovasc Pharmacol. 2007;49:13–19. doi: 10.1097/FJC.0b013e31802b6530. [DOI] [PubMed] [Google Scholar]
- Asaba K, Tojo A, Onozato ML, Goto A, Quinn MT, Fujita T, Wilcox CS. Effects of NADPH oxidase inhibitor in diabetic nephropathy. Kidney Int. 2005;67:1890–1898. doi: 10.1111/j.1523-1755.2005.00287.x. [DOI] [PubMed] [Google Scholar]
- Asghar M, Lokhandwala MF. Antioxidant supplementation normalizes elevated protein kinase C activity in the proximal tubules of old rats. Exp Biol Med (Maywood) 2004;229:270–275. doi: 10.1177/153537020422900308. [DOI] [PubMed] [Google Scholar]
- Asghar M, Lokhandwala MF. Antioxidant tempol lowers age-related increases in insulin resistance in Fischer 344 rats. Clin Exp Hypertens. 2006;28:533–541. doi: 10.1080/10641960600798697. [DOI] [PubMed] [Google Scholar]
- Awe SO, Tsakadze NL, D’Souza SE, Adeagbo AS. tert-Butyl hydroperoxide-mediated vascular responses in DOCA-salt hypertensive rats. Vascul Pharmacol. 2003;40:51–57. doi: 10.1016/s1537-1891(02)00309-9. [DOI] [PubMed] [Google Scholar]
- Babilonia E, Li D, Wang Z, Sun P, Lin DH, Jin Y, Wang WH. Mitogen-activated protein kinases inhibit the ROMK (Kir 1.1)-like small conductance K channels in the cortical collecting duct. J Am Soc Nephrol. 2006;17:2687–2696. doi: 10.1681/ASN.2006050426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babilonia E, Wei Y, Sterling H, Kaminski P, Wolin M, Wang WH. Superoxide anions are involved in mediating the effect of low K intake on c-Src expression and renal K secretion in the cortical collecting duct. J Biol Chem. 2005;280:10790–10796. doi: 10.1074/jbc.M414610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badzyńska B, Grzelec-Mojzesowicz M, Sadowski J. Effect of exogenous angiotensin II on renal tissue nitric oxide and intrarenal circulation in anaesthetized rats. Acta Physiol Scand. 2004;182:313–318. doi: 10.1111/j.1365-201X.2004.01364.x. [DOI] [PubMed] [Google Scholar]
- Bai P, Bakondi E, Szabó E, Gergely P, Szabó C, Virág L. Partial protection by poly(ADP-ribose) polymerase inhibitors from nitroxyl-induced cytotoxicity in thymocytes. Free Radic Biol Med. 2001;31:1616–1623. doi: 10.1016/s0891-5849(01)00756-0. [DOI] [PubMed] [Google Scholar]
- Banday AA, Fazili FR, Lokhandwala MF. Oxidative stress causes renal dopamine D1 receptor dysfunction and hypertension via mechanisms that involve nuclear factor-κB and protein kinase C. J Am Soc Nephrol. 2007a;18:1446–1457. doi: 10.1681/ASN.2006121373. [DOI] [PubMed] [Google Scholar]
- Banday AA, Fazili FR, Marwaha A, Lokhandwala MF. Mitogen-activated protein kinase upregulation reduces renal D1 receptor affinity and G-protein coupling in obese rats. Kidney Int. 2007b;71:397–406. doi: 10.1038/sj.ki.5002055. [DOI] [PubMed] [Google Scholar]
- Banday AA, Lau YS, Lokhandwala MF. Oxidative stress causes renal dopamine D1 receptor dysfunction and salt-sensitive hypertension in Sprague-Dawley rats. Hypertension. 2007c;51:367–375. doi: 10.1161/HYPERTENSIONAHA.107.102111. [DOI] [PubMed] [Google Scholar]
- Banday AA, Marwaha A, Tallam LS, Lokhandwala MF. Tempol reduces oxidative stress, improves insulin sensitivity, decreases renal dopamine D1 receptor hyperphosphorylation, and restores D1 receptor-G-protein coupling and function in obese Zucker rats. Diabetes. 2005;54:2219–2226. doi: 10.2337/diabetes.54.7.2219. [DOI] [PubMed] [Google Scholar]
- Banday AA, Muhammad AB, Fazili FR, Lokhandwala M. Mechanisms of oxidative stress-induced increase in salt sensitivity and development of hypertension in Sprague-Dawley rats. Hypertension. 2007d;49:664–671. doi: 10.1161/01.HYP.0000255233.56410.20. [DOI] [PubMed] [Google Scholar]
- Baudry M, Etienne S, Bruce A, Palucki M, Jacobsen E, Malfroy B. Salen-manganese complexes are superoxide dismutase-mimics. Biochem Biophys Res Commun. 1993;192:964–968. doi: 10.1006/bbrc.1993.1509. [DOI] [PubMed] [Google Scholar]
- Bayorh MA, Mann G, Walton M, Eatman D. Effects of enalapril, tempol, and eplerenone on salt-induced hypertension in Dahl salt-sensitive rats. Clin Exp Hypertens. 2006;28:121–132. doi: 10.1080/10641960500468276. [DOI] [PubMed] [Google Scholar]
- Behringer W, Safar P, Kentner R, Wu X, Kagan VE, Radovsky A, Clark RS, Kochanek PM, Subramanian M, Tyurin VA, et al. Antioxidant tempol enhances hypothermic cerebral preservation during prolonged cardiac arrest in dogs. J Cereb Blood Flow Metab. 2002;22:105–117. doi: 10.1097/00004647-200201000-00013. [DOI] [PubMed] [Google Scholar]
- Bek MJ, Eisner GM, Felder RA, Jose PA. Dopamine receptors in hypertension. Mt Sinai J Med. 2001;68:362–369. [PubMed] [Google Scholar]
- Bell D, Zhao Y, McCoy FP, Devine AB, McDermott BJ. Differential effects of an anti-oxidant intervention on cardiomyocyte expression of adrenomedullin and intermedin and their receptor components in chronic nitric oxide deficiency. Cell Physiol Biochem. 2007;20:269–282. doi: 10.1159/000107513. [DOI] [PubMed] [Google Scholar]
- Beltowski J, Jamroz-Wiśniewska A, Borkowska E, Nazar J, Marciniak A. Antioxidant treatment normalizes renal Na+, K+-ATPase activity in leptin-treated rats. Pharmacol Rep. 2005;57:219–228. [PubMed] [Google Scholar]
- Beltowski J, Marciniak A, Jamroz-Wiśniewska A, Borkowska E. Nitric oxide-superoxide cooperation in the regulation of renal Na+, K+-ATPase. Acta Biochim Pol. 2004;51:933–942. [PubMed] [Google Scholar]
- Beswick RA, Zhang H, Marable D, Catravas JD, Hill WD, Webb RC. Long-term antioxidant administration attenuates mineralocorticoid hypertension and renal inflammatory response. Hypertension. 2001;37:781–786. doi: 10.1161/01.hyp.37.2.781. [DOI] [PubMed] [Google Scholar]
- Bidani AK, Griffin KA. Pathophysiology of hypertensive renal damage: implications for therapy. Hypertension. 2004;44:595–601. doi: 10.1161/01.HYP.0000145180.38707.84. [DOI] [PubMed] [Google Scholar]
- Bidani AK, Schwartz MM, Lewis EJ. Renal autoregulation and vulnerability to hypertensive injury in remnant kidney. Am J Physiol. 1987;252:F1003–F1010. doi: 10.1152/ajprenal.1987.252.6.F1003. [DOI] [PubMed] [Google Scholar]
- Bieri VG, Wallach DF, Lin PS. Focal erythrocyte membrane perturbations caused by nitroxide lipid analogues. Proc Natl Acad Sci U S A. 1974;71:4797–4801. doi: 10.1073/pnas.71.12.4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Rivero J, Sagredo A, Balfagón G, Ferrer M. Protein kinase C activation increases endothelial nitric oxide release in mesenteric arteries from orchidectomized rats. J Endocrinol. 2007;192:189–197. doi: 10.1677/joe.1.07079. [DOI] [PubMed] [Google Scholar]
- Bobko AA, Kirilyuk IA, Grigor’ev IA, Zweier JL, Khramtsov VV. Reversible reduction of nitroxides to hydroxylamines: roles for ascorbate and glutathione. Free Radic Biol Med. 2007;42:404–412. doi: 10.1016/j.freeradbiomed.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonini MG, Mason RP, Augusto O. The mechanism by which 4-hydroxy-2,2,6,6-tetramethylpiperidene-1-oxyl (tempol) diverts peroxynitrite decomposition from nitrating to nitrosating species. Chem Res Toxicol. 2002;15:506–511. doi: 10.1021/tx015571z. [DOI] [PubMed] [Google Scholar]
- Borisenko GG, Martin I, Zhao Q, Amoscato AA, Kagan VE. Nitroxides scavenge myeloperoxidase-catalyzed thiyl radicals in model systems and in cells. J Am Chem Soc. 2004;126:9221–9232. doi: 10.1021/ja0495157. [DOI] [PubMed] [Google Scholar]
- Branca M, Denurra T, Turrini F. Reduction of nitroxide free radical by normal and G6PD deficient red blood cells. Free Radic Biol Med. 1988;5:7–11. doi: 10.1016/0891-5849(88)90057-3. [DOI] [PubMed] [Google Scholar]
- Brands MW, Bell TD, Gibson B. Nitric oxide may prevent hypertension early in diabetes by counteracting renal actions of superoxide. Hypertension. 2004;43:57–63. doi: 10.1161/01.HYP.0000104524.25807.EE. [DOI] [PubMed] [Google Scholar]
- Buehler PW, Haney CR, Gulati A, Ma L, Hsia CJ. Polynitroxyl hemoglobin: a pharmacokinetic study of covalently bound nitroxides to hemoglobin platforms. Free Radic Biol Med. 2004;37:124–135. doi: 10.1016/j.freeradbiomed.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Buehler PW, Mehendale S, Wang H, Xie J, Ma L, Trimble CE, Hsia CJ, Gulati A. Resuscitative effects of polynitroxylated αα-cross-linked hemoglobin following severe hemorrhage in the rat. Free Radic Biol Med. 2000;29:764–774. doi: 10.1016/s0891-5849(00)00383-x. [DOI] [PubMed] [Google Scholar]
- Burlando B, Viarengo A. Ca2+ is mobilized by hydroxyl radical but not by superoxide in RTH-149 cells: the oxidative switching-on of Ca2+ signaling. Cell Calcium. 2005;38:507–513. doi: 10.1016/j.ceca.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Cai H, Griendling KK, Harrison DG. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci. 2003;24:471–478. doi: 10.1016/S0165-6147(03)00233-5. [DOI] [PubMed] [Google Scholar]
- Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- Campese VM, Krol E. Neurogenic factors in renal hypertension. Curr Hypertens Rep. 2002;4:256–260. doi: 10.1007/s11906-002-0016-3. [DOI] [PubMed] [Google Scholar]
- Campese VM, Ye S, Zhong H, Yanamadala V, Ye Z, Chiu J. Reactive oxygen species stimulate central and peripheral sympathetic nervous system activity. Am J Physiol Heart Circ Physiol. 2004;287:H695–H703. doi: 10.1152/ajpheart.00619.2003. [DOI] [PubMed] [Google Scholar]
- Cappuccio FP. Dietary salt reduction. In: Wilcox CS, editor. Therapy in Nephrology and Hypertension. 3. Saunders Elsevier; Philadelphia: 2008. [Google Scholar]
- Carroll RT, Galatsis P, Borosky S, Kopec KK, Kumar V, Althaus JS, Hall ED. 4-Hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (tempol) inhibits peroxynitrite-mediated phenol nitration. Chem Res Toxicol. 2000;13:294–300. doi: 10.1021/tx990159t. [DOI] [PubMed] [Google Scholar]
- Cash TP, Pan Y, Simon MC. Reactive oxygen species and cellular oxygen sensing. Free Radic Biol Med. 2007;43:1219–1225. doi: 10.1016/j.freeradbiomed.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabrashvili T, Kitiyakara C, Blau J, Karber A, Aslam S, Welch WJ, Wilcox CS. Effects of Ang II type 1 and 2 receptors on oxidative stress, renal NADPH oxidase, and SOD expression. Am J Physiol Regul Integr Comp Physiol. 2003;285:R117–R124. doi: 10.1152/ajpregu.00476.2002. [DOI] [PubMed] [Google Scholar]
- Chabrashvili T, Tojo A, Onozato ML, Kitiyakara C, Quinn MT, Fujita T, Welch WJ, Wilcox CS. Expression and cellular localization of classic NADPH oxidase subunits in the spontaneously hypertensive rat kidney. Hypertension. 2002;39:269–274. doi: 10.1161/hy0202.103264. [DOI] [PubMed] [Google Scholar]
- Chade AR, Krier JD, Rodriguez-Porcel M, Breen JF, McKusick MA, Lerman A, Lerman LO. Comparison of acute and chronic antioxidant interventions in experimental renovascular disease. Am J Physiol Renal Physiol. 2004;286:F1079–F1086. doi: 10.1152/ajprenal.00385.2003. [DOI] [PubMed] [Google Scholar]
- Champion D, Simatos D, Kalogianni EP, Cayot P, Le Meste M. Ascorbic acid oxidation in sucrose aqueous model systems at subzero temperatures. J Agric Food Chem. 2004;52:3399–3404. doi: 10.1021/jf035184q. [DOI] [PubMed] [Google Scholar]
- Chan SH, Hsu KS, Huang CC, Wang LL, Ou CC, Chan JY. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced pressor effect via activation of p38 mitogen-activated protein kinase in the rostral ventrolateral medulla. Circ Res. 2005;97:772–780. doi: 10.1161/01.RES.0000185804.79157.C0. [DOI] [PubMed] [Google Scholar]
- Charloux C, Paul M, Loisance D, Astier A. Inhibition of hydroxyl radical production by lactobionate, adenine, and tempol. Free Radic Biol Med. 1995;19:699–704. doi: 10.1016/0891-5849(95)00079-d. [DOI] [PubMed] [Google Scholar]
- Chateauneuf J, Lusztyk J, Ingold KU. Absolute rate constants for the reactions of some carbon-centered radicals with 2,2,6,6-tetramethylpiperidine-N-oxyl. J Org Chem. 1988;53:1629–1632. [Google Scholar]
- Chen K, Glockner JF, Morse PD, 2nd, Swartz HM. Effects of oxygen on the metabolism of nitroxide spin labels in cells. Biochemistry. 1989;28:2496–2501. doi: 10.1021/bi00432a022. [DOI] [PubMed] [Google Scholar]
- Chen X, Patel K, Connors SG, Mendonca M, Welch WJ, Wilcox CS. Acute antihypertensive action of tempol in the spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol. 2007a;293:H3246–H3253. doi: 10.1152/ajpheart.00957.2007. [DOI] [PubMed] [Google Scholar]
- Chen Y, Pearlman A, Luo Z, Wilcox CS. Hydrogen peroxide mediates a transient vasorelaxation with tempol during oxidative stress. Am J Physiol Heart Circ Physiol. 2007b;293:H2085–H2092. doi: 10.1152/ajpheart.00968.2006. [DOI] [PubMed] [Google Scholar]
- Chen YF, Cowley AW, Jr, Zou AP. Increased H2O2 counteracts the vasodilator and natriuretic effects of superoxide dismutation by tempol in renal medulla. Am J Physiol Regul Integr Comp Physiol. 2003;285:R827–R833. doi: 10.1152/ajpregu.00636.2002. [DOI] [PubMed] [Google Scholar]
- Chen YF, Li PL, Zou AP. Oxidative stress enhances the production and actions of adenosine in the kidney. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1808–R1816. doi: 10.1152/ajpregu.2001.281.6.R1808. [DOI] [PubMed] [Google Scholar]
- Chen YJ, Li J, Quilley J. Effect of inhibition of nitric oxide synthase on renal cyclooxygenase in the diabetic rat. Eur J Pharmacol. 2006;541:80–86. doi: 10.1016/j.ejphar.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Cheng CM, Hong HJ, Liu JC, Shih NL, Juan SH, Loh SH, Chan P, Chen JJ, Cheng TH. Crucial role of extracellular signal-regulated kinase pathway in reactive oxygen species-mediated endothelin-1 gene expression induced by endothelin-1 in rat cardiac fibroblasts. Mol Pharmacol. 2003;63:1002–1011. doi: 10.1124/mol.63.5.1002. [DOI] [PubMed] [Google Scholar]
- Christensen FH, Hansen T, Stankevicius E, Buus NH, Simonsen U. Elevated pressure selectively blunts flow-evoked vasodilatation in rat mesenteric small arteries. Br J Pharmacol. 2007a;150:80–87. doi: 10.1038/sj.bjp.0706965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen FH, Stankevicius E, Hansen T, Jørgensen MM, Valverde VL, Simonsen U, Buus NH. Flow- and acetylcholine-induced dilatation in small arteries from rats with renovascular hypertension—effect of tempol treatment. Eur J Pharmacol. 2007b;566:160–166. doi: 10.1016/j.ejphar.2007.03.058. [DOI] [PubMed] [Google Scholar]
- Chumakov VM, Ivanov VP, Yaguzhinskii LS, Rozantsev EG, Kalmanson AE. EPR investigation of various nitroxide free radicals in biological and artificial membranes. Mol Biol. 1972;6:188–192. [PubMed] [Google Scholar]
- Chumakov VM, Yaguzhinskii LS, Panin VI, Krinitskaya LA, Ivanov VP, Rozantsev EG, Kalmanson AE. Two types of spin-labels and their interaction with phospholipid micelles and mitochondrial enzymes. Mol Biol. 1974;8:102–106. [PubMed] [Google Scholar]
- Coppey LJ, Gellett JS, Davidson EP, Yorek MA. Preventing superoxide formation in epineurial arterioles of the sciatic nerve from diabetic rats restores endothelium-dependent vasodilation. Free Radic Res. 2003;37:33–40. doi: 10.1080/1071576021000028442. [DOI] [PubMed] [Google Scholar]
- Cosentino F, Eto M, De Paolis P, van der Loo B, Bachschmid M, Ullrich V, Kouroedov A, Delli Gatti C, Joch H, Volpe M, et al. High glucose causes upregulation of cyclooxygenase-2 and alters prostanoid profile in human endothelial cells: role of protein kinase C and reactive oxygen species. Circulation. 2003;107:1017–1023. doi: 10.1161/01.cir.0000051367.92927.07. [DOI] [PubMed] [Google Scholar]
- Couet WR, Brasch RC, Sosnovsky G, Tozer TN. Factors affecting nitroxide reduction in ascorbate solution and tissue homogenates. Magn Reson Imaging. 1985;3:83–88. doi: 10.1016/0730-725x(85)90012-8. [DOI] [PubMed] [Google Scholar]
- Cowley AW, Jr, Mori T, Mattson D, Zou AP. Role of renal NO production in the regulation of medullary blood flow. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1355–R1369. doi: 10.1152/ajpregu.00701.2002. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, McDonald MC, Mazzon E, Filipe HM, Centorrino T, Lepore V, Terranova ML, Ciccolo A, Caputi AP, Thiemermann C. Beneficial effects of tempol, a membrane-permeable radical scavenger, on the multiple organ failure induced by zymosan in the rat. Crit Care Med. 2001;29:102–111. doi: 10.1097/00003246-200101000-00022. [DOI] [PubMed] [Google Scholar]
- Dabrowska A, Jacewicz D, Lapińska A, Banecki B, Figarski A, Szkatula M, Lehman J, Krajewski J, Kubasik-Juraniec J, WoŸniak M, et al. Pivotal participation of nitrogen dioxide in L-arginine induced acute necrotizing pancreatitis: protective role of superoxide scavenger 4-OH-TEMPO. Biochem Biophys Res Commun. 2005;326:313–320. doi: 10.1016/j.bbrc.2004.11.032. [DOI] [PubMed] [Google Scholar]
- Damiani E, Astolfi P, Cionna L, Ippoliti F, Greci L. Synthesis and application of a novel sunscreen-antioxidant. Free Radic Res. 2006;40:485–494. doi: 10.1080/10715760600590065. [DOI] [PubMed] [Google Scholar]
- Damiani E, Belaid C, Carloni P, Greci L. Comparison of antioxidant activity between aromatic indolinonic nitroxides and natural and synthetic antioxidants. Free Radic Res. 2003;37:731–741. doi: 10.1080/1071576031000102169. [DOI] [PubMed] [Google Scholar]
- Damiani E, Carloni P, Biondi C, Greci L. Increased oxidative modification of albumin when illuminated in vitro in the presence of a common sunscreen ingredient: protection by nitroxide radicals. Free Radic Biol Med. 2000a;28:193–201. doi: 10.1016/s0891-5849(99)00221-x. [DOI] [PubMed] [Google Scholar]
- Damiani E, Carloni P, Stipa P, Greci L. Reactivity of an indolinonic aminoxyl with superoxide anion and hydroxyl radicals. Free Radic Res. 1999a;31:113–121. doi: 10.1080/10715769900301621. [DOI] [PubMed] [Google Scholar]
- Damiani E, Greci L, Parsons R, Knowland J. Nitroxide radicals protect DNA from damage when illuminated in vitro in the presence of dibenzoylmethane and a common sunscreen ingredient. Free Radic Biol Med. 1999b;26:809–816. doi: 10.1016/s0891-5849(98)00292-5. [DOI] [PubMed] [Google Scholar]
- Damiani E, Kalinska B, Canapa A, Canestrari S, Wozniak M, Olmo E, Greci L. The effects of nitroxide radicals on oxidative DNA damage. Free Radic Biol Med. 2000b;28:1257–1265. doi: 10.1016/s0891-5849(00)00242-2. [DOI] [PubMed] [Google Scholar]
- Damiani E, Paganga G, Greci L, Rice-Evans C. Inhibition of copper-mediated low density lipoprotein peroxidation by quinoline and indolinone nitroxide radicals. Biochem Pharmacol. 1994;48:1155–1161. doi: 10.1016/0006-2952(94)90152-x. [DOI] [PubMed] [Google Scholar]
- Day BJ, Crapo JD. A metalloporphyrin superoxide dismutase mimetic protects against paraquat-induced lung injury in vivo. Toxicol Appl Pharmacol. 1996;140:94–100. doi: 10.1006/taap.1996.0201. [DOI] [PubMed] [Google Scholar]
- De Matteo R, Head GA, Mayorov DN. Tempol in the dorsomedial hypothalamus attenuates the hypertensive response to stress in rabbits. Am J Hypertens. 2006;19:396–402. doi: 10.1016/j.amjhyper.2005.09.024. [DOI] [PubMed] [Google Scholar]
- de Richelieu LT, Sorensen CM, Holstein-Rathlou NH, Salomonsson M. NO-independent mechanism mediates tempol-induced renal vasodilation in SHR. Am J Physiol Renal Physiol. 2005;289:F1227–F1234. doi: 10.1152/ajprenal.00116.2005. [DOI] [PubMed] [Google Scholar]
- Deffner U, Schimmack W. Letter: Radiation effects on aqueous solutions of the nitroxyl free radical TMPN (2,2,6,6-tetramethyl-4-piperidinol-N-oxyl) Int J Radiat Biol Relat Stud Phys Chem Med. 1976;29:71–75. doi: 10.1080/09553007614551571. [DOI] [PubMed] [Google Scholar]
- Devaux PF, Bienvenüe A, Lauquin G, Brisson AD, Vignais PM, Vignais PV. Interaction between spin-labeled acyl-coenzyme A and the mitochondrial adenosine diphosphate carrier. Biochemistry. 1975;14:1272–1280. doi: 10.1021/bi00677a028. [DOI] [PubMed] [Google Scholar]
- Didion SP, Kinzenbaw DA, Schrader LI, Faraci FM. Heterozygous CuZn superoxide dismutase deficiency produces a vascular phenotype with aging. Hypertension. 2006;48:1072–1079. doi: 10.1161/01.HYP.0000247302.20559.3a. [DOI] [PubMed] [Google Scholar]
- Dikalov S, Grigor’ev IA, Voinov M, Bassenge E. Detection of superoxide radicals and peroxynitrite by 1-hydroxy-4-phosphonooxy-2,2,6,6-tetramethylpiperidine: quantification of extracellular superoxide radicals formation. Biochem Biophys Res Commun. 1998;248:211–215. doi: 10.1006/bbrc.1998.8936. [DOI] [PubMed] [Google Scholar]
- Dikalova A, Clempus R, Lassègue B, Cheng G, McCoy J, Dikalov S, San Martin A, Lyle A, Weber DS, Weiss D, et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112:2668–2676. doi: 10.1161/CIRCULATIONAHA.105.538934. [DOI] [PubMed] [Google Scholar]
- Dobesová Z, Kunes J, Zicha J. The altered balance between sympathetic nervous system and nitric oxide in salt hypertensive Dahl rats: ontogenetic and F2 hybrid studies. J Hypertens. 2002;20:945–955. doi: 10.1097/00004872-200205000-00030. [DOI] [PubMed] [Google Scholar]
- Dobrian AD, Schriver SD, Prewitt RL. Role of angiotensin II and free radicals in blood pressure regulation in a rat model of renal hypertension. Hypertension. 2001;38:361–366. doi: 10.1161/01.hyp.38.3.361. [DOI] [PubMed] [Google Scholar]
- Drenjancevic-Peric I, Lombard JH. Reduced angiotensin II and oxidative stress contribute to impaired vasodilation in Dahl salt-sensitive rats on low-salt diet. Hypertension. 2005;45:687–691. doi: 10.1161/01.HYP.0000154684.40599.03. [DOI] [PubMed] [Google Scholar]
- Drummond GR, Cai H, Davis ME, Ramasamy S, Harrison DG. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression by hydrogen peroxide. Circ Res. 2000;86:347–354. doi: 10.1161/01.res.86.3.347. [DOI] [PubMed] [Google Scholar]
- Dutta UK, Lane J, Roberts LJ, Majid DS. Superoxide formation and interaction with nitric oxide modulate systemic arterial pressure and renal function in salt-depleted dogs. Exp Biol Med (Maywood) 2006;231:269–276. doi: 10.1177/153537020623100305. [DOI] [PubMed] [Google Scholar]
- Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 2001;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- El-Remessy AB, Khalil IE, Matragoon S, Abou-Mohamed G, Tsai NJ, Roon P, Caldwell RB, Caldwell RW, Green K, Liou GI. Neuroprotective effect of (−)Δ9-tetrahydrocannabinol and cannabidiol in N-methyl-D-aspartate-induced retinal neurotoxicity: involvement of peroxynitrite. Am J Pathol. 2003;163:1997–2008. doi: 10.1016/s0002-9440(10)63558-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eligini S, Habib A, Lebret M, Créminon C, Lévy-Toledano S, Maclouf J. Induction of cyclo-oxygenase-2 in human endothelial cells by SIN-1 in the absence of prostaglandin production. Br J Pharmacol. 2001;133:1163–1171. doi: 10.1038/sj.bjp.0704163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmarakby AA, Loomis ED, Pollock JS, Pollock DM. NADPH oxidase inhibition attenuates oxidative stress but not hypertension produced by chronic ET-1. Hypertension. 2005;45:283–287. doi: 10.1161/01.HYP.0000153051.56460.6a. [DOI] [PubMed] [Google Scholar]
- Elmarakby AA, Williams JM, Imig JD, Pollock JS, Pollock DM. Synergistic actions of enalapril and tempol during chronic angiotensin II-induced hypertension. Vascul Pharmacol. 2007;46:144–151. doi: 10.1016/j.vph.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmedal B, de Dam MY, Mulvany MJ, Simonsen U. The superoxide dismutase mimetic, tempol, blunts right ventricular hypertrophy in chronic hypoxic rats. Br J Pharmacol. 2004;141:105–113. doi: 10.1038/sj.bjp.0705580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmerson PT, Howard-Flanders P. Sensitization of anoxic bacteria to x-rays by D-t-butyl nitroxide and analogues. Nature. 1964;204:1005–1006. doi: 10.1038/2041005a0. [DOI] [PubMed] [Google Scholar]
- Emmerson PT, Howard-Flanders P. Preferential sensitization of anoxic bacteria to x-rays by organic nitroxide-free radicals. Radiat Res. 1965;26:54–62. [PubMed] [Google Scholar]
- Eriksson UG, Brasch RC, Tozer TN. Nonenzymatic bioreduction in rat liver and kidney of nitroxyl spin labels, potential contrast agents in magnetic resonance imaging. Drug Metab Dispos. 1987;15:155–160. [PubMed] [Google Scholar]
- Faraci FM, Modrick ML, Lynch CM, Didion LA, Fegan PE, Didion SP. Selective cerebral vascular dysfunction in Mn-SOD-deficient mice. J Appl Physiol. 2006;100:2089–2093. doi: 10.1152/japplphysiol.00939.2005. [DOI] [PubMed] [Google Scholar]
- Fardoun RZ, Asghar M, Lokhandwala M. Role of oxidative stress in defective renal dopamine D1 receptor-G protein coupling and function in old Fischer 344 rats. Am J Physiol Renal Physiol. 2006;291:F945–F951. doi: 10.1152/ajprenal.00111.2006. [DOI] [PubMed] [Google Scholar]
- Felder RA, Jose PA. Mechanisms of disease: the role of GRK4 in the etiology of essential hypertension and salt sensitivity. Nat Clin Pract Nephrol. 2006;2:637–650. doi: 10.1038/ncpneph0301. [DOI] [PubMed] [Google Scholar]
- Feldman A, Wildman E, Bartolinini G, Piette LH. In vivo electron spin resonance in rats. Phys Med Biol. 1975;20:602–612. doi: 10.1088/0031-9155/20/4/007. [DOI] [PubMed] [Google Scholar]
- Fellner SK, Parker L. Endothelin-1, superoxide and adeninediphosphate ribose cyclase in shark vascular smooth muscle. J Exp Biol. 2005;208:1045–1052. doi: 10.1242/jeb.01506. [DOI] [PubMed] [Google Scholar]
- Feng MG, Dukacz SAW, Kline RL. Selective effect of tempol on renal medullary hemodynamics in spontaneously hypertensive rats. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1420–R1425. doi: 10.1152/ajpregu.2001.281.5.R1420. [DOI] [PubMed] [Google Scholar]
- Fernandes DC, Medinas DB, Alves MJ, Augusto O. Tempol diverts peroxynitrite/carbon dioxide reactivity toward albumin and cells from protein-tyrosine nitration to protein-cysteine nitrosation. Free Radic Biol Med. 2005;38:189–200. doi: 10.1016/j.freeradbiomed.2004.09.027. [DOI] [PubMed] [Google Scholar]
- Figueroa XF, Isakson BE, Duling BR. Vascular gap junctions in hypertension. Hypertension. 2006;48:804–811. doi: 10.1161/01.HYP.0000242483.03361.da. [DOI] [PubMed] [Google Scholar]
- Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- Finkelstein E, Rosen GM, Rauckman EJ. Superoxide-dependent reduction of nitroxides by thiols. Biochim Biophys Acta. 1984;802:90–98. [Google Scholar]
- Fischer AE, Carpenter TA, Tyler JA, Hall LD. Visualisation of mass transport of small organic molecules and metal ions through articular cartilage by magnetic resonance imaging. Magn Reson Imaging. 1995;13:819–826. doi: 10.1016/0730-725x(95)00040-n. [DOI] [PubMed] [Google Scholar]
- Fortepiani LA, Reckelhoff JF. Role of oxidative stress in the sex differences in blood pressure in spontaneously hypertensive rats. J Hypertens. 2005;23:801–805. doi: 10.1097/01.hjh.0000163149.05083.13. [DOI] [PubMed] [Google Scholar]
- Fortepiani LA, Zhang H, Racusen L, Roberts LJ, 2nd, Reckelhoff JF. Characterization of an animal model of postmenopausal hypertension in spontaneously hypertensive rats. Hypertension. 2003;41:640–645. doi: 10.1161/01.HYP.0000046924.94886.EF. [DOI] [PubMed] [Google Scholar]
- Fujii T, Takaoka M, Ohkita M, Matsumura Y. Tempol protects against ischemic acute renal failure by inhibiting renal noradrenaline overflow and endothelin-1 overproduction. Biol Pharm Bull. 2005;28:641–645. doi: 10.1248/bpb.28.641. [DOI] [PubMed] [Google Scholar]
- Fujita M, Ando K, Nagae A, Fujita T. Sympathoexcitation by oxidative stress in the brain mediates arterial pressure elevation in salt-sensitive hypertension. Hypertension. 2007;50:360–367. doi: 10.1161/HYPERTENSIONAHA.107.091009. [DOI] [PubMed] [Google Scholar]
- Fujita M, Kuwaki T, Ando K, Fujita T. Sympatho-inhibitory action of endogenous adrenomedullin through inhibition of oxidative stress in the brain. Hypertension. 2005;45:1165–1172. doi: 10.1161/01.HYP.0000165690.85505.37. [DOI] [PubMed] [Google Scholar]
- Gadjeva V, Kuchukova D, Tolekova A, Tanchev S. Beneficial effects of spin-labelled nitrosourea on CCNU-induced oxidative stress in rat blood compared with vitamin E. Pharmazie. 2005;60:530–532. [PubMed] [Google Scholar]
- Gallez B, Demeure R, Debuyst R, Leonard D, Dejehet F, Dumont P. Evaluation of nonionic nitroxyl lipids as potential organ-specific contrast agents for magnetic resonance imaging. Magn Reson Imaging. 1992;10:445–455. doi: 10.1016/0730-725x(92)90516-3. [DOI] [PubMed] [Google Scholar]
- Gao YJ, Hirota S, Zhang DW, Janssen LJ, Lee RM. Mechanisms of hydrogenperoxide-induced biphasic response in rat mesenteric artery. Br J Pharmacol. 2003;138:1085–1092. doi: 10.1038/sj.bjp.0705147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Lee RMKW. Hydrogen peroxide induces a greater concentration in mesenteric arteries of spontaneously hypertensive rats through thromboxane A2 production. Br J Pharmacol. 2001;134:1639–1646. doi: 10.1038/sj.bjp.0704420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García NH, Plato CF, Stoos BA, Garvin JL. Nitric oxide-induced inhibition of transport by thick ascending limbs from Dahl salt-sensitive rats. Hypertension. 1999;34:508–513. doi: 10.1161/01.hyp.34.3.508. [DOI] [PubMed] [Google Scholar]
- Garvin JL, Hong NJ. Nitric oxide inhibits sodium/hydrogen exchange activity in the thick ascending limb. Am J Physiol. 1999;277:F377–F382. doi: 10.1152/ajprenal.1999.277.3.F377. [DOI] [PubMed] [Google Scholar]
- Garvin JL, Hong NJ. Cellular stretch increases superoxide production in the thick ascending limb. Hypertension. 2008;51:488–493. doi: 10.1161/HYPERTENSIONAHA.107.102228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebremedhin D, Yamaura K, Harder DR. Role of 20-hete in the hypoxia-induced activation of Ca2+-activated K+ channel currents in rat cerebral arterial muscle cells. Am J Physiol Heart Circ Physiol. 2008;294:H107–H120. doi: 10.1152/ajpheart.01416.2006. [DOI] [PubMed] [Google Scholar]
- Ghatta S, Hemmer RB, Uppala S, O’Rourke ST. Role of endogenous hydrogen peroxide in the development of nitrate tolerance. Vascul Pharmacol. 2007;46:247–252. doi: 10.1016/j.vph.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Ghosh M, Wang HD, McNeill JR. Tiron exerts effects unrelated to its role as a scavenger of superoxide anion: effects on calcium binding and vascular responses. Can J Physiol Pharmacol. 2002;80:755–760. doi: 10.1139/y02-106. [DOI] [PubMed] [Google Scholar]
- Ghosh M, Wang HD, McNeill JR. Role of oxidative stress and nitric oxide in regulation of spontaneous tone in aorta of DOCA-salt hypertensive rats. Br J Pharmacol. 2004;141:562–573. doi: 10.1038/sj.bjp.0705557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilani M, Kaiser D, Bratteli C, Alinder C, Rajala S, Bank A, Cohn J. Role of nitric oxide deficiency and its detection as a risk factor in pre-hypertension. J Am Soc Hypertens. 2007;1:45–55. doi: 10.1016/j.jash.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Girouard H, de Champlain J. Inhibitory effect of melatonin on α1-adrenergic-induced vasoconstriction in mesenteric beds of spontaneously hypertensive rats. Am J Hypertens. 2004;17:339–346. doi: 10.1016/j.amjhyper.2003.12.013. [DOI] [PubMed] [Google Scholar]
- Glebska J, Pulaski L, Gwoździński K, Skolimowski J. Structure-activity relationship studies of protective function of nitroxides in Fenton system. Biometals. 2001;14:159–170. doi: 10.1023/a:1016689607579. [DOI] [PubMed] [Google Scholar]
- Glebska J, Skolimowski J, Kudzin Z, Gwoździński K, Grzelak A, Bartosz G. Pro-oxidative activity of nitroxides in their reactions with glutathione. Free Radic Biol Med. 2003;35:310–316. doi: 10.1016/s0891-5849(03)00306-x. [DOI] [PubMed] [Google Scholar]
- Goldstein S, Merenyi G, Russo A, Samuni A. The role of oxoammonium cation in the SOD-mimic activity of cyclic nitroxides. J Am Chem Soc. 2003;125:789–795. doi: 10.1021/ja028190w. [DOI] [PubMed] [Google Scholar]
- Grebenshchikov YuB, Likhtenshtein GI, Ivanov VP, Rozantsev EG. Investigation of electrostatic charges in proteins by the paramagnetic probe method. Mol Biol. 1972;6:400–406. [PubMed] [Google Scholar]
- Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury: Part I: basic mechanisms and in vivo. Circulation. 2003;108:1912–1916. doi: 10.1161/01.CIR.0000093660.86242.BB. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Ushio-Fukai M. Reactive oxygen species as mediators of angiotensin II signaling. Regul Pept. 2000;91:21–27. doi: 10.1016/s0167-0115(00)00136-1. [DOI] [PubMed] [Google Scholar]
- Griffin KA, Picken M, Bidani AK. Method of renal mass reduction is a critical modulator of subsequent hypertension and glomerular injury. J Am Soc Nephrol. 1994;4:2023–2031. doi: 10.1681/ASN.V4122023. [DOI] [PubMed] [Google Scholar]
- Griffin KA, Picken MM, Bidani AK. Blood pressure lability and glomerulosclerosis after normotensive 5/6 renal mass reduction in the rat. Kidney Int. 2004;65:209–218. doi: 10.1111/j.1523-1755.2004.00356.x. [DOI] [PubMed] [Google Scholar]
- Griffith OH, McConnell HM. A nitroxide-maleimide spin label. Proc Natl Acad Sci U S A. 1966;55:8–11. doi: 10.1073/pnas.55.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, Willgoss DA, Matthias A, Manley SW, Crozier S, Gobe G, Endre ZH. Facilitation of renal autoregulation by angiotensin II is mediated through modulation of nitric oxide. Acta Physiol Scand. 2003;179:189–201. doi: 10.1046/j.1365-201X.2003.01125.x. [DOI] [PubMed] [Google Scholar]
- Guo P, Nishiyama A, Rahman M, Nagai Y, Noma T, Namba T, Ishizawa M, Murakami K, Miyatake A, Kimura S, et al. Contribution of reactive oxygen species to the pathogenesis of left ventricular failure in Dahl salt-sensitive hypertensive rats: effects of angiotensin II blockade. J Hypertens. 2006;24:1097–1104. doi: 10.1097/01.hjh.0000226200.73065.5d. [DOI] [PubMed] [Google Scholar]
- Guron GS, Grimberg ES, Basu S, Herlitz H. Acute effects of the superoxide dismutase mimetic tempol on split kidney function in two-kidney one-clip hypertensive rats. J Hypertens. 2006;24:387–394. doi: 10.1097/01.hjh.0000200511.02700.99. [DOI] [PubMed] [Google Scholar]
- Gutterman DD. Mitochondria and reactive oxygen species: an evolution in function. Circ Res. 2005;97:302–304. doi: 10.1161/01.RES.0000179773.18195.12. [DOI] [PubMed] [Google Scholar]
- Guyton AC, Coleman TG. Quantitative analysis of the pathophysiology of hypertension. J Am Soc Nephrol. 1999;10:2248–2258. [PubMed] [Google Scholar]
- Guyton AC, Hall JE, Coleman TG, Manning RD, Jr, Norman RA. The dominant role of the kidneys in long-term arterial pressure regulation in normal and hypertensive states. In: Laragh JH, Brenner BM, editors. Hypertension: Pathophysiology, Diagnosis and Management. Raven Press; New York: 1995. pp. 1311–1326. [Google Scholar]
- Gwoździński K. Effect of cupric ions on the permeability of erythrocyte membrane to non-electrolyte spin labels. Physiol Chem Phys Med NMR. 1985;17:431–434. [PubMed] [Google Scholar]
- Gwoździński K. Effect of thiol reactive reagents and ionizing radiation on the permeability of erythrocyte membrane for non-electrolyte spin labels. Radiat Environ Biophys. 1986;25:107–111. doi: 10.1007/BF01211734. [DOI] [PubMed] [Google Scholar]
- Gwoździński K, Bartosz G, Leyko W. Effect of thiol reagents and ionizing radiation on the permeability of erythrocyte membrane for spin-labeled non-electrolytes. Radiat Environ Biophys. 1983;22:53–59. doi: 10.1007/BF01323760. [DOI] [PubMed] [Google Scholar]
- Hacimuftuoglu A, Handy CR, Goettl VM, Lin CG, Dane S, Stephens RL., Jr Antioxidants attenuate multiple phases of formalin-induced nociceptive response in mice. Behav Brain Res. 2006;173:211–216. doi: 10.1016/j.bbr.2006.06.030. [DOI] [PubMed] [Google Scholar]
- Hahn SM, DeLuca AM, Coffin D, Krishna CM, Mitchell JB. In vivo radioprotection and effects on blood pressure of the stable free radical nitroxides. Int J Radiat Oncol Biol Phys. 1998;42:839–842. doi: 10.1016/s0360-3016(98)00317-4. [DOI] [PubMed] [Google Scholar]
- Hahn SM, Krishna MC, DeLuca AM, Coffin D, Mitchell JB. Evaluation of the hydroxylamine Tempol-H as an in vivo radioprotector. Free Radic Biol Med. 2000;28:953–958. doi: 10.1016/s0891-5849(00)00176-3. [DOI] [PubMed] [Google Scholar]
- Hahn SM, Lepinski DL, DeLuca AM, Mitchell JB, Pellmar TC. Neurophysiological consequences of nitroxide antioxidants. Can J Physiol Pharmacol. 1995;73:399–403. doi: 10.1139/y95-051. [DOI] [PubMed] [Google Scholar]
- Hahn SM, Sullivan FJ, DeLuca AM, Bacher JD, Liebmann J, Krishna MC, Coffin D, Mitchell JB. Hemodynamic effect of the nitroxide superoxide dismutase mimics. Free Radic Biol Med. 1999;27:529–535. doi: 10.1016/s0891-5849(99)00099-4. [DOI] [PubMed] [Google Scholar]
- Hahn SM, Tochner Z, Krishna CM, Glass J, Wilson L, Samuni A, Sprague M, Venzon D, Glatstein E, Mitchell JB, et al. Tempol, a stable free radical, is a novel murine radiation protector. Cancer Research. 1992a;52:1750–1753. [PubMed] [Google Scholar]
- Hahn SM, Wilson L, Krishna CM, Liebmann J, DeGraff W, Gamson J, Samuni A, Venzon D, Mitchell JB. Identification of nitroxide radioprotectors. Radiat Res. 1992b;132:87–93. [PubMed] [Google Scholar]
- Haj-Yehia AI, Nassar T, Assaf P, Nassar H, Anggård EE. Effects of the superoxide dismutase-mimic compound TEMPOL on oxidant stress-mediated endothelial dysfunction. Antioxid Redox Signal. 1999;1:221–232. doi: 10.1089/ars.1999.1.2-221. [DOI] [PubMed] [Google Scholar]
- Han Y, Zhang Y, Wang HJ, Gao XY, Wang W, Zhu GQ. Reactive oxygen species in paraventricular nucleus modulates cardiac sympathetic afferent reflex in rats. Brain Res. 2005;1058:82–90. doi: 10.1016/j.brainres.2005.07.055. [DOI] [PubMed] [Google Scholar]
- Hanna ST, Cao K, Sun X, Wang R. Mediation of the effect of nicotine on Kir6.1 channels by superoxide anion production. J Cardiovasc Pharmacol. 2005;45:447–455. doi: 10.1097/01.fjc.0000159046.35241.4e. [DOI] [PubMed] [Google Scholar]
- Harrison D, Gongora MC, Guzik TJ, Widder J. Oxidative stress and hypertension. Hypertension. 2007;1:30–44. doi: 10.1016/j.jash.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Hasdan G, Benchetrit S, Rashid G, Green J, Bernheim J, Rathaus M. Endothelial dysfunction and hypertension in 5/6 nephrectomized rats are mediated by vascular superoxide. Kidney Int. 2002;61:586–590. doi: 10.1046/j.1523-1755.2002.00166.x. [DOI] [PubMed] [Google Scholar]
- Hattori Y, Akimoto K, Gross SS, Hattori S, Kasai K. Angiotensin-II-induced oxidative stress elicits hypoadiponectinaemia in rats. Diabetologia. 2005;46:1066–1074. doi: 10.1007/s00125-005-1766-7. [DOI] [PubMed] [Google Scholar]
- Herrera M, Ortiz PA, Garvin JL. Regulation of thick ascending limb transport: role of nitric oxide. Am J Physiol Renal Physiol. 2006;290:F1279–F1284. doi: 10.1152/ajprenal.00465.2005. [DOI] [PubMed] [Google Scholar]
- Herrling T, Fuchs J, Groth N. Kinetic measurements using EPR imaging with a modulated field gradient. J Magn Reson. 2002;154:6–14. doi: 10.1006/jmre.2001.2459. [DOI] [PubMed] [Google Scholar]
- Heumüller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schröder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- Hewett SJ, Espey MG, Uliasz TF, Wink DA. Neurotoxicity of nitroxyl: insights into HNO and NO biochemical imbalance. Free Radic Biol Med. 2005;39:1478–1488. doi: 10.1016/j.freeradbiomed.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Higashi Y, Sasaki S, Nakagawa K, Matsuura H, Oshima T, Chayama K. Endothelial function and oxidative stress in renovascular hypertension. N Engl J Med. 2002;346:1954–1962. doi: 10.1056/NEJMoa013591. [DOI] [PubMed] [Google Scholar]
- Himmelfarb J. Linking oxidative stress and inflammation in kidney disease: which is the chicken and which is the egg? Semin Dial. 2004;17:449–454. doi: 10.1111/j.0894-0959.2004.17605.x. [DOI] [PubMed] [Google Scholar]
- Himmelfarb J, Stenvinkel P, Ikizler TA, Hakim RM. The elephant in uremia: oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2002;62:1524–1538. doi: 10.1046/j.1523-1755.2002.00600.x. [DOI] [PubMed] [Google Scholar]
- Hinojosa-Laborde C, Frohlich BH, Cowley AW., Jr Whole body autoregulation in reduced renal mass hypertension. Hypertension. 1992;20:659–665. doi: 10.1161/01.hyp.20.5.659. [DOI] [PubMed] [Google Scholar]
- Hirono Y, Yoshimoto T, Suzuki N, Sugiyama T, Sakurada M, Takai S, Kobayashi N, Shichiri M, Hirata Y. Angiotensin II receptor type 1-mediated vascular oxidative stress and proinflammatory gene expressions in aldosterone-induced hypertension: the possible role of local renin-angiotensin system. Endocrinology. 2007;148:1688–1696. doi: 10.1210/en.2006-1157. [DOI] [PubMed] [Google Scholar]
- Hisaki R, Fujita H, Saito F, Kushiro T. Tempol attenuates the development of hypertensive renal injury in Dahl salt-sensitive rats. Am J Hypertens. 2005;18:707–713. doi: 10.1016/j.amjhyper.2004.11.045. [DOI] [PubMed] [Google Scholar]
- Hoagland KM, Maier KG, Roman RJ. Contributions of 20-HETE to the antihypertensive effects of Tempol in Dahl salt-sensitive rats. Hypertension. 2003;41:697–702. doi: 10.1161/01.HYP.0000047881.15426.DC. [DOI] [PubMed] [Google Scholar]
- Hollon TR, Bek MJ, Lachowicz JE, Ariano MA, Mezey E, Ramachandran R, Wersinger SR, Soares-da-Silva P, Liu ZF, Grinberg A, et al. Mice lacking D5 dopamine receptors have increased sympathetic tone and are hypertensive. J Neurosci. 2002;22:10801–10810. doi: 10.1523/JNEUROSCI.22-24-10801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong NJ, Garvin JL. Flow increases superoxide production by NADPH oxidase via activation of Na/K/2Cl cotransport and mechanical stress in thick ascending limbs. Am J Physiol Renal Physiol. 2007;292:F993–F998. doi: 10.1152/ajprenal.00383.2006. [DOI] [PubMed] [Google Scholar]
- Howard LL, Patterson ME, Mullins JJ, Mitchell KD. Salt-sensitive hypertension develops after transient induction of ANG II-dependent hypertension in Cyp1a1-Ren2 transgenic rats. Am J Physiol Renal Physiol. 2005;288:F810–F815. doi: 10.1152/ajprenal.00148.2004. [DOI] [PubMed] [Google Scholar]
- Hucks D, Thuraisingham RC, Raftery MJ, Yaqoob MM. Homocysteine induced impairment of nitric oxide-dependent vasorelaxation is reversible by the superoxide dismutase mimetic TEMPOL. Nephrol Dial Transplant. 2004;19:1999–2005. doi: 10.1093/ndt/gfh270. [DOI] [PubMed] [Google Scholar]
- Hussain MB, Puntmann VO, Mayr M, Khong T, Singer DR. The role of oxidant stress in angiotensin II-mediated contraction of human resistance arteries in the state of health and the presence of cardiovascular disease. Vascul Pharmacol. 2006;45:395–399. doi: 10.1016/j.vph.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Hyodo F, Matsumoto K, Matsumoto A, Mitchell JB, Krishna MC. Probing the intracellular redox status of tumors with magnetic resonance imaging and redox-sensitive contrast agents. Cancer Res. 2006;66:9921–9928. doi: 10.1158/0008-5472.CAN-06-0879. [DOI] [PubMed] [Google Scholar]
- Iannone A, Bini A, Swartz HM, Tomasi A, Vannini V. Metabolism in rat liver microsomes of the nitroxide spin probe tempol. Biochem Pharmacol. 1989a;38:2581–2586. doi: 10.1016/0006-2952(89)90541-8. [DOI] [PubMed] [Google Scholar]
- Iannone A, Hu HP, Tomasi A, Vannini V, Swartz HM. Metabolism of aqueous soluble nitroxides in hepatocytes: effects of cell integrity, oxygen, and structure of nitroxides. Biochim Biophys Acta. 1989b;991:90–96. doi: 10.1016/0304-4165(89)90033-0. [DOI] [PubMed] [Google Scholar]
- Ibrahim HN, Hostetter TH. The renin-aldosterone axis in two models of reduced renal mass in the rat. J Am Soc Nephrol. 1998;9:72–76. doi: 10.1681/ASN.V9172. [DOI] [PubMed] [Google Scholar]
- Ichihara A, Hayashi M, Hirota N, Saruta T. Superoxide inhibits neuronal nitric oxide synthase influences on afferent arterioles in spontaneously hypertensive rats. Hypertension. 2001;37:630–634. doi: 10.1161/01.hyp.37.2.630. [DOI] [PubMed] [Google Scholar]
- Iglarz M, Touyz RM, Viel EC, Amiri F, Schiffrin EL. Involvement of oxidative stress in the profibrotic action of aldosterone: interaction with the renin-angiotensin system. Am J Hypertens. 2004;17:597–603. [PubMed] [Google Scholar]
- Imanishi T, Kobayashi K, Kuroi A, Mochizuki S, Goto M, Yoshida K, Akasaka T. Effects of angiotensin II on NO bioavailability evaluated using a catheter-type NO sensor. Hypertension. 2006;48:1058–1065. doi: 10.1161/01.HYP.0000248920.16956.d8. [DOI] [PubMed] [Google Scholar]
- Imanishi T, Kuroi A, Ikejima H, Mochizuki S, Goto M, Akasaka T. Evaluation of pharmacological modulation of nitroglycerin-induced impairment of nitric oxide bioavailability by a catheter-type nitric oxide sensor. Circ J. 2007;71:1473–1479. doi: 10.1253/circj.71.1473. [DOI] [PubMed] [Google Scholar]
- Israeli A, Patt M, Oron M, Samuni A, Kohen R, Goldstein S. Kinetics and mechanism of the comproportionation reaction between oxoammonium cation and hydroxylamine derived from cyclic nitroxides. Free Radic Biol Med. 2005;38:317–324. doi: 10.1016/j.freeradbiomed.2004.09.037. [DOI] [PubMed] [Google Scholar]
- Itoh T, Kajikuri J, Hattori T, Kusama N, Yamamoto T. Involvement of H2O2 in superoxide-dismutase-induced enhancement of endothelium-dependent relaxation in rabbit mesenteric resistance artery. Br J Pharmacol. 2003;139:444–456. doi: 10.1038/sj.bjp.0705255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson TC, Mi Z, Jackson EK. Modulation of cyclic AMP production by signal transduction pathways in preglomerular microvessels and microvascular smooth muscle cells. J Pharmacol Exp Ther. 2004;310:349–358. doi: 10.1124/jpet.103.063081. [DOI] [PubMed] [Google Scholar]
- Jaimes EA, Tian RX, Pearse D, Raij L. Up-regulation of glomerular COX-2 by angiotensin II: role of reactive oxygen species. Kidney Int. 2005;68:2143–2153. doi: 10.1111/j.1523-1755.2005.00670.x. [DOI] [PubMed] [Google Scholar]
- Jaimes EA, Zhou MS, Pearse DD, Puzis L, Raij L. Up-regulation of cortical COX-2 in salt sensitive hypertension: role of angiotensin II and reactive oxygen species. Am J Physiol Renal Physiol. 2008;294:F385–F392. doi: 10.1152/ajprenal.00302.2007. [DOI] [PubMed] [Google Scholar]
- Jang S, Huon T, Kim K, Um E, Han O. Regiochemical and stereochemical evidence for enzyme-initiated catalysis in dual positional specific maize lipoxygenase-1. Org Lett. 2007;9:3113–3116. doi: 10.1021/ol0712024. [DOI] [PubMed] [Google Scholar]
- Jankov RP, Kantores C, Pan J, Belik J. Contribution of xanthine oxidase-derived superoxide to chronic hypoxic pulmonary hypertension in neonatal rats. Am J Physiol Lung Cell Mol Physiol. 2008;294:L233–L245. doi: 10.1152/ajplung.00166.2007. [DOI] [PubMed] [Google Scholar]
- Jerez S, Peral de Bruno M, Coviello A. Nitric oxide modulates angiotensin II-induced endothelial vasoconstrictor prostanoid release. Eur J Pharmacol. 2005;520:127–134. doi: 10.1016/j.ejphar.2005.07.030. [DOI] [PubMed] [Google Scholar]
- Jin L, Ying Z, Webb RC. Activation of Rho/Rho kinase signaling pathway by reactive oxygen species in rat aorta. Am J Physiol Heart Circ Physiol. 2004;287:H1495–H1500. doi: 10.1152/ajpheart.01006.2003. [DOI] [PubMed] [Google Scholar]
- Johnstone PA, DeGraff WG, Mitchell JB. Protection from radiation-induced chromosomal aberrations by the nitroxide Tempol. Cancer. 1995;75:2323–2327. doi: 10.1002/1097-0142(19950501)75:9<2323::aid-cncr2820750922>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Jozwiak Z, Gwoździński K, Helszer Z. Effect of adenine nucleotides and gamma radiation on the transport of TEMPOL across the erythrocyte membrane. Int J Radiat Biol Relat Stud Phys Chem Med. 1983;44:301–305. doi: 10.1080/09553008314551181. [DOI] [PubMed] [Google Scholar]
- Juncos R, Garvin JL. Superoxide enhances Na-K-2Cl cotransporter activity in the thick ascending limb. Am J Physiol Renal Physiol. 2005;288:F982–F987. doi: 10.1152/ajprenal.00348.2004. [DOI] [PubMed] [Google Scholar]
- Juncos R, Hong NJ, Garvin JL. Differential effects of superoxide on luminal and basolateral Na+/H+ exchange in the thick ascending limb. Am J Physiol Regul Integr Comp Physiol. 2006;290:R79–R83. doi: 10.1152/ajpregu.00447.2005. [DOI] [PubMed] [Google Scholar]
- Jung K, Ristori S, Gallori E, Martini G. Stability of water-soluble and lipid-soluble paramagnetic probes in Bacillus subtilis. Biochim Biophys Acta. 1998;1425:387–397. doi: 10.1016/s0304-4165(98)00091-9. [DOI] [PubMed] [Google Scholar]
- Just A, Olson AJ, Whitten CL, Arendshorst WJ. Superoxide mediates acute renal vasoconstriction produced by angiotensin II and catecholamines by a mechanism independent of nitric oxide. Am J Physiol Heart Circ Physiol. 2007;292:H83–H92. doi: 10.1152/ajpheart.00715.2006. [DOI] [PubMed] [Google Scholar]
- Kagiyama S, Tsuchihashi T, Abe I, Matsumura K, Fujishima M. Central infusion of L-arginine or superoxide dismutase does not alter arterial pressure in SHR. Hypertens Res. 2000;23:339–343. doi: 10.1291/hypres.23.339. [DOI] [PubMed] [Google Scholar]
- Kamataria M, Yasui H, Ogata T, Sakurai H. Local pharmacokinetic analysis of a stable spin probe in mice by in vivo L-band ESR with surface-coil-type resonators. Free Radic Res. 2002;36:1115–1125. doi: 10.1080/1071576021000028352. [DOI] [PubMed] [Google Scholar]
- Kang KT, Sullivan JC, Sasser JM, Imig JD, Pollock JS. Novel nitric oxide synthase-dependent mechanism of vasorelaxation in small arteries from hypertensive rats. Hypertension. 2007;49:893–901. doi: 10.1161/01.HYP.0000259669.40991.1e. [DOI] [PubMed] [Google Scholar]
- Kawada N, Dennehy K, Solis G, Modlinger P, Hamel R, Kawada JT, Aslam S, Moriyama T, Imai E, Welch WJ, et al. TP receptors regulate renal hemodynamics during angiotensin II slow pressor response. Am J Physiol. 2004;287:F753–F759. doi: 10.1152/ajprenal.00423.2003. [DOI] [PubMed] [Google Scholar]
- Kawada N, Imai E, Karber A, Welch WJ, Wilcox CS. A mouse model of angiotensin II slow pressor response: role of oxidative stress. J Am Soc Nephrol. 2002;13:2860–2868. doi: 10.1097/01.asn.0000035087.11758.ed. [DOI] [PubMed] [Google Scholar]
- Keana JF, Pou S, Rosen GM. Nitroxides as potential contrast enhancing agents for MRI application: influence of structure on the rate of reduction by rat hepatocytes, whole liver homogenate, subcellular fractions, and ascorbate. Magn Reson Med. 1987;5:525–536. doi: 10.1002/mrm.1910050603. [DOI] [PubMed] [Google Scholar]
- Khramtsov VV, Yelinova VI, Weiner LM, Berezina TA, Martin VV, Volodarsky LB. Quantitative determination of SH groups in low- and high-molecular-weight compounds by an electron spin resonance method. Anal Biochem. 1989;182:58–63. doi: 10.1016/0003-2697(89)90718-5. [DOI] [PubMed] [Google Scholar]
- Kimura S, Zhang GX, Nagai Y, Miyata K, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Fujisawa Y, et al. Time-dependent transition of tempol-sensitive reduction of blood pressure in angiotensin II-induced hypertension. J Hypertens. 2004;22:2161–2168. doi: 10.1097/00004872-200411000-00019. [DOI] [PubMed] [Google Scholar]
- Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Abe Y. Mitochondria-derived reactive oxygen species and vascular MAP kinases: comparison of angiotensin II and diazoxide. Hypertension. 2005a;45:438–444. doi: 10.1161/01.HYP.0000157169.27818.ae. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Hirooka Y, Sagara Y, Ito K, Kishi T, Shimokawa H, Takeshita A, Sunagawa K. Overexpression of inducible nitric oxide synthase in rostral ventrolateral medulla causes hypertension and sympathoexcitation via an increase in oxidative stress. Circ Res. 2005b;96:252–260. doi: 10.1161/01.RES.0000152965.75127.9d. [DOI] [PubMed] [Google Scholar]
- Kiritoshi S, Nishikawa T, Sonoda K, Kukidome D, Senokuchi T, Matsuo T, Matsumura T, Tokunaga H, Brownlee M, Araki E. Reactive oxygen species from mitochondria induce cyclooxygenase-2 gene expression in human mesangial cells: potential role in diabetic nephropathy. Diabetes. 2003;52:2570–2577. doi: 10.2337/diabetes.52.10.2570. [DOI] [PubMed] [Google Scholar]
- Kishi T, Hirooka Y, Kimura Y, Ito K, Shimokawa H, Takeshita A. Increased reactive oxygen species in rostral ventrolateral medulla contribute to neutral mechanisms of hypertension in stroke-prone spontaneously hypertensive rats. Circulation. 2004;109:2357–2362. doi: 10.1161/01.CIR.0000128695.49900.12. [DOI] [PubMed] [Google Scholar]
- Kitayama J, Yi C, Faraci FM, Heistad DD. Modulation of dilator responses of cerebral arterioles by extracellular superoxide dismutase. Stroke. 2006;37:2802–2806. doi: 10.1161/01.STR.0000245134.12145.ae. [DOI] [PubMed] [Google Scholar]
- Kitiyakara C, Chabrashvili T, Chen Y, Blau J, Karber A, Aslam S, Welch WJ, Wilcox CS. Salt intake, oxidative stress and renal expression of NADPH oxidase and superoxide dismutase. J Am Soc Nephrol. 2003;14:2775–2782. doi: 10.1097/01.asn.0000092145.90389.65. [DOI] [PubMed] [Google Scholar]
- Kobori H, Nishiyama A. Effects of tempol on renal angiotensinogen production in Dahl salt-sensitive rats. Biochem Biophys Res Commun. 2004;315:746–750. doi: 10.1016/j.bbrc.2004.01.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobori H, Nishiyama A, Abe Y, Navar LG. Enhancement of intrarenal angiotensinogen in Dahl salt-sensitive rats on high salt diet. Hypertension. 2003;41:592–597. doi: 10.1161/01.HYP.0000056768.03657.B4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobori H, Ozawa Y, Suzaki Y, Prieto-Carrasquero MC, Nishiyama A, Shoji T, Cohen EP, Navar LG. Young Scholars Award Lecture: Intratubular angiotensinogen in hypertension and kidney diseases. Am J Hypertens. 2006;19:541–550. doi: 10.1016/j.amjhyper.2005.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocherginsky N, Swartz HM. Nitroxide Spin Labels: Reactions in Biology and Chemistry. CRC Press; Boca Raton, FL: 1995. [Google Scholar]
- Komarov AM, Joseph J, Lai CS. In vivo pharmacokinetics of nitroxides in mice. Biochem Biophys Res Commun. 1994;201:1035–1042. doi: 10.1006/bbrc.1994.1806. [DOI] [PubMed] [Google Scholar]
- Komers R, Lindsley JN, Oyama TT, Schutzer WE, Reed JF, Mader SL, Anderson S. Immunohistochemical and functional correlations of renal cycooxygenase-2 in experimental diabetes. J Clin Invest. 2001;107:889–898. doi: 10.1172/JCI10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konorev EA, Tarpey MM, Joseph J, Baker JE, Kalyanaraman B. Nitronyl nitroxides as probes to study the mechanism of vasodilatory action of nitrovasodilators, nitrone spin traps, and nitroxides: role of nitric oxide. Free Rad Biol Med. 1995;18:169–177. doi: 10.1016/0891-5849(94)00112-w. [DOI] [PubMed] [Google Scholar]
- Kopkan L, Castillo A, Navar LG, Majid DS. Enhanced superoxide generation modulates renal function in ANG II-induced hypertensive rats. Am J Physiol Renal Physiol. 2006;290:F80–F86. doi: 10.1152/ajprenal.00090.2005. [DOI] [PubMed] [Google Scholar]
- Kopkan L, Husková Z, Vanourková Z, Thumová M, Skaroupková P, Cervenka L, Majid DS. Superoxide and its interaction with nitric oxide modulates renal function in prehypertensive Ren-2 transgenic rats. J Hypertens. 2007;25:2257–2265. doi: 10.1097/HJH.0b013e3282efb195. [DOI] [PubMed] [Google Scholar]
- Kopkan L, Majid DS. Superoxide contributes to development of salt sensitivity and hypertension induced by nitric oxide deficiency. Hypertension. 2005;46:1026–1031. doi: 10.1161/01.HYP.0000174989.39003.58. [DOI] [PubMed] [Google Scholar]
- Kotchen TA, Piering AW, Cowley AW, Grim CE, Gaudet D, Hamet P, Kaldunski ML, Kotchen JM, Roman RJ. Glomerular hyperfiltration in hypertensive African Americans. Hypertension. 2000;35:822–826. doi: 10.1161/01.hyp.35.3.822. [DOI] [PubMed] [Google Scholar]
- Krishna MC, DeGraff W, Hankovsky O, Sár CP, Kálai T, Jeko J, Russo A, Mitchell JB. Studies of structure-activity relationship of nitroxide free radicals and their precursors as modifiers against oxidative damage. J Med Chem. 1998;41:3477–3492. doi: 10.1021/jm9802160. [DOI] [PubMed] [Google Scholar]
- Krishna MC, Grahame DA, Samuni A, Mitchell JB, Russo A. Oxoammonium cation intermediate in the nitroxide-catalyzed dismutation of superoxide. Proc Natl Acad Sci U S A. 1992;89:5537–5541. doi: 10.1073/pnas.89.12.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna MC, Russo A, Mitchell JB, Goldstein S, Dafni H, Samuni A. Do nitroxide antioxidants act as scavengers of or as SOD mimics? J Biol Chem. 1996a;271:26026–26031. doi: 10.1074/jbc.271.42.26026. [DOI] [PubMed] [Google Scholar]
- Krishna MC, Samuni A, Taira J, Goldstein S, Mitchell JB, Russo A. Stimulation by nitroxides of catalase-like activity of hemeproteins. J Biol Chem. 1996b;271:26018–26025. doi: 10.1074/jbc.271.42.26018. [DOI] [PubMed] [Google Scholar]
- Kroll C, Borchert HH. Metabolism of the stable nitroxyl radical 4-oxo-2,2,6, 6-tetramethylpiperidine-N-oxyl (TEMPONE) Eur J Pharm Sci. 1999;8:5–9. doi: 10.1016/s0928-0987(98)00047-5. [DOI] [PubMed] [Google Scholar]
- Kroll C, Langner A, Borchert HH. Nitroxide metabolism in the human keratinocyte cell line HaCaT. Free Radic Biol Med. 1999;26:850–857. doi: 10.1016/s0891-5849(98)00268-8. [DOI] [PubMed] [Google Scholar]
- Kruglov AG, Subbotina KB, Saris NE. Redox-cycling compounds can cause the permeabilization of mitochondrial membranes by mechanisms other than ROS production. Free Radic Biol Med. 2008;44:646–656. doi: 10.1016/j.freeradbiomed.2007.10.049. [DOI] [PubMed] [Google Scholar]
- Kunes J, Dobesová Z, Zicha J. Altered balance of main vasopressor and vasodepressor systems in rats with genetic hypertension and hypertriglyceridaemia. Clin Sci. 2002;102:269–277. [PubMed] [Google Scholar]
- Kuo ML, Lee KC, Lin JK, Huang TS. Pronounced activation of protein kinase C, ornithine decarboxylase and c-jun proto-oncogene by paraquat-generated active oxygen species in WI-38 human lung cells. Biochim Biophys Acta. 1995;1268:229–236. doi: 10.1016/0167-4889(95)00076-5. [DOI] [PubMed] [Google Scholar]
- Kuppusamy P, Li H, Ilangovan G, Cardounel AJ, Zweier JL, Yamada K, Krishna MC, Mitchell JB. Noninvasive imaging of tumor redox status and its modification by tissue glutathione levels. Cancer Res. 2002;62:307–312. [PubMed] [Google Scholar]
- Kuppusamy P, Wang P, Shankar RA, Ma L, Trimble CE, Hsia CJ, Zweier JL. In vivo topical EPR spectroscopy and imaging of nitroxide free radicals and polynitroxyl-albumin. Magn Reson Med. 1998;40:806–811. doi: 10.1002/mrm.1910400604. [DOI] [PubMed] [Google Scholar]
- Kurihara N, Yanagisawa H, Sato M, Tien CK, Wada O. Increased renal vascular resistance in zinc-deficient rats: role of nitric oxide and superoxide. Clin Exp Pharmacol Physiol. 2002;29:1096–1104. doi: 10.1046/j.1440-1681.2002.03783.x. [DOI] [PubMed] [Google Scholar]
- Kutala VK, Khan M, Mandal R, Potaraju V, Colantuono G, Kumbala D, Kuppusamy P. Prevention of postischemic myocardial reperfusion injury by the combined treatment of NCX-4016 and Tempol. J Cardiovasc Pharmacol. 2006;48:79–87. doi: 10.1097/01.fjc.0000242050.16790.65. [DOI] [PubMed] [Google Scholar]
- Kwan J, Wang H, Munk S, Xia L, Goldberg HJ, Whiteside CI. In high glucose protein kinase C-ζ activation is required for mesangial cell generation of reactive oxygen species. Kidney Int. 2005;68:2526–2541. doi: 10.1111/j.1523-1755.2005.00660.x. [DOI] [PubMed] [Google Scholar]
- Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic Biol Med. 2007;43:332–347. doi: 10.1016/j.freeradbiomed.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambeth JD, Kawahara T, Diebold B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic Biol Med. 2007;43:319–331. doi: 10.1016/j.freeradbiomed.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen JB, Rajagopalan S, Galis Z, Tarpey M, Freeman BA, Harrison DG. Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation. 1997;95:588–593. doi: 10.1161/01.cir.95.3.588. [DOI] [PubMed] [Google Scholar]
- Lavi S, Yang EH, Prasad A, Mathew V, Barsness GW, Rihal CS, Lerman LO, Lerman A. The interaction between coronary endothelial dysfunction, local oxidative stress, and endogenous nitric oxide in humans. Hypertension. 2008;51:127–133. doi: 10.1161/HYPERTENSIONAHA.107.099986. [DOI] [PubMed] [Google Scholar]
- Lee YM, Cheng PY, Hong SF, Chen SY, Lam KK, Sheu JR, Yen MH. Oxidative stress induces vascular heme oxygenase-1 expression in ovariectomized rats. Free Radic Biol Med. 2005;39:108–117. doi: 10.1016/j.freeradbiomed.2005.02.033. [DOI] [PubMed] [Google Scholar]
- Lewinska A, Wnuk M, Slota E, Bartosz G. The nitroxide antioxidant Tempol affects metal-induced cyto- and genotoxicity in human lymphocytes in vitro. Mutat Res. 2008;649:7–14. doi: 10.1016/j.mrgentox.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Li CJ, Higashiyama K, Yoshimura Y, Nagai T, Takayama K, Obata Y. Promoting mechanism of menthol derivative, 1-O-ethyl-3-buthylcyclohexanol, on the percutaneous absorption of ketoprofen. Biol Pharm Bull. 2001;24:1044–1048. doi: 10.1248/bpb.24.1044. [DOI] [PubMed] [Google Scholar]
- Li H, Ma L, Hsia CJ, Zweier JL, Kuppusamy P. Polynitroxyl-albumin (PNA) enhances myocardial infarction therapeutic effect of tempol in rat hearts subjected to regional ischemia-reperfusion. Free Radic Biol Med. 2002;32:712–719. doi: 10.1016/s0891-5849(02)00762-1. [DOI] [PubMed] [Google Scholar]
- Li J, Chen YJ, Quilley J. Effect of tempol on renal cyclooxygenase expression and activity in experimental diabetes in the rat. J Pharmacol Exp Ther. 2005a;314:818–824. doi: 10.1124/jpet.104.076927. [DOI] [PubMed] [Google Scholar]
- Li L, Watts SW, Banes AK, Galligan JJ, Fink GD, Chen AF. NADPH oxidase-derived superoxide augments endothelin-1-induced venoconstriction in mineralocorticoid hypertension. Hypertension. 2003;42:316–321. doi: 10.1161/01.HYP.0000084853.47326.F2. [DOI] [PubMed] [Google Scholar]
- Li LP, Li BS, Storey P, Fogelson L, Li W, Prasad P. Effect of free radical scavenger (tempol) on intrarenal oxygenation in hypertensive rats as evaluated by BOLD MRI. J Magn Reson Imaging. 2005b;21:245–248. doi: 10.1002/jmri.20260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Mendonca M, Welch WJ, Wilcox CS. Salt-sensitive hypertension in a model of chronic renal failure is ameliorated by Tempol (Abstract) J Am Soc Nephrol. 2007;18:846A. [Google Scholar]
- Li WG, Zhang XY, Wu YJ, Gao MT, Zheng RL. The relationship between structure and antioxidative activity of piperidine nitroxides. J Pharm Pharmacol. 2006;58:941–949. doi: 10.1211/jpp.58.7.0009. [DOI] [PubMed] [Google Scholar]
- Lin L, Mistry M, Stier CT, Jr, Nasjletti A. Role of prostanoids in renin-dependent and renin-independent hypertension. Hypertension. 1991;17:517–525. doi: 10.1161/01.hyp.17.4.517. [DOI] [PubMed] [Google Scholar]
- Liu R, Garvin JL, Ren Y, Pagano PJ, Carretero OA. Depolarization of the macula densa induces superoxide production via NAD(P)H oxidase. Am J Physiol Renal Physiol. 2007a;292:F1867–F1872. doi: 10.1152/ajprenal.00515.2006. [DOI] [PubMed] [Google Scholar]
- Liu R, Ren Y, Garvin JL, Carretero OA. Superoxide enhances tubuloglomerular feedback by constricting the afferent arteriole. Kidney Int. 2004;66:268–274. doi: 10.1111/j.1523-1755.2004.00727.x. [DOI] [PubMed] [Google Scholar]
- Liu S, Ma X, Gong M, Shi L, Lincoln T, Wang S. Glucose down-regulation of cGMP-dependent protein kinase I expression in vascular smooth muscle cells involves NAD(P)H oxidase-derived reactive oxygen species. Free Radic Biol Med. 2007b;42:852–863. doi: 10.1016/j.freeradbiomed.2006.12.025. [DOI] [PubMed] [Google Scholar]
- Liu Y, Gutterman DD. Oxidative stress and potassium channel function. Clin Exp Pharmacol Physiol. 2002;29:305–311. doi: 10.1046/j.1440-1681.2002.03649.x. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res. 2003;93:573–580. doi: 10.1161/01.RES.0000091261.19387.AE. [DOI] [PubMed] [Google Scholar]
- Long LS, Zaharia LC, Connors S, Jackson EK, Welch WJ. Ang II-induced oxidative stress stimulates adenosine release in the renal cortex (Abstract) FASEB J. 2007;21:A1246. [Google Scholar]
- Loomis ED, Sullivan JC, Osmond DA, Pollock DM, Pollock JS. Endothelin mediates superoxide production and vasoconstriction through activation of NADPH oxidase and uncoupled nitric-oxide synthase in the rat aorta. J Pharmacol Exp Ther. 2005;315:1058–1064. doi: 10.1124/jpet.105.091728. [DOI] [PubMed] [Google Scholar]
- López B, Salom MG, Arregui B, Valero F, Fenoy FJ. Role of superoxide in modulating the renal effects of angiotensin II. Hypertension. 2003;42:1150–1156. doi: 10.1161/01.HYP.0000101968.09376.79. [DOI] [PubMed] [Google Scholar]
- Lu N, Helwig BG, Fels RJ, Parimi S, Kenney MJ. Central tempol alters basal sympathetic nerve discharge and attenuates sympathetic excitation to central Ang II. Am J Physiol Heart Circ Physiol. 2004;287:H2626–H2633. doi: 10.1152/ajpheart.00030.2004. [DOI] [PubMed] [Google Scholar]
- Lu X, Kassab GS. Nitric oxide is significantly reduced in ex vivo porcine arteries during reverse flow because of increased superoxide production. J Physiol. 2004;561:575–582. doi: 10.1113/jphysiol.2004.075218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luft FC, Wilcox CS, Unger T, Kühn R, Demmert G, Rohmeiss P, Ganten D, Sterzel RB. Angiotensin-induced hypertension in the rat: sympathetic nerve activity and prostaglandins. Hypertension. 1989;14:396–403. doi: 10.1161/01.hyp.14.4.396. [DOI] [PubMed] [Google Scholar]
- Luo Z, Chen Y, Chen SY, Welch WJ, Jose PA, Wilcox CS. Comparative effects of antioxidants on Ang II-induced superoxide generation by SHR preglomerular vascular smooth muscle cells (Abstract) FASEB J. 2007;21:A820. [Google Scholar]
- MacKenzie A, Filippini S, Martin W. Effects of superoxide dismutase mimetics on the activity of nitric oxide in rat aorta. Br J Pharmacol. 1999;127:1159–1164. doi: 10.1038/sj.bjp.0702670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie A, Martin W. Loss of endothelium-derived nitric oxide in rabbit aorta by oxidant stress: restoration by superoxide dismutase mimetics. Br J Pharmacol. 1998;124:719–728. doi: 10.1038/sj.bjp.0701899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majid DS, Kopkan L. Nitric oxide and superoxide interactions in the kidney and their implication in the development of salt-sensitive hypertension. Clin Exp Pharmacol Physiol. 2007;34:946–952. doi: 10.1111/j.1440-1681.2007.04642.x. [DOI] [PubMed] [Google Scholar]
- Majid DS, Navar LG. Nitric oxide in the control of renal hemodynamics and excretory function. Am J Hypertens. 2001;14:74S–82S. doi: 10.1016/s0895-7061(01)02073-8. [DOI] [PubMed] [Google Scholar]
- Majid DS, Nishiyama A. Nitric oxide blockade enhances renal responses to superoxide dismutase inhibition in dogs. Hypertension. 2002;39:293–297. doi: 10.1161/hy0202.104137. [DOI] [PubMed] [Google Scholar]
- Majid DS, Nishiyama A, Jackson KE, Castillo A. Inhibition of nitric oxide synthase enhances superoxide activity in canine kidney. Am J Physiol Regul Integr Comp Physiol. 2004;287:R27–R32. doi: 10.1152/ajpregu.00073.2004. [DOI] [PubMed] [Google Scholar]
- Majid DS, Nishiyama A, Jackson KE, Castillo A. Superoxide scavenging attenuates renal responses to Ang II during nitric oxide synthesis inhibition in anesthetized dogs. Am J Physiol Renal Physiol. 2005;288:F412–F419. doi: 10.1152/ajprenal.00294.2004. [DOI] [PubMed] [Google Scholar]
- Makino A, Skelton MM, Zou AP, Cowley AW., Jr Increased renal medullary H2O2 leads to hypertension. Hypertension. 2003;42:25–30. doi: 10.1161/01.HYP.0000074903.96928.91. [DOI] [PubMed] [Google Scholar]
- Manevich Y, Sweitzer T, Pak JH, Feinstein SI, Muzykantov V, Fisher AB. 1-Cys peroxiredoxin overexpression protects cells against phospholipid peroxidation-mediated membrane damage. Proc Natl Acad Sci U S A. 2002;99:11599–11604. doi: 10.1073/pnas.182384499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann B, Hartner A, Jensen BL, Hilgers KF, Höcherl K, Krämer BK, Kurtz A. Acute upregulation of COX-2 by renal artery stenosis. Am J Physiol Renal Physiol. 2001;280:F119–F125. doi: 10.1152/ajprenal.2001.280.1.F119. [DOI] [PubMed] [Google Scholar]
- Manning RD, Jr, Meng S, Tian N. Renal and vascular oxidative stress and salt-sensitivity of arterial pressure. Acta Physiol Scand. 2003;179:243–250. doi: 10.1046/j.0001-6772.2003.01204.x. [DOI] [PubMed] [Google Scholar]
- Manning RD, Jr, Tian N, Meng S. Oxidative stress and antioxidant treatment in hypertension and the associated renal damage. Am J Nephrol. 2005;25:311–317. doi: 10.1159/000086411. [DOI] [PubMed] [Google Scholar]
- Marwaha A, Lokhandwala MF. Tempol reduces oxidative stress and restores renal dopamine D1-like receptor-G protein coupling and function in hyperglycemic rats. Am J Physiol Renal Physiol. 2006;291:F58–F66. doi: 10.1152/ajprenal.00362.2005. [DOI] [PubMed] [Google Scholar]
- Marx L, Chiarelli R, Guiberteau T, Rassat A. A comparative study of the reduction by ascorbate of 1,1,3,3-tetraethylisoindolin-2-yloxyl and of 1,1,3,3-tetramethylisoindolin-2-yloxyl. J Chem Soc Perkin Trans. 2000;1:1181–1182. [Google Scholar]
- Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, Kanaide H, Takeshita A. Hydrogen peroxide is an endothelium-derived hyper-polarizing factor in mice. J Clin Invest. 2000;106:1521–1530. doi: 10.1172/JCI10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K, Krishna MC, Mitchell JB. Novel pharmacokinetic measurement using electron paramagnetic resonance spectroscopy and simulation of in vivo decay of various nitroxyl spin probes in mouse blood. J Pharmacol Exp Ther. 2004;310:1076–1083. doi: 10.1124/jpet.104.066647. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Yahiro T, Yamada K, Utsumi H. In vivo EPR spectroscopic imaging for a liposomal drug delivery system. Magn Reson Med. 2005;53:1158–1165. doi: 10.1002/mrm.20460. [DOI] [PubMed] [Google Scholar]
- Matsumoto S, Mori N, Tsuchihashi N, Ogata T, Lin Y, Yokoyama H, Ishida S. Enhancement of nitroxide-reducing activity in rats after chronic administration of vitamin E, vitamin C, and idebenone examined by an in vivo electron spin resonance technique. Magn Reson Med. 1998;40:330–333. doi: 10.1002/mrm.1910400219. [DOI] [PubMed] [Google Scholar]
- May JM, Qu ZC, Juliao S, Cobb CE. Ascorbic acid decreases oxidant stress in endothelial cells caused by the nitroxide tempol. Free Radic Res. 2005;39:195–202. doi: 10.1080/10715760400019661. [DOI] [PubMed] [Google Scholar]
- May JM, Qu ZC, Mendiratta S. Protection and recycling of α-tocopherol in human erythrocytes by intracellular ascorbic acid. Arch Biochem Biophys. 1998;349:281–289. doi: 10.1006/abbi.1997.0473. [DOI] [PubMed] [Google Scholar]
- Mayorov DN. Selective sensitization by nitric oxide of sympathetic baroreflex in rostral ventrolateral medulla of conscious rabbits. Hypertension. 2005;45:901–906. doi: 10.1161/01.HYP.0000160322.83725.6b. [DOI] [PubMed] [Google Scholar]
- Mayorov DN, Head GA, De Matteo R. Tempol attenuates excitatory actions of angiotensin II in the rostral ventrolateral medulla during emotional stress. Hypertension. 2004;44:101–106. doi: 10.1161/01.HYP.0000131290.12255.04. [DOI] [PubMed] [Google Scholar]
- Mazzali M, Jefferson JA, Ni Z, Vaziri ND, Johnson RJ. Microvascular and tubulointerstitial injury associated with chronic hypoxia-induced hypertension. Kidney Int. 2003;63:2088–2093. doi: 10.1046/j.1523-1755.2003.00011.x. [DOI] [PubMed] [Google Scholar]
- McCord JM, Edeas MA. SOD, oxidative stress and human pathologies: a brief history and a future vision. Biomed Pharmacother. 2005;59:139–142. doi: 10.1016/j.biopha.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Mehta SH, Webb RC, Ergul A, Tawfik A, Dorrance AM, Tawak A. Neuroprotection by tempol in a model of iron-induced oxidative stress in acute ischemic stroke. Am J Physiol Regul Integr Comp Physiol. 2004;286:R283–R288. doi: 10.1152/ajpregu.00446.2002. [DOI] [PubMed] [Google Scholar]
- Meng S, Cason GW, Gannon AW, Racusen LC, Manning RD., Jr Oxidative stress in Dahl salt-sensitive hypertension. Hypertension. 2003;41:1346–1352. doi: 10.1161/01.HYP.0000070028.99408.E8. [DOI] [PubMed] [Google Scholar]
- Mikuni T, Tatsuta M. Slow rate of free radical scavenging in the gastric antral mucosa of male Wistar rats: a possible mechanism of gastric carcinogenesis induced by N-methyl-N′-nitro-N-nitrosoguanidine. Int J Cancer. 1998;76:228–231. doi: 10.1002/(sici)1097-0215(19980413)76:2<228::aid-ijc10>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Miura H, Bosnjak JJ, Ning G, Saito T, Miura M, Gutterman DD. Role for hydrogen peroxide in flow-induced dilation of human coronary arterioles. Circ Res. 2003;92:e31–e40. doi: 10.1161/01.res.0000054200.44505.ab. [DOI] [PubMed] [Google Scholar]
- Miura Y, Utsumi H, Hamada A. Effects of inspired oxygen concentration on in vivo redox reaction of nitroxide radicals in whole mice. Biochem Biophys Res Commun. 1992;182:1108–1114. doi: 10.1016/0006-291x(92)91846-i. [DOI] [PubMed] [Google Scholar]
- Miura Y, Utsumi H, Kashiwagi M, Hamada A. Effects of oxygen on the membrane structure and the metabolism of lipophilic nitroxide in rat liver microsomes. J Biochem (Tokyo) 1990;108:516–518. doi: 10.1093/oxfordjournals.jbchem.a123233. [DOI] [PubMed] [Google Scholar]
- Modlinger P, Chabrashvili T, Gill PS, Mendonca M, Harrison DG, Griendling KK, Li M, Raggio J, Wellstein A, Chen Y, et al. RNA silencing in vivo reveals role of p22phox in rat angiotensin slow pressor response. Hypertension. 2006;47:238–244. doi: 10.1161/01.HYP.0000200023.02195.73. [DOI] [PubMed] [Google Scholar]
- Modlinger PS, Wilcox CS, Aslam S. Nitric oxide, oxidative stress and progression of chronic renal failure. Semin Nephrol. 2004;24:354–365. doi: 10.1016/j.semnephrol.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Mok JS, Paisley K, Martin W. Inhibition of nitrergic neurotransmission in the bovine retractor penis muscle by an oxidant stress: effects of superoxide dismutase mimetics. Br J Pharmacol. 1998;124:111–118. doi: 10.1038/sj.bjp.0701809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollace V, Iannone M, Muscoli C, Palma E, Granato T, Rispoli V, Nisticò R, Rotiroti D, Salvemini D. The role of oxidative stress in paraquat-induced neurotoxicity in rats: protection by non peptidyl superoxide dismutase mimetic. Neurosci Lett. 2003;335:163–166. doi: 10.1016/s0304-3940(02)01168-0. [DOI] [PubMed] [Google Scholar]
- Monti E, Cova D, Guido E, Morelli R, Oliva C. Protective effect of the nitroxide tempol against the cardiotoxicity of Adriamycin. Free Radic Biol Med. 1996;21:463–470. doi: 10.1016/0891-5849(96)00124-4. [DOI] [PubMed] [Google Scholar]
- Moore KL, Moronne MM, Mehlhorn RJ. Electron spin resonance study of peroxidase activity and kinetics. Arch Biochem Biophys. 1992;299:47–56. doi: 10.1016/0003-9861(92)90242-o. [DOI] [PubMed] [Google Scholar]
- Moreau P, d’Uscio LV, Shaw S, Takase H, Barton M, Lüscher TF. Angiotensin II increases tissue endothelin and induces vascular hypertrophy. Circulation. 1997;96:1593–1597. doi: 10.1161/01.cir.96.5.1593. [DOI] [PubMed] [Google Scholar]
- Moreno JM, Rodríguez Gómez I, Wangensteen R, Osuna A, Bueno P, Vargas F. Cardiac and renal antioxidant enzymes and effects of Tempol in hyperthyroid rats. Am J Physiol Endocrinol Metab. 2005;289:E776–E783. doi: 10.1152/ajpendo.00611.2004. [DOI] [PubMed] [Google Scholar]
- Morikawa K, Shimokawa H, Matoba T, Kubota H, Akaike T, Talukder MA, Hatanaka M, Fujiki T, Maeda H, Takahashi S, et al. Pivotal role of Cu, Zn-superoxide dismutase in endothelium-dependent hyperpolarization. J Clin Invest. 2003;112:1871–1879. doi: 10.1172/JCI19351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mügge A, Elwell JH, Peterson TE, Hofmeyer TG, Heistad DD, Harrison DG. Chronic treatment with polyethylene-glycolated superoxide dismutase partially restores endothelium-dependent vascular relaxations in cholesterol-fed rabbits. Circ Res. 1991;69:1293–1300. doi: 10.1161/01.res.69.5.1293. [DOI] [PubMed] [Google Scholar]
- Murakami K, Haneda M, Qiao S, Naruse M, Yoshino M. Prooxidant action of rosmarinic acid: transition metal-dependent generation of reactive oxygen species. Toxicol In Vitro. 2007;21:613–617. doi: 10.1016/j.tiv.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Murakami K, Haneda M, Yoshino M. Prooxidant action of xanthurenic acid and quinoline compounds: role of transition metals in the generation of reactive oxygen species and enhanced formation of 8-hydroxy-2′-deoxyguanosine in DNA. Biometals. 2006a;19:429–435. doi: 10.1007/s10534-005-4528-6. [DOI] [PubMed] [Google Scholar]
- Murakami K, Ishida K, Watakabe K, Tsubouchi R, Haneda M, Yoshino M. Prooxidant action of maltol: role of transition metals in the generation of reactive oxygen species and enhanced formation of 8-hydroxy-2′-deoxyguanosine formation in DNA. Biometals. 2006b;19:253–257. doi: 10.1007/s10534-005-6998-y. [DOI] [PubMed] [Google Scholar]
- Murakami K, Ishida K, Watakabe K, Tsubouchi R, Naruse M, Yoshino M. Maltol/iron-mediated apoptosis in HL60 cells: participation of reactive oxygen species. Toxicol Lett. 2006c;161:102–107. doi: 10.1016/j.toxlet.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Murakami K, Ohara Y, Haneda M, Tsubouchi R, Yoshino M. Prooxidant action of hinokitiol: hinokitiol-iron dependent generation of reactive oxygen species. Basic Clin Pharmacol Toxicol. 2005;97:392–394. doi: 10.1111/j.1742-7843.2005.pto_214.x. [DOI] [PubMed] [Google Scholar]
- Musiek ES, Gao L, Milne GL, Han W, Everhart MB, Wang D, Backlund MG, DuBois RN, Zanoni G, Vidari G, et al. Cyclopentenone isoprostanes inhibit the inflammatory response in macrophages. J Biol Chem. 2005;280:35562–35570. doi: 10.1074/jbc.M504785200. [DOI] [PubMed] [Google Scholar]
- Nabha L, Garbern JC, Buller CL, Charpie JR. Vascular oxidative stress precedes high blood pressure in spontaneously hypertensive rats. Clin Exp Hypertens. 2005;27:71–82. doi: 10.1081/ceh-200044267. [DOI] [PubMed] [Google Scholar]
- Nakajima Y, Nakashima T, Inaba K, Sumida Y, Yoh T, Ishikawa H, Mitsuyoshi H, Shima T, Senmaru H. Effects of nitric oxide on the redox status of liver microsomes-electron spin resonance monitoring using nitroxide probes. Hepatol Res. 2002;24:72. doi: 10.1016/s1386-6346(02)00011-6. [DOI] [PubMed] [Google Scholar]
- Nakano D, Itoh C, Ishii F, Kawanishi H, Takaoka M, Kiso Y, Tsuruoka N, Tanaka T, Matsumura Y. Effects of sesamin on aortic oxidative stress and endothelial dysfunction in deoxycorticosterone acetate-salt hypertensive rats. Biol Pharm Bull. 2003;26:1701–1705. doi: 10.1248/bpb.26.1701. [DOI] [PubMed] [Google Scholar]
- Nakazono K, Watanabe N, Matsuno K, Sasaki J, Sato T, Inoue M. Does superoxide underlie the pathogenesis of hypertension? Proc Natl Acad Sci U S A. 1991;88:10045–10048. doi: 10.1073/pnas.88.22.10045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson SK, Bose SK, McCord JM. The toxicity of high-dose superoxide dismutase suggests that superoxide can both initiate and terminate lipid peroxidation in the reperfused heart. Free Radic Biol Med. 1994;16:195–200. doi: 10.1016/0891-5849(94)90143-0. [DOI] [PubMed] [Google Scholar]
- Nilsson UA, Olsson LI, Carlin G, Bylund-Fellenius AC. Inhibition of lipid peroxidation by spin labels: relationships between structure and function. J Biol Chem. 1989;264:11131–11135. [PubMed] [Google Scholar]
- Nishiyama A, Abe Y. Aldosterone and renal injury. Nippon Yakurigaku Zasshi. 2004;124:101–109. doi: 10.1254/fpj.124.101. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Fukui T, Fujisawa Y, Rahman M, Tian RX, Kimura S, Abe Y. Systemic and regional hemodynamic responses to tempol in angiotensin II-infused hypertensive rats. Hypertension. 2001;37:77–83. doi: 10.1161/01.hyp.37.1.77. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Kobori H, Fukui T, Zhang GX, Yao L, Rahman M, Hitomi H, Kiyomoto H, Shokoji T, Kimura S, et al. Role of angiotensin II and reactive oxygen species in cyclosporine A-dependent hypertension. Hypertension. 2003;42:754–760. doi: 10.1161/01.HYP.0000085195.38870.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama A, Yao L, Nagai Y, Miyata K, Yoshizumi M, Kagami S, Kondo S, Kiyomoto H, Shokoji T, Kimura S, et al. Possible contributions of reactive oxygen species and mitogen-activated protein kinase to renal injury in aldosterone/salt-induced hypertensive rats. Hypertension. 2004a;43:841–848. doi: 10.1161/01.HYP.0000118519.66430.22. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Yoshizumi M, Hitomi H, Kagami S, Kondo S, Miyatake A, Fukunaga M, Tamaki T, Kiyomoto H, Kohno M, et al. The SOD mimetic tempol ameliorates glomerular injury and reduces mitogen-activated protein kinase activity in Dahl salt-sensitive rats. J Am Soc Nephrol. 2004b;15:306–315. doi: 10.1097/01.asn.0000108523.02100.e0. [DOI] [PubMed] [Google Scholar]
- Nisoli E, Clementi E, Carruba MO, Moncada S. Defective mitochondrial biogenesis: a hallmark of the high cardiovascular risk in the metabolic syndrome? Circ Res. 2007;100:795–806. doi: 10.1161/01.RES.0000259591.97107.6c. [DOI] [PubMed] [Google Scholar]
- Nothiglaslo V, Bobst AM. Reinvestigation of the oxidation properties of nitroxides. Croat Chem Acta. 1991;64:1–8. [Google Scholar]
- Nouri P, Chen Y, Wilcox CS. Tempol prevents the attenuation of endothelium-dependent relaxation response to acetylcholine caused by intravenous injection of iron dextran (Abstract) J Am Soc Nephrol. 2007;18:638A. [Google Scholar]
- Oberley TD, Coursin DB, Cihla HP, Oberley LW, el-Sayyad N, Ho YS. Immunolocalization of manganese superoxide dismutase in normal and transgenic mice expressing the human enzyme. Histochem J. 1993;25:267–279. doi: 10.1007/BF00159118. [DOI] [PubMed] [Google Scholar]
- O’Connell DP, Ragsdale NV, Boyd DG, Felder RA, Carey RM. Differential human renal tubular responses to dopamine type 1 receptor stimulation are determined by blood pressure status. Hypertension. 1997;29:115–122. doi: 10.1161/01.hyp.29.1.115. [DOI] [PubMed] [Google Scholar]
- Offer T, Russo A, Samuni A. The pro-oxidative activity of SOD and nitroxide SOD mimics. FASEB J. 2000;14:1215–1223. doi: 10.1096/fasebj.14.9.1215. [DOI] [PubMed] [Google Scholar]
- Offer T, Samuni A. Nitroxides inhibit peroxyl radical-mediated DNA scission and enzyme inactivation. Free Radic Biol Med. 2002;32:872–881. doi: 10.1016/s0891-5849(02)00750-5. [DOI] [PubMed] [Google Scholar]
- Ogihara T, Asano T, Ando K, Chiba Y, Sakoda H, Anai M, Shojima N, Ono H, Onishi Y, Fujishiro M, et al. Angiotensin II-induced insulin resistance is associated with enhanced insulin signaling. Hypertension. 2002;40:872–879. doi: 10.1161/01.hyp.0000040262.48405.a8. [DOI] [PubMed] [Google Scholar]
- Okajo A, Matsumoto K, Mitchell JB, Krishna MC, Endo K. Competition of nitroxyl contrast agents as an in vivo tissue redox probe: comparison of pharmacokinetics by the bile flow monitoring (BFM) and blood circulating monitoring (BCM) methods using X-band EPR and simulation of decay profiles. Magn Reson Med. 2006;56:422–431. doi: 10.1002/mrm.20958. [DOI] [PubMed] [Google Scholar]
- Omar BA, Gad NM, Jordan MC, Striplin SP, Russell WJ, Downey JM, McCord JM. Cardioprotection by Cu, Zn-superoxide dismutase is lost at high doses in the reoxygenated heart. Free Radic Biol Med. 1990;9:465–471. doi: 10.1016/0891-5849(90)90123-z. [DOI] [PubMed] [Google Scholar]
- Omar BA, McCord JM. The cardioprotective effect of Mn-superoxide dismutase is lost at high doses in the postischemic isolated rabbit heart. Free Radic Biol Med. 1990;9:473–478. doi: 10.1016/0891-5849(90)90124-2. [DOI] [PubMed] [Google Scholar]
- Onishi H, Morales MF. States of myosin subfragment-1 studied by catalyzed ascorbate reduction of bound spin label. Arch Biochem Biophys. 1976;172:12–19. doi: 10.1016/0003-9861(76)90042-4. [DOI] [PubMed] [Google Scholar]
- Onuma S, Nakanishi K. Superoxide dismutase mimetic tempol decreases blood pressure by increasing renal medullary blood flow in hyperinsulinemic-hypertensive rats. Metabolism. 2004;53:1305–1308. doi: 10.1016/j.metabol.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Ortiz MC, Manriquez MC, Romero JC, Juncos LA. Antioxidants block angiotensin II-induced increases in blood pressure and endothelin. Hypertension. 2001a;38:655–659. doi: 10.1161/01.hyp.38.3.655. [DOI] [PubMed] [Google Scholar]
- Ortiz PA, Garvin JL. NO inhibits NaCl absorption by rat thick ascending limb through activation of cGMP-stimulated phosphodiesterase. Hypertension. 2001;37:467–471. doi: 10.1161/01.hyp.37.2.467. [DOI] [PubMed] [Google Scholar]
- Ortiz PA, Garvin JL. Interaction of and NO in the thick ascending limb. Hypertension. 2002a;39:591–596. doi: 10.1161/hy0202.103287. [DOI] [PubMed] [Google Scholar]
- Ortiz PA, Garvin JL. Superoxide stimulates NaCl absorption by the thick ascending limb. Am J Physiol Renal Physiol. 2002b;283:F957–F962. doi: 10.1152/ajprenal.00102.2002. [DOI] [PubMed] [Google Scholar]
- Ortiz PA, Hong NJ, Garvin JL. NO decreases thick ascending limb chloride absorption by reducing Na+-K+-2Cl− cotransporter activity. Am J Physiol Renal Physiol. 2001b;281:F819–F825. doi: 10.1152/ajprenal.2001.281.5.F819. [DOI] [PubMed] [Google Scholar]
- Oteki T, Nagase S, Yokoyama H, Ohya H, Akatsuka T, Tada M, Ueda A, Hirayama A, Koyama A. Evaluation of Adriamycin nephropathy by an in vivo electron paramagnetic resonance. Biochem Biophys Res Commun. 2005;332:326–331. doi: 10.1016/j.bbrc.2005.04.134. [DOI] [PubMed] [Google Scholar]
- Ozawa Y, Hayashi K, Kanda T, Homma K, Takamatsu I, Tatematsu S, Yoshioka K, Kumagai H, Wakino S, Saruta T. Impaired nitric oxide- and endothelium-derived hyperpolarizing factor-dependent dilation of renal afferent arteriole in Dahl salt-sensitive rats. Nephrology (Carlton) 2004;9:272–277. doi: 10.1111/j.1440-1797.2004.00292.x. [DOI] [PubMed] [Google Scholar]
- Paller MS, Eaton JW. Hazards of antioxidant combinations containing superoxide dismutase. Free Radic Biol Med. 1995;18:883–890. doi: 10.1016/0891-5849(94)00222-6. [DOI] [PubMed] [Google Scholar]
- Palm F, Onozato ML, Luo Z, Wilcox CS. Dimethylarginine dimethylaminohydrolase (DDAH): expression, regulation, and function in the cardiovascular and renal systems. Am J Physiol Heart Circ Physiol. 2007;293:H3227–H3245. doi: 10.1152/ajpheart.00998.2007. [DOI] [PubMed] [Google Scholar]
- Park JB, Touyz RM, Chen X, Schiffrin EL. Chronic treatment with a superoxide dismutase mimetic prevents vascular remodeling and progression of hypertension in salt-loaded stroke-prone spontaneously hypertensive rats. Am J Hypertens. 2002;15:78–84. doi: 10.1016/s0895-7061(01)02233-6. [DOI] [PubMed] [Google Scholar]
- Patel K, Chen Y, Dennehy K, Blau J, Connors S, Mendonca M, Tarpey M, Krishna M, Mitchell JB, Welch WJ, et al. Acute antihypertensive action of nitroxides in the spontaneously hypertensive rat. Am J Physiol Regul Integr Comp Physiol. 2006;290:R37–R43. doi: 10.1152/ajpregu.00469.2005. [DOI] [PubMed] [Google Scholar]
- Patterson ME, Mouton CR, Mullins JJ, Mitchell KD. Interactive effects of superoxide anion and nitric oxide on blood pressure and renal hemodynamics in transgenic rats with inducible malignant hypertension. Am J Physiol Renal Physiol. 2005;289:F754–F759. doi: 10.1152/ajprenal.00419.2004. [DOI] [PubMed] [Google Scholar]
- Payne JA, Reckelhoff JF, Khalil RA. Role of oxidative stress in age-related reduction of NO-cGMP-mediated vascular relaxation in SHR. Am J Physiol Regul Integr Comp Physiol. 2003;285:R542–R551. doi: 10.1152/ajpregu.00056.2003. [DOI] [PubMed] [Google Scholar]
- Persichini T, Percario Z, Mazzon E, Colasanti M, Cuzzocrea S, Musci G. Copper activates the NF-κB pathway in vivo. Antioxid Redox Signal. 2006;8:1897–1904. doi: 10.1089/ars.2006.8.1897. [DOI] [PubMed] [Google Scholar]
- Pikula S, Hayden JB, Awasthi S, Awasthi YC, Zimniak P. Organic anion-transporting ATPase of rat liver. II. Functional reconstitution of active transport and regulation by phosphorylation. J Biol Chem. 1994;269:27574–27579. [PubMed] [Google Scholar]
- Pinaud F, Bocquet A, Dumont O, Retailleau K, Baufreton C, Andriantsitohaina R, Loufrani L, Henrion D. Paradoxical role of angiotensin II type 2 receptors in resistance arteries of old rats. Hypertension. 2007;50:96–102. doi: 10.1161/HYPERTENSIONAHA.106.085035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock DM. Endothelin, angiotensin, and oxidative stress in hypertension. Hypertension. 2005;45:477–480. doi: 10.1161/01.HYP.0000158262.11935.d0. [DOI] [PubMed] [Google Scholar]
- Pollock DM, Pollock JS. Evidence for endothelin involvement in the response to high salt. Am J Physiol Renal Physiol. 2001;281:F144–F150. doi: 10.1152/ajprenal.2001.281.1.F144. [DOI] [PubMed] [Google Scholar]
- Prabhutendolkar A, Liu X, Mathias EV, Ba Y, Kornfield JA. Synthesis of chlorambucil-tempol adduct and its delivery using fluoroalkyl double-ended poly (ethylene glycol) micelles. Drug Deliv. 2006;13:433–440. doi: 10.1080/10717540600559452. [DOI] [PubMed] [Google Scholar]
- Preti SC, da Cunha V, Vassallo DV, Stefanon I. The superoxide dismutase mimetic, tempol, reduces the bioavailability of nitric oxide and does not alter L-NAME-induced hypertension in rats. Basic Clin Pharmacol Toxicol. 2005;97:29–34. doi: 10.1111/j.1742-7843.2005.pto_97105.x. [DOI] [PubMed] [Google Scholar]
- Püntmann VO, Hussain MB, Mayr M, Xu Q, Singer DR. Role of oxidative stress in angiotensin-II mediated contraction of human conduit arteries in patients with cardiovascular disease. Vascul Pharmacol. 2005;43:277–282. doi: 10.1016/j.vph.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Qamirani E, Ren Y, Kuo L, Hein TW. C-reactive protein inhibits endothelium-dependent NO-mediated dilation in coronary arterioles by activating p38 kinase and NAD(P)H oxidase. Arterioscler Thromb Vasc Biol. 2005;25:995–1001. doi: 10.1161/01.ATV.0000159890.10526.1e. [DOI] [PubMed] [Google Scholar]
- Racasan S, Braam B, Koomans HA, Joles JA. Brief perinatal inducible NO synthase inhibition and NO supplementation both lead to sustained reduction in blood pressure (Abstract) J Am Soc Nephrol. 2002;13:52A–53A. [Google Scholar]
- Racasan S, Braam B, Koomans HA, Joles JA. Programming blood pressure in adult SHR by shifting perinatal balance of NO and reactive oxygen species toward NO: the inverted Barker phenomenon. Am J Physiol Renal Physiol. 2005;288:F626–F636. doi: 10.1152/ajprenal.00314.2004. [DOI] [PubMed] [Google Scholar]
- Randall MD, Kendall DA. Involvement of a cannabinoid in endothelium-derived hyperpolarizing factor-mediated coronary vasorelaxation. Eur J Pharmacol. 1997;335:205–209. doi: 10.1016/s0014-2999(97)01237-5. [DOI] [PubMed] [Google Scholar]
- Ren Y, Carretero OA, Garvin JL. Mechanism by which superoxide potentiates tubuloglomerular feedback. Hypertension. 2002;39:624–628. doi: 10.1161/hy0202.103299. [DOI] [PubMed] [Google Scholar]
- Risso-de Faverney C, Lafaurie M, Girard JP, Rahmani R. The nitroxide stable radical tempo prevents metal-induced inhibition of CYP1A1 expression and induction. Toxicol Lett. 2000;111:219–227. doi: 10.1016/s0378-4274(99)00183-6. [DOI] [PubMed] [Google Scholar]
- Romanko OP, Stepp DW. Reduced constrictor reactivity balances impaired vasodilation in the mesenteric circulation of the obese Zucker rat. Am J Physiol Heart Circ Physiol. 2005;289:H2097–H2102. doi: 10.1152/ajpheart.00213.2005. [DOI] [PubMed] [Google Scholar]
- Rosenberger C, Khamaisi M, Abassi Z, Shilo V, Weksler-Zangen S, Goldfarb M, Shina A, Zibertrest F, Eckardt KU, Rosen S, et al. Adaptation to hypoxia in the diabetic rat kidney. Kidney Int. 2008;73:34–42. doi: 10.1038/sj.ki.5002567. [DOI] [PubMed] [Google Scholar]
- Ross AH, McConnell HM. Permeation of a spin-label phosphate into the human erythrocyte. Biochemistry. 1975;14:2793–2798. doi: 10.1021/bi00684a001. [DOI] [PubMed] [Google Scholar]
- Rozantsev EG. Free Nitroxyl Radicals. Plenum Press; New York: 1970. [Google Scholar]
- Rozantsev EG, Sholle VD. Organic Chemistry of Free Radicals. Khimiya; Moscow: 1979. [Google Scholar]
- Rozantsev EG, Zhdanov RI. Nitroxyl Radicals. Synthesis, Chemistry and Applications. Nauka; Moscow: 1987. [Google Scholar]
- Rubanyi GM, Vanhoutte PM. Superoxide anion and hyperoxia inactivate endothelium-derived relaxing factor. Am J Physiol. 1986;250:H822–H827. doi: 10.1152/ajpheart.1986.250.5.H822. [DOI] [PubMed] [Google Scholar]
- Ruef J, Moser M, Kübler W, Bode C. Induction of endothelin-1 expression by oxidative stress in vascular smooth muscle cells. Cardiovasc Pathol. 2001;10:311–315. doi: 10.1016/s1054-8807(01)00095-3. [DOI] [PubMed] [Google Scholar]
- Sainz J, Wangensteen R, Rodríguez Gómez I, Moreno JM, Chamorro V, Osuna A, Bueno P, Vargas F. Antioxidant enzymes and effects of tempol on the development of hypertension induced by nitric oxide inhibition. Am J Hypertens. 2005;18:871–877. doi: 10.1016/j.amjhyper.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Saito K, Takeshita K, Ueda J, Ozawa T. Two reaction sites of a spin label, TEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl), with hydroxyl radical. J Pharm Sci. 2003;92:275–280. doi: 10.1002/jps.10304. [DOI] [PubMed] [Google Scholar]
- Samai M, Sharpe MA, Gard PR, Chatterjee PK. Comparison of the effects of the superoxide dismutase mimetics EUK-134 and tempol on paraquat-induced nephrotoxicity. Free Radic Biol Med. 2007;43:528–534. doi: 10.1016/j.freeradbiomed.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Samuni A, Godinger D, Aronovitch J, Russo A, Mitchell JB. Nitroxides block DNA scission and protect cells from oxidative damage. Biochemistry. 1991a;30:555–561. doi: 10.1021/bi00216a033. [DOI] [PubMed] [Google Scholar]
- Samuni A, Karmeli F, Moshen M, Rachmilewitz D. Mechanisms underlying gastric antiulcerative activity of nitroxides in rats. Free Radic Res. 1999;30:133–140. doi: 10.1080/10715769900300141. [DOI] [PubMed] [Google Scholar]
- Samuni A, Krishna CM, Mitchell JB, Collins CR, Russo A. Superoxide reaction with nitroxides. Free Radic Res Commun. 1990a;9:241–249. doi: 10.3109/10715769009145682. [DOI] [PubMed] [Google Scholar]
- Samuni A, Krishna CM, Riesz P, Finkelstein E, Russo A. A novel metal-free low molecular weight superoxide dismutase mimic. J Biol Chem. 1988;263:17921–17924. [PubMed] [Google Scholar]
- Samuni A, Min A, Krishna CM, Mitchell JB, Russo A. SOD-like activity of 5-membered ring nitroxide spin labels. Adv Exp Med Biol. 1990b;264:85–92. doi: 10.1007/978-1-4684-5730-8_12. [DOI] [PubMed] [Google Scholar]
- Samuni A, Mitchell JB, DeGraff W, Krishna CM, Samuni U, Russo A. Nitroxide SOD-mimics: mode of action. Free Radic Res Commun. 1991b;12–13:187–194. doi: 10.3109/10715769109145785. [DOI] [PubMed] [Google Scholar]
- Samuni AM, Barenholz Y. Stable nitroxide radicals protect lipid acyl chains from radiation damage. Free Radic Biol Med. 1997;22:1165–1174. doi: 10.1016/s0891-5849(96)00509-6. [DOI] [PubMed] [Google Scholar]
- Samuni AM, Barenholz Y. Site-activity relationship of nitroxide radical’s antioxidative effect. Free Radic Biol Med. 2003;34:177–185. doi: 10.1016/s0891-5849(02)01238-8. [DOI] [PubMed] [Google Scholar]
- Samuni AM, Barenholz Y, Crommelin DJ, Zuidam NJ. Gamma-irradiation damage to liposomes differing in composition and their protection by nitroxides. Free Radic Biol Med. 1997;23:972–979. doi: 10.1016/s0891-5849(97)00123-8. [DOI] [PubMed] [Google Scholar]
- Samuni AM, DeGraff W, Krishna MC, Mitchell JB. Cellular sites of H2O2-induced damage and their protection by nitroxides. Biochim Biophys Acta. 2001;1525:70–76. doi: 10.1016/s0304-4165(00)00172-0. [DOI] [PubMed] [Google Scholar]
- Samuni AM, DeGraff W, Krishna MC, Mitchell JB. Nitroxides as antioxidants: Tempol protects against EO9 cytotoxicity. Mol Cell Biochem. 2002;234/235:327–333. [PubMed] [Google Scholar]
- Samuni AM, Lipman A, Barenholz Y. Damage to liposomal lipids: protection by antioxidants and cholesterol-mediated dehydration. Chem Phys Lipids. 2000;105:121–134. doi: 10.1016/s0009-3084(99)00136-x. [DOI] [PubMed] [Google Scholar]
- Samuni Y, Gamson J, Samuni A, Yamada K, Russo A, Krishna MC, Mitchell JB. Factors influencing nitroxide reduction and cytotoxicity in vitro. Antioxid Redox Signal. 2004;6:587–595. doi: 10.1089/152308604773934341. [DOI] [PubMed] [Google Scholar]
- Saphier O, Silberstein T, Shames AI, Likhtenshtein GI, Maimon E, Mankuta D, Mazor M, Katz M, Meyerstein D, Meyerstein N. The reduction of a nitroxide spin label as a probe of human blood antioxidant properties. Free Radic Res. 2003;37:301–308. doi: 10.1080/1071576021000050410. [DOI] [PubMed] [Google Scholar]
- Sartori-Valinotti JC, Iliescu R, Fortepiani LA, Yanes LL, Reckelhoff JF. Sex differences in oxidative stress and the impact on blood pressure control and cardiovascular disease. Clin Exp Pharmacol Physiol. 2007;34:938–945. doi: 10.1111/j.1440-1681.2007.04643.x. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Lin LR, Yokoyama T, Sevilla MD, Reddy VN, Giblin FJ. TEMPOL protects against lens DNA strand breaks and cataract in the x-rayed rabbit. Invest Ophthalmol Vis Sci. 1998;39:544–552. [PubMed] [Google Scholar]
- Sasser JM, Pollock JS, Pollock DM. Renal endothelin in chronic angiotensin II hypertension. Am J Physiol Regul Integr Comp Physiol. 2002;283:R243–R248. doi: 10.1152/ajpregu.00086.2002. [DOI] [PubMed] [Google Scholar]
- Sato M, Yanagisawa H, Nojima Y, Tamura J, Wada O. Zn deficiency aggravates hypertension in spontaneously hypertensive rats: possible role of Cu/Zn-superoxide dismutase. Clin Exp Hypertens. 2002;24:355–370. doi: 10.1081/ceh-120004797. [DOI] [PubMed] [Google Scholar]
- Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- Schnackenberg CG, Welch WJ, Wilcox CS. Normalization of blood pressure and renal vascular resistance in SHR with a membrane-permeable superoxide dismutase mimetic: role of nitric oxide. Hypertension. 1998;32:59–64. doi: 10.1161/01.hyp.32.1.59. [DOI] [PubMed] [Google Scholar]
- Schnackenberg CG, Welch WJ, Wilcox CS. TP receptor-mediated vasoconstriction in microperfused afferent arterioles: role O2− and NO. Am J Physiol Renal Physiol. 2000;279:F302–F308. doi: 10.1152/ajprenal.2000.279.2.F302. [DOI] [PubMed] [Google Scholar]
- Schnackenberg CG, Wilcox CS. Two-week administration of tempol attenuates both hypertension and renal excretion of 8-isoprostaglandin F2α. Hypertension. 1999;33:424–428. doi: 10.1161/01.hyp.33.1.424. [DOI] [PubMed] [Google Scholar]
- Schrammel A, Gorren AC, Schmidt K, Pfeiffer S, Mayer B. S-nitrosation of glutathione by nitric oxide, peroxynitrite, and . Free Radic Biol Med. 2003;34:1078–1088. doi: 10.1016/s0891-5849(03)00038-8. [DOI] [PubMed] [Google Scholar]
- Sedeek MH, Llinas MT, Drummond H, Fortepiani L, Abram SR, Alexander BT, Reckelhoff JF, Granger JP. Role of reactive oxygen species in endothelin-induced hypertension. Hypertension. 2003;42:806–810. doi: 10.1161/01.HYP.0000084372.91932.BA. [DOI] [PubMed] [Google Scholar]
- Sharma K, Cook A, Smith M, Valancius C, Inscho EW. TGF-β impairs renal autoregulation via generation of ROS. Am J Physiol Renal Physiol. 2005;288:F1069–F1077. doi: 10.1152/ajprenal.00345.2004. [DOI] [PubMed] [Google Scholar]
- Sharpe MA, Ollosson R, Stewart VC, Clark JB. Oxidation of nitric oxide by oxomanganese-salen complexes: a new mechanism for cellular protection by superoxide dismutase/catalase mimetics. Biochem J. 2002;366:97–107. doi: 10.1042/BJ20020154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shastri S, Gopalakrishnan V, Poduri R, Di Wang H. Tempol selectively attenuates angiotensin II evoked vasoconstrictor responses in spontaneously hypertensive rats. J Hypertens. 2002;20:1381–1391. doi: 10.1097/00004872-200207000-00025. [DOI] [PubMed] [Google Scholar]
- Shen B, Scaiano JC, English AM. Zeolite encapsulation decreases TiO2-photosensitized ROS generation in cultured human skin fibroblasts. Photochem Photobiol. 2006;82:5–12. doi: 10.1562/2005-05-29-RA-551. [DOI] [PubMed] [Google Scholar]
- Shibata S, Nagase M, Yoshida S, Kawachi H, Fujita T. Podocyte as the target for aldosterone: roles of oxidative stress and Sgk1. Hypertension. 2007;49:355–364. doi: 10.1161/01.HYP.0000255636.11931.a2. [DOI] [PubMed] [Google Scholar]
- Shokoji T, Fujisawa Y, Kimura S, Rahman M, Kiyomoto H, Matsubara K, Moriwaki K, Aki Y, Miyatake A, Kohno M, et al. Effects of local administrations of tempol and diethyldithio-carbamic on peripheral nerve activity. Hypertension. 2004;44:236–243. doi: 10.1161/01.HYP.0000136393.26777.63. [DOI] [PubMed] [Google Scholar]
- Shokoji T, Nishiyama A, Fujisawa Y, Hitomi H, Kiyomoto H, Takahashi N, Kimura S, Kohno M, Abe Y. Renal sympathetic nerve responses to tempol in spontaneously hypertensive rats. Hypertension. 2003;41:266–273. doi: 10.1161/01.hyp.0000049621.85474.cf. [DOI] [PubMed] [Google Scholar]
- Skórko-Glonek J, Zurawa D, Kuczwara E, Wozniak M, Wypych Z, Lipinska B. The Escherichia coli heat shock protease HtrA participates in defense against oxidative stress. Mol Gen Genet. 1999;262:342–350. doi: 10.1007/s004380051092. [DOI] [PubMed] [Google Scholar]
- Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J Biol Chem. 1996;271:33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- Smith WL, Marnett LJ. Prostaglandin endoperoxide synthase: structure and catalysis. Biochim Biophys Acta. 1991;1083:1–17. doi: 10.1016/0005-2760(91)90119-3. [DOI] [PubMed] [Google Scholar]
- Song P, Wu Y, Xu J, Xie Z, Dong Y, Zhang M, Zou MH. Reactive nitrogen species induced by hyperglycemia suppresses Akt signaling and triggers apoptosis by upregulating phosphatase PTEN (phosphatase and tensin homologue deleted on chromosome 10) in an LKB1-dependent manner. Circulation. 2007;116:1585–1595. doi: 10.1161/CIRCULATIONAHA.107.716498. [DOI] [PubMed] [Google Scholar]
- Song WZ, Chen AF, Wang DH. Increased salt sensitivity induced by sensory denervation: role of superoxide. Acta Pharmacol Sin. 2004;25:1626–1632. [PubMed] [Google Scholar]
- Soule BP, Hyodo F, Matsumoto K, Simone NL, Cook JA, Krishna MC, Mitchell JB. The chemistry and biology of nitroxide compounds. Free Radic Biol Med. 2007;42:1632–1650. doi: 10.1016/j.freeradbiomed.2007.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart T, Jung FF, Manning J, Vehaskari VM. Kidney immune cell infiltration and oxidative stress contribute to prenatally programmed hypertension. Kidney Int. 2005;68:2180–2188. doi: 10.1111/j.1523-1755.2005.00674.x. [DOI] [PubMed] [Google Scholar]
- Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. 1994;11:95–102. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]
- Stoos BA, Carretero OA, Farhy RD, Scicli G, Garvin JL. Endothelium-derived relaxing factor inhibits transport and increases cGMP content in cultured mouse cortical collecting duct cells. J Clin Invest. 1992;89:761–765. doi: 10.1172/JCI115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoos BA, Carretero OA, Garvin JL. Endothelial-derived nitric oxide inhibits sodium transport by affecting apical membrane channels in cultured collecting duct cells. J Am Soc Nephrol. 1994;4:1855–1860. doi: 10.1681/ASN.V4111855. [DOI] [PubMed] [Google Scholar]
- Stoos BA, Garcia NH, Garvin JL. Nitric oxide inhibits sodium reabsorption in the isolated perfused cortical collecting duct. J Am Soc Nephrol. 1995;6:89–94. doi: 10.1681/ASN.V6189. [DOI] [PubMed] [Google Scholar]
- Suganami T, Mori K, Tanaka I, Mukoyama M, Sugawara A, Makino H, Muro S, Yahata K, Ohuchida S, Maruyama T, et al. Role of prostaglandin E receptor EP1 subtype in the development of renal injury in genetically hypertensive rats. Hypertension. 2003;42:1183–1190. doi: 10.1161/01.HYP.0000101689.64849.97. [DOI] [PubMed] [Google Scholar]
- Sullivan JC, Pollock JS, Pollock DM. Superoxide-dependent hypertension in male and female endothelin B receptor-deficient rats. Exp Biol Med (Maywood) 2006;231:818–823. [PubMed] [Google Scholar]
- Sun C, Sellers KW, Sumners C, Raizada MK. NAD(P)H oxidase inhibition attenuates neuronal chronotropic actions of angiotensin II. Circ Res. 2005;96:659–666. doi: 10.1161/01.RES.0000161257.02571.4b. [DOI] [PubMed] [Google Scholar]
- Swartz HM. Principles of the metabolism of nitroxides and their implications for spin trapping. Free Radic Res Commun. 1990;9:399–405. doi: 10.3109/10715769009145700. [DOI] [PubMed] [Google Scholar]
- Swartz HM, Khan N, Khramtsov VV. Use of electron paramagnetic resonance spectroscopy to evaluate the redox state in vivo. Antioxid Redox Signal. 2007;9:1757–1771. doi: 10.1089/ars.2007.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada M, Yokoyama H, Ito O, Ohya H, Ogata T. Evaluation of the hepatic reduction of a nitroxide radical in rats receiving ascorbic acid, glutathione or ascorbic acid oxidase by in vivo electron spin resonance study. J Gastroenterol Hepatol. 2004;19:99–105. doi: 10.1111/j.1440-1746.2004.03201.x. [DOI] [PubMed] [Google Scholar]
- Tada M, Yokoyama H, Toyoda Y, Ohya H, Ogata T, Kamada H. In vivo ESR study on hepatic reduction of a nitroxide radical after administration of glucose in rats. IUBMB Life. 2001;51:45–48. doi: 10.1080/15216540117208. [DOI] [PubMed] [Google Scholar]
- Takechi K, Tamura H, Yamaoka K, Sakurai H. Pharmacokinetic analysis of free radicals by in vivo BCM (blood circulation monitoring)-ESR method. Free Radic Res. 1997;26:483–496. doi: 10.3109/10715769709097819. [DOI] [PubMed] [Google Scholar]
- Takeshita K, Saito K, Ueda J, Anzai K, Ozawa T. Kinetic study on ESR signal decay of nitroxyl radicals, potent redox probes for in vivo ESR spectroscopy, caused by reactive oxygen species. Biochim Biophys Acta. 2002;1573:156–164. doi: 10.1016/s0304-4165(02)00420-8. [DOI] [PubMed] [Google Scholar]
- Tatchum-Talom R, Martin DS. Tempol improves vascular function in the mesenteric vascular bed of senescent rats. Can J Physiol Pharmacol. 2004;82:200–207. doi: 10.1139/y04-010. [DOI] [PubMed] [Google Scholar]
- Taylor L, Menconi MJ, Polgar P. The participation of hydroperoxides and oxygen radicals in the control of prostaglandin synthesis. J Biol Chem. 1983;258:6855–6857. [PubMed] [Google Scholar]
- Tesfamariam B, Ogletree ML. Dissociation of endothelial cell dysfunction and blood pressure in SHR. Am J Physiol. 1995;269:H189–H194. doi: 10.1152/ajpheart.1995.269.1.H189. [DOI] [PubMed] [Google Scholar]
- Thakali KM, Lau Y, Fink GD, Galligan JJ, Chen AF, Watts SW. Mechanisms of hypertension induced by nitric oxide (NO) deficiency: focus on venous function. J Cardiovasc Pharmacol. 2006;47:742–750. doi: 10.1097/01.fjc.0000211789.37658.e4. [DOI] [PubMed] [Google Scholar]
- Thomas SR, Chen K, Keaney JF., Jr Hydrogen peroxide activates endothelial nitric-oxide synthase through coordinated phosphorylation and dephosphorylation via a phosphoinositide 3-kinase-dependent signaling pathway. J Biol Chem. 2002;277:6017–6024. doi: 10.1074/jbc.M109107200. [DOI] [PubMed] [Google Scholar]
- Timoshin AA, Ruuge EK. Interaction of rat myocardial tissue with nitroxide radicals. Biofizika. 1994;39:709–712. [PubMed] [Google Scholar]
- Tojo A, Asaba K, Onozato ML. Suppressing renal NADPH oxidase to treat diabetic nephropathy. Expert Opin Ther Targets. 2007;11:1011–1018. doi: 10.1517/14728222.11.8.1011. [DOI] [PubMed] [Google Scholar]
- Touyz RM. Reactive oxygen species in vascular biology: role in arterial hypertension. Expert Rev Cardiovasc Ther. 2003;1:91–106. doi: 10.1586/14779072.1.1.91. [DOI] [PubMed] [Google Scholar]
- Touyz RM. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension. 2004;44:248–252. doi: 10.1161/01.HYP.0000138070.47616.9d. [DOI] [PubMed] [Google Scholar]
- Touyz RM. Apocynin, NADPH oxidase, and vascular cells: a complex matter. Hypertension. 2008;51:172–174. doi: 10.1161/HYPERTENSIONAHA.107.103200. [DOI] [PubMed] [Google Scholar]
- Touyz RM, Pu Q, He G, Chen X, Yao G, Neves MF, Viel E. Effects of low dietary magnesium intake on development of hypertension in stroke-prone spontaneously hypertensive rats: role of reactive oxygen species. J Hypertens. 2002;20:2221–2232. doi: 10.1097/00004872-200211000-00022. [DOI] [PubMed] [Google Scholar]
- Touyz RM, Tabet F, Schiffrin EL. Redox-dependent signalling by angiotensin II and vascular remodelling in hypertension. Clin Exp Pharmacol Physiol. 2003;30:860–866. doi: 10.1046/j.1440-1681.2003.03930.x. [DOI] [PubMed] [Google Scholar]
- Touyz RM, Yao G, Viel E, Amiri F, Schiffrin EL. Angiotensin II and endothelin-1 regulate MAP kinases through different redox-dependent mechanisms in human vascular smooth muscle cells. J Hypertens. 2004;22:1141–1149. doi: 10.1097/00004872-200406000-00015. [DOI] [PubMed] [Google Scholar]
- Troncoso Brindeiro CM, da Silva AQ, Allahdadi KJ, Youngblood V, Kanagy NL. Reactive oxygen species contribute to sleep apnea-induced hypertension in rats. Am J Physiol Heart Circ Physiol. 2007;293:H2971–H2976. doi: 10.1152/ajpheart.00219.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udassin R, Haskel Y, Samuni A. Nitroxide radical attenuates ischaemia/reperfusion injury to the rat small intestine. Gut. 1998;42:623–627. doi: 10.1136/gut.42.5.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda A, Nagase S, Yokoyama H, Tada M, Noda H, Ohya H, Kamada H, Hirayama A, Koyama A. Importance of renal mitochondria in the reduction of TEMPOL, a nitroxide radical. Mol Cell Biochem. 2003;244:119–124. [PubMed] [Google Scholar]
- Ueda A, Nagase S, Yokoyama H, Tada M, Ohya H, Kamada H, Hirayama A, Koyama A. Identification by an EPR technique of decreased mitochondrial reducing activity in puromycin aminonucleoside-induced nephrosis. Free Radic Biol Med. 2002;33:1082–1088. doi: 10.1016/s0891-5849(02)00997-8. [DOI] [PubMed] [Google Scholar]
- Valentin F, Field MC, Tippins JR. The mechanism of oxidative stress stabilization of the thromboxane receptor in COS-7 cells. J Biol Chem. 2004;279:8316–8324. doi: 10.1074/jbc.M306761200. [DOI] [PubMed] [Google Scholar]
- van der Poel C, Edwards JN, Macdonald WA, Stephenson DG. Mitochondrial superoxide production in skeletal muscle fibers of the rat and decreased fiber excitability. Am J Physiol Cell Physiol. 2006;292:C1353–C1360. doi: 10.1152/ajpcell.00469.2006. [DOI] [PubMed] [Google Scholar]
- Van Dyke K, Ghareeb E, Van Dyke M, Van Thiel DH. Ultrasensitive peroxynitrite-based luminescence with L-012 as a screening system for antioxidative/antinitrating substances, e.g. Tylenol (acetaminophen), 4-OH tempol, quercetin and carboxy-PTIO. Luminescence. 2007;22:267–274. doi: 10.1002/bio.959. [DOI] [PubMed] [Google Scholar]
- Varela M, Herrera M, Garvin JL. Inhibition of Na-K-ATPase in thick ascending limbs by NO depends on O2− and is diminished by a high-salt diet. Am J Physiol Renal Physiol. 2004;287:F224–F230. doi: 10.1152/ajprenal.00427.2003. [DOI] [PubMed] [Google Scholar]
- Vaziri ND, Dicus M, Ho ND, Boroujerdi-Rad L, Sindhu RK. Oxidative stress and dysregulation of superoxide dismutase and NADPH oxidase in renal insufficiency. Kidney Int. 2003a;63:179–185. doi: 10.1046/j.1523-1755.2003.00702.x. [DOI] [PubMed] [Google Scholar]
- Vaziri ND, Ding Y. Effect of lead on nitric oxide synthase expression in coronary endothelial cells: role of superoxide. Hypertension. 2001;37:223–226. doi: 10.1161/01.hyp.37.2.223. [DOI] [PubMed] [Google Scholar]
- Vaziri ND, Ding Y, Ni Z. Compensatory up-regulation of nitric-oxide synthase isoforms in lead-induced hypertension; reversal by a superoxide dismutase-mimetic drug. J Pharmacol Exp Ther. 2001;298:678–685. [PubMed] [Google Scholar]
- Vaziri ND, Lin CY, Farmand F, Sindhu RK. Superoxide dismutase, catalase, glutathione peroxidase and NADPH oxidase in lead-induced hypertension. Kidney Int. 2003b;63:186–194. doi: 10.1046/j.1523-1755.2003.00711.x. [DOI] [PubMed] [Google Scholar]
- Vejrazka M, Mícek R, Stípek S. Apocynin inhibits NADPH oxidase in phagocytes but stimulates ROS production in non-phagocytic cells. Biochim Biophys Acta. 2005;1722:143–147. doi: 10.1016/j.bbagen.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Vislisel JM, Schafer FQ, Buettner GR. A simple and sensitive assay for ascorbate using a plate reader. Anal Biochem. 2007;365:31–39. doi: 10.1016/j.ab.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanad B, Srinivasan K, Kaul CL, Ramarao P. Effect of tempol on altered angiotensin II and acetylcholine-mediated vascular responses in thoracic aorta isolated from rats with insulin resistance. Pharmacol Res. 2006;53:209–215. doi: 10.1016/j.phrs.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Wakade C, Khan MM, De Sevilla LM, Zhang QG, Mahesh VB, Brann DW. Tamoxifen neuroprotection in cerebral ischemia involves attenuation of kinase activation and superoxide production and potentiation of mitochondrial superoxide dismutase. Endocrinology. 2008;149:367–379. doi: 10.1210/en.2007-0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther FJ, Wade AB, Warburton D, Forman HJ. Augmentation of superoxide dismutase and catalase activity in alveolar type II cells. Am J Respir Cell Mol Biol. 1991;4:364–368. doi: 10.1165/ajrcmb/4.4.364. [DOI] [PubMed] [Google Scholar]
- Wang D, Borrego-Conde LJ, Falck JR, Sharma KK, Wilcox CS, Umans JG. Contributions of NO, EDHF and EETs to endothelium-dependent relaxation in rabbit renal afferent arterioles. Kidney Int. 2003a;63:2187–2193. doi: 10.1046/j.1523-1755.2003.00036.x. [DOI] [PubMed] [Google Scholar]
- Wang D, Chabrashvili T, Borrego L, Aslam S, Umans JG. Angiotensin II infusion alters vascular function in mouse resistance vessels: roles of and endothelium. J Vasc Res. 2006a;43:109–119. doi: 10.1159/000089969. [DOI] [PubMed] [Google Scholar]
- Wang D, Chabrashvili T, Wilcox CS. Enhanced contractility of renal afferent arterioles from angiotensin-infused rabbits: roles of oxidative stress, thromboxane-prostanoid receptors and endothelium. Circ Res. 2004;94:1436–1442. doi: 10.1161/01.RES.0000129578.76799.75. [DOI] [PubMed] [Google Scholar]
- Wang D, Chen Y, Chabrashvili T, Aslam S, Borrego Conde LJ, Umans JG, Wilcox CS. Role of oxidative stress in endothelial dysfunction and enhanced responses to Ang II of afferent arterioles from rabbits infused with Ang II. J Am Soc Nephrol. 2003b;14:2783–2789. doi: 10.1097/01.asn.0000090747.59919.d2. [DOI] [PubMed] [Google Scholar]
- Wang D, Jose P, Wilcox CS. β1 receptors protect the renal afferent arteriole of angiotensin-infused rabbits from norepinephrine-induced oxidative stress. J Am Soc Nephrol. 2006b;17:3347–3354. doi: 10.1681/ASN.2006030212. [DOI] [PubMed] [Google Scholar]
- Wang D, Wang X, Welch WJ, Ji H, Sandberg K, Wu X, Jose P, Wilcox CS. Endothelin is a potent endothelium-derived contracting factor released by the renal afferent arterioles and mesenteric resistance vessels of mice with prolonged oxidative stress due to EC-SOD knockout (Abstract) J Am Soc Nephrol. 2006c;17:716A. [Google Scholar]
- Wang JL, Cheng HF, Zhang MZ, McKanna JA, Harris RC. Selective increase of cyclooxygenase-2 expression in a model of renal ablation. Am J Physiol. 1998;275:F613–F622. doi: 10.1152/ajprenal.1998.275.4.F613. [DOI] [PubMed] [Google Scholar]
- Wang T. Nitric oxide regulates HCO3− and Na+ transport by a cGMP-mediated mechanism in the kidney proximal tubule. Am J Physiol. 1997;272:F242–F248. doi: 10.1152/ajprenal.1997.272.2.F242. [DOI] [PubMed] [Google Scholar]
- Wang T. Role of nitric oxide synthase (nNOS, iNOS, and eNOS) in modulating proximal tubule bicarbonate transport and acid-base balance (Abstract) J Am Soc Nephrol. 2000;11:11A. [Google Scholar]
- Wang T. Role of iNOS and eNOS in modulating proximal tubule transport and acid-base balance. Am J Physiol Renal Physiol. 2002;283:F658–F662. doi: 10.1152/ajprenal.00243.2001. [DOI] [PubMed] [Google Scholar]
- Wang T, Inglis FM, Kalb RG. Defective fluid and HCO3− absorption in proximal tubule of neuronal nitric oxide synthase-knockout mice. Am J Physiol Renal Physiol. 2000;279:F518–F524. doi: 10.1152/ajprenal.2000.279.3.F518. [DOI] [PubMed] [Google Scholar]
- Wang Z, Armando I, Asico LD, Escano C, Wang X, Lu Q, Felder RA, Schnackenberg CG, Sibley DR, Eisner GM, et al. The elevated blood pressure of human GRK4γ A142V transgenic mice is not associated with increased ROS production. Am J Physiol Heart Circ Physiol. 2007;292:H2083–H2092. doi: 10.1152/ajpheart.00944.2006. [DOI] [PubMed] [Google Scholar]
- Wei Y, Whaley-Connell AT, Chen K, Habibi J, Uptergrove GM, Clark SE, Stump CS, Ferrario CM, Sowers JR. NADPH oxidase contributes to vascular inflammation, insulin resistance, and remodeling in the transgenic (mRen2) rat. Hypertension. 2007;50:384–391. doi: 10.1161/HYPERTENSIONAHA.107.089284. [DOI] [PubMed] [Google Scholar]
- Weiss RH, Flickinger AG, Rivers WJ, Hardy MM, Aston KW, Ryan US, Riley DP. Evaluation of activity of putative superoxide dismutase mimics: direct analysis by stopped-flow kinetics. J Biol Chem. 1993;268:23049–23054. [PubMed] [Google Scholar]
- Welch WJ. Adenosine type 1 receptor antagonists in fluid retaining disorders. Expert Opin Investig Drugs. 2002;11:1553–1562. doi: 10.1517/13543784.11.11.1553. [DOI] [PubMed] [Google Scholar]
- Welch WJ, Blau J, Xie H, Chabrashvili T, Wilcox CS. Angiotensin-induced defects in renal oxygenation: role of oxidative stress. Am J Physiol Heart Circ Physiol. 2005a;288:H22–H28. doi: 10.1152/ajpheart.00626.2004. [DOI] [PubMed] [Google Scholar]
- Welch WJ, Chabrashvili T, Solis G, Chen Y, Gill PS, Aslam S, Wang X, Ji H, Sandberg K, Jose P, et al. Role of extracellular superoxide dismutase in the mouse angiotensin slow pressor response. Hypertension. 2006;48:934–941. doi: 10.1161/01.HYP.0000242928.57344.92. [DOI] [PubMed] [Google Scholar]
- Welch WJ, Mendonca M, Aslam S, Wilcox CS. Roles of oxidative stress and AT1 receptors in renal hemodynamics and oxygenation in the post-clipped 2K,1C kidney. Hypertension. 2003;41:692–696. doi: 10.1161/01.HYP.0000052945.84627.8F. [DOI] [PubMed] [Google Scholar]
- Welch WJ, Mendonca M, Blau J, Karber A, Dennehy K, Patel K, Lao YS, José PA, Wilcox CS. Antihypertensive response to prolonged tempol in the spontaneously hypertensive rat. Kidney Int. 2005b;68:179–187. doi: 10.1111/j.1523-1755.2005.00392.x. [DOI] [PubMed] [Google Scholar]
- Welch WJ, Patel K, Modlinger P, Mendonca M, Kawada N, Dennehy K, Aslam S, Wilcox CS. Roles of vasoconstrictor prostaglandins, COX-1 and -2, AT1, AT2, and TP receptors in a rat model of early 2K,1C hypertension. Am J Physiol Heart Circ Physiol. 2007;293:H2644–H2649. doi: 10.1152/ajpheart.00748.2007. [DOI] [PubMed] [Google Scholar]
- Welch WJ, Wilcox CS. AT1 receptor antagonist combats oxidative stress and restores nitric oxide signaling in the SHR. Kidney Int. 2001;59:1257–1263. doi: 10.1046/j.1523-1755.2001.0590041257.x. [DOI] [PubMed] [Google Scholar]
- Whaley-Connell A, Govindarajan G, Habibi J, Hayden MR, Cooper SA, Wei Y, Ma L, Qazi M, Link D, Karuparthi PR, et al. Angiotensin II-mediated oxidative stress promotes myocardial tissue remodeling in the transgenic (mRen2) 27 Ren2 rat. Am J Physiol Endocrinol Metab. 2007;293:E355–E363. doi: 10.1152/ajpendo.00632.2006. [DOI] [PubMed] [Google Scholar]
- Wilcox CS. L-Arginine-NO pathway. In: Seldin DW, Giebisch G, editors. The Kidney: Physiology and Pathophysiology. Raven Press; New York: 2000. pp. 849–871. [Google Scholar]
- Wilcox CS. Reactive oxygen species: role in blood pressure and kidney function. Curr Hypertens Rep. 2002;4:160–166. doi: 10.1007/s11906-002-0041-2. [DOI] [PubMed] [Google Scholar]
- Wilcox CS. Redox regulation of the afferent arteriole and tubuloglomerular feedback. Acta Physiol Scand. 2003;179:217–223. doi: 10.1046/j.0001-6772.2003.01205.x. [DOI] [PubMed] [Google Scholar]
- Wilcox CS. Oxidative stress and nitric oxide deficiency in the kidney: a critical link to hypertension? Am J Physiol Regul Integr Comp Physiol. 2005;289:R913–R935. doi: 10.1152/ajpregu.00250.2005. [DOI] [PubMed] [Google Scholar]
- Wilcox CS, Gutterman D. Focus on oxidative stress in the cardiovascular and renal system. Am J Physiol Heart Circ Physiol. 2005;288:H3–H6. doi: 10.1152/ajpheart.00854.2004. [DOI] [PubMed] [Google Scholar]
- Wilcox CS, Lin L. Vasoconstrictor prostaglandins in angiotensin-dependent and renovascular hypertension. J Nephrol. 1993;6:124–133. [Google Scholar]
- Wilcox CS, Welch WJ. Interaction between nitric oxide and oxygen radicals in regulation of tubuloglomerular feedback. Acta Physiol Scand. 2000;168:119–124. doi: 10.1046/j.1365-201x.2000.00668.x. [DOI] [PubMed] [Google Scholar]
- Wilcox CS, Welch WJ. Oxidative stress: cause or consequence of hypertension? Exp Biol Med (Maywood) 2001;226:619–620. doi: 10.1177/153537020222600702. [DOI] [PubMed] [Google Scholar]
- Wilcox CS, Welch WJ, Murad F, Gross SS, Taylor G, Levi R, Schmidt HH. Nitric oxide synthase in macula densa regulates glomerular capillary pressure. Proc Natl Acad Sci U S A. 1992;89:11993–11997. doi: 10.1073/pnas.89.24.11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JM, Pollock JS, Pollock DM. Arterial pressure response to the antioxidant tempol and ETB receptor blockade in rats on a high-salt diet. Hypertension. 2004;44:770–775. doi: 10.1161/01.HYP.0000144073.42537.06. [DOI] [PubMed] [Google Scholar]
- Wink DA, Feelisch M, Fukuto J, Chistodoulou D, Jourd’heuil D, Grisham MB, Vodovotz Y, Cook JA, Krishna M, DeGraff WG, et al. The cytotoxicity of nitroxyl: possible implications for the pathophysiological role of NO. Arch Biochem Biophys. 1998;351:66–74. doi: 10.1006/abbi.1997.0565. [DOI] [PubMed] [Google Scholar]
- Wolin MS, Xie YW, Hintze TH. Nitric oxide as a regulator of tissue oxygen consumption. Curr Opin Nephrol Hypertens. 1999;8:97–103. doi: 10.1097/00041552-199901000-00015. [DOI] [PubMed] [Google Scholar]
- Wu XC, Johns EJ. Interactions between nitric oxide and superoxide on the neural regulation of proximal fluid reabsorption in hypertensive rats. Exp Physiol. 2004;89:255–261. doi: 10.1113/expphysiol.2003.002640. [DOI] [PubMed] [Google Scholar]
- Wu YJ, Li WG, Zhang ZM, Tian X. Antioxidative activity of 4-oxy- and 4-hydroxy-nitroxides in tissues and erythrocytes from rats. Zhongguo Yao Li Xue Bao. 1997;18:150–154. [PubMed] [Google Scholar]
- Xia L, Wang H, Goldberg HJ, Munk S, Fantus IG, Whiteside CI. Mesangial cell NADPH oxidase upregulation in high glucose is protein kinase C dependent and required for collagen IV expression. Am J Physiol Renal Physiol. 2006;290:F345–F356. doi: 10.1152/ajprenal.00119.2005. [DOI] [PubMed] [Google Scholar]
- Ximenes VF, Kanegae MP, Rissato SR, Galhiane MS. The oxidation of apocynin catalyzed by myeloperoxidase: proposal for NADPH oxidase inhibition. Arch Biochem Biophys. 2007;457:134–141. doi: 10.1016/j.abb.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Xu H, Bian X, Watts SW, Hlavacova A. Activation of vascular BK channel by tempol in DOCA-salt hypertensive rats. Hypertension. 2005;46:1154–1162. doi: 10.1161/01.HYP.0000186278.50275.fa. [DOI] [PubMed] [Google Scholar]
- Xu H, Fink GD, Chen A, Watts S, Galligan JJ. Nitric oxide independent effects of tempol on sympathetic nerve activity and blood pressure in normotensive rats. Am J Physiol Heart Circ Physiol. 2001;281:H975–H980. doi: 10.1152/ajpheart.2001.281.2.H975. [DOI] [PubMed] [Google Scholar]
- Xu H, Fink GD, Galligan JJ. Nitric-oxide-independent effects of tempol on sympathetic nerve activity and blood pressure in DOCA-salt rats. Am J Physiol Heart Circ Physiol. 2002;283:H885–H892. doi: 10.1152/ajpheart.00134.2002. [DOI] [PubMed] [Google Scholar]
- Xu H, Fink GD, Galligan JJ. Tempol lowers blood pressure and sympathetic nerve activity but not vascular in DOCA-salt rats. Hypertension. 2004;43:329–334. doi: 10.1161/01.HYP.0000112304.26158.5c. [DOI] [PubMed] [Google Scholar]
- Xu H, Jackson WF, Fink GD, Galligan JJ. Activation of potassium channels by tempol in arterial smooth muscle cells from normotensive and deoxy-corticosterone acetate-salt hypertensive rats. Hypertension. 2006;48:1080–1087. doi: 10.1161/01.HYP.0000249511.96555.57. [DOI] [PubMed] [Google Scholar]
- Xu J, Wu Y, Song P, Zhang M, Wang S, Zou MH. Proteasome-dependent degradation of guanosine 5′-triphosphate cyclohydrolase I causes tetrahydrobiopterin deficiency in diabetes mellitus. Circulation. 2007;116:944–953. doi: 10.1161/CIRCULATIONAHA.106.684795. [DOI] [PubMed] [Google Scholar]
- Xue B, Johnson AK, Hay M. Sex differences in angiotensin II-induced hypertension. Braz J Med Biol Res. 2007;40:727–734. doi: 10.1590/s0100-879x2007000500018. [DOI] [PubMed] [Google Scholar]
- Yada T, Shimokawa H, Morikawa K, Takaki A, Shinozaki Y, Mori H, Goto M, Ogasawara Y, Kajiya F. Role of Cu, Zn-SOD in the synthesis of endogenous vasodilator hydrogen peroxide during reactive hyperemia in mouse mesenteric microcirculation in vivo. Am J Physiol Heart Circ Physiol. 2008;294:H441–H448. doi: 10.1152/ajpheart.01021.2007. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Nakano T, Kimoto E. Oxidation of nitroxide radicals by the reaction of hemoglobin with hydrogen peroxide. Biochem Biophys Res Commun. 1984;120:534–539. doi: 10.1016/0006-291x(84)91287-7. [DOI] [PubMed] [Google Scholar]
- Yanagisawa H, Sato M, Nodera M, Wada O. Excessive zinc intake elevates systemic blood pressure levels in normotensive rats—potential role of superoxide-induced oxidative stress. J Hypertens. 2004;22:543–550. doi: 10.1097/00004872-200403000-00017. [DOI] [PubMed] [Google Scholar]
- Yanes L, Romero D, Iliescu R, Cucchiarelli VE, Fortepiani LA, Santacruz F, Bell W, Zhang H, Reckelhoff JF. Systemic arterial pressure response to two weeks of tempol therapy in SHR: involvement of NO, the RAS, and oxidative stress. Am J Physiol Regul Integr Comp Physiol. 2005;288:R903–R908. doi: 10.1152/ajpregu.00530.2004. [DOI] [PubMed] [Google Scholar]
- Yang T, Zhang A, Honeggar M, Kohan DE, Mizel D, Sanders K, Hoidal JR, Briggs JP, Schnermann JB. Hypertonic induction of COX-2 in collecting duct cells by reactive oxygen species of mitochondrial origin. J Biol Chem. 2005;280:34966–34973. doi: 10.1074/jbc.M502430200. [DOI] [PubMed] [Google Scholar]
- Yang Z, Asico LD, Yu P, Wang Z, Jones JE, Escano CS, Wang X, Quinn MT, Sibley DR, Romero GG, et al. D5 dopamine receptor regulation of reactive oxygen species production, NADPH oxidase, and blood pressure. Am J Physiol Regul Integr Comp Physiol. 2006;290:R96–R104. doi: 10.1152/ajpregu.00434.2005. [DOI] [PubMed] [Google Scholar]
- Yang ZZ, Zhang AY, Yi FX, Li PL, Zou AP. Redox regulation of HIF-1α levels and expression in renal medullary interstitial cells. Am J Physiol Renal Physiol. 2003;284:F1207–F1215. doi: 10.1152/ajprenal.00017.2002. [DOI] [PubMed] [Google Scholar]
- Yasunari K, Kohno M, Kano H, Minami M, Yoshikawa J. Dopamine as a novel antioxidative agent for rat vascular smooth muscle cells through dopamine D1-like receptors. Circulation. 2000;101:2302–2308. doi: 10.1161/01.cir.101.19.2302. [DOI] [PubMed] [Google Scholar]
- Ye S, Zhong H, Yanamadala S, Campese VM. Oxidative stress mediates the stimulation of sympathetic nerve activity in the phenol renal injury model of hypertension. Hypertension. 2006;48:309–315. doi: 10.1161/01.HYP.0000231307.69761.2e. [DOI] [PubMed] [Google Scholar]
- Yin W, Doss GA, Stearns RA, Chaudhary AG, Hop CE, Franklin RB, Kumar S. A novel P450-catalyzed transformation of the 2,2,6,6-tetramethyl piperidine moiety to a 2,2-dimethyl pyrrolidine in human liver microsomes: characterization by high resolution quadrupole-time-of-flight mass spectrometry and 1H-NMR. Drug Metab Dispos. 2003;31:215–223. doi: 10.1124/dmd.31.2.215. [DOI] [PubMed] [Google Scholar]
- Yin W, Mitra K, Stearns RA, Baillie TA, Kumar S. Conversion of the 2,2,6,6-tetramethylpiperidine moiety to a 2,2-dimethylpyrrolidine by cytochrome P450: evidence for a mechanism involving nitroxide radicals and heme iron. Biochemistry. 2004;43:5455–5466. doi: 10.1021/bi035944q. [DOI] [PubMed] [Google Scholar]
- Ylitalo P, Hepp R, Möhring J, Gross F. Effects of varying sodium intake on blood pressure and renin-angiotensin system in subtotally nephrectomized rats. J Lab Clin Med. 1976;88:807–816. [PubMed] [Google Scholar]
- Yoshino F, Shoji H, Lee MC. Vascular effects of singlet oxygen (1O2) generated by photo-excitation on adrenergic neurotransmission in isolated rabbit mesenteric vein. Redox Rep. 2002;7:266–270. doi: 10.1179/135100002125000767. [DOI] [PubMed] [Google Scholar]
- Yu L, Bao HF, Self JL, Eaton DC, Helms MN. Aldosterone-induced increases in superoxide production counters nitric oxide inhibition of epithelial Na channel activity in A6 distal nephron cells. Am J Physiol Renal Physiol. 2007;293:F1666–F1677. doi: 10.1152/ajprenal.00444.2006. [DOI] [PubMed] [Google Scholar]
- Yura T, Fukunaga M, Khan R, Nassar GN, Badr KF, Montero A. Free-radical-generated F2-isoprostane stimulates cell proliferation and endothelin-1 expression on endothelial cells. Kidney Int. 1999;56:471–478. doi: 10.1046/j.1523-1755.1999.00596.x. [DOI] [PubMed] [Google Scholar]
- Zamir E, Zhang R, Samuni A, Kogan M, Pe’er J. Nitroxide stable radical suppresses autoimmune uveitis in rats. Free Radic Biol Med. 1999;27:7–15. doi: 10.1016/s0891-5849(99)00026-x. [DOI] [PubMed] [Google Scholar]
- Zeltcer G, Berenshtein E, Kitrossky N, Chevion M, Samuni A. Time window of nitroxide effect on myocardial ischemic-reperfusion injury potentiated by iron. Free Radic Biol Med. 2002;32:912–919. doi: 10.1016/s0891-5849(02)00783-9. [DOI] [PubMed] [Google Scholar]
- Zeltcer G, Berenshtein E, Samuni A, Chevion M. Nitroxide radicals prevent metal-aggravated reperfusion injury in isolated rat heart. Free Radic Res. 1997;27:627–635. doi: 10.3109/10715769709097866. [DOI] [PubMed] [Google Scholar]
- Zeng C, Yang Z, Wang Z, Jones J, Wang X, Altea J, Mangrum AJ, Hopfer U, Sibley DR, Eisner GM, et al. Interaction of angiotensin II type 1 and D5 dopamine receptors in renal proximal tubule cells. Hypertension. 2005;45:804–810. doi: 10.1161/01.HYP.0000155212.33212.99. [DOI] [PubMed] [Google Scholar]
- Zhang C, Hein TW, Wang W, Kuo L. Divergent roles of angiotensin II AT1 and AT2 receptors in modulating coronary microvascular function. Circ Res. 2003a;92:322–329. doi: 10.1161/01.res.0000056759.53828.2c. [DOI] [PubMed] [Google Scholar]
- Zhang GX, Kimura S, Nishiyama A, Shokoji T, Rahman M, Abe Y. ROS during the acute phase of Ang II hypertension participates in cardiovascular MAPK activation but not vasoconstriction. Hypertension. 2004a;43:117–124. doi: 10.1161/01.HYP.0000105110.12667.F8. [DOI] [PubMed] [Google Scholar]
- Zhang GX, Nagai Y, Nakagawa T, Miyanaka H, Fujisawa Y, Nishiyama A, Izuishi K, Ohmori K, Kimura S. Involvement of endogenous nitric oxide in angiotensin II-induced activation of vascular mitogen-activated protein kinases. Am J Physiol Heart Circ Physiol. 2007;293:H2403–H2408. doi: 10.1152/ajpheart.00288.2007. [DOI] [PubMed] [Google Scholar]
- Zhang R, Goldstein S, Samuni A. Kinetics of superoxide-induced exchange among nitroxide antioxidants and their oxidized and reduced forms. Free Radic Biol Med. 1999;26:1245–1252. doi: 10.1016/s0891-5849(98)00328-1. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Croft KD, Mori TA, Schyvens CG, McKenzie KU, Whitworth JA. The antioxidant tempol prevents and partially reverses dexamethasone-induced hypertension in the rat. Am J Hypertens. 2004b;17:260–265. doi: 10.1016/j.amjhyper.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Jang R, Mori TA, Croft KD, Schyvens CG, McKenzie KU, Whitworth JA. The antioxidant tempol reverses and partially prevents adrenocorticotrophic hormone-induced hypertension in the rat. J Hypertens. 2003b;21:1513–1518. doi: 10.1097/00004872-200308000-00015. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Rhinehart K, Kwon W, Weinman E, Pallone TL. ANG II signaling in vasa recta pericytes by PKC and reactive oxygen species. Am J Physiol Heart Circ Physiol. 2004c;287:H773–H781. doi: 10.1152/ajpheart.01135.2003. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Rhinehart K, Solis G, Pittner J, Lee-Kwon W, Welch WJ, Wilcox CS, Pallone TL. Chronic Ang II infusion increases NO generation by rat descending vasa recta. Am J Physiol Heart Circ Physiol. 2005;288:H29–H36. doi: 10.1152/ajpheart.00623.2004. [DOI] [PubMed] [Google Scholar]
- Zheng JS, Yang XQ, Lookingland KJ, Fink GD, Hesslinger C, Kapatos G, Kovesdi I, Chen AF. Gene transfer of human guanosine 5′-triphosphate cyclohydrolase I restores vascular tetrahydrobiopterin level and endothelial function in low renin hypertension. Circulation. 2003;108:1238–1245. doi: 10.1161/01.CIR.0000089082.40285.C3. [DOI] [PubMed] [Google Scholar]
- Zhu J, Drenjancevic-Peric I, McEwen S, Friesema J, Schulta D, Yu M, Roman RJ, Lombard JH. Role of superoxide and angiotensin II suppression in salt-induced changes in endothelial Ca2+ signaling and NO production in rat aorta. Am J Physiol Heart Circ Physiol. 2006;291:H929–H938. doi: 10.1152/ajpheart.00692.2005. [DOI] [PubMed] [Google Scholar]
- Zhu J, Huang T, Lombard JH. Effect of high-salt diet on vascular relaxation and oxidative stress in mesenteric resistance arteries. J Vasc Res. 2007;44:382–390. doi: 10.1159/000102955. [DOI] [PubMed] [Google Scholar]
- Zhu J, Mori T, Huang T, Lombard JH. Effect of high-salt diet on NO release and superoxide production in rat aorta. Am J Physiol Heart Circ Physiol. 2004;286:H575–H583. doi: 10.1152/ajpheart.00331.2003. [DOI] [PubMed] [Google Scholar]
- Zicha J, Dobesová Z, Kunes J. Relative deficiency of nitric oxide-dependent vasodilation in salt-hypertensive Dahl rats: the possible role of superoxide anions. J Hypertens. 2001;19:247–254. doi: 10.1097/00004872-200102000-00011. [DOI] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res. 2004;95:210–216. doi: 10.1161/01.RES.0000135483.12297.e4. [DOI] [PubMed] [Google Scholar]
- Zöllner S, Haseloff RF, Kirilyuk IA, Blasig IE, Rubanyi GM. Nitroxides increase the detectable amount of nitric oxide released from endothelial cells. J Biol Chem. 1997;272:23076–23080. doi: 10.1074/jbc.272.37.23076. [DOI] [PubMed] [Google Scholar]
- Zou AP, Li N, Cowley AW., Jr Production and actions of superoxide in the renal medulla. Hypertension. 2001;37:547–553. doi: 10.1161/01.hyp.37.2.547. [DOI] [PubMed] [Google Scholar]
- Zou M, Martin C, Ullrich V. Tyrosine nitration as a mechanism of selective inactivation of prostacyclin synthase by peroxynitrite. J Biol Chem. 1997;378:707–713. doi: 10.1515/bchm.1997.378.7.707. [DOI] [PubMed] [Google Scholar]
- Zs-Nagy I. Chemistry, toxicology, pharmacology and pharmacokinetics of idebenone: a review. Arch Gerontol Geriatr. 1990;11:177–186. doi: 10.1016/0167-4943(90)90063-c. [DOI] [PubMed] [Google Scholar]