

Abstract
We recently reported that zidovudine (AZT) selected for the Q509L mutation in the ribonuclease H (RNase H) domain of HIV-1 reverse transcriptase (RT), which increases resistance to AZT in combination with the thymidine analogue mutations D67N, K70R, and T215F. In the current study, we have defined the mechanism by which Q509L confers AZT resistance by performing in-depth biochemical analyses of wild type, D67N/K70R/T215F and D67N/K70R/T215F/Q509L HIV-1 RT. Our results show that Q509L increases AZT-monophosphate (AZT-MP) excision activity of RT on RNA/DNA template/primers (T/Ps) but not DNA/DNA T/Ps. This increase in excision activity on the RNA/DNA T/P is due to Q509L decreasing a secondary RNase H cleavage event that reduces the RNA/DNA duplex length to 10 nucleotides and significantly impairs the enzyme's ability to excise the chain-terminating nucleotide. Presteady-state kinetic analyses indicate that Q509L does not affect initial rates of the polymerase-directed RNase H activity but only polymerase-independent cleavages that occur after a T/P dissociation event. Furthermore, competition binding assays suggest that Q509L decreases the affinity of the enzyme to bind T/P with duplex lengths less than 18 nucleotides in the polymerase-independent RNase H cleavage mode, while not affecting the enzyme's affinity to bind the same T/P in an AZT-MP excision competent mode. Taken together, this study provides the first mechanistic insights into how a mutation in the RNase H domain of RT increases AZT resistance and highlights how the polymerase and RNase H domains of RT function in concert to confer drug resistance.
HIV-1 reverse transcriptase (RT1) catalyzes the conversion of the viral RNA genome into double-stranded DNA through its DNA polymerase and ribonuclease H (RNase H) activities. RT is a heterodimer consisting of a catalytically active 66 kDa subunit containing DNA polymerase, connection and RNase H domains, and a 51 kDa subunit that contains only the DNA polymerase and connection domains (1). Because of its essential role in HIV-1 replication, RT is a primary target for antiretroviral drug development and two therapeutic classes of RT inhibitors (RTIs) have been approved for the treatment of HIV-1 infection. These include the nucleoside RT inhibitors (NRTI) and the nonnucleoside RT inhibitors (NNRTI). All NRTI, including zidovudine (AZT), lack a 3′-hydroxyl group on the ribose pseudosugar. Once metabolized by host cell kinases to their active triphosphate (TP) forms, the NRTI-TP act as chain-terminators of viral DNA synthesis. Although antiretroviral therapies that include NRTI have profoundly reduced morbidity and mortality from HIV-1 infection, their long-term efficacy is limited by the selection of drug-resistant variants of HIV-1.
Mutations known to confer resistance to NRTIs have been identified by in vitro passage experiments and from viral isolates or sequences amplified from patients experiencing virologic failure on RTI therapy. All of the RTI mutations included in the most widely used resistance tables, such as that from the International AIDS Society-U.S.A. expert panel (2), map to the DNA polymerase domain of HIV-1 RT. However, most genotypic assays do not analyze the connection and RNase H domains of RT, despite the fact that these regions contain key residues that are essential for RT structure-function. Recently, however, compelling evidence has emerged that implicates mutations in the RNase H domain of RT in NRTI resistance. For example, Nikolenko et al. demonstrated that the mutations H539N and D549N in the RNase domain significantly increase AZT resistance alone and in combination with thymidine analogue mutation (TAMs) (3). H539N and D549N, however, have not been reported to be selected by AZT (4). More recently, we carried out in vitro selections of HIV-1 with AZT and examined the entire coding sequence of RT for the emergence of resistance-related mutations (5). This study identified two novel mutations in the connection domain (A371V) and RNase H domain (Q509L) of RT that were selected in combination with D67N, K70R, and T215F. Site-directed mutagenesis studies demonstrated that Q509L increased AZT resistance 7.4-fold when combined with D67N, K70R, and T215F. By comparison, A371V in combination with the same TAMs did not augment AZT resistance, although high-level AZT resistance was observed with viruses containing D67N, K70R, T215F, A371V, and Q509L (5).
The molecular mechanisms by which TAMs confer AZT resistance have been well defined (see refs 6 and 7 for recent reviews). By contrast, the biochemical mechanisms by which mutations outside of the DNA polymerase domain of RT augment AZT resistance have not been thoroughly evaluated. Recent studies by our group and other groups have investigated novel RTI resistance mutations in the connection domain of HIV-1 RT (8-11). To our knowledge, no studies have investigated the mechanism(s) by which mutations in the RNase H domain of RT confer drug resistance. In the current study, we sought to address this issue by determining the mechanism(s) by which Q509L in HIV-1 RT increases AZT resistance when combined with D67N, K70R, and T215F.
EXPERIMENTAL PROCEDURES
Reagents
Wild type (WT) and mutant HIV-1 RTs were constructed, overexpressed in bacteria, and purified to homogeneity, as reported previously (12, 13). The protein concentration of the purified enzymes was determined spectrophotometrically at 280 nm using an extinction coefficient (ε280) of 260450 M-1 cm-1, and by Bradford protein assays (Sigma-Aldrich, St. Louis, MO). The RNA- and DNA-dependent DNA polymerase activities of the purified WT and mutant enzymes were essentially identical (data not shown). 3′-Azido-2′,3′-dideoxythymidine triphosphate (AZTTP) was purchased from TriLink Biotechnologies (San Diego, CA). [3H]dTTP and dNTPs were purchased from GE Healthcare, and [γ-32P]ATP was acquired from PerkinElmer Life Sciences. RNA and DNA oligonucleotides were synthesized by IDT (Coralville, IA).
Inhibition of WT and Mutant RT by AZT-TP
Fixed time point assays were used to determine HIV-1 RT-associated RNA-dependent DNA polymerase activity, as reported previously (14). Briefly, reactions were carried out in 50 mM Tris-HCl at pH 7.5 (37 °C), 50 mM KCl, 10 mM MgCl2, 600 nM of poly(rA)-oligo(dT)18 (the oligo(dT)18 primer was biotinylated on the 5′-end), 25 μM [3H]TTP, and variable concentrations of AZT-TP (0-500 nM). Reactions were initiated by the addition of 25 nM of RT, incubated for 20 min at 37 °C and then quenched with 0.5 M EDTA. Streptavidin Scintillation Proximity Assay beads (GE Healthcare, Piscataway, NJ) were then added to each reaction, and the extent of radionucleotide incorporation was determined by scintillation spectrometry using a 1450 Microbeta Liquid Scintillation Counter (Perkin-Elmer, Waltham, MA).
AZT-monophosphate (AZT-MP) Excision Assays
A 26 nucleotide DNA primer (P; 5′-CCTGTTCGGGCGCCACTGCTAGAGAT-3′) was 5′-radiolabeled with [γ-32P]ATP and chain-terminated with AZT-MP to generate PAZT as reported previously (15, 16). PAZT was then annealed to either a 35 nucleotide DNA (TDNA; 5′-AGAATGGAAAATCTCTAGCAGTGG CGCCCGAACAG-3′) or RNA (TRNA: 5′-AGAAUGGAAAAUCUCUAGCAGUGGCGCCCG AACAG-3′) template. ATP-mediated AZT-MP excision assays were carried out by first incubating 20 nM TRNA/PAZT or TDNA/PAZT with varying concentrations of ATP, 10 mM MgCl2, 1 μM dTTP, and 10 μM ddCTP in a buffer containing 50 mM Tris-HCl (pH 7.5) and 50 mM KCl. Reactions were initiated by the addition of 200 nM WT or mutant RT. Aliquots were removed at defined times, quenched with sample loading buffer (98% deionized formamide, 1 mg/mL each of bromophenol blue and xylenecyanol), denatured at 95 °C for 8 min, and then the product was resolved from substrate by denaturing polyacylamide gel electrophoresis and analyzed, as reported previously (15, 16). Excision assays using the PAZT primer annealed to a series of 3′-recessed templates were also carried out as described previously (17).
Assay for RT RNase H Activity
WT and mutant RT RNase H activity was evaluated using the same AZT-MP chain-terminated RNA/DNA T/P substrate described above, except that the 5′-end of the RNA was 32P-end-labeled. Assays were carried out using 20 nM TRNA/PAZT, 0.3 mM ATP, and 10 mM MgCl2 in a buffer containing 50 mM Tris-HCl (pH 7.5) and 50 mM KCl. Reactions were initiated by the addition of 200 nM WT or mutant HIV-1 RT. Aliquots were removed, quenched at varying times, and analyzed as described above.
Polyacrylamide Gel Electrophoresis Analysis of RT Polymerization Products Formed under Continuous DNA Polymerization Conditions
Heteropolymeric RNA-dependent or DNA-dependent DNA polymerase T/Ps were prepared as reported previously (18, 19). DNA polymerization reactions were carried out by incubating 20 nM heteropolymeric T/P complex with 1 μM concentration of each dNTP, 2 μM of AZT-TP, 3 mM ATP, and 10 mM MgCl2 in buffer containing 50 mM Tris-HCl (pH 7.5) and 50 mM KCl. Reactions were initiated by the addition of 200 nM WT or mutant RT. After defined incubation periods, aliquots were removed from the reaction tube and quenched with equal volumes of gel loading dye. Products were separated by denaturing gel electrophoresis and quantified, as described above.
Presteady-State Kinetic Analyses of RNase H Cleavage
Transient kinetic analyses were used to determine the initial rates of WT and mutant RT RNase H activity. All reactions described below were carried out using an RQF-3 Rapid Quench Instrument (Kintek Corporation, Clarence, PA). A 20 μL solution of 40 nM of TRNA/PAZT and 400 nM WT or mutant RT in 50 mM Tris-HCl (pH7.5), 50 mM KCl, and 2 mM EDTA was rapidly mixed with a 20 μL solution of 50 mM Tris-HCl (pH 7.5) and 50 mM KCl containing 22 mM MgCl2. The final concentrations of RT, T/P, and MgCl2 in the reaction were 200 nM, 20 nM, and 10 mM, respectively. Reactions were quenched by mixing with 50 μL of 50 mM EDTA at times ranging from 17.5 ms to 30 s. Products were separated from substrates and then processed as described above.
Competition Assays for T/P Binding to WT or Mutant RT
Competition binding assays were used to evaluate the affinity of WT or mutant HIV-1 RT for an TRNA/PAZT T/P that has a duplex length of 16 nucleotides (TRNA16/PAZT; see Figure 2C for sequence). Either PAZT or TRNA16 was 5′-end labeled with [γ-32P]-ATP prior to T/P annealing to allow detection of excision or RNase H activity, respectively. WT (200 nM) or mutant RT was first preincubated at 37 °C with 20 nM TRNA 16/PAZT in 50 mM Tris-HCl (pH 7.5), 50 mM KCl, and 0.5 mM EDTA for 15 min before the addition of 10.5 mM MgCl2 and 3 mM ATP, and varying concentrations (0-8 μM) of a nucleic acid trap (TRNA 16/P that was not radioactively labeled or AZT-MP terminated). The residual RNase H or AZT-MP excision activities were evaluated after 20 or 120 min, respectively. Samples were processed as described above.
Figure 2.

(A) Representative autoradiogram of RNase H activity of WT and mutant HIV-1 RT during ATP-mediated AZT-MP excision. The time points in the experiments were 10, 20, 30, 45, 60, 90, 120, 150, and 180 min, respectively. (B) Isotherm for the rate of appearance of the TRNA 10/PAZT RNase H cleavage product generated by WT and mutant HIV-1 during ATP-mediated AZTMP excision. The intensity of the TRNA 10/PAZT RNase H cleavage product was determined by densitometric analyses using Bio-Rad GS525 Molecular Imager FX software. WT, AZTR, and AZTR/Q509L RT are represented by the symbols □ ▽ and Ο respectively. Data was fit to the equation [cleavage product]) A[1 -exp(-kRNaseHt)], where A represents the amplitude for product formation, and kRNaseH is the apparent rate for RNase H cleavage. Error bars represent standard errors from 3 separate experiments. (C) Ability of WT and mutant RT to excise AZT-MP on RNA/DNA T/P with decreasing duplex lengths. The assay incubation time was 30 min. WT, AZTR, and AZTR/Q509L RT are represented by black, white, and gray bars, respectively. Error bars represent standard errors from 3 repeated experiments.
RESULTS
Two distinct mechanisms of HIV-1 resistance to NRTIs have been described (6, 7). The mutations K65R, K70E, L74V, Q151 M, and M184V increase the selectivity of RT for incorporation of the natural dNTP substrate versus the NRTI-triphosphate (NRTI-TP) (16, 20-23). By comparison, TAMs, which include M41L, D67N, K70R, L210W, T215F/Y, and K219Q/E, increase the ability of HIV-1 RT to excise a chain-terminating NRTI-monophosphate (NRTI-MP) from a DNA chain (19, 24). In the experiments described below, we examined both the discrimination and excision pheno-types to elucidate the mechanism(s) by which Q509L confers zidovudine resistance. The enzymes included in this study were WT RT, D67N/K70R/T215F (AZTR) RT, and AZTR/Q509L RT.
Incorporation of AZT-TP by WT and Mutant HIV-1 RT
To determine whether the Q509L mutation affects the ability of RT to discriminate against the incoming nucleotide analogue, we determined the concentration of AZT-TP required to inhibit the incorporation of dTTP into the homopolymeric poly(rA)-oligo(dT)18 T/P by WT or mutant enzymes under steady-state assay conditions. The data show that each of the three recombinant enzymes was equally sensitive to inhibition by AZT-TP (Table 1), indicating that Q509L does not confer zidovudine resistance via a discrimination phenotype.
Table 1.
Inhibition of WT, AZTR, and AZTR/Q509L HIV-1 RT by AZT-TP
| enzyme | IC50 (nM)a | fold-resistance (range)b | p-valuec |
|---|---|---|---|
| WT | 87 ± 12 | ||
| AZTR | 125 ± 16 | 1.4 (1.2-1.7) | 0.11 |
| AZTR/Q509L | 113 ± 11 | 1.3 (1.1-1.6) | 0.18 |
Reported values are the mean and standard error of 3-6 independent experiments.
Upper and lower limits were determined for fold-resistance on the basis of standard errors calculated for IC50 values.
Mutant RT IC50 values were compared to WT IC50 for statistically significant differences using a two-sided Student's t-test.
Excision of AZT-MP by WT and Mutant HIV-1 RT from Chain-Terminated T/Ps
To determine whether Q509L directly altered the efficiency of AZT-MP excision, we investigated the ability of WT and mutant RTs to excise AZT-MP from chain-terminated DNA/DNA and RNA/DNA T/P at both high (3 mM) and low (0.3 mM) concentrations of ATP (Figure 1). These experiments allowed the determination of an apparent rate constant for AZT-MP excision (kexcision) and also the burst or total concentration of excision product generated during the reaction (Table 2).
Figure 1.

(A) ATP-mediated excision of AZT-MP from DNA/DNA T/P by WT and mutant HIV-1 RT at 0.3 mM and 3 mM ATP. WT, AZTR, and AZTR/Q509L RT are represented by the symbols 0, 3, and O, respectively. Error bars represent standard errors from 2-5 repeated experiments. Some error bars are smaller than the size of the symbol. (B) ATP-mediated excision of AZT-MP from RNA/DNA T/P by WT and mutant HIV-1 RT at 0.3 mM and 3 mM ATP. The symbols for all three enzymes are the same as in (A). Apparent excision rate constants (kexcision; shown in Table 2) were determined by fitting the excision isotherms to the equation: [product] = A[1 - exp(-kexcisiont)], where A represents the amplitude for product formation.
Table 2.
Kinetic Rate Constants for AZT-MP Excision by WT and Mutant HIV-1 RT at 0.3 mM and 3 mM ATPa
| 0.3 mM ATP |
3 mM ATP |
|||||||
|---|---|---|---|---|---|---|---|---|
| DNA/DNA T/P |
RNA/DNA T/P |
DNA/DNA T/P |
RNA/DNA T/P |
|||||
| enzyme | burst (nM) | kexcision (min-1) | burst (nM) | kexcision (min-1) | burst (nM) | kexcision (min-1) | burst (nM) | kexcision (min-1) |
| wild type | 13.4 ± 4.6 | 0.021 ± 0.018 | 7.7 ± 4.1 | 0.022 ± 0.008 | 18.6 ± 2.2 | 0.051 ± 0.012 | 14.3 ± 1.6 | 0.031 ± 0.016 |
| AZTR | 17.9 ± 1.0 | 0.060 ± 0.011 | 12.8 ± 1.2 | 0.036 ± 0.006 | 19.1 ± 0.6 | 0.24 ± 0.09 | 19.1 ± 0.7 | 0.109 ± 0.020 |
| AZTR/Q509L | 18.1 ± 0.9 | 0.059 ± 0.005 | 16.1 ± 0.7 | 0.032 ± 0.006 | 19.0 ± 0.2 | 0.31 ± 0.05 | 19.0 ± 0.3 | 0.135 ± 0.013 |
Apparent excision rate constants (kexcision) were determined by fitting the excision isotherms in Figure 1 to the equation: [product] = A[1-exp(-kexcisiont)], where A represents the amplitude for product formation (i.e., burst). Data is the mean ± standard deviation from 3-4 independent experiments.
Consistent with prior reports, AZTR RT was more efficient than WT enzyme in excising AZT-MP from the 3′-end of the primer on both DNA/DNA and RNA/DNA T/P(8,9,15,25). In comparison with the WT enzyme, the rates of AZT-MP excision for AZTR RT were increased 2.9- and 4.7-fold at 0.3 mM and 3.0 mM ATP, respectively, on DNA/DNA T/P and 1.6- and 3.5-fold at 0.3 mM and 3 mM ATP, respectively, on RNA/DNA T/P (Figure 1; Table 2). The efficiency of AZT-MP excision by AZTR/Q509L RT was identical to that of the AZTR enzyme on DNA/DNA T/P, and this result was independent of the ATP concentration used in the assay (Figure 1A; Table 2). By contrast, differences in AZT-MP excision between the AZTR and AZTR/Q509L enzymes were evident on the RNA/DNA T/P at low but not high concentrations of ATP (Figure 1B). At 0.3 mM ATP, the AZTR/Q509L RT was more efficient at excising AZT-MP than the AZTR enzyme, and this increase in excision efficiency was driven predominantly by an increase in the burst concentration and not by an increase in rate (Table 2). At 3 mM ATP, there was no difference in excision activity between the AZTR/Q509L and AZTR enzymes.
RNA Template Degradation by WT and Mutant HIV-1 RT during the AZT-MP Excision Reaction
The data described above suggested to us that Q509L did not exert a direct effect on ATP-mediated excision because there was no change in the rates at which HIV-1 RT unblocked the AZT-MP chain-terminated primer on either DNA/DNA or RNA/DNA T/P. Instead, we only observed increased excision on RNA/DNA T/P at low ATP concentrations where the rates of ATP-mediated excision are slow (Table 2). Because Nikolenko et al. hypothesized that mutations that decrease RNase H activity of RT may increase AZT resistance by limiting RNA template degradation (3), we next evaluated the RNase H activity that occurred during the ATP-mediated excision reaction by WT and mutant RTs, and also determined whether these cleavage events affected the efficiency of the excision reaction.
Figure 2A shows autoradiograms of the RNase H products generated during ATP-mediated excision assays by WT, AZTR, and AZTR/Q509L RT. In comparison with the other two enzymes, AZTR/Q509L RT accumulated more cleavage product with RNA/DNA duplex length of 15 or 16 nucleotides. There was also a significant decrease in the rate of appearance of a cleavage event that reduces the RNA/DNA duplex length to 10 nucleotides (Figure 2A,B); the apparent rate constants for this RNase cleavage event were calculated to be 0.034 min-1, 0.036 min-1, and 0.016 min-1for the WT, AZTR, and AZTR/Q509L RTs, respectively.
To explore the relationship between the efficiency of AZTMP excision and RNase H activity, we next evaluated the ability of WT and mutant HIV-1 RT to excise AZT-MP from a chain-terminated DNA primer that was annealed to different RNA templates that were recessed from the 3′-end, mimicking the T/P products generated by RNase H cleavage, as described previously (17). Consistent with our previous findings ((17) Figure 2C), these analyses demonstrated that the efficiency of AZT-MP excision by WT and mutant RT was severely reduced when the RNA/DNA duplex length was decreased to 10 nucleotides, the duplex length arising from the secondary RNase H cleavage event described in Figure 2A. The finding that Q509L significantly decreases the formation of this 10 nucleotide duplex provides one mechanism by which this mutation enhances AZT-MP excision. It should also be noted that the efficiency of AZTMP excision by AZTR/Q509L RT on the 10 nucleotide duplex was significantly greater than that of either WT or AZTR enzymes (Figure 2C), indicating a second mechanism whereby Q509L enhances AZT-MP excision.
Cumulative Effect of Q509L in Assays That Evaluate Multiple AZT-TP Incorporation and AZT-MP Excision Events
In the experiments described above, we evaluated the AZT-MP excision and RNase H activity of the WT and mutant enzymes on a defined (in terms of sequence and length) T/P. Because both the excision and RNase H activities of RT are likely affected by nucleic acid sequence and length, we next evaluated the ability of WT and mutant enzymes to synthesize DNA in the presence of AZT-TP and ATP using a long heteropolymeric RNA or DNA template, corresponding to the HIV-1 sequence used for (—) strong stop DNA synthesis, primed with a DNA oligonucleotide(18,19). The 173-nucleotide incorporation events needed to produce the full-length DNA product in this assay system allow for multiple AZT-TP incorporation and AZT-MP excision events during the formation of the full-length final product. In the presence of 3 mM ATP, the AZTR/Q509L was significantly more efficient than the AZTR enzyme in synthesizing the full-length product on the RNA/DNA T/P (Figure 3) but not DNA/DNA T/P (data not shown). These results reinforce the findings described in Figure 1 and further demonstrate that the Q509L mutation augments zidovudine resistance on an RNA/DNA T/P but not a DNA/DNA T/P.
Figure 3.
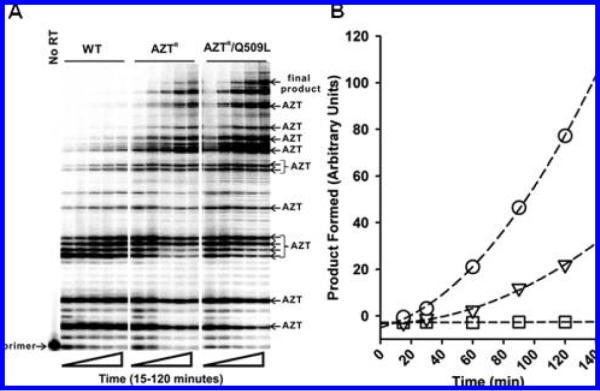
(A) Autoradiogram of steady-state DNA synthesis by WT and mutant HIV-1 RT in the presence of AZT-TP and 3 mM ATP. Experiments were carried out as described in Experimental Procedures. The primer, final product, and AZT-MP chaintermination sites are indicated. (B) Quantitative evaluation of the formation of final product in (A) by WT (0), AZTR (▽), and AZTR/Q509L (○) HIV-1 RT. The intensity of the final product was determined by densiometric analyses using Bio-Rad GS525 Molecular Imager FX software and is reported as arbitrary units.
Presteady-State Kinetic Analyses of RNase H CleaVage by WT and Mutant RT
Our analyses of RNase H activity (Figure 2) demonstrated that Q509L in RT decreases the formation of a secondary RNase H cleavage product that reduces the RNA template to 19 nucleotides (nt) and the RNA/DNA T/P duplex length to 10 nt. However, these assays were carried out using AZT-MP excision reaction conditions, and RNase H cleavage was monitored only in the minute time range. Thus, these studies did not inform if Q509L also affected the primary RNase H cleavage event, which occurs in the millisecond time range, or the sequence of cleavage and T/P dissociation events that generate the 19 nt RNA product. Accordingly, we used presteady-state kinetics to evaluate the rates of initial and secondary RNase H cleavage. The data from these experiments show that the initial rates of RNase H cleavage were similar for the WT, AZTR, and AZTR/Q509L enzymes (Figure 4A). However, as described above, the rate of appearance of the TRNA10/PAZT significantly decreased for AZTR/Q509L RT compared with the WT and AZTR enzymes (Figure 4B).
Figure 4.

(A) Representative autoradiogram and isotherm for the rates of primary polymerase-directed RNase H cleavage by WT (□), AZTR (▽), and AZTR/Q509L (○) HIV-1 RT. Experiments were carried out as described in Experimental Procedures. Primary cleavage was determined by densiometric analyses of the full-length 35 nt RNA template band (or RNA/DNA duplex length of 26 nts) as a function of time (0.035, 0.050, 0.075, 0.15, 0.25, 0.5, 0.75, and 1 s, respectively). Data was fit to the equation [cleavage product]) A[exp(—kRNaseHt)], where A represents the amplitude for product formation, and kRNaseH is the rate for primary RNase H cleavage. (B) Representative autoradiogram and isotherm for the rate of appearance of the final secondary RNase H cleavage product by WT (□), AZTR (▽), and AZTR/Q59L (○) HIV-1 RT. Secondary cleavage was determined by densiometric analyses of the formation of TRNA10/PAZT as a function of time (0.5, 0.75, 1, 2, 4, 8, 16, 20, and 30 min, respectively). Data was fit to the equation [cleavage product]) A[1 - exp(—kRNaseHt)], where A represents the amplitude for product formation, and kRNaseH is the rate for secondary RNase H cleavage. (C) Representative autoradiogram of RNase H cleavage by WT HIV-1 RT in the absence and presence of a nucleic acid trap. The time points were 0.0355 s, 0.050 s, 0.15 s, 1 s, 30 s, 2 min, 4 min, 8 min, and 20 min, respectively. Experiment was carried out as described in Experimental Procedures.
It should be noted that in these RNase H assays, the primary cleavage is polymerase-dependent (i.e., the 3′-end of the DNA primer resides in the polymerase active site), but all subsequent cleavages are polymerase-independent (the 3′-end of the primer cannot reside in the polymerase active site if the enzyme is poised for secondary RNase H cleavage). Because the RT-nucleic acid binding interactions must be different between these 2 modes of binding, we next determined whether RT dissociated from the T/P as it transitioned from one mode to the other by adding a nucleic acid trap to the RNase H reaction upon mixing. The results show that no cleavage products are formed that have RNA/DNA duplexes less than 15-nt for either the WT RT (Figure 4C) or for the AZTR and AZTR/Q509L enzymes (data not shown). These data suggest that RT dissociates from TRNA15/16/PAZT and that it must rebind to this substrate in a polymerase-independent, RNase H competent mode to generate the TRNA10/PAZT cleavage product.
RT-T/P Dissociation from Polymerase-Dependent and Polymerase-Independent RNase H Cleavage Binding Modes
Because RT dissociates and rebinds to TRNA16/PAZT to generate TRNA10/PAZT, we hypothesized that Q509L might decrease the efficiency of this cleavage by directly affecting the binding interactions involved. Accordingly, we next assessed the ability of WT or mutant RTs to bind TRNA16/PAZT in an excision-competent mode or polymerase-independent, RNase H-competent mode by measuring AZTMP excision or RNase H cleavage at defined times after the addition of trap to a preformed RT-T/P complex.
For the RNase H competition binding assays, WT or mutant RT was preincubated with TRNA16/PAZT for 15 min before 10 mM MgCl2 was added to initiate RNase H activity as well as varying concentrations of a nucleic acid trap. Reactions were terminated after 20 min, and the amount of TRNA10/PAZT formed for each trap concentration was compared to a no trap control. The concentrations of trap required to inhibit 50% of the enzyme's RNase H activity were calculated to be 4.5 ± 0.4 μM, 4.2 ± 1.4 μM, and 2.2 (0.8 μM trap for the WT, AZTR, and AZTR/Q509L RTs, respectively (Figure 5). The lower IC50 value for the AZTR/Q509L RT implies that this enzyme is more sensitive to inhibition by trap and therefore likely dissociates more readily from the TRNA16/PAZT substrate.
Figure 5.
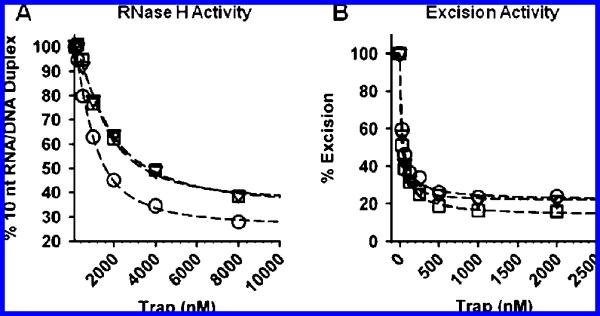
(A) Sensitivity of WT (□), AZTR (▽) and AZTR/Q509L (○) RT to nucleic acid trap when bound to TRNA16/PAZT in an RNase H competent mode. The concentration of trap required to inhibit 50% of the RNase H activity of WT, AZTR, and AZTR/Q509L RT was calculated to be 4.5 ± 0.4 μM, 4.2 ± 1.4 μM, and 2.2 (0.8 μM, respectively. (B) Sensitivity of WT (□), AZTR (▽), and AZTR/Q509L (○) RT to nucleic acid trap when bound to TRNA16/PAZT in an excision competent mode. The concentration of trap required to inhibit 50% of the ATP-mediated AZT-MP excision activity of WT, AZTR, and AZTR/Q509L RT was calculated to be 47 ± 35 nM, 58 ± 37 nM, and 65 ± 20 nM, respectively. Data from both experiments are an average ± standard deviation of at least 2 independent experiments.
For the AZT-MP excision competition binding assays, WT or mutant RT was preincubated with TRNA16/PAZT for 15 min before the reaction was initiated with 3 mM ATP and 10 mM Mg2+, and varying concentrations of trap. Reactions were terminated after 120 min, and the amount of AZT-MP excision product for each trap concentration was again compared to a no trap control. The concentrations of trap required to inhibit 50% of the enzyme's excision activity were calculated to be 47 ± 35 nM, 58 ± 37 nM, and 65 ± 20 nM trap for the WT, AZTR, and AZTR/Q509L RTs, respectively (Figure 5). Since all three enzymes exhibited similar IC50 values, we must conclude that Q509L does not affect the binding interaction between RT and TRNA16/PAZT when the enzyme is bound in a polymerase- or excision-competent mode.
DISCUSSION
Recent studies of HIV sequence databases (4, 9, 26) and genotypic/phenotypic analyses of clinical isolates from patients failing NRTI therapies (27, 28) have identified several mutations in the connection domain of HIV-1 RT that are strongly associated with RTI resistance. Biochemical studies have demonstrated that these mutations impact NRTI sensitivity by several distinct mechanisms. For example, G333D in HIV-1 RT allows the enzyme to effectively discriminate between the normal substrate dCTP and lamivudine-triphosphate (8). It also enhances the ability of RT containing TAMs and M184V to bind AZT-MP terminated T/P, thereby restoring ATP-mediated excision of AZT-MP (8). We and others have also demonstrated that mutations, such as N348I, may increase AZT resistance by decreasing RNA template degradation (9, 11). In addition, some of these mutations may also directly affect the excision activity of RT by an RNase H independent mechanism (10, 11).
In contrast to the identification of novel resistance mutations in the connection domain of RT, very few mutations in the RNase H domain have been identified that impact RTI resistance. This is likely due to the limited availability of sequence data for this domain from laboratory or clinical isolates. Recently, however, we carried out in Vitro selections of HIV-1 with AZT and identified the Q509L mutation in the RNase H domain of RT that was selected in combination with D67N, K70R, and T215F (5). Site-directed mutagenesis studies confirmed the role of this mutation in AZT resistance (5), and the goal of the current study was to define the biochemical mechanisms involved.
Our studies demonstrate that the Q509L mutation confers AZT resistance by increasing the AZT-MP excision activity of the enzyme via an RNase H-dependent mechanism. By using a well-defined RNA/DNA T/P, we show that AZTR/Q509L increases the efficiency of AZT-MP excision by decreasing the frequency of a secondary RNase H cleavage event that reduces the RNA/DNA duplex to 10 nts. The resultant product (TRNA10/PAZT) serves as an inefficient substrate for ATP-mediated excision of AZT-MP by RT. However, even on this T/P, the AZTR/Q509L enzyme has a significantly better ability to unblock the chain-terminated primer than WT or AZTR. It should be noted that under our assay conditions, increased excision was only observed at 0.3 mM ATP and not at 3 mM ATP. This is because at 3 mM ATP, the rate of formation of the TRNA10/PAZT product by AZTR RT is much slower (0.03 min-1) than the rate of AZT-MP excision (0.10 min-1); whereas at 0.3 mM ATP, the rates of T10/PAZT product formation and AZT-MP excision (0.03 min-1) are comparable, allowing competition between these two distinct activities. Because the Q509L mutation decreases the rate of TRNA10/PAZT product formation (to 0.01 min-1), it favors AZT-MP excision at 0.3 mM ATP. The fact that the increase in excision efficiency for the AZTR/Q509L RT, observed in Figure 1B, is driven by an increase in burst concentration and not rate is entirely consistent with a model of competition between RNA template degradation and AZT-MP excision. It is also important to understand that the kinetics of AZT-MP excision are dependent on nucleic acid sequence and structure (29), and therefore, Q509L will likely show a greater effect at positions where AZT-MP excision is slow. This hypothesis is supported by the results showing that AZTR/Q509L RT exhibits a significant advantage over the AZTR enzyme at 3 mM ATP in assays that evaluate multiple AZT-TP and AZT-MP excision events (Figure 3).
We also addressed the mechanism by which Q509L decreases the RNase H activity of RT. In this regard, Nikolenko et al. hypothesized that any mutation in RT that directly decreases the RNase H activity of the enzyme will increase AZT resistance by preventing RNA template degradation (3). However, the replication fitness of viruses with decreased RNase H activities are likely to be compromised due to the important role of this activity in reverse transcription. In our study, we show by transient kinetic analyses that Q509L does not impact the rates of the initial polymerase-directed RNase H cleavage, but only polymerase-independent cleavages that occur after a T/P dissociation event (Figure 4). By contrast, Delviks-Frankenberry et al. reported that NRTI-associated mutations in the connection domain of RT affect both the primary and secondary RNase H cleavages of RT (10). However, these assays were carried out under steady-state assay conditions (i.e., [T/P] ⪢ [RT]) in which the rate-limiting step of the reaction is T/P dissociation, and as such, they do not have the ability to resolve cleavage events that occur in the millisecond time scale.
In Figure 6, we propose a model, based on our findings, to explain how Q509L decreases the RNase H activity of RT. The initial RNase H cleavages reduce the RNA/DNA duplex to 15 to18 nts in length (Figure 2). Because the distance between the DNA polymerase and RNase H active sites is 18 nts (30), RT has to bind the resultant T/P products in one of two distinct conformations to carry out either excision or RNase H activity. Thus, after the primary RNase H cleavages and T/P dissociation event, an equilibrium forms in which RT binds the T/P in either of these configurations (Figure 6). Our data show that Q509L selectively decreases the affinity of RT binding to TRNA16/PAZT in an RNase H competent mode but not in an excision competent mode. The net effect of this is to decrease RNA template degradation and ultimately favor AZT-MP excision. Ehteshami et al. have proposed a similar model for the N348I and A360V connection domain mutations (11).
Figure 6.
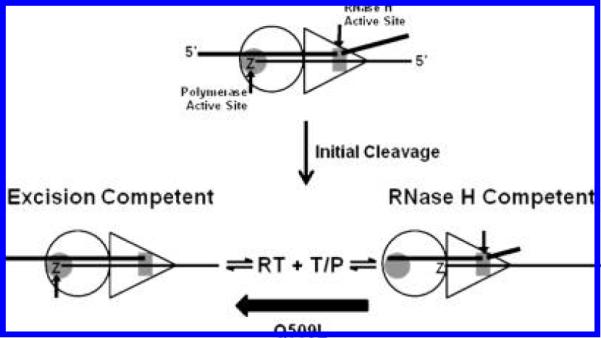
Proposed model for how Q509L in RT affects the balance between AZT-MP excision and RNase H activity on RNA/DNA T/P. The initial RNase H cleavages reduce the RNA/DNA duplex to 15 to 18 nts in length. Because the distance between the DNA polymerase and RNase H active sites is 18 nts, RT has to bind the resultant T/P products in one of two distinct conformations to carry-out either excision or RNase H activity. Therefore, after the primary RNase H cleavages and T/P dissociation event, an equilibrium forms in which RT binds the T/P in both of these configurations. Q509L selectively decreases the affinity of RT to bind the TRNA16/PAZT in an RNase H competent mode but not an excision competent mode.
In conclusion, our data provide evidence that Q509L in HIV-1 RT confers AZT resistance by affecting the balance between AZT-MP excision and RNase H activities of RT on RNA/DNA T/P. Q509L does not appear to directly decrease the RNase H activity of RT, rather it affects the enzyme's ability to bind T/P with short RNA/DNA duplexes in a polymerase-independent RNase H cleavage mode. Furthermore, this study, together with other biochemical studies on NRTI resistance mutations in the connection domain of RT, clearly demonstrates that the entire RT molecule functions in concert to confer drug resistance. Consequently, the inclusion of the C-terminal domains of RT in clinical genotype and phenotype assays could lead to a more accurate determination of NRTI drug resistance.
ACKNOWLEDGMENT
We thank Jessica Radzio for establishing many of the protocols used in this study and Brian Herman for technical assistance with the rapid-quench instrument.
This work was supported by grants from the University of Pittsburgh Clinical and Translational Science Institute (CTSI RR024153) to N.S.-C., the National Cancer Institute 325 (SAIC contract 20XS190A), and the National Institute of Allergy and Infectious Diseases 326 (Virology Support Subcontract from the AACTG Central Group Grant U01AI38858) to J.W.M and the Pitt AIDS Research Training Program (NIH/NIAID 5T32AI065380-03) to J.H.B.
Footnotes
Abbreviations: RT, reverse transcriptase; RNase H, ribonuclease H; RTI, RT inhibitor; NRTI, nucleoside reverse transcriptase inhibitor; NNRTI, nonnucleoside reverse transcriptase inhibitor; AZT, zidovudine or 3′-azido-3′-dideoxythymidine; TP, triphosphate; TAMs, thymidine analogue mutations; WT, wild type; MP, monophosphate; T/P, template/primer; AZTR, D67N/K70R/T215F; nt, nucleotide.
REFERENCES
- 1.Wang J, Smerdon SJ, Jager J, Kohlstaedt LA, Rice PA, Friedman JM, Steitz TA. Structural basis of asymmetry in the human immunodeficiency virus type 1 reverse transcriptase heterodimer. Proc. Natl. Acad. Sci. U.S.A. 1994;91:7242–7246. doi: 10.1073/pnas.91.15.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson VA, Brun-Vezinet F, Clotet B, Gunthard HF, Kuritzkes DR, Pillay D, Schapiro JM, Richman DD. Update of the Drug Resistance Mutations in HIV-1: Spring 2008. Top. HIV Med. 2008;16:62–68. doi: 10.1007/s11750-007-0034-z. [DOI] [PubMed] [Google Scholar]
- 3.Nikolenko GN, Palmer S, Maldarelli F, Mellors JW, Coffin JM, Pathak VK. Mechanism for nucleoside analogmediated abrogation of HIV-1 replication: balance between RNase H activity and nucleotide excision. Proc. Natl. Acad. Sci. U.S.A. 2005;102:2093–2098. doi: 10.1073/pnas.0409823102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roquebert B, Wirden M, Simon A, Deval J, Katlama C, Calvez V, Marelin AG. Relationship between mutations in HIV-1 RNase H domain and nucleoside reverse transcriptase inhibitors resistance mutations in naive and pre-treated HIV infected patients. J. Med. Virol. 2007;79:207–211. doi: 10.1002/jmv.20788. [DOI] [PubMed] [Google Scholar]
- 5.Brehm JH, Koontz D, Meteer JD, Pathak V, Sluis-Cremer N, Mellors JW. Selection of mutations in the connection and RNase H domains of human immunodeficiency virus type 1 reverse transcriptase that increase resistance to 3′-azido-3′-dideoxythymidine. J. Virol. 2007;81:7852–7859. doi: 10.1128/JVI.02203-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sluis-Cremer N. Molecular mechanisms of HIV-1 resistance to nucleoside and nucleotide reverse transcriptase inhibitors. Future HIV Ther. 2007;1:191–201. doi: 10.1007/PL00000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menendez-Arias L. Mechanisms of resistance to nucleoside analogue inhibitors of HIV-1 reverse transcriptase. Virus Res. 2008;134:124–146. doi: 10.1016/j.virusres.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 8.Zelina S, Sheen CW, Radzio J, Mellors JW, Sluis-Cremer N. Mechanisms by which the G333D mutation in human immunodeficiency virus type 1 reverse transcriptase facilitates dual resistance to zidovudine and lamivudine. Antimicrob. Agents Chemother. 2008;52:157–163. doi: 10.1128/AAC.00904-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yap SH, Sheen CW, Fahey J, Zanin M, Tyssen D, Lima VD, Wynhoven B, Kuiper M, Sluis-Cremer N, Harrigan PR, Tachedjian G. N348I in the connection domain of HIV-1 reverse transcriptase confers zidovudine and nevirapine resistance. PLoS Mes. 2007;4:e335. doi: 10.1371/journal.pmed.0040335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delviks-Frankenberry KA, Nikolenko GN, Boyer PL, Hughes SH, Coffin JM, Jere A, Pathak VK. HIV-1 reverse transcriptase connection subdomain mutations reduce template RNA degradation and enhance AZT excision. Proc. Natl. Acad. Sci. U.S.A. 2008;105:10943–10948. doi: 10.1073/pnas.0804660105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ehteshami M, Beilhartz GL, Scarth BJ, Tchesnokov EP, McCormick S, Wynhoven B, Harrigan PR, Gotte M. Connection domain mutations N348I and A360V in HIV-1 reverse transcriptase enhance resistance to 3′-azido-3′-deoxythymidine through both RNase H-dependent and -independent mechanisms. J. Biol. Chem. 2008;283:22222–22232. doi: 10.1074/jbc.M803521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le Grice SF, Gruninger-Leitch F. Rapid purification of homodimer and heterodimer HIV-1 reverse transcriptase by metal chelate affinity chromatography. Eur. J. Biochem. 1990;187:307–314. doi: 10.1111/j.1432-1033.1990.tb15306.x. [DOI] [PubMed] [Google Scholar]
- 13.Le Grice SF, Cameron CE, Benkovic SJ. Purification and characterization of human immunodeficiency virus type 1 reverse transcriptase. Methods Enzymol. 1995;262:130–144. doi: 10.1016/0076-6879(95)62015-x. [DOI] [PubMed] [Google Scholar]
- 14.Nissley DV, Radzio J, Ambrose Z, Sheen CW, Hamamouch N, Moore KL, Tachedjian G, Sluis-Cremer N. Characterization of novel non-nucleoside reverse transcriptase (RT) inhibitor resistance mutations at residues 132 and 135 in the 51 kDa subunit of HIV-1 RT. Biochem. J. 2007;404:151–157. doi: 10.1042/BJ20061814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sluis-Cremer N, Arion D, Parikh U, Koontz D, Schinazi RF, Mellors JW, Parniak MA. The 3′-azido group is not the primary determinant of 3′-azido-3′-deoxythymidine (AZT) responsible for the excision phenotype of AZT-resistant HIV-1. J. Biol. Chem. 2005;280:29047–29052. doi: 10.1074/jbc.M503166200. [DOI] [PubMed] [Google Scholar]
- 16.Sluis-Cremer N, Sheen CW, Zelina S, Torres PS, Parikh UM, Mellors JW. Molecular mechanism by which the K70E mutation in human immunodeficiency virus type 1 reverse transcriptase confers resistance to nucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 2007;51:48–53. doi: 10.1128/AAC.00683-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Radzio J, Sluis-Cremer N. Efavirenz accelerates HIV-1 reverse transcriptase ribonuclease H cleavage, leading to diminished zidovudine excision. Mol. Pharmacol. 2008;73:601–606. doi: 10.1124/mol.107.038596. [DOI] [PubMed] [Google Scholar]
- 18.Arts EJ, Li X, Gu Z, Kleiman L, Parniak MA, Wainberg MA. Comparison of deoxyoligonucleotide and tRNA(Lys-3) as primers in an endogenous human immunodeficiency virus-1 in vitro reverse transcription/template-switching reaction. J. Biol. Chem. 1994;269:14672–14680. [PubMed] [Google Scholar]
- 19.Arion D, Kaushik N, McCormick S, Borkow G, Parniak MA. Phenotypic mechanism of HIV-1 resistance to 3′-azido-3′-deoxythymidine (AZT): increased polymerization processivity and enhanced sensitivity to pyrophosphate of the mutant viral reverse transcriptase. Biochemistry. 1998;37:15908–15917. doi: 10.1021/bi981200e. [DOI] [PubMed] [Google Scholar]
- 20.Feng JY, Anderson KS. Mechanistic studies examining the efficiency and fidelity of DNA synthesis by the 3TCresistant mutant (184V) of HIV-1 reverse transcriptase. Biochemistry. 1999;38:9440–9448. doi: 10.1021/bi990709m. [DOI] [PubMed] [Google Scholar]
- 21.Selmi B, Boretto J, Sarfati SR, Guerreiro C, Canard B. Mechanism-based suppression of dideoxynucleotide resistance by K65R human immunodeficiency virus reverse transcriptase using an alpha-boranophosphate nucleoside analogue. J. Biol. Chem. 2001;276:48466–48472. doi: 10.1074/jbc.M107003200. [DOI] [PubMed] [Google Scholar]
- 22.Deval J, Selmi B, Boretto J, Egloff MP, Guerreiro C, Sarfati S, Canard B. The molecular mechanism of multidrug resistance by the Q151M human immunodeficiency virus type 1 reverse transcriptase and its suppression using alpha-boranophosphate nucleotide analogues. J. Biol. Chem. 2002;277:42097–42104. doi: 10.1074/jbc.M206725200. [DOI] [PubMed] [Google Scholar]
- 23.Deval J, Navarro JM, Selmi B, Courcambeck J, Boretto J, Halfon P, Garrido-Urbani S, Sire J, Canard B. A loss of viral replicative capacity correlates with altered DNA polymerization kinetics by the human immunodeficiency virus reverse transcriptase bearing the K65R and L74V dideoxynucleoside resistance substitutions. J. Biol. Chem. 2004;279:25489–25496. doi: 10.1074/jbc.M313534200. [DOI] [PubMed] [Google Scholar]
- 24.Meyer PR, Matsuura SE, Mian AM, So AG, Scott WA. A mechanism of AZT resistance: an increase in nucleotide-dependent primer unblocking by mutant HIV-1 reverse transcriptase. Mol. Cell. 1999;4:35–43. doi: 10.1016/s1097-2765(00)80185-9. [DOI] [PubMed] [Google Scholar]
- 25.Parikh UM, Zelina S, Sluis-Cremer N, Mellors JW. Molecular mechanisms of bidirectional antagonism between K65R and thymidine analog mutations in HIV-1 reverse transcriptase. AIDS. 2007;21:1405–1414. doi: 10.1097/QAD.0b013e3281ac229b. [DOI] [PubMed] [Google Scholar]
- 26.Santos AF, Lengruber RB, Soares EA, Jere A, Sprinz E, Martinez AM, Silveira J, Sion FS, Pathak VK, Soares MA. Conservation patterns of HIV-1 RT connection and RNase H domains: identification of new mutations in NRTI-treated patients. PLoS One. 2008;3:e1781. doi: 10.1371/journal.pone.0001781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kemp SD, Shi C, Bloor S, Harrigan PR, Mellors JW, Larder BA. A novel polymorphism at codon 333 of human immunodeficiency virus type 1 reverse transcriptase can facilitate dual resistance to zidovudine and L-2′,3′-dideoxy-3′-thiacytidine. J. Virol. 1998;72:5093–5098. doi: 10.1128/jvi.72.6.5093-5098.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nikolenko GN, Delviks-Frankenberry KA, Palmer S, Maldarelli F, Fivash MJ, Jr., Coffin JM, Pathak VK. Mutations in the connection domain of HIV-1 reverse transcriptase increase 3′-azido-3′-deoxythymidine resistance. Proc. Natl. Acad. Sci. U.S.A. 2007;104:317–322. doi: 10.1073/pnas.0609642104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meyer PR, Smith AJ, Matsuura SE, Scott WA. Effects of primer-template sequence on ATP-dependent removal of chain-terminating nucleotide analogues by HIV-1 reverse transcriptase. J. Biol. Chem. 2004;279:45389–45398. doi: 10.1074/jbc.M405072200. [DOI] [PubMed] [Google Scholar]
- 30.Gotte M, Maier G, Gross HJ, Heumann H. Localization of the active site of HIV-1 reverse transcriptaseassociated RNase H domain on a DNA template using site-specific generated hydroxyl radicals. J. Biol. Chem. 1998;273:10139–10146. doi: 10.1074/jbc.273.17.10139. [DOI] [PubMed] [Google Scholar]