

Summary
In this article, the history of the LDL receptor is recounted by its co-discoverers. Their early work on the LDL receptor explained a genetic cause of heart attacks and led to new ways of thinking about cholesterol metabolism. The LDL receptor discovery also introduced three general concepts to cell biology: receptor-mediated endocytosis, receptor recycling, and feedback regulation of receptors. The latter concept provides the mechanism by which statins selectively lower plasma LDL, reducing heart attacks and prolonging life.
In 1972 the two of us, working as newly appointed Assistant Professors in Medicine at the University of Texas Southwestern Medical Center in Dallas, set out to unravel a human genetic disease called familial hypercholesterolemia (FH). In patients with FH, the concentration of cholesterol in blood is elevated many times above normal, and heart attacks occur early in life. FH is inherited as an autosomal dominant trait. When we began our work 37 years ago, the underlying defects in about 35 inborn errors of metabolism were known. All of these diseases were recessive, and all were caused by deficiencies in enzymes (e.g., phenylketonuria, galactosemia). This observation gave rise to the general notion that a 50% deficiency of an enzyme, as in heterozygotes for enzyme deficiencies, would not be sufficient to cause disease.
Because the FH mutation caused disease in the heterozygous state, we suspected that the basic defect in FH would not be an enzyme deficiency. Rather, we hypothesized that FH would be a defect in regulation. A 50% deficiency of a regulatory gene product might disrupt a homeostatic mechanism and produce disease. Cholesterol synthesis had already been shown by others to be subject to end-product feedback regulation, and we speculated that FH subjects had a defective protein that participates in this regulation. The possibility excited us because genetic defects in feedback regulation had not been observed, and we hoped that study of this disease might expose a fundamental regulatory mechanism.
Cholesterol is an essential component of the plasma membrane of animal cells, where it maintains the barrier function between cells and environment, modulates fluidity, and creates “rafts” that concentrate signaling molecules. Cholesterol is also the precursor for the manufacture of all steroid hormones and bile acids, and it plays a crucial role in the formation of the myelin sheath that surrounds axons. In the bloodstream of humans and other vertebrates, cholesterol is transported in lipoprotein particles. In the 1950's and 1960's, physiologists delineated the two major cholesterol-carrying blood lipoproteins: low density lipoprotein (LDL) and high density lipoprotein (HDL). Epidemiologists observed that elevated concentrations of LDL predispose to heart attacks, whereas elevated amounts of HDL are protective; and physicians learned that the excess circulating cholesterol in FH patients is contained in LDL, not HDL.
Our approach to unraveling the genetic defect in FH was to apply the techniques of cell culture. Our studies led to the discovery of a cell surface receptor for LDL and to the elucidation of the mechanism by which this receptor carries LDL particles into cells through coated pits and coated vesicles. Within the cell, LDL-derived cholesterol elicits several regulatory functions, including feedback inhibition of cholesterol synthesis. We soon found that FH is caused by genetic defects in the LDL receptor. These defects disrupt the normal regulation of cholesterol metabolism. Moreover, the LDL receptor studies provided clear evidence for selective uptake of macromolecules into cells, giving rise to the concept of receptor-mediated endocytosis.
Background
In 1938 Carl Müller, a Norwegian clinician, described FH as an “inborn error of metabolism” that produces high blood cholesterol and myocardial infarctions (heart attacks) in young people 1. Müller concluded that FH is transmitted as an autosomal dominant trait. In 1964 Khachadurian, at the American University in Beirut, showed that FH exists in two forms: the less severe heterozygous form and the more severe homozygous form 2.
FH heterozygotes are now known to carry a single copy of a mutant LDL receptor gene. They are quite common, accounting for 1 out of every 500 persons among most ethnic groups throughout the world 3. FH heterozygotes have a 50% deficiency of LDL receptors, which produces a two-fold increase in the number of LDL particles in blood from birth. These individuals begin to have heart attacks at 30 years of age. Among people under age 60 who suffer myocardial infarctions, about 5% have heterozygous FH.
The attractiveness of FH as an experimental model stems from the existence of homozygotes (Figure 1). These individuals, who number about 1 in 1 million, inherit two mutant LDL receptor genes, one from each parent. Their disease is severe. They have six to ten-fold elevations in plasma LDL from birth, and they often have heart attacks in early childhood 3. The early atherosclerosis in homozygous FH children who do not have any other risk factors (e.g., smoking, hypertension, diabetes, type A personality) provides formal genetic proof that elevated LDL alone can produce atherosclerosis in humans. From an experimental viewpoint, the availability of FH homozygotes permits study of the manifestations of the mutant allele in its purest form without any confounding effects from the normal allele.
Figure 1.
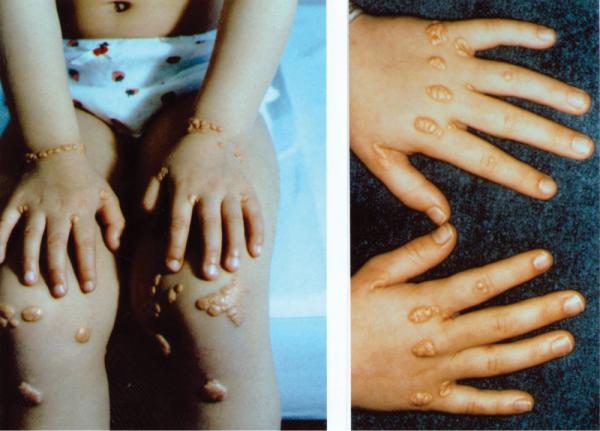
A 10-year-old girl with homozygous FH. Note the elevated orange-yellow xanthomas lying superficially over the knees, the wrists, and interdigital webs. These xanthomas arise from the deposition of plasma LDL-derived cholesterol into macrophages of the skin. The rate of deposition is proportional to the severity and duration of the elevation in plasma LDL. A similar deposition of LDL-derived cholesterol occurred in the coronary arteries of this girl, producing atheromas of artery wall, which led to her first myocardial infarction at age 8.
When we began our studies in 1972, it was felt that all of the important events in cholesterol metabolism take place in the liver or intestine. It was impossible to perform meaningful studies in livers of humans with FH. Our only chance depended on the mutant phenotype being faithfully manifest in cells, such as skin fibroblasts, that can be obtained from human biopsies and cultured in vitro. Techniques for growing such cells had been established over the preceding two decades. Moreover, a handful of inherited enzyme defects were known to be manifest in cultured fibroblasts from patients with rare recessive diseases, such as galactosemia and the Lesch-Nyhan syndrome.
There was some reason to believe that the FH derangement might be manifest in cultured fibroblasts. Studies in the 1960's by Bailey 4 and by Rothblat 5 had demonstrated that cultured mouse lymphoblasts and L cells synthesize cholesterol and that this synthesis is subject to feedback regulation. They showed that when whole serum, containing lipoproteins, is present in the medium, cultured cells produce little cholesterol from radioactive acetate. When serum lipoproteins are removed from the culture medium, cholesterol synthesis increases. Incubation of cells with radiolabeled acetate is a convenient way to monitor the overall action of the 26 enzymes that are needed to convert the simple 2-carbon precursor (acetate) to the complex 27-carbon, four-ring structure of cholesterol. This biosynthetic pathway was delineated in the 1950's largely through the studies of Konrad Bloch, who received the 1964 Nobel Prize in Physiology or Medicine for this tour de force of biochemistry.
Discovery of the LDL Receptor
When we began in 1972, both of us had been trained in enzyme biochemistry. Instead of measuring cholesterol synthesis from acetate, we decided to measure the activity of a single enzyme, 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG CoA reductase), in extracts of cultured fibroblasts 6. Earlier studies in rat livers had shown that HMG CoA reductase catalyzes a rate-limiting step in cholesterol production, and its activity is reduced when rats ingest cholesterol 7. We established a micro-assay for HMG CoA reductase using the trace amounts of material available from cultured fibroblasts 6. Immediately, we observed feedback regulation. When normal human fibroblasts were grown in the presence of serum, HMG CoA reductase activity was low. As shown in Figure 2A, when the cholesterol-carrying lipoproteins were removed from the culture medium, the activity of HMG CoA reductase rose by 50-fold over a 24 hour period. The induced enzyme was rapidly suppressed when LDL was added back to the medium (Figure 2B).
Figure 2.
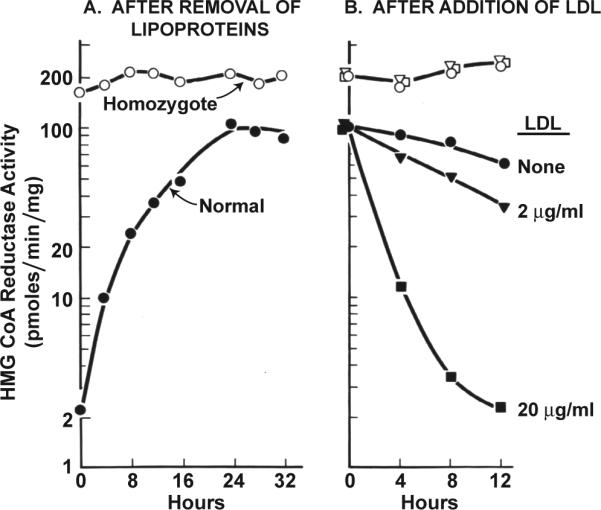
Regulation of HMG CoA reductase activity in fibroblasts from a normal subject and from an FH homozygote. A, Monolayers of cells were grown in dishes containing 10% fetal calf serum. On day 6 of cell growth (zero time), the medium was replaced with fresh medium containing 5% human serum from which the lipoproteins had been removed. At the indicated time, extracts were prepared, and HMG CoA reductase activity was measured. B, 24 hours after addition of 5% human lipoprotein-deficient serum, human LDL was added to give the indicated cholesterol concentration. HMG CoA reductase activity was measured in cell free extracts at the indicated time. (Modified from 8).
Not all lipoproteins could suppress HMG CoA reductase activity. Of the two major cholesterol-carrying lipoproteins in human plasma, LDL and HDL, only LDL was effective 6. This specificity was our first clue that a receptor might be involved. Our second clue was the concentration of LDL that was required. The lipoprotein was active at concentrations as low as 5 μg of protein per ml, which is ∼ 10−9 molar. A high affinity receptor mechanism seemed likely. Study of receptors was in its infancy, and none had been characterized at a molecular level.
The key to the receptor mechanism emerged in 1973 from studies of cells from patients with homozygous FH 8. When grown in serum containing lipoproteins, the homozygous FH cells had HMG CoA reductase activities that were 50 to 100-fold above normal (Figure 2A). This activity did not increase significantly when the lipoproteins were removed from the serum, and there was no suppression when LDL was added back (Figure 2B).
The simplest interpretation of these results was that FH homozygotes have a defect in the gene encoding HMG CoA reductase that renders the enzyme resistant to feedback regulation by LDL-derived cholesterol. This working hypothesis was immediately disproved by our next experiment. We delivered cholesterol in ethanol instead of in LDL. When mixed with albumin-containing solutions, cholesterol forms a quasi-soluble emulsion that enters cells by adsorption to the plasma membrane. When cholesterol was added in this form, the HMG CoA reductase activities of normal and FH homozygote fibroblasts were equally suppressed 9.
Clearly, the defect in the FH homozygote cells must reside in their ability to extract cholesterol from the lipoprotein, and not in the ability of the cholesterol, once extracted by the cells, to act. But how do normal cells extract the cholesterol of LDL? The high affinity action of LDL suggested that a cell surface receptor was involved. The existence of cell surface receptors for hormones (such as epinephrine and glucagon) had been postulated for many years. It was generally thought that these receptors act by binding the ligand at the surface and then generating a “second messenger” on the intracellular side of the plasma membrane. In 1973 the classic “second messenger” was cyclic AMP. We gave some thought to the hypothesis that LDL acted by binding to a receptor and generating some second messenger that suppresses HMG CoA reductase.
The existence of the postulated LDL receptor was formally demonstrated in 1974 when we radiolabeled LDL with 125Iodine and showed that normal cells have high affinity binding sites for 125I-LDL, whereas FH homozygote cells lack these sites 10. This seemed to explain the genetic defect in FH, but it did not reveal how LDL releases its cholesterol so as to suppress HMG CoA reductase. The answer came from studies of the fate of the surface-bound 125I-LDL. Techniques were developed to distinguish surface-bound from intracellular 125I-LDL 11, and these revealed that the receptor-bound LDL remained on the surface of normal cells for less than 10 min on average (Figure 3A). Within this time most of the surface-bound LDL particles entered the cell; within another 60 min the protein component of 125I-LDL was digested completely to amino acids and the 125I, which had been attached to tyrosine residues on LDL, was released into the culture medium as 125I-monoiodotyrosine (Figure 3A) 10, 11. Meanwhile, the cholesteryl esters of LDL were hydrolyzed, generating unesterified cholesterol that remained within the cell 12.
Figure 3.

Internalization and degradation at 37° C of 125I-LDL by fibroblasts from a normal subject (A) and from J.D., a patient with the internalization-defective form of FH (B). Each cell monolayer was allowed to bind 125I-LDL (10 μg protein/ml) at 4° C for 2 hours, after which the cells were washed extensively. In one set of dishes, the amount of 125I-LDL bound was determined by measuring the amount of 125I-LDL that could be released from the surface by treatment with heparin. Replicate dishes then received warm medium and were incubated at 37° C. After the indicated interval, the dishes were rapidly chilled to 4° C, and the amounts of surface-bound (heparin-releasable) 125I-LDL, internalized (heparin-resistant) 125I-LDL, and degraded (trichloroacetic acid-soluble) 125I-LDL were measured. (Modified from 27).
Our laboratory purified the LDL receptor from bovine adrenal glands in 1982 13, cloned its human cDNA shortly thereafter 14, and isolated the gene in 1985 15. These advances laid the groundwork for the molecular analysis of the mutations underlying FH 3. As of January 2009, more than 1100 mutations in the LDL receptor gene have been identified in FH patients.
The LDL Receptor Pathway and Cholesterol Homeostasis
Having demonstrated the receptor-mediated internalization of LDL, we next sought to determine where in the cell the LDL was degraded and how this degradation suppressed HMG CoA reductase activity and cholesterol synthesis. The only cellular organelle in which LDL could have been degraded so completely and rapidly is the lysosome, which contains acid hydrolases that could easily digest all of the components of LDL (Figure 4). We confirmed the lysosomal digestion of LDL through the use of chloroquine 16, which raises the pH of lysosomes and inhibits lysosomal enzymes, and through studies of cultured fibroblasts from patients with a genetic deficiency of lysosomal acid lipase 17. Cells from these patients bound and internalized LDL but failed to hydrolyze its cholesteryl esters, even though they did degrade the protein component.
Figure 4.

Sequential steps in the LDL receptor pathway of mammalian cells (already defined in text). (Modified from 58).
The cholesterol that is generated from LDL within the lysosome proved to be the agent responsible for suppressing HMG CoA reductase activity (Figure 4). We now know that LDL-derived cholesterol acts at several levels, including suppression of transcription of the HMG CoA reductase gene through the sterol regulatory element-binding protein (SREBP) pathway (discussed below) 18 and acceleration of the degradation of the enzyme protein 19. The LDL-derived cholesterol also regulates other processes in a coordinated action that stabilizes the cell's cholesterol content. It activates a cholesterol-esterifying enzyme, acyl CoA: cholesterol acyltransferase (ACAT), so that excess cholesterol can be stored as cholesteryl ester droplets in the cytoplasm 20. By inhibiting the SREBP pathway, LDL also suppresses transcription of the LDL receptor gene 18. This latter action allows cells to adjust the number of LDL receptors to provide sufficient cholesterol for metabolic needs without producing cholesterol over accumulation 21. Through these regulatory mechanisms, cells keep the level of unesterified cholesterol in membranes remarkably constant despite wide fluctuations in cholesterol requirements and exogenous supply.
Figure 4 summarizes the sequential steps in the LDL receptor pathway as deduced from the biochemical and genetic studies performed between 1972 and 1977 22. Figure 5 shows the striking “allor-none” biochemical differences in the metabolism of LDL and its regulatory actions in fibroblasts derived from a normal subject and from an FH homozygote with a complete deficiency of LDL receptors.
Figure 5.

Actions attributable to the LDL receptor in fibroblasts from a normal subject and from a homozygote with the receptor-negative form of FH . Cells were incubated with varying concentrations of 125I-LDL or unlabeled LDL at 37° C for 5 hours and assayed as described 58. All data are normalized to 1 mg of total cell protein. The units for each assay are as follows: Binding, μg of 125I-LDL bound to cell surface; Internalization, μg of 125I-LDL contained within the cell; Hydrolysis of apoprotein B-100, μg of 125I-LDL degraded to 125I-monoiodotyrosine per hour; Hydrolysis of cholesteryl esters, nmol of [3H]cholesterol formed per hour from the hydrolysis of LDL labeled with [3H]cholesteryl linoleate; Cholesterol synthesis, nmol of [14C]acetate incorporated into [14C]cholesterol per hour by intact cells; Cholesterol esterification, nmol of [14C]oleate incorporated into cholesteryl [14C]oleate per hour by intact cells. (Modified from 58).
Receptor-Mediated Endocytosis: Origin of the Concept
The rapidity of internalization and degradation of receptor-bound LDL (Figure 3A) implied that cells possess a special mechanism for transport of receptor-bound LDL from the cell surface to the lysosome. The likely mechanism was endocytosis, the process by which surface membranes pouch inward and pinch off to form vesicles. Endocytosis was first demonstrated by cinematography of phagocytic cells in the 1930's, and its universal occurrence was established in the 1950's by the electron microscopic studies of Palade. Prior to 1975, endocytosis was felt to be a nonspecific process that transported bulk fluid and its contents into cells. There was no precedent for selective entry of specific receptors into cells by this route.
To determine whether endocytosis was involved in LDL uptake, we began in 1975 a collaboration with Richard G.W. Anderson, then a young cell biologist at our medical school in Dallas. Through the use of LDL coupled to electron-dense ferritin, we found that receptor-bound LDL is internalized by endocytosis. More importantly, these morphological studies explained the efficiency of internalization: efficiency is contingent upon the clustering of the LDL receptors in coated pits 23, 24. Coated pits had been described in 1964 by Roth and Porter 25 during electron microscopic studies of the uptake of yolk proteins by mosquito oocytes. These investigators showed that coated pits pinch off from the surface to form coated endocytic vesicles that carry extracellular fluid and its contents into the cell. In 1976, Pearse 26 purified coated vesicles and discovered that the cytoplasmic coat was composed predominantly of one protein, clathrin. The finding that LDL receptors are clustered in clathrin-coated pits raised the general possibility that these structures serve as gathering-places for cell surface receptors that are destined for endocytosis 24. Other cell surface proteins, being excluded from coated pits, could not rapidly enter the cell.
The interpretation of coated pit function was strengthened by study of fibroblasts from a unique FH homozygote. Cells from most FH homozygotes simply fail to bind LDL (Figure 5). But cells from one FH patient, whose initials are J.D., bound LDL, but failed to internalize it (Figure 3B) 27, 28. In collaboration with Anderson, we showed that the receptors in these mutant cells are excluded from coated pits 29. This was an important finding, for it established the essential role of coated pits in the high efficiency uptake of receptor-bound molecules 30.
Later molecular studies showed that the J.D. mutation results from a single base pair change in the LDL receptor gene, resulting in the substitution of a cysteine for a tyrosine in the receptor's cytoplasmic domain 31, 32. This observation stimulated a series of in vitro mutagenesis experiments, which revealed that this tyrosine is part of a tetrameric sequence NPVY (Asn-Pro-Val-Tyr) that directs LDL receptors to clathrin-coated pits for rapid internalization 33. A version of this sequence, NPxY (where x can be any amino acid), is present in one or more copies in the cytoplasmic tails of other members of the LDL receptor gene family 34. An NPxY sequence is also present in the cytoplasmic domains of other cell-surface receptors, including the amyloid precursor protein (APP) and several receptors with tyrosine kinase domains (EGF, c-erb-B/neu, insulin, IGF-1) 33. In addition to roles in endocytosis, NPxY sequence serves as a binding site for proteins that transmit regulatory signals into the cell 34. Such a secondary signaling function has never been demonstrated for the LDL receptor, whose sole function seems to be to carry LDL and related lipoproteins into cells.
The LDL receptor studies exposed another general feature of receptor-mediated endocytosis -namely, that receptors can be recycled 35, 36. After internalization, the receptors dissociate from their ligands when they are exposed to a decrease in pH in endosomes. After dissociation, the receptors find their way back to the cell surface. The LDL receptor makes one round trip into and out of the cell every 10 min for a total of several hundred trips in its 20-hour lifespan 36. Inasmuch as each LDL particle contains ∼1600 molecules of cholesterol, this rapid recycling of LDL receptors provides an efficient mechanism for delivery of cholesterol to cells.
Regulation of LDL Receptors
Shortly after the discovery of LDL receptors, we found that the receptors were themselves subject to feedback regulation. When cell cholesterol increases, the production of LDL receptors is reduced 21. Together with the reduction in HMG CoA reductase, this regulatory response decreases cholesterol input from plasma as well as from endogenous synthesis. The mechanism for this dual regulation was clarified two decades later when we discovered a pair of sterol-regulated, membrane-bound transcription factors called SREBPs 18, 37. The long quest that led to the discovery of SREBPs was recently reviewed elsewhere 38, and it will be summarized briefly here.
Unlike other transcription factors, the SREBPs are synthesized as membrane-bound proteins attached to the endoplasmic reticulum (ER). In cholesterol-depleted cells, the SREBPs are transported to the Golgi complex where they are processed by two proteases to release a soluble fragment that enters the nucleus where it activates transcription of the genes encoding HMG CoA reductase and all the other enzymes of cholesterol biosynthesis as well as the LDL receptor 39. When LDL-derived cholesterol enters cells, it blocks the transport of SREBPs to the Golgi complex, thereby blocking the proteolytic release of the active fragment SREBPs from membranes. Transcription of the target genes declines, and the cells produce less cholesterol, thus preventing cholesterol overload.
SREBP-mediated regulation of LDL receptors is essential for the action of statin drugs in lowering plasma LDL-cholesterol levels in individuals at risk for coronary heart disease. When a statin is ingested, the drug is routed primarily to the liver where it binds and inhibits HMG CoA reductase, lowering cholesterol production. This decrease in liver cholesterol activates SREBP processing, thereby increasing the number of LDL receptors displayed on liver cell membranes. The SREBPs also increase the amount of HMG CoA reductase, but this does not increase cholesterol synthesis because the enzyme is inhibited by the statin. The newly produced LDL receptors remove LDL from the blood, and deliver it to the interior of the cell where the LDL is digested and its released cholesterol becomes available for metabolic purposes. The net effect is that the amount of cholesterol in the liver is maintained at a normal level while at the same time the level of LDL-cholesterol in blood is kept low 40. Fortunately, the LDL receptors don't bind HDL so the blood level of this beneficial lipoprotein doesn't drop. The remarkable efficiency and safety of statins derive from the specificity of their effect in selectively lowering plasma LDL.
LDL Receptor Story: The Latest Chapter
A major gap in our understanding of cellular cholesterol metabolism lies in our limited insight into the ways in which cholesterol is transported from one organelle to another, eventually leading to its remarkably constant concentration in the plasma membrane. Recently, some progress has been made through study of another genetic disease called Niemann-Pick C (NPC). Homozygotes for this recessive disease exhibit profound accumulation of cholesterol in lysosomes of cells throughout the body, especially within liver cells and neurons of the central nervous system. This leads to the degeneration of these organs, causing death within the first or second decade 41.
Lysosomal cholesterol accumulation is manifest in cultured fibroblasts from NPC patients where it can be shown to result from the receptor-mediated uptake of LDL 42. In these cells LDL is taken up normally, and the cholesteryl esters are hydrolyzed in lysosomes, but the liberated cholesterol cannot leave the lysosome. As a result, SREBP processing is uninhibited, and the cells continue to produce cholesterol and to take it up from LDL despite the massive accumulation in lysosomes 43.
The extensive studies of Pentchev and colleagues identified the first defective gene in NPC patients, and they named it NPC1. The gene encodes a large polytopic membrane protein of 1228 amino acids with 13 membrane-spanning regions that is routed to endosomes and lysosomes 44. Soon thereafter, a second defective gene was identified. The encoded protein, designated NPC2, is a small soluble protein of only 132 amino acids 45. NPC2 is also found in endosomes and lysosomes, and a fraction is secreted from the cell. Homozygous defects in either NPC1 or NPC2 produce the identical biochemical and phenotypic defect, suggesting that both proteins are required in order for LDL-derived cholesterol to exit lysosomes: loss of either protein interrupts this process 42, 46.
Inasmuch as NPC2 is a soluble protein, progress in understanding its function occurred relatively quickly. Indeed, NPC2 had earlier been identified in seminal fluid where it was shown to bind cholesterol 47. Xu, et al. 48 made an important advance when they determined the crystal structure of NPC2 with bound cholesterol sulfate. The sterol is bound in a directional orientation with its hydrophobic isooctyl side chain buried deep within a hydrophobic pocket. At the other end of the molecule, the 3ß-hydroxyl group is exposed on the surface. These findings account for the ability of NPC2 to bind a sterol with a bulky sulfate group attached to the 3ß-hydroxyl and its inability to bind a sterol like 25-hydroxycholesterol with a hydrophilic group attached to the side chain 49. NPC2 was also shown to donate its bound cholesterol to phospholipid liposomes, which are surrogates for membranes 50-52.
Progress in understanding the complex, insoluble NPC1 protein has been much slower. T. Y. Chang and co-workers 53 added a photoactivated derivative of cholesterol to cultured cells and showed that it could be crosslinked to the NPC1 protein, implying that NPC1 is also a cholesterol-binding protein, but the specificity of binding could not be demonstrated by this method. The difficulty in purifying a polytopic lipid binding protein from cell membranes is notorious, and prior to last year no one had succeeded in this difficult task.
Our laboratory entered this field inadvertently. A graduate student, Rodney Infante, endeavored to purify a membrane-bound protein that could bind 25-hydroxycholesterol and other oxysterols with hydroxyl groups on the side chain. Our intent was not to study NPC disease, but rather to identify a postulated oxysterol-binding protein of the ER that would play a role in regulating SREBP processing 54. After heroic effort, Infante purified an oxysterol-binding protein from a crude membrane fraction of rabbit liver. Astonishingly, mass spectrometric analysis revealed that this protein was NPC1. Infante quickly showed that this purified NPC1 could bind cholesterol as well as oxysterols. Thus, NPC1, like NPC2, is a cholesterol-binding protein 43, 49.
The next observation was even more surprising. The binding site for cholesterol was not in the hydrophobic membrane-spanning portion of NPC1. Rather, it was contained in a hydrophilic domain comprising the first 240 amino acids. This domain projects into the lumen of the lysosome, and it is connected to the membranous portion of NPC1 by a short proline-rich linker. This domain is referred to as the N-terminal domain of NPC1 or NPC1(NTD). Infante prepared a cDNA encoding this domain as a soluble truncated species, and he found that it is secreted into the culture medium of transfected cells 43. This finding allowed him to prepare large amounts of NPC1(NTD) for biochemical studies. He quickly showed that NPC1(NTD) has a binding specificity that is opposite from NPC2. In contrast to NPC2, NPC1(NTD) shows strict specificity for the ß-orientation of the 3-hydroxyl group. On the other hand, NPC1(NTD) does not require complete hydrophobicity in the side chain. Thus, NPC1(NTD), but not NPC2, can bind 25-hydroxycholesterol and 27-hydroxycholesterol as well as cholesterol 49.
In addition to the differential orientation of the binding site, NPC1(NTD) and NPC2 differ in the ease with which they bind and release cholesterol 52. When [3H]cholesterol was delivered to both proteins in detergent at 4°C, the sterol bound to NPC2 within minutes, and it also dissociated quickly. On the other hand, binding to NPC1(NTD) was extremely slow, requiring several hours to reach equilibrium. The dissociation rate was equally slow, so that the eventual equilibrium constants were similar for NPC2 and NPC1(NTD). Binding and release of cholesterol from NPC1(NTD) could be accelerated by two orders of magnitude when cholesterol was delivered by NPC2, raising the possibility that NPC2 somehow opened the binding site on NPC1(NTD) so that cholesterol could enter the protein more rapidly. NPC2 could also accept cholesterol from NPC1(NTD). Moreover, when NPC1(NTD) was preloaded with [3H]cholesterol, the sterol could be transferred to phospholipid liposomes, but only when NPC2 was present 49. These data led us to suggest a working model in which NPC2 extracts cholesterol from LDL in the lysosome and then transfers it to the N-terminal domain of membrane-bound NPC1 for insertion into the lysosomal membrane. This model is currently being tested in our laboratory through high resolution structural analysis of NPC1(NTD) together with functional analysis of NPC1(NTD) and NPC2 proteins that have been subjected to an alanine scan mutagenesis.
The Legacy
The early work on the LDL receptor, summarized above, led to a new way of thinking about cholesterol homeostasis and introduced three general concepts to biology and medicine.
The concept of selective sorting of proteins within the plasma membrane, which forms the basis for receptor clustering in coated pits, a prerequisite for receptor-mediated endocytosis.
The concepts of receptor-mediated endocytosis and receptor recycling, which provide a mechanism by which cells selectively and efficiently internalize macromolecules, including transport proteins, hormones, growth factors, lysosomal enzymes, and certain viruses.
The concept of feedback regulation of receptors, which explains the cholesterol-lowering effects of the statins, a class of drugs that are remarkably effective in lowering plasma LDL levels, reducing heart attacks, and prolonging life 55-57.
Acknowledgments
We thank the National Institutes of Health (HL20948) for their long-standing support of our research. The Perot Family Foundation has also provided generous support.
Biography
The Authors
Michael S. Brown and Joseph L. Goldstein (Figure 6) are currently Regental Professors in the Department of Molecular Genetics at the University of Texas Southwestern Medical Center in Dallas where they have worked together since 1972. Brown received his M.D. from University of Pennsylvania in 1966. Goldstein received his M.D. from the University of Texas Southwestern Medical School in 1966. From 1966−68, both Brown and Goldstein were interns and residents in Medicine at the Massachusetts General Hospital in Boston. Brown and Goldstein then did postdoctoral research at the NIH, Brown working in the laboratory of Earl Stadtman and Goldstein in the laboratory of Marshall Nirenberg.
Figure 6.

Joseph L. Goldstein (left) and Michael S. Brown on the day of announcement of their Nobel Prize in Physiology or Medicine on October 15, 1985.
Brown and Goldstein are members of the U.S. National Academy of Sciences, American Philosophical Society, and the Institute of Medicine. They are also Foreign Members of the Royal Society (London). Among their awards are the Lasker Award in Basic Medical Research (1985), Nobel Prize in Physiology or Medicine (1985), U.S. National Medal of Science (1988), the Albany Medical Center Prize (2003), and the Woodrow Wilson Award for Public Service (2005). Both have received honorary degrees from numerous institutions, including the University of Chicago, University of Paris, and Rockefeller University.
Footnotes
This article is modified from a paper by M.S. Brown and J.L. Goldstein that originally appeared online in Great Experiments (http://www.ergito.com/main.jsp?bcs=EXP.6.7&bhcp=1) and is reprinted here with permission from Jones and Bartlett Publishers, LLC.
References
- 1.Muller C. Xanthomata, hypercholesterolemia, angina pectoris. Acta Med Scand. 1938;89:75–84. [Google Scholar]
- 2.Khachadurian AK. The inheritance of essential familial hypercholesterolemia. Am J Med. 1964;37:402–7. doi: 10.1016/0002-9343(64)90196-2. [DOI] [PubMed] [Google Scholar]
- 3.Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. McGraw-Hill, Inc.; New York: 2001. pp. 2863–913. [Google Scholar]
- 4.Bailey JM. Regulation of cell cholesterol content. In: Porter R, Knight J, editors. Atherogenesis: Initiating Factors. 12 ed. Ciba Foundation Symposium; Elsevier, Amsterdam: 1973. pp. 63–92. [Google Scholar]
- 5.Rothblat GH. Lipid metabolism in tissue culture cells. Adv Lipid Res. 1969;7:l35–l62. [PubMed] [Google Scholar]
- 6.Brown MS, Dana SE, Goldstein JL. Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts by lipoproteins. Proc Natl Acad Sci USA. 1973;70:2l62–6. doi: 10.1073/pnas.70.7.2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siperstein MD. Regulation of cholesterol biosynthesis in normal and malignant tissues. Curr Topics Cell Reg. 1970;2:65–l00. [Google Scholar]
- 8.Goldstein JL, Brown MS. Familial hypercholesterolemia: Identification of a defect in the regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity associated with overproduction of cholesterol. Proc Natl Acad Sci USA. 1973;70:2804–8. doi: 10.1073/pnas.70.10.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown MS, Dana SE, Goldstein JL. Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in cultured human fibroblasts: Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. J Biol Chem. 1974;249:789–96. [PubMed] [Google Scholar]
- 10.Goldstein JL, Brown MS. Binding and degradation of low density lipoproteins by cultured human fibroblasts: Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. J Biol Chem. 1974;249:5l53–62. [PubMed] [Google Scholar]
- 11.Goldstein JL, Basu SK, Brunschede GY, Brown MS. Release of low density lipoprotein from its cell surface receptor by sulfated glycosaminoglycans. Cell. 1976;7:85–95. doi: 10.1016/0092-8674(76)90258-0. [DOI] [PubMed] [Google Scholar]
- 12.Brown MS, Dana SE, Goldstein JL. Receptor-dependent hydrolysis of cholesteryl esters contained in plasma low density lipoprotein. Proc Natl Acad Sci USA. 1975;72:2925–9. doi: 10.1073/pnas.72.8.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schneider WJ, Beisiegel U, Goldstein JL, Brown MS. Purification of the low density lipoprotein receptor, an acidic glycoprotein of 164,000 molecular weight. J Biol Chem. 1982;257:2664–73. [PubMed] [Google Scholar]
- 14.Yamamoto T, Davis CG, Brown MS, Schneider WJ, Casey ML, Goldstein JL, Russell DW. The human LDL receptor: A cysteine-rich protein with multiple Alu sequences in its mRNA. Cell. 1984;39:27–38. doi: 10.1016/0092-8674(84)90188-0. [DOI] [PubMed] [Google Scholar]
- 15.Südhof TC, Goldstein JL, Brown MS, Russell DW. The LDL receptor gene: A mosaic of exons shared with different proteins. Science. 1985;228:815–22. doi: 10.1126/science.2988123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldstein JL, Brunschede GY, Brown MS. Inhibition of the proteolytic degradation of low density lipoprotein in human fibroblasts by chloroquine, concanavalin A, and Triton WR l339. J Biol Chem. 1975;250:7854–62. [PubMed] [Google Scholar]
- 17.Goldstein JL, Dana SE, Faust JR, Beaudet AL, Brown MS. Role of lysosomal acid lipase in the metabolism of plasma low density lipoprotein: Observations in cultured fibroblasts from a patient with cholesteryl ester storage disease. J Biol Chem. 1975;250:8487–95. [PubMed] [Google Scholar]
- 18.Brown MS, Goldstein JL. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci USA. 1999;96:11041–8. doi: 10.1073/pnas.96.20.11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gil G, Faust JR, Chin DJ, Goldstein JL, Brown MS. Membrane-bound domain of HMG CoA reductase is required for sterol-enhanced degradation of the enzyme. Cell. 1985;41:249–58. doi: 10.1016/0092-8674(85)90078-9. [DOI] [PubMed] [Google Scholar]
- 20.Brown MS, Dana SE, Goldstein JL. Cholesteryl ester formation in cultured human fibroblasts: Stimulation by oxygenated sterols. J Biol Chem. 1975;250:4025–7. [PubMed] [Google Scholar]
- 21.Brown MS, Goldstein JL. Regulation of the activity of the low density lipoprotein receptor in human fibroblasts. Cell. 1975;6:307–l6. doi: 10.1016/0092-8674(75)90182-8. [DOI] [PubMed] [Google Scholar]
- 22.Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 23.Anderson RGW, Goldstein JL, Brown MS. Localization of low density lipoprotein receptors on plasma membrane of normal human fibroblasts and their absence in cells from a familial hypercholesterolemia homozygote. Proc Natl Acad Sci USA. 1976;73:2434–8. doi: 10.1073/pnas.73.7.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson RGW, Brown MS, Goldstein JL. Role of the coated endocytic vesicle in the uptake of receptor-bound low density lipoprotein in human fibroblasts. Cell. 1977;l0:35l–64. doi: 10.1016/0092-8674(77)90022-8. [DOI] [PubMed] [Google Scholar]
- 25.Roth TF, Porter KR. Yolk protein uptake in the oocyte of the mosquito Aedes aegypti L. J Cell Biol. 1964;20:3l3–32. doi: 10.1083/jcb.20.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pearse BMF. Clathrin: A unique protein associated with intracellular transfer of membrane by coated vesicles. Proc Natl Acad Sci USA. 1976;73:l255–l259. doi: 10.1073/pnas.73.4.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown MS, Goldstein JL. Analysis of a mutant strain of human fibroblasts with a defect in the internalization of receptor-bound low density lipoprotein. Cell. 1976;9:663–74. doi: 10.1016/0092-8674(76)90130-6. [DOI] [PubMed] [Google Scholar]
- 28.Goldstein JL, Brown MS, Stone NJ. Genetics of the LDL receptor: Evidence that the mutations affecting binding and internalization are allelic. Cell. 1977;l2:629–4l. doi: 10.1016/0092-8674(77)90263-x. [DOI] [PubMed] [Google Scholar]
- 29.Anderson RGW, Goldstein JL, Brown MS. A mutation that impairs the ability of lipoprotein receptors to localise in coated pits on the cell surface of human fibroblasts. Nature. 1977;270:695–9. doi: 10.1038/270695a0. [DOI] [PubMed] [Google Scholar]
- 30.Goldstein JL, Anderson RGW, Brown MS. Coated pits, coated vesicles, and receptor-mediated endocytosis. Nature. 1979;279:679–85. doi: 10.1038/279679a0. [DOI] [PubMed] [Google Scholar]
- 31.Lehrman MA, Goldstein JL, Brown MS, Russell DW, Schneider WJ. Internalization-defective LDL receptors produced by genes with nonsense and frameshift mutations that truncate the cytoplasmic domain. Cell. 1985;41:735–43. doi: 10.1016/s0092-8674(85)80054-4. [DOI] [PubMed] [Google Scholar]
- 32.Davis CG, Lehrman MA, Russell DW, Anderson RGW, Brown MS, Goldstein JL. The J.D. mutation in familial hypercholesterolemia: Amino acid substitution in cytoplasmic domain impedes internalization of LDL receptors. Cell. 1986;45:15–24. doi: 10.1016/0092-8674(86)90533-7. [DOI] [PubMed] [Google Scholar]
- 33.Chen W-J, Goldstein JL, Brown MS. NPXY, a sequence often found in cytoplasmic tails, is required for coated pit-mediated internalization of the low density lipoprotein receptor. J Biol Chem. 1990;265:3116–23. [PubMed] [Google Scholar]
- 34.Herz J, Bock HH. Lipoprotein receptors in the nervous system. Ann Rev Biochem. 2002;71:405–34. doi: 10.1146/annurev.biochem.71.110601.135342. [DOI] [PubMed] [Google Scholar]
- 35.Basu SK, Goldstein JL, Anderson RGW, Brown MS. Monensin interrupts the recycling of low density lipoprotein receptors in human fibroblasts. Cell. 1981;24:493–502. doi: 10.1016/0092-8674(81)90340-8. [DOI] [PubMed] [Google Scholar]
- 36.Brown MS, Anderson RGW, Goldstein JL. Recycling receptors: The round-trip itinerary of migrant membrane proteins. Cell. 1983;32:663–7. doi: 10.1016/0092-8674(83)90052-1. [DOI] [PubMed] [Google Scholar]
- 37.Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 38.Brown MS, Goldstein JL. Cholesterol feedback: from Schoenheimer's bottle to Scap's MELADL. J Lipid Res. 2009 doi: 10.1194/jlr.R800054-JLR200. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–31. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown MS, Goldstein JL. A tribute to Akira Endo, discoverer of a “Penicillin” for cholesterol. Atherosclerosis Suppl. 2004;5:13–6. [Google Scholar]
- 41.Pentchev PG, Vanier MT, Suzuki K, Patterson MC. Niemann-Pick disease type C: A cellular cholesterol lipidosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. 7 ed. McGraw-Hill Inc.; New York: 1995. pp. 2625–39. [Google Scholar]
- 42.Pentchev PG. Niemann-Pick C research from mouse to gene. Biochim Biophys Acta. 2004;1685:3–7. doi: 10.1016/j.bbalip.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 43.Infante RE, Abi-Mosleh L, Radhakrishnan A, Dale JD, Brown MS, Goldstein JL. Purified NPC1 protein: I. Binding of cholesterol and oxysterols to a 1278-amino acid membrane protein. J Biol Chem. 2008;283:1052–63. doi: 10.1074/jbc.M707943200. [DOI] [PubMed] [Google Scholar]
- 44.Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, Gu J, Rosenfeld MA, Pavan WJ, Krizman DB, Nagle J, Polymeropoulos MH, Sturley SL, Ioannou YA, Higgins ME, Comly M, Cooney A, Brown A, Kaneski CR, Blanchette-Mackie J, Dwyer NK, Neufeld EB, Chang T-Y, Liscum L, Strauss JF, III, Ohno K, Zeigler M, Carmi R, Sokol J, Markie D, O'Neill RR, van Diggelen OP, Elleder M, Patterson MC, Brady RO, Vanier MT, Pentchev PG, Tagle DA. Niemann-Pick C1 disease gene: Homology to mediators of cholesterol homeostasis. Science. 1997;277:228–31. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- 45.Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, Jadot M, Lobel P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290:2298–301. doi: 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- 46.Sleat DE, Wiseman JA, El-Banna M, Price SM, Verot L, Shen MM, Tint GS, Vanier MT, Walkley SU, Lobel P. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc Natl Acad Sci USA. 2004;101:5886–91. doi: 10.1073/pnas.0308456101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okamura N, Kiuchi S, Tamba M, Kashima T, Hiramoto S, Baba T, Dacheux F, Dacheux J-L, Sugita Y, Jin Y-Z. A porcine homolog of the major secretory protein of human epididymis, HE1, specifically binds cholesterol. Biochim Biophys Acta. 1999;1438:377–87. doi: 10.1016/s1388-1981(99)00070-0. [DOI] [PubMed] [Google Scholar]
- 48.Xu S, Benoff B, Liou H-L, Lobel P, Stock AM. Structural basis of sterol binding by NPC2, a lysosomal protein deficient in Niemann-Pick type C2 disease. J Biol Chem. 2007;282:23525–31. doi: 10.1074/jbc.M703848200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Infante RE, Radhakrishnan A, Abi-Mosleh L, Kinch LN, Wang ML, Grishin NV, Goldstein JL, Brown MS. Purified NPC1 protein: II. Localization of sterol binding to a 240-amino acid soluble luminal loop. J Biol Chem. 2008;283:1064–75. doi: 10.1074/jbc.M707944200. [DOI] [PubMed] [Google Scholar]
- 50.Cheruku SR, Xu Z, Dutia R, Lobel P, Storch J. Mechanism of cholesterol transfer from the Niemann-Pick type C2 protein to model membranes supports a role in lysosomal cholesterol transport. J Biol Chem. 2006;281:31594–604. doi: 10.1074/jbc.M602765200. [DOI] [PubMed] [Google Scholar]
- 51.Babalola JO, Wendeler M, Breiden B, Arenz C, Schwarzmann G, Locatelli-Hoops S, Sandhoff K. Development of an assay for the intermembrane transfer of cholesterol by Niemann-Pick protein. Biol Chem. 2007;388:617–26. doi: 10.1515/BC.2007.063. [DOI] [PubMed] [Google Scholar]
- 52.Infante RE, Wang ML, Radhakrishnan A, Kwon HJ, Brown MS, Goldstein JL. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc Natl Acad Sci USA. 2008;105:15287–92. doi: 10.1073/pnas.0807328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ohgami N, Ko DC, Thomas M, Scott MP, Chang CCY, Chang T-Y. Binding between the Niemann-Pick C1 protein and a photoactivatable cholesterol analog requires a functional sterol-sensing domain. Proc Natl Acad Sci USA. 2004;101:12473–8. doi: 10.1073/pnas.0405255101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adams CM, Reitz J, DeBrabander JK, Feramisco JD, Brown MS, Goldstein JL. Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J Biol Chem. 2004;279:52772–80. doi: 10.1074/jbc.M410302200. [DOI] [PubMed] [Google Scholar]
- 55.Brown MS, Goldstein JL. Lowering LDL - not only how low, but how long? Science. 2006;311:1721–3. doi: 10.1126/science.1125884. [DOI] [PubMed] [Google Scholar]
- 56.LaRosa JC, He J, Vupputuri S. Effect of statins on risk of coronary disease. JAMA. 1999;282:2340–6. doi: 10.1001/jama.282.24.2340. [DOI] [PubMed] [Google Scholar]
- 57.Cholesterol Treatment Trialists (CTT) Collaborators Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90 056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–78. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 58.Brown MS, Goldstein JL. Receptor-mediated endocytosis: Insights from the lipoprotein receptor system. Proc Natl Acad Sci USA. 1979;76:3330–7. doi: 10.1073/pnas.76.7.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]