

Abstract
Uranium is a naturally occurring radioactive material present everywhere in the environment. It is toxic because of its chemical or radioactive properties. Uranium enters environment mainly from mines and industry and cause threat to human health by accumulating in lungs as a result of inhalation. In our previous study, we have shown the effectiveness of antioxidant system response to the oxidative stress induced by uranyl acetate (UA) in rat lung epithelial (LE) cells. As part of our continuing studies; here, we investigated the mechanism underlying when LE cells are exposed to different concentration of UA. Oxidative stress may lead to apoptotic signaling pathways. LE cells treated with 0.25, 0.5 and 1 mM of UA results in dose and time-dependent increase in activity of both caspases-3 and -8. Increase in the concentration of cytochrome-c oxidase in cytosol was seen in LE cells treated with 1 mM UA as a result of mitochondria membrane permeability. The cyto-chrome-c leakage may trigger the apoptotic pathway. TUNEL assay performed in LE cells treated with 1 mM of UA showed significant incorporation of dNTPs in the nucleus after 24 h. In the presence of the caspase inhibitors, we observed the significant decrease in the activity of caspases-8 and -3 in 0.5 and 1 mM UA-treated LE cells.
Keywords: Uranium, Apoptosis, Cytochrome-c, Caspase
Introduction
Uranium is causing major international concern because of its potential threat to public health (The Royal Society 2002; Bleise et al. 2003; WHO 2001; U S Dept. H & H Services 1999). Established literature suggests that depleted uranium (DU) can cause embryo toxicity, reproductive and developmental damage, genomic instability and single-stranded DNA breakage (Sandra et al. 2007). Although these toxicities have been shown to occur because of exposure to uranyl acetate (UA), very little has been studied regarding its mechanism of toxic inductions. The major route of exposure to UA is through inhalation (The Royal Society 2002; Bleise et al. 2003; WHO 2001; US Dept. H & H Services 1999). Human bronchial cells and lung epithelial (LE) cells are therefore the major target of UA toxicity. Studies with human bronchial cells have shown neoplastic transformation upon chronic exposure with DU (Yang et al. 2002). While other reports suggested induction of lipid peroxidation and micronuclei formation in bronchial epithelial cell (Susumu et al. 1998).
As mentioned earlier, the route of exposure to uranium is largely through inhalation and therefore it makes the lung tissues as one of its target organs. DU of size <5 μm can lodge deep into the lung alveoli, thus producing toxicological impact at the site of contact (Marjorie et al. 2006). Several studies and epidemiological data for UA-induced toxicity have been reported. These studies have shown increased risk of cancer in the population exposed to DU (Grosche et al. 2006; Klervi et al. 2007; Chobanova et al. 2003), gene mutations (Joachim et al. 2007) and cytotoxicity (Sandra et al. 2007; Periyakaruppan et al. 2007). In macrophage cell line increased secretion of TNF-α in association with JNK and P38 activation has been reported (Vincent et al. 2004). The occurrence of a genomic instability in lymphocytes of uranium miners, especially those who developed cancer were observed by studying the cytogenetic endpoint marker micronuclei (Mn) (Kryscio et al. 2001). Further in vitro analysis with uranium acetate has shown significant increase in DNA single strand breaks in the presence of ascorbic acid (Monica et al. 2003). Treatment of DU to J774 macrophage cells resulted in the apoptotic death of these cells as confirmed by both DNA ladder and annexin-V staining (John et al. 2002). A mechanistic approach to study the effect of uranium on genomic DNA in CHO–EM9 cells has suggested DNA strand breaks or cross-links as UA-induced mutagenic lesions. These unique mutagenic lesion-induced mutation spectrum elicited by exposure to uranium suggested that it generates mutations in ways that are different from spontaneous and that are mediated by free radical as well as through radiological mechanisms (Virginia and Diane 2006). In addition to the mutagenic potential of uranium, it is also observed to alter the proteome in human lung cells (Veronique et al. 2005). The proteomic analysis showed cytokeratin and peroxire-doxin-1, of which the latter is involved in protection against oxidative injury. There is also growing number of reports documenting the possibility of genotoxicity by uranium possibly through a free radical mechanism (Kryscio et al. 2001). We and others have earlier reported inhibition of cell proliferation by UA in lung cells and these effects could be reverted in the presence of glutathione (Periyakaruppan et al. 2007). Cell proliferation can be inhibited by apoptosis and metals are known to induce apoptosis through a number of mechanisms (Valko et al. 2006; Ivan 2006; Bertin and Averbeck 2006). UA also follows similar toxic response and induces apoptosis in normal rat kidney cells (Levine and Saltzman 1994). Involvements of caspase have been argued in the mechanism of UA-induced apoptosis in these cells.
In the present study, we made an attempt to study UA-induced apoptosis in rat LE cells and report the involvement of caspases in this process.
Materials and methods
Rat LE cell line (RL-65, CRL-10354) obtained from ATCC, was grown in Dulbecco’s minimum essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin in 95% humidified air at 37°C with 5% CO2.
Uranium solutions were prepared as described by Prat et al. (2005). In brief, all experiments were performed using 0.1 M stock solution of uranyl (VI) acetate (Amresco, Catalog No: 541093, OH, USA) pH 4, in water. For cell treatments, 100 mM uranyl bicarbonate was prepared using appropriate dilutions of the stock solution in aqueous 0.1 M bicarbonate solutions and filtered through 0.22 μm filter.
Activity of caspase-8 and -3
The assay was performed according to manufacturer’s protocol. (Caspase Apoptosis Detection Kit, Santa Cruz Bio-technology, Inc., Catalog No: sc-4263 AK and sc-4267 AK). In brief 2 × 104 cells/well were seeded in a 96-well plate containing DMEM with 10% of FBS and grown under standard conditions at 37°C. Following overnight, incubation cells were starved for 24 h and treated with 0.25, 0.5 and 1 mM of uranium acetate and allowed to grow 8 h and 3 h for caspases-3 and -8 activities, respectively. Assay was performed by the addition of 5 μl of IETD-AFC substrate for caspase-8 and DEVD-AFC substrate caspase-3 to the cell lysate in separate wells and incubated for 1 h at 37°C. Free AFC levels formed were measured in a plate reader with a 400 nm excitation and a 505 nm emission. The results of experimental samples were compared with control and expressed as fluorescence units/milligram of protein. To confirm the induction of caspase enzymes, the assay for caspase-8 and 3 was performed in LE cells treated with 0.5 and 1 mM UA in the presence caspase inhibitors at the concentration of 1:1,000 (EMD Chemicals, Inc., Catalog No: 627610, San Diego, CA, USA).
Cytochrome-c oxidase leakage
Cells (2 × 104/well) were seeded in a six well plate and grown under standard conditions and starved for 18 h. Following treatment with 1 mM concentration of UA, the cells were scraped and homogenized in phosphate buffered saline (PBS) at 24, 48, and 72 h. The mitochondrial and cytosol fractions were separated from whole cell lysate according to manufacture’s protocol (Proteo Extract Cyto-sol/Mitochondria ELISA kit, EMD chemicals, Inc., QIA88, San Diego). Using cytochrome-c ELISA Kit (EMD chemicals, Inc., Catalog No: QIA74) the presence of cytochrome-c oxidase in whole cell lysate, mitochondrial and cytosol fractions were assayed and the results were normalized to the total protein.
TUNEL assay
In brief (2 × 105/well) cells were seeded on cover slip and allowed to grow overnight under standard condition. Following starvation LE cells were treated with 1 mM UA incubated for 37°C for 24 h. Assay was performed according to manufacture’s protocol (Promega Corporation., Part No: TB235, Madison). Briefly LE cells were fixed with cold methanol and permeabilized using 0.01% TritonX-100 in PBS for 10 min and incubated with fluorescently labeled dNTPs for 2 h. Cells were then stained with propidium iodide and imaged using Nikon Fluorescence microscope (Nis Element, Nikon Instruments Inc, Melville).
Statistical analysis
Data were expressed as mean ± SD and statistical significance was assessed by student t test. Difference between control and uranium-treated cell samples were considered significant if the level was P < 0.05.
Results
LE cells treated with 1 mM of UA showed significant incorporation of dNTPs in the nucleus after 24 h of treatment (Fig. 1). These results do suggest that UA induces apoptosis in these cells and this agrees with the results of apoptotic induction in other cellular models (John et al. 2002; Celine et al. 2007). No significant induction of apoptosis at lower doses of UA was observed. The inhibition of cell proliferation happens at doses lower than 1 mM (0.25, 0.5, and 0.75 mM) as reported earlier (Periyakaruppan et al. 2007). The ambiguity of the loss of co-relation between cell death by apoptosis and proliferation remains unexplained, and to be investigated. However, the results have shown cell death due to necrosis by UA in liver of whitefish (Coregonus clupeaformis) and this could explain the partial response of UA in LE cells. It is possible that necrosis takes over as a major cause of cell death by UA in these cells.
Fig. 1.

TUNEL assay of UA-treated LE cells. a Control, b 1 mM concentration of UA, and c 1 mM concentration of UA + caspase inhibitor. The fluorographs represent nuclear staining, TUNEL staining, and merging of both nuclear and TUNEL staining. The arrows indicate damage to the nuclear membrane
Caspase play a central role in the induction of apoptosis induced by a variety of stimulus, so we investigated the involvement of the caspases in UA-induced apoptosis in LE cells. After 8 h of treatment with various concentrations of UA a dose-dependent increase in the caspase-3 activities was observed in LE cells (Fig. 2). Moreover, the activation of caspase-3 is dependent on the early activation of either caspases-8 or 9. Hence, we further determined the activity of caspase-8 after 3 h of treatment with various concentration of UA. As shown in Fig. 2, there was a dose-dependent increase in the activity of caspase-8 in cells treated with UA. These results indicate that UA activates both caspase-8 and -3 in a dose-dependent manner. To confirm the induction of caspase enzymes we further used caspase inhibitors to block the activation of these caspases by UA. As shown in Fig. 3 in presence of the inhibitors there was a significant decrease in the activity of both the caspases-8 and -3 in UA-treated cells. Thus, UA-induced activation of caspases was very specific in this cell line. We also found decrease in apoptosis after treatment with the inhibitors. Suggesting that the induction of apoptosis by UA in LE cells was via the activation of at least the caspases-8 and -3.
Fig. 2.
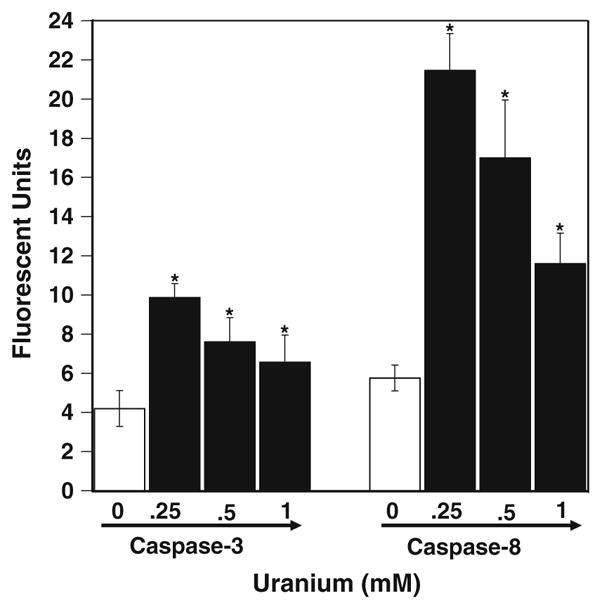
Caspases-8 and -3 activities in UA-treated LE cells. The results of different concentration of UA-treated LE cells were found to be significantly increased compared with the control in both caspase-8 and -3. Values are mean ± SD of three treatments and done experiments independently. * P < 0.01 significance as estimated by student t test
Fig. 3.
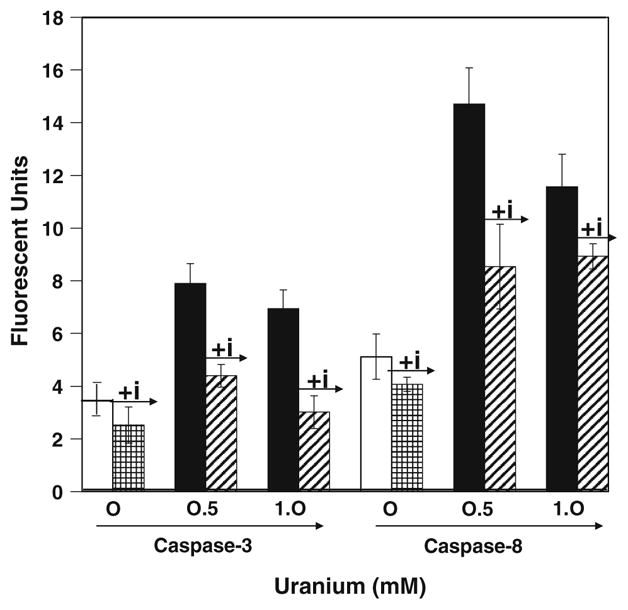
Effect of caspase inhibitor on caspases-8 and -3 activities. Inhibitor and enzyme in the ratio of 1:1,000 were used. AFC formed was measured in a plate reader with a 400 nm excitation and 505 nm emissions. The results were expressed as fluorescence units per microgram of protein. UA-treated LE cells both 0.5 and 1.0 mM exhibited increase in caspases-8 and -3 activities, whereas in the presence of caspase inhibitor the activities decreased. The figure is a representation of three experiments performed independently
Caspase-8 is an initiator of the death receptor pathway and also functions as a caspase-3 target independent of the death receptor pathway (Kanno et al. 2008). The activation of caspase-8 induces pro-apoptotic protein Bid which then can regulate the mitochondria by inducing the release of cytochrome-c oxidase into the cytosol. Apoptosis is thus triggered by leakage of cytochrome-c oxidase from the mitochondria into the cytosol and this can be measured separately by estimating the enzyme activity in mitochondrial and cytosolic fraction of the cells. We first determined in the whole cell lysate the total cytochrome-c oxidase activity and found no significant change in the total activity of the enzyme (Table 1). This result suggests that UA did not change the total amount of protein in the cells and therefore no transcriptional alteration was induced by the metal in the cells. Cytosolic fractions of similarly treated cells showed an increase in the cytochrome-c oxidase activity after 24 h (Table 1). The increased activity remained till the end of 72 h. Conversely, we also found that there was a significant decrease in the mitochondrial cytochrome-c oxidase activity suggesting the loss of this protein from the mitochondria into the cytosol (Table 1). Thus, one of the mechanisms of UA-induced apoptosis was activation of caspase-8 and -3 thereby increasing the membrane permeability of the mitochondria which resulting in the leakage of the cytochrome-c into the cytosol.
Table 1.
The increase in caspase-8 and -3 level results in leakage of cytochrome-c oxidase in to the cytosol
| Cell fractions | pg of Cytochrome/mg protein at different time (h) |
|||
|---|---|---|---|---|
| 0 | 24 | 48 | 72 | |
| Whole cell | 1,087 ± 12 | 1,188 ± 98 | 1,191 ± 132 | 1,205 ± 38 |
| Cytosol | 11 ± 3 | 329 ± 6* | 526 ± 148* | 593 ± 146* |
| Mitochondrial | 1,039 ± 38 | 979 ± 92 | 569 ± 28* | 713 ± 44* |
The table represents the activity of cytochrome-c oxidase after treatment with UA in whole cell lysate, cytosolic fraction, and mitochondrial fraction
Values are mean ± SD of three treatments and done experiments independently
P < 0.01 significance as estimated by student t test
Discussion
Apoptosis-induced cell death results as a consequence of DNA damage generated from free radicals or through extrinsic pathways involving death receptor (Kendra and Geoffrey 2007; James and Andrew 2007). We have shown earlier that UA significantly increases reactive oxygen species in LE cells (Periyakaruppan et al. 2007) and following those findings, we now report that the induction of apoptosis in these cells is a consequence of oxidative stress. In support of our present findings Thiebault et al. recently showed the induction of apoptosis by UA in NRK cells (Celine et al. 2007). In response to UA, LE cells showed significant increase in apoptotic nuclei at 1 mM concentrations of UA. Earlier studies with mice kidney and human renal cells exposed to UA indicated significant modification in apoptosis-related genes. In this study, we show that UA increased caspases-8 and -3 activities in a dose dependence. Moreover, at higher concentration there is decrease in the activities of caspases, may be because of the cell death at higher concentration of UA. Increase in the activity of these two enzymes by UA do indicates that apoptosis induction might be through the mitochondria thereby activating the intrinsic pathway of apoptosis. This pathway is activated by a variety of stimuli that include not only cytotoxic drugs and irradiation (Padmavati and Arora 1976) but also metals such as arsenic and cadmium (Bradley and Caldwell 1978; Seon–Hee et al. 2004; Seon–Hee and Sung–Chul 2006). The induction of apoptosis activated caspase-8 and -3 are evident from the increase in the activity of these enzymes after UA treatment. The activity of both of these enzymes decreased with increasing dosage of UA suggesting that at high concentrations of UA the cells undergo necrosis. Similar findings have been reported in NRK cells treated with UA (Celine et al. 2007). Further 10–20% cells undergo apoptosis by UA at low concentrations and have been demonstrated in CD4 + T cells and NRK cells (Celine et al. 2007; Bin et al. 2006). Caspase-8 was induced approximately twofold more than caspase-3 at all the doses of UA treatments in LE cells. In addition, the use of specific inhibitors completely blocked the activation of the enzymes suggesting the specificity of the assay. Induction of apoptosis by heavy metals like Cd and involvement of caspases are known in HepG2 cells (Seon–Hee et al. 2004), thus this may be a generalized phenomenon for apoptosis by heavy metals. The activation of caspases results in the release of cytochrome-c from the mitochondria. UA did not change the amount of total cytochrome-c but leakage of the protein from the mitochondria into the cytosol was easily detected in the cytosolic fraction. These results may help to understand the consequence of oxidative stress induced by UA in LE cells as reported by us earlier. Our results indicate and also support earlier demonstrations that UA can induce apoptosis in cells and this might be one of the causes of cell death by UA.
Acknowledgments
This work was supported by NASA funding NCC 9-165: NCC-1-02038: NAG 9-1414: NIH 1P20MD001822-1 (GR).
Contributor Information
Adaikkappan Periyakaruppan, Environmental Toxicology Program, Department of Chemistry, Texas Southern University, Houston, TX 77004, USA.
Shubhashish Sarkar, Environmental Toxicology Program, Department of Chemistry, Texas Southern University, Houston, TX 77004, USA.
Prabakaran Ravichandran, Molecular Toxicology Laboratory, Department of Biology, Center for Biotechnology and Biomedical Sciences, Norfolk State University, Norfolk, VA 23504, USA.
Bindu Sadanandan, Environmental Toxicology Program, Department of Chemistry, Texas Southern University, Houston, TX 77004, USA.
Chidananda S. Sharma, Molecular Toxicology Laboratory, Department of Biology, Center for Biotechnology and Biomedical Sciences, Norfolk State University, Norfolk, VA 23504, USA
Vani Ramesh, Molecular Toxicology Laboratory, Department of Biology, Center for Biotechnology and Biomedical Sciences, Norfolk State University, Norfolk, VA 23504, USA.
Joseph C. Hall, Molecular Toxicology Laboratory, Department of Biology, Center for Biotechnology and Biomedical Sciences, Norfolk State University, Norfolk, VA 23504, USA
Renard Thomas, Environmental Toxicology Program, Department of Chemistry, Texas Southern University, Houston, TX 77004, USA.
Bobby L. Wilson, Environmental Toxicology Program, Department of Chemistry, Texas Southern University, Houston, TX 77004, USA
Govindarajan T. Ramesh, Molecular Toxicology Laboratory, Department of Biology, Center for Biotechnology and Biomedical Sciences, Norfolk State University, Norfolk, VA 23504, USA e-mail: gtramesh@nsu.edu
References
- Bertin G, Averbeck D. Cadmium: cellular effects, modifications of biomolecules, modulation of DNA repair and genotoxic consequences (a review) Biochimie. 2006;88:1549–1559. doi: 10.1016/j.biochi.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Bin W, James FT, Terry SW, Gary SS. In vitro immune toxicity of depleted uranium: effects on murine macrophages, CD4 + T cells, and gene expression profiles. Environ Health Perspect. 2006;114(1):85–91. doi: 10.1289/ehp.8085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleise A, Danesi PR, Burkart W. Properties, use and health effects of depleted uranium (DU): a general overview. J Environ Radioact. 2003;64:93–112. doi: 10.1016/s0265-931x(02)00041-3. [DOI] [PubMed] [Google Scholar]
- Bradley RH, Caldwell BM. Screening the environment. Am J Orthopsychiatry. 1978;48(1):114–130. doi: 10.1111/j.1939-0025.1978.tb01293.x. [DOI] [PubMed] [Google Scholar]
- Celine T, Marie C, Sarah M, Angelique S, Laure A, Barbara G. Uranium induces apoptosis and is genotoxic to normal rat kidney (NRK-52E) proximal cells. Toxicol Sci. 2007;98(2):479–487. doi: 10.1093/toxsci/kfm130. [DOI] [PubMed] [Google Scholar]
- Chobanova N, Genchev G, Yagova A, Georgieva L. Cancer in populations living in regions with radio ecological problems in Bulgaria. J BUON (official journal of the Balkan Union of Oncology) 2003;8(2):143–146. [PubMed] [Google Scholar]
- Grosche B, Kreuzer M, Kreisheimer M, Schnelzer M, Tschense A. Lung cancer risk among German male uranium miners: a cohort study, 1946–1998. Br J Cancer. 2006;95:1280–1287. doi: 10.1038/sj.bjc.6603403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan S. Common mechanisms in nephropathy induced by toxic metals. Nephron Physiol. 2006;104:107–114. doi: 10.1159/000095539. [DOI] [PubMed] [Google Scholar]
- James KA, Andrew GP. Apoptosis commitment—translating survival signals into decisions on mitochondria. Cell Res. 2007;17:976–984. doi: 10.1038/cr.2007.101. [DOI] [PubMed] [Google Scholar]
- Joachim S, Monika P, Paria Y, Thilo D, Hans-Joachim W. ATM gene mutations in former uranium miners of SDAG Wismut: a pilot study. Oncol Rep. 2007;17:477–482. [PubMed] [Google Scholar]
- John KF, Narayani R, Vilmar V, David ME. Depleted uranium/uranyl chloride induces apoptosis in mouse J774 macrophages. Toxicology. 2002;179:105–114. doi: 10.1016/s0300-483x(02)00318-9. [DOI] [PubMed] [Google Scholar]
- Kanno S, Kitajima Y, Kakuta M, Osanai Y, Kurauchi K, Ujibe M, Is-hikawa M. Costunolide-induced apoptosis is caused by receptor-mediated pathway and inhibition of telomerase activity in NALM-6 cells. Biol Pharm Bull. 2008;5:1024–1028. doi: 10.1248/bpb.31.1024. [DOI] [PubMed] [Google Scholar]
- Kendra CL, Geoffrey HG. Regulation of the cellular DNA double-strand break response. Biochem Cell Biol. 2007;85:663–674. doi: 10.1139/O07-135. [DOI] [PubMed] [Google Scholar]
- Klervi L, Solenne B, Dominique B, Margot T, Sylvaine C, Benoıt Q, Dominique L. Lung cancer risk associated to exposure to radon and smoking in a case–control study of French uranium miners. Health Phys. 2007;92(4):371–378. doi: 10.1097/01.HP.0000252259.72683.2a. [DOI] [PubMed] [Google Scholar]
- Kryscio A, Ulrich Muller WU, Wojcik A, Kotschy N, Grobelny S, Streffer C. A cytogenetic analysis of the long-term effect of uranium mining on peripheral lymphocytes using the micronucleus—centromere assay. Int J Radiat Biol. 2001;77(11):1087–1093. doi: 10.1080/09553000110070289. [DOI] [PubMed] [Google Scholar]
- Levine S, Saltzman A. The balance between lymphatic and systemic absorption determines the outcome of sensitization for anaphylaxis in rats. Int Arch Allergy Immunol. 1994;105(1):91–95. doi: 10.1159/000236808. [DOI] [PubMed] [Google Scholar]
- Marjorie M, Michel MD, Francxois P, Valerie C, Gerard D, Marie D. Genotoxic and inflammatory effects of depleted uranium particles inhaled by rats. Toxicol Sci. 2006;89(1):287–295. doi: 10.1093/toxsci/kfj010. [DOI] [PubMed] [Google Scholar]
- Monica Y, Shania GL, Edgar CR, Diane SM. Uranyl acetate causes DNA single strand breaks in-vitro in the presence of ascorbate (Vitamin C) Chem Res Toxicol. 2003;16(4):524–530. doi: 10.1021/tx025685q. [DOI] [PubMed] [Google Scholar]
- Padmavati S, Arora R. Beta-adrenergic blocking agents in cardiology. J Assoc Physicians of India. 1976;24(11):725–728. [PubMed] [Google Scholar]
- Periyakaruppan A, Kumar F, Sarkar S, Sharma CS, Ramesh GT. Uranium induces oxidative stress in lung epithelial cells. Arch Toxicol. 2007;81(6):389–395. doi: 10.1007/s00204-006-0167-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat O, Berenguer F, Malard V, Tavan E, Sage N, Steinmetz G, Quemeneur E. Transcriptomic and proteomic responses of human renal HEK293 cells to uranium toxicity. Proteomics. 2005;5:297–306. doi: 10.1002/pmic.200400896. [DOI] [PubMed] [Google Scholar]
- Sandra WS, Douglas TW, AbouEl–Makarim A, Michael MD, John WP., Sr Particulate depleted uranium is cytotoxic and clastogenic to human lung cells. Chem Res Toxicol. 2007;20(5):815–820. doi: 10.1021/tx700026r. [DOI] [PubMed] [Google Scholar]
- Seon–Hee O, Sung–Chul L. A rapid and transient ROS generation by cadmium triggers apoptosis via caspase-dependent pathway in HepG2 cells and this is inhibited through N-acetylcysteine-mediated catalase upregulation. Toxicol Appl Pharmacol. 2006;212:212–223. doi: 10.1016/j.taap.2005.07.018. [DOI] [PubMed] [Google Scholar]
- Seon–Hee O, Byung-Hoon L, Sung-Chul L. Cadmium induces apoptotic cell death in WI 38 cells via caspase-dependent Bid cleavage and calpain-mediated mitochondrial Bax cleavage by Bcl-2-independent pathway. Biochem Pharmacol. 2004;68:1845–1855. doi: 10.1016/j.bcp.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Susumu O, Ying X, Motohide T. Effects of uranium ore dust on cultured human lung cells. Environ Toxicol Pharmacol. 1998;5(4):267–271. doi: 10.1016/s1382-6689(98)00010-6. [DOI] [PubMed] [Google Scholar]
- The Royal Society Working Group on the Health Hazards of Depleted Uranium Munitions. The health effects of depleted uranium munitions: a summary. J Radiol Prot. 2002;22:131–139. doi: 10.1088/0952-4746/22/2/301. [DOI] [PubMed] [Google Scholar]
- US Department of Health and Human Services. Toxicological profile for uranium. Agency for Toxic Substances and Disease Registry. 1999:462. [PubMed] [Google Scholar]
- Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Veronique M, Odette P, Elisabeth D, Frederic B, Nicole S, Eric Q. Proteomic analysis of the response of human lung cells to uranium. Proteomics. 2005;5:4568–4580. doi: 10.1002/pmic.200402038. [DOI] [PubMed] [Google Scholar]
- Vincent G, Saadia K, Gerard G, Marc P, Herve R. Uranium induces TNF-α secretion and MAPK activation in a rat alveolar macrophage cell line. Toxicol Appl Pharmacol. 2004;194:49–59. doi: 10.1016/j.taap.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Virginia CH, Diane SM. Molecular analysis of hprt mutations generated in Chinese hamster ovary EM9 cells by uranyl acetate, by hydrogen peroxide, and spontaneously. Mol Carcinog. 2006;45:60–72. doi: 10.1002/mc.20155. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Health effects of depleted uranium. Fifty-fourth World Health Assembly. 2001;A54(19):Add.1. [Google Scholar]
- Yang ZH, Fan BX, Lu Y, Cao ZS, Yu S, Fan FY, Zhu MX. Malignant transformation of human bronchial epithelial cell (BE-AS-2B) induced by depleted uranium. Ai-Zheng (Chin J Cancer) 2002;21(9):944–948. [PubMed] [Google Scholar]