

Abstract
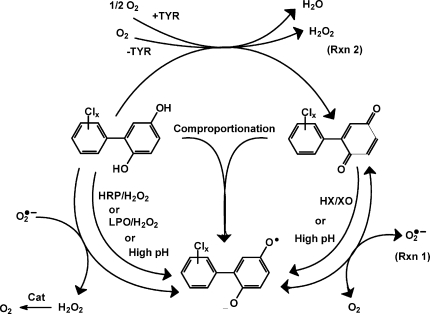
Polychlorinated biphenyls (PCBs) can be oxygenated to form very reactive hydroquinone and quinone products. A guiding hypothesis in the PCB research community is that some of the detrimental health effects of some PCBs are a consequence of these oxygenated forms undergoing one-electron oxidation or reduction, generating semiquinone radicals (SQ•−). These radicals can enter into a futile redox cycle resulting in the formation of reactive oxygen species, that is, superoxide and hydrogen peroxide. Here, we examine some of the properties and chemistry of these semiquinone free radicals. Using electron paramagnetic resonance (EPR) to detect SQ•− formation, we observed that (i) xanthine oxidase can reduce quinone PCBs to the corresponding SQ•−; (ii) the heme-containing peroxidases (horseradish and lactoperoxidase) can oxidize hydroquinone PCBs to the corresponding SQ•−; (iii) tyrosinase acting on PCB ortho-hydroquinones leads to the formation of SQ•−; (iv) mixtures of PCB quinone and hydroquinone form SQ•− via a comproportionation reaction; (v) SQ•− are formed when hydroquinone-PCBs undergo autoxidation in high pH buffer (≈>pH 8); and, surprisingly, (vi) quinone-PCBs in high pH buffer can also form SQ•−; (vii) these observations along with EPR suggest that hydroxide anion can add to the quinone ring; (viii) H2O2 in basic solution reacts rapidly with PCB-quinones; and (ix) at near-neutral pH SOD can catalyze the oxidization of PCB-hydroquinone to quinone, yielding H2O2. However, using 5,5-dimethylpyrroline-1-oxide (DMPO) as a spin-trapping agent, we did not trap superoxide, indicating that generation of superoxide from SQ•− is not kinetically favorable. These observations demonstrate multiple routes for the formation of SQ•− from PCB-quinones and hydroquinones. Our data also point to futile redox cycling as being one mechanism by which oxygenated PCBs can lead to the formation of reactive oxygen species, but this is most efficient in the presence of SOD.
Introduction
Polychlorinated biphenyls (PCBs) are a class of environmental pollutants that have from one to 10 chlorines substituted on the phenyl rings. PCBs have been widely used from the 1930s to the 1960s in diverse industrial applications, including cooling and insulating fluids for industrial transformers and capacitors, hydraulic fluids, and sealants (1,2). PCBs are known to elicit various adverse effects, including carcinogenicity, neuroendocrine disturbances, developmental and reproductive toxicity, and immunotoxicity (3,4). PCBs have been implicated in or related to cancer such as malignant melanoma, breast, and lung cancers in exposed populations (5). The production of PCBs was banned in the 1970s due to the high toxicity of most PCB congeners and mixtures. However, because of their physical and chemical properties, PCBs are quite stable; thus, they remain as ubiquitous environmental contaminants, which are frequently found as complex mixtures of isomers and congeners in air, water, soil, and dust on surfaces in homes and in factories (6).
Lower halogenated PCBs have been shown to be metabolized by rat microsomes to phenol and dihydroxybiphenyl metabolites (7–9). Robertson et al. have reported that the primary metabolites of 4-monochlorobiphenyl are 4′-chloro-2-hydroxybiphenyl, 4′-chloro-3-hydroxybiphenyl, and 4′-chloro-4-hydroxybiphenyl; these phenolic compounds can then subsequently undergo a second hydroxylation yielding 4′-chloro-3,4-dihydroxybiphenyl, 4′-chloro-2,3-dihydroxybiphenyl, and 4′-chloro-2,5-dihydroxybiphenyl (10). These para-dihydroxy PCBs and catechol type ortho-dihydroxy PCBs can be further oxidized to reactive quinones.
Early research suggests that the intracellular activation of hydroquinones and quinones producing relatively stable semiquinone free radicals is an important step to account for their cytotoxicity (11). Radicals produced “downstream”, such as superoxide anion radical (O2•−), will lead to hydrogen peroxide and hydroxyl radical formation (HO•); these active oxygen species will deplete antioxidants and potentially lead to an increase in oxidative stress (12–15). These reactive oxygen species can oxidize lipids, proteins, and DNA (16–18).
In the work reported here, we used EPR to examine the many routes that can lead to the formation of PCB-semiquinone radicals. We also examined the different spectral patterns of these various semiquinone radicals and used the changes observed in the EPR spectra to understand their chemistry.
Materials and Methods
Caution:PCB derivatives should be handled as hazardous compounds in accordance with NIH guidelines.
Materials
Hypoxanthine (HX),1 xanthine oxidase (XO), horseradish peroxidase (HRP), tyrosinase (TYR), lactoperoxidase (LPO), catalase (Cat), superoxide dismutase (SOD), and diethylenetriaminepentaacetic acid (DETAPAC or DTPA) were from Sigma (St. Louis, MO); benzoquinone, hydroquinone, phenyl-quinone, sodium hydroxide, and H2O2 were from Fisher; and quinhydrone was from Alfa Aesar (Ward Hill, MA). DMPO was from Dojindo Laboratories (Japan). Materials were used without further purification. The various PCB hydroquinones and quinones were synthesized as previously described (19–21). Stock solutions (200 mM each) of PCBs were prepared in DMSO. Stock solutions of HX (10 mM), HRP (0.2 U/µL), TYR (1 U/µL), H2O2 (20 mM), SOD (100 U/µL), and Cat (20 U/µL) were prepared with Nanopure water just before use; XO (10.8 mU/µL) and LPO (208 mU/µL) were used as received. All experiments were carried out in 100 mM phosphate buffer containing 250 µM DETAPAC. When needed, adjustment of the pH was done with 5 M sodium hydroxide solution. All of the PCB quinones and hydroquinones studied are shown in Figure 1.
Figure 1.

Chemical structures and nomenclature of the PCB quinones and hydroquinones used.
EPR Spectroscopy
EPR spectroscopy was done using a Bruker EMX spectrometer equipped with a high-sensitivity cavity and an Aqua-X sample holder. Spectra were obtained at room temperature. Typical EPR parameters were as follows: 3511 G center field; 15 or 80 G sweep width (for the spin trapping experiments with DMPO); 9.854 GHz microwave frequency; 20 mW power; 2 × 105 receiver gain; modulation frequency of 100 kHz; modulation amplitude of 1 or 0.1 G; with the conversion time and time constant both being 40.96 ms with 5 X-scans for each 1024 point spectrum.
The final concentrations of PCB derivatives were 100 µM unless specifically mentioned. Alkaline solutions (pH 10) were prepared by adjusting with 5 M sodium hydroxide solution.
Spectral simulations of EPR spectra were performed using the WinSim program developed at the NIEHS by Duling (22). Correlation coefficients of simulated spectra were typically >0.99.
Oxygen Uptake
Oxygen uptake was determined with a Clark type electrode using an YSI model 5300 Biological Oxygen Monitor (Yellow Springs Instrument Co., Yellow Springs, OH). All measurements were made at room temperature; 3.00 mL of PBS buffer was loaded in the chamber of the oxygen monitor and aerated by stirring for 5 min, and then, the electrode was placed in contact with the solution, leaving no headspace for air and ensuring that all air bubbles were removed. After a 2 min equilibration, reagents were introduced through the access slot, and then, oxygen consumption was monitored. Each experiment was repeated at least three times. Results of replicate experiments varied less than ±10%.
UV−Vis Spectroscopy
UV spectra were recorded with a Hewlett-Packard 8453 diode array spectrometer. All measurements were made at room temperature; 1.00 mL of PBS buffer was loaded in the cuvette, stirring with a small stirring bar to ensure that the solution mixed well. Using the kinetics setting, after the introduction of reagents, the change in absorbance at 249 nm was recorded vs time.
Results and Discussion
The generation of semiquinone radicals by oxidation/reduction of hydroquinones and quinones is well-documented (23–25). Polychlorinated biphenyls can be oxidized to hydroquinones (H2Q) and quinones (Q). These species can be converted to their corresponding semiquinone free radicals (SQ•−) that will have a quite different reactivity than the parent quinone or hydroquinone. Here, we examine the formation of these SQ•− radicals.
PCB Semiquinone Radicals Can Be Generated by Several Approaches
SQ•− Via Xanthine Oxidase
The one-electron reduction of a quinone will yield its corresponding semiquinone radical. To determine if typical reducing enzymes can facilitate such a reaction, we examined the potential formation of SQ•− from quinones by the molybdoflavin enzyme xanthine oxidase. Xanthine oxidase has been shown to reduce by one-electron compounds, such as adriamycin, to their corresponding SQ•−(26) (Scheme 1). To demonstrate the usefulness of the hypoxanthine/xanthine oxidase system (HX/XO) in our experiments with PCB-derived quinones, we examined its ability to reduce 1,4-benzoquinone to its corresponding semiquinone radical. As seen in the EPR spectrum of Figure 2a, the well-studied five-line semiquinone free radical (1:4:6:4:1 intensity ratio, aH(4) = 2.37 G) of 1,4-benzoquinone (27,28) was obtained immediately after the introduction of 1,4-benzoquinone to the HX/XO enzyme system. This demonstrates the utility of HX/XO, a flavoenzyme, in studying the reductive formation of SQ•−from Q.
Scheme 1. Reaction Pathways of PCB Hydroquinone, Semiquinone, And Quinone.

High pH implies values greater than 8 or 9, as seen in Figure 7. Tyrosinase only acts on ortho-hydroquinones.
Figure 2.
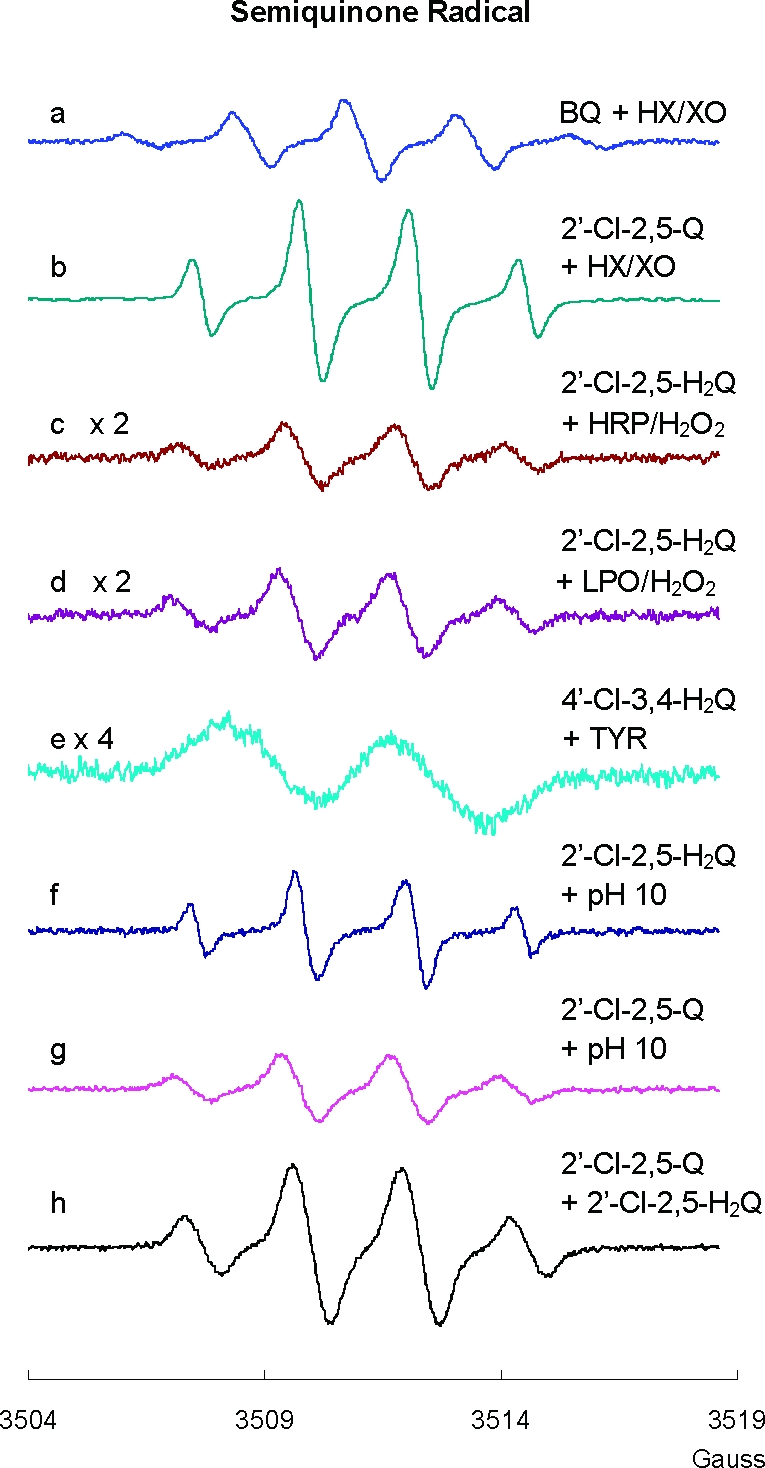
Semiquinone radical (SQ•−) can be formed by many routes: (a) SQ•− generated from BQ reacted with HX/XO, [HX] = 50 µM, [XO] = 20 mU/mL; (b) SQ•− generated from 2′-Cl-2,5-Q reacted with HX/XO, [HX] = 50 µM, [XO] = 20 mU/mL; (c) SQ•− generated from 2′-Cl-2,5-H2Q reacted with HRP/H2O2, [HRP] = 10 mU/mL, [H2O2] = 10 µM; (d) SQ•− generated from 2′-Cl-2,5-H2Q reacted with LPO/H2O2 system, [LPO] = 0.1 U/mL, [H2O2] = 10 µM; (e) SQ•− generated from 4′-Cl-3,4-H2Q reacted with tyrosinase, [TYR] = 2.5 U/mL; (f) SQ•− generated from 2′-Cl-2,5-H2Q at pH 10; (g) SQ•− generated from 2′-Cl-2,5-Q at pH 10; and (h) SQ•− generated from 2′-Cl-2,5-Q and 2′-Cl-2,5-H2Q mixed 1:1, pH 7.4. The EPR modulation amplitude was 1 G for spectra a−e and h and 0.1 G for spectra f and g.
PCB-quinones can be viewed as substituted benzoquinone (Figure 1). Addition of the PCB-quinone 2′-Cl-2,5-Q to the HX/XO system (pH 7.4) resulted in a distinct four-line spectrum with aH3 = aH4 = 2.1 G and aH6 = 2.5 G (spectrum b of Figure 2). The spectrum of this radical appeared rapidly upon mixing (<130 s); its intensity decreased with time lasting for several minutes under our experimental conditions (an observed half-life of about 6 min). The WinSim program provided the best fit when the hydrogens and chlorine on the second ring made a contribution to the simulated spectrum (Table 1). These splitting constants are all very small; thus, their contribution is in essence only to the observed line width.
Table 1. Hyperfine splitting Constants from the Various PCB-SQ•−a.
| compd | 2 | 3 | 4 | 5 | 6 | 2′ | 3′ | 4′ | 5′ | 6′ | line widthb |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2′-Cl-2,5-Q | (OH) | 2.2 (H) | 2.4 (H) | (OH) | 2.2 (H) | 0.1 (Cl) | 0.1 (H) | 0.1 (H) | 0.1 (H) | 0.2 (H) | 0.17 |
| 3′-Cl-2,5-Q | (OH) | 2.1 (H) | 2.5 (H) | (OH) | 2.1 (H) | 0.1 (H) | 0.1 (Cl) | 0.1 (H) | 0.2 (H) | 0.2 (H) | 0.37 |
| 4′-Cl-2,5-Q | (OH) | 2.1 (H) | 2.5 (H) | (OH) | 2.1 (H) | 0.2 (H) | 0.1 (H) | 0.1 (Cl) | 0.1 (H) | 0.2 (H) | 0.32 |
| 3′,4′-Cl-2,5-Q | (OH) | 2.1 (H) | 2.5 (H) | (OH) | 2.1 (H) | 0.1 (H) | 0.17 (Cl) | 0.1 (Cl) | 0.1 (H) | 0.3 (H) | 0.32 |
| 2′-Cl-2,5-H2Q | (OH) | 2.2 (H) | 2.5 (H) | (OH) | 2.2 (H) | 0.1 (Cl) | 0.1 (H) | 0.1 (H) | 0.1 (H) | 0.2 (H) | 0.11 |
| 3′-Cl-2,5-H2Q | (OH) | 2.1 (H) | 2.5 (H) | (OH) | 2.1 (H) | 0.1 (H) | 0.1 (Cl) | 0.2 (H) | 0.2 (H) | 0.3 (H) | 0.09 |
| 4′-Cl-2,5-H2Q | (OH) | 2.1 (H) | 2.5 (H) | (OH) | 2.1 (H) | 0.2 (H) | 0.1 (H) | 0.1 (Cl) | 0.1 (H) | 0.2 (H) | 0.19 |
| 4′-Cl-2,3-H2Q | (OH) | (OH) | 0.5 (H) | 3.9 (H) | 3.2 (H) | 0.1 (H) | 0.1 (H) | 0.1 (Cl) | 0.1 (H) | 0.1 (H) | 0.10 |
| 3′,4′-Cl-2,3-H2Q | (OH) | (OH) | 0.6 (H) | 3.8 (H) | 3.3 (H) | 0.1 (H) | 0.1 (Cl) | 0.1 (Cl) | 0.1 (H) | 0.1 (H) | 0.14 |
| 4′-Cl-3,4-H2Q | 0.1 (H) | (OH) | (OH) | 1.2 (H) | 3.2 (H) | 0.7 (H) | 0.3 (H) | 0.1 (Cl) | 0.3 (H) | 0.7 (H) | 0.11 |
| 2′,5′-Cl-3,4-H2Q | 0.7 (H) | (OH) | (OH) | 1.0 (H) | 3.3 (H) | 0.2 (Cl) | 0.4 (H) | 0.2 (H) | 0.2 (Cl) | 0.6 (H) | 0.20 |
| 2′,3′-Cl-3,4-H2Q | 0.8 (H) | (OH) | (OH) | 1.1 (H) | 3.3 (H) | 0.3 (Cl) | 0.2 (Cl) | 0.1 (H) | 0.2 (H) | 0.4 (H) | 0.07 |
These hyperfine splittings (in Gauss) are the results from simulations of the experimental spectra. Assignment of hyperfine splittings to particular hydrogens on the semiquinone ring followed examples of previously published work (36–38). On the secondary ring, we make no definite assignments. The numbering pattern is for the structure:
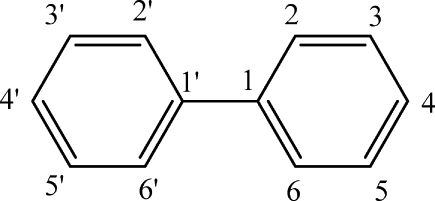
In Gauss.
HRP/LPO with H2O2
Horseradish peroxidase (HRP) and lactoperoxidase (LPO) are the enzymes known to catalyze H2O2-dependent one-electron oxidations; these systems will oxidize hydroquinones to the corresponding SQ•−. When 2′-Cl-2,5-H2Q was introduced into these systems, the spectra observed were consistent with the formation of 2′-Cl-2,5-SQ•− (Figure 2c,d). When 3′-Cl-2,5-H2Q or 4′-Cl-2,5-H2Q was introduced, their corresponding SQ•− radicals were observed (not shown). These spectra were identical (except for intensity) to those observed when their respective quinones were exposed to the HX/XO system. Thus, the same SQ•− can be derived from either the hydroquinone or the quinone with appropriate one-electron oxidation or reduction.
Tyrosinase
Tyrosinase (TYR) is a copper-containing enzyme present in plant and animal tissues. It catalyzes the two-electron oxidation of ortho-hydroquinones (not para-hydroquinones) to their corresponding quinones (29,30). In our reaction, tyrosinase will catalyze the oxidation of one molecule of PCB ortho-hydroquinone to its quinone with the reduction of 1/2 molecule of oxygen to form one H2O (Scheme 1). This introduction of quinone will allow the formation of SQ•− via the comproportionation of hydroquinone and quinone. In air-saturated buffer at pH 7.4, 100 µM ortho-hydroquinone 4′-Cl-3,4-H2Q produced a small background EPR signal of 4′-Cl-3,4-SQ•− (at the noise level), due to the slow oxidation of the hydroquinone (not shown). However, in the presence of TYR, a much stronger EPR spectrum of SQ•− from 4′-Cl-3,4-H2Q was observed (Figure 2e). When a para-hydroquinone was exposed to TYR, no increase in SQ•− was observed (not shown). In the EPR experiments, if too much TYR was introduced into the incubation such that all of the hydroquinone was very rapidly oxidized to quinone, the EPR signal of SQ•− was quite weak or below the limit of detection.
If reaction mixtures identical to those used for EPR were monitored for oxygen consumption, the predicted stoichiometric loss of oxygen was observed with ortho-hydroquinones, while no oxygen was lost with para-hydroquinones. These results demonstrate that ortho-hydroquinone-PCBs have additional mechanisms for radical formation as compared to para-hydroquinone PCBs.
Autoxidation in Aerobic Solutions
When the phenolic OH groups of hydroquinones ionize, the anions are prone to rapid autoxidation. Indeed, the semiquinone radical of hydroquinone was observed by EPR spectroscopy in the 1950s when it was introduced to oxygenated, alkaline media (31). We hypothesized that hydroquinone derivatives of PCBs would also undergo a parallel autoxidation reaction and generate corresponding PCB semiquinones (Scheme 2A). To examine this hypothesis, we introduced 2′-Cl-2,5-H2Q to an alkaline environment (pH 10); we obtained the corresponding SQ•− radical (Figure 2f and Table 1). It has been suggested that the generation of SQ•− in alkaline solution is only the first stage of the autoxidation of dihydroxy phenols (32–34). The radical can continue to oxidize to quinone, and then, a comproportionation reaction will turn one molecule of hydroquinone and one molecule of quinone into two molecules of SQ•− (Scheme 2B). Increasing the pH from 7 to 12 dramatically increased the concentration of the initially formed SQ•−; this is because high [OH−] increases the rate of autoxidation of hydroquinone and decreases the rate of decay of SQ•−, principally by disproportionation (Scheme 1), which is dependent on [H+]2(35).
Scheme 2. Mechanisms for the Autoxidation of PCB Hydroquinone and Quinone To Generate SQ•− and SQ(II)•−.

Using the HX/XO system, we have shown above that PCB quinones can be reduced by one electron to corresponding PCB semiquinone radicals. In addition, H2Q can be oxidized to form SQ•−; high pH can increase this rate. However, a most surprising observation is that 2′-Cl-2,5-Q in high pH solution also generates a strong SQ•− signal (Figure 2g). To generate SQ•− from Q, there must be a source of electrons. In this experiment, we observed that the EPR signal for the initial SQ•− (≈1:3:3:1) radical gives way to a 1:2:1 spectrum, consistent with the formation of a secondary radical SQ(II)•−. A possible mechanism for the continued autoxidation PCB quinone is described in Scheme 2C. The evolution of the EPR spectrum from a ≈1:3:3:1 pattern to a 1:2:1 pattern indicates the loss of a hydrogen from the quinone ring (Figure 3). This suggests a base-catalyzed Michael addition of an OH− to the quinone ring (OH− can in principle substitute for any of the three hydrogens on the quinone ring; only one possibility is shown) (Scheme 2C). When quinone is introduced to an alkaline solution, we initially see only the primary radical, SQ•−; the secondary radical SQ(II)•− is observed after some time has passed, indicating that the reaction is somewhat slow and a significant amount of the trihydroxy compound must be formed before the 1:2:1 spectrum can be observed. The addition of OH− to the quinone ring provides the reducing equivalents to form the initial SQ•− observed as well as SQ(II)•− (Scheme 2C).
Figure 3.
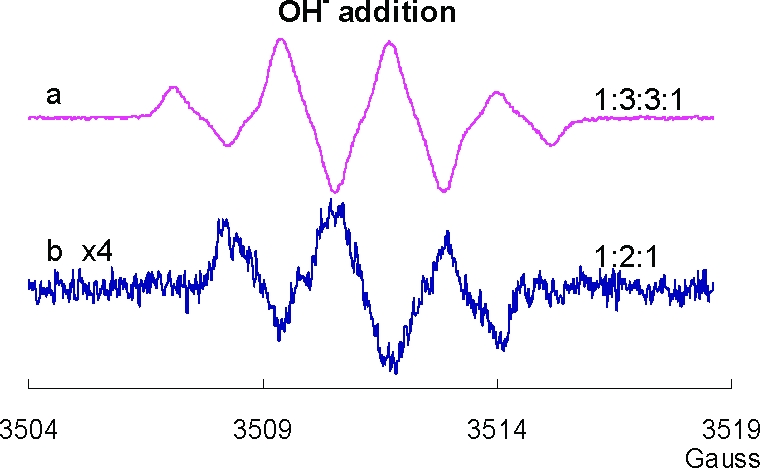
EPR spectra from dihydroxyl and trihydroxyl PCB: Changes in the EPR spectra of 2′-Cl-2,5-H2Q after introduction of NaOH (15 µL of 5 M NaOH add into 2000 µL of 100 µM 2′-Cl-2,5-H2Q in buffer): (a) before introduction of NaOH and (b) 5 min after the introduction of NaOH. The EPR modulation amplitude was 1 G.
SQ•− Via Comproportionation
To demonstrate that SQ•− can be formed by the comproportionation reaction of H2Q and Q, we examined by EPR solutions containing both species (Scheme 2B). We used quinhydrone, a charge transfer complex consisting of equal parts of hydroquinone interacting with benzoquinone through ring stacking, as a test system; a strong EPR spectrum for the benzosemiquinone radical was observed in neutral solution consistent with an equilibrium comproportionation reaction (spectrum not shown; it has the same characteristics as the spectrum of Figure 2a). When 2′-Cl-2,5-Q and 2′-Cl-2,5-H2Q were mixed (1:1 ratio, 50 µM each) in neutral solution, the corresponding SQ•− radical was observed by EPR (Figure 2h). When the ratio of H2Q:Q was varied over the range of 1:9 to 9:1 (keeping [H2Q] + [Q] = 100 µM), SQ•− was observed at all ratios with approximately the same intensity. This is consistent with comproportionation and disproportionation reactions achieving equilibrium rapidly, on the time scale of the EPR observations, with an accompanying autoxidation reaction removing the radical. As expected, the intensity of the EPR signal increased with increasing pH. At high pH, the hydroquinone will ionize, leading to a more rapid autoxidation; in addition, the disproportionation reaction is slowed, as protons are needed, thus a higher EPR signal intensity.
Characterization of SQ•− Generated from a Wide Range of Oxygenated PCBs
Because semiquinone radicals are more stable in alkaline solution, we generated a series of PCB semiquinone radicals at pH 10 to investigate the influence of the chemical structure on the EPR spectra (Figure 4). Here, we focus on the initially formed, primary radicals that are observed upon raising the pH to 10.
Figure 4.

Semiquinone radical generated from PCB with different substitution patterns at pH 10 and computer simulations. Hyperfine splittings and spectral line widths derived by simulation of experimental spectra are presented in Table 1. (A) Semiquinone radicals generated from different 2,5-Q at pH 10: (a) SQ•− generated from 4′-Cl-2,5-Q; (b) SQ•− generated from 3′-Cl-2,5-Q; (c) SQ•− generated from 3′, 4′-Cl-2,5-Q; and (d) SQ•− generated from PQ. (B) Semiquinone radicals generated from different 2,5-H2Q at pH 10: (a) SQ•− generated from 4′-Cl-2,5-H2Q; (b) simulation of 4′-Cl-2,5-SQ•−; and (c) SQ•− generated from 3′-Cl-2,5-H2Q. (C) Semiquinone radicals generated from different 2,3-H2Q at pH 10: (a) SQ•− generated from 4′-Cl-2,3-H2Q; (b) SQ•− generated from 3′,4′-Cl-2,3-H2Q; and (c) simulation of 3′,4′-Cl-2,3- SQ•−. (D) Semiquinone radicals generated from different 3,4-H2Q at pH 10: (a) SQ•− generated from 4′-Cl-3,4-H2Q; (b) simulation of 4′-Cl-3,4-SQ•−; (c) SQ•− generated from 2′,5′-Cl-3,4-H2Q; (d) simulation of 2′,5′-Cl-3,4-SQ•−; (e) SQ•− generated from 2′,3′-Cl-3,4-H2Q; and (f) simulation of 2′,3′-Cl-3,4-SQ•−. All experimental spectra were collected using a 0.1 G modulation amplitude.
The EPR spectra of the SQ•− generated from different xn′ -Cl-2,5-Q and xn′-Cl-2,5-H2Q are quite similar (Figure 4A,B); starting with either the quinone or the hydroquinone of a specific xn′ -Cl-2,5-Q, the same semiquinone spectrum was observed. Hyperfine splitting constants, derived from simulation (Table 1), showed three nearly identical hydrogens, which yield a 1:3:3:1 four-line spectrum. These results show that the primary interactions are with the hydrogens on the semiquinone ring; the other ring provides no unique splittings, perhaps only contributing to the line width.
There are two different families of PCB ortho-hydroquinones, 2,3-H2Q and 3,4-H2Q. When oxidized to their corresponding SQ•−, each member of a family produced a similar EPR spectral pattern (Figure 4C,D and Table 1). Successful simulations of spectra from 2,5-SQ•− or 2,3-SQ•− did not require any contributions of hyperfine splittings from the other phenyl ring. However, for the 3,4-SQ•− (Figure 4D), hyperfine splittings from the second phenyl ring contributed markedly. Definitive assignments of hyperfine splittings to specific H or Cl atoms from the second phenyl ring (the phenyl ring with Cl) have not been made (Table 1). We have used the literature (36–38) to guide our assignment of the hyperfine splitting constants for the hydrogens on the semiquinone ring. The simulation results are consistent with low spin density on the secondary ring. However, the xn′-Cl-3,4-SQ•− radicals show considerable spin density on the secondary ring, especially 4′-Cl-3,4-SQ•−.
Spin Trapping with DMPO
Semiquinones can transfer an electron to oxygen generating the superoxide radical (18,39). Superoxide can play a key role in the generation of oxidative damage under various pathophysiological conditions. However, O2•− is very short-lived, and in most cases, the sensitivity of EPR spectroscopy is insufficient for direct detection. Thus, we used EPR spin trapping to probe for the formation of superoxide radicals (40–42). A spin trap reacts with short-lived radicals to form much longer-lived spin adducts that accumulate to a level detectable by EPR.
In Figure 5, spectrum “a” is a control demonstrating the absence of artifactual signals from DMPO. Spectrum “b” represents the typical DMPO/•OOH spin adduct generated from HX/XO system and trapped by DMPO; some DMPO/HO• spin adduct is also present. When 2′-Cl-2,5-Q was added to DMPO in neutral solution, both SQ•− and DMPO/HO• were detected (Figure 5c). If SOD was included, there was no significant change in the intensity of the spectrum of SQ•− (spectrum “d”), but the signal of the DMPO/HO• radical increased. Because no evidence for DMPO/•OOH radical was observed in “c” and SOD did not decrease the DMPO/HO• signal, there appears to be no significant superoxide formation from 2′-Cl-2,5-Q.
Figure 5.
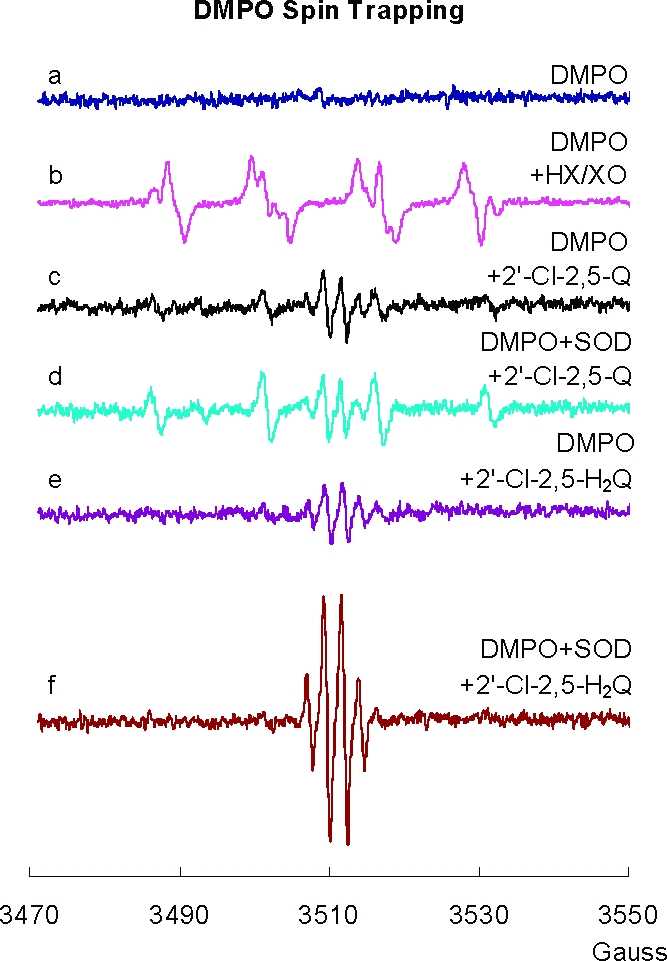
Spin trapping with DMPO indicates that superoxide formation is below the limit of detection. (a) EPR spectrum by DMPO (50 mM) in PBS with DETAPAC (250 µM); (b) superoxide and hydroxyl adducts of DMPO (50 mM) generated with HX/XO at pH 7.4 (aN = 14.1 G, aH = 11.3 G with an additional hydrogen splitting of 1.3 G for DMPO/•OOH, and aN = aH = 14.9 G for DMPO/•OH); (c) spectrum generated from 2′-Cl-2,5-Q (1 mM) and DMPO (100 mM) at pH 7.4; (d) spectrum generated from 2′-Cl-2,5-Q (1 mM), DMPO (100 mM), and 500 U SOD/mL at pH 7.4; (e) spectrum generated from 2′-Cl-2,5- H2Q (1 mM) and DMPO (100 mM) at pH 7.4; and (f) spectrum generated from 2′-Cl-2,5-H2Q (1 mM), DMPO (100 mM), and 500 U SOD/mL at pH 7.4. All experimental spectra were collected using a 1.0 G modulation amplitude.
When 2′-Cl-2,5-H2Q was substituted for 2′-Cl-2,5-Q, the addition of SOD stimulated the generation of SQ•− (Figure 5e,f); the intensity of the SQ•− spectrum increased over time, reached a steady state, and then decreased (data not shown). Cat had no effect on the generation of SQ•− or DMPO/HO• radical (not shown). These observations indicate that generation of superoxide from SQ•−, reaction 1, is not kinetically or thermodynamically favorable.
The equilibrium for this reaction lies far to the left. Using benzoquinone and various substituted quinones as references, the rate constant for the forward reaction will be approximately 104−105 M−1 s−1, while the rate constant for the reverse reaction is on the order of 109 M−1 s−118,43,44. This rapid back reaction explains why our spin trapping experiment cannot detect superoxide formation.
SOD Catalyzes the Autoxidation of Hydroquinone as Seen by UV Spectroscopy
The increase in [SQ•−] observed in Figure 5f suggests that SOD can catalyze the autoxidation of 2′-Cl-2,5-H2Q to 2′-Cl-2,5-Q. To observe if the rate of formation of 2′-Cl-2,5-Q increases when SOD is added to a near-neutral solution of 2′-Cl-2,5-H2Q, we used UV spectroscopy to follow the rate of formation of 2′-Cl-2,5-Q. In the absence of SOD, the autoxidation of 2′-Cl-2,5-H2Q (100 µM) to quinone is relatively slow (Figure 6a,b). When SOD is present at time zero, there is no change in the initial rate of autoxidation; however, after about 10 min or so, autoxidation accelerates (Figure 6c). If a trace of 2′-Cl-2,5-Q (1 µM) is introduced at time zero along with SOD, there is no lag time for a much more rapid rate of autoxidation. These observations are parallel to those observed by Eyer in a study of hydroquinone autoxidation (44); this same process appears to hold with coenzyme Q semiquinone radical in mitochondria with changes in MnSOD (45). Superoxide is formed by reaction 1; however, the equilibrium lies far to the left. SOD pulls this equilibrium by removing superoxide before it enters the back reaction due to its very rapid dismutation of superoxide, reaction 2.
In the experiment of Figure 6c, the rate of autoxidation remains low until significant 2′-Cl-2,5-Q is generated, allowing for the comproportionation reaction of 2′-Cl-2,5-Q with 2′-Cl-2,5-H2Q to form SQ•−, reaction 3. However, if a trace of 2′-Cl-2,5-Q is introduced at time zero (Figure 6d), no lag time is observed because all of the ingredients are present to make significant SQ•−.
These observations suggest that oxygen will be consumed in the autoxidation reaction, giving rise to the formation of H2O2, the net reaction being
Figure 6.
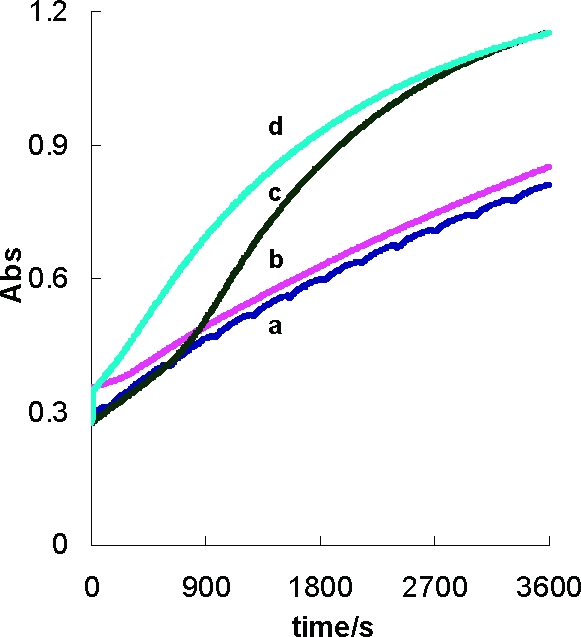
SOD accelerates the autoxidation of hydroquinone. The formation of 2′-Cl-2,5-Q during the autoxidation of 2′-Cl-2,5-H2Q (0.1 mM) was followed at 249 nm. (a) 2′-Cl-2,5-H2Q (0.1 mM); (b) 2′-Cl-2,5-H2Q (0.1 mM) mixed with 2′-Cl-2,5-Q (1 µM); (c) 2′-Cl-2,5-H2Q (0.1 mM) with 100 U/mL SOD; and (d) 2′-Cl-2,5-H2Q (0.1 mM) mixed with 2′-Cl-2,5-Q (1 µM) plus 100 U/mL SOD.
Oxygen Consumption
Oxygen Uptake at Near-Neutral pH
If SOD accelerates the rate of oxidation of 2′-Cl-2,5-H2Q as seen by formation of quinone, then the rate of oxygen consumption should also increase. Indeed, when 2′-Cl-2,5-H2Q (500 µM) was introduced into pH 7.4 PBS, the rate of oxygen consumption paralleled all of the results of Figure 6 (data not shown). The introduction of Cat to a partially oxidized solution of 2′-Cl-2,5-H2Q demonstrated that nearly all of the oxygen consumed was present as H2O2. These observations support reactions 1–4 as being operative in this system.
Oxygen Uptake in Alkaline pH Solutions
Autoxidation of 2′-Cl-2,5-H2Q in alkaline pH is accelerated due to ionization of the phenolic hydroxyls and subsequent one-electron oxidation to 2′-Cl-2,5-SQ•−, followed by loss of a second electron yielding 2′-Cl-2,5-Q. One might predict that 1 equiv of H2Q would consume 1 equiv of dioxygen, yielding 1 equiv of H2O2 (reaction 4). Interestingly, the amount of oxygen consumed is a function of pH (Figure 7). We observed a sharp increase in the amount of oxygen consumed as the pH was increased from pH 8 to pH 13. At higher pH values, we observed that H2Q consumed almost 2 equiv of oxygen, rather than the 1 equiv anticipated; unexpectedly, we observed that the quinone also consumed oxygen in a pH-dependent manner. The amounts of oxygen consumed by H2Q and Q paralleled each other; the quinone form always consumed one-half equiv less oxygen than H2Q at a particular pH. These observations are consistent with oxidation of hydroquinone to quinone, followed by the nucleophilic addition of OH− to the quinone forming a trihydroxy compound that can be oxidized in the high pH environment (Scheme 2C). When starting with the quinone, there would be no initial oxidation; thus, less oxygen would be consumed.
Figure 7.
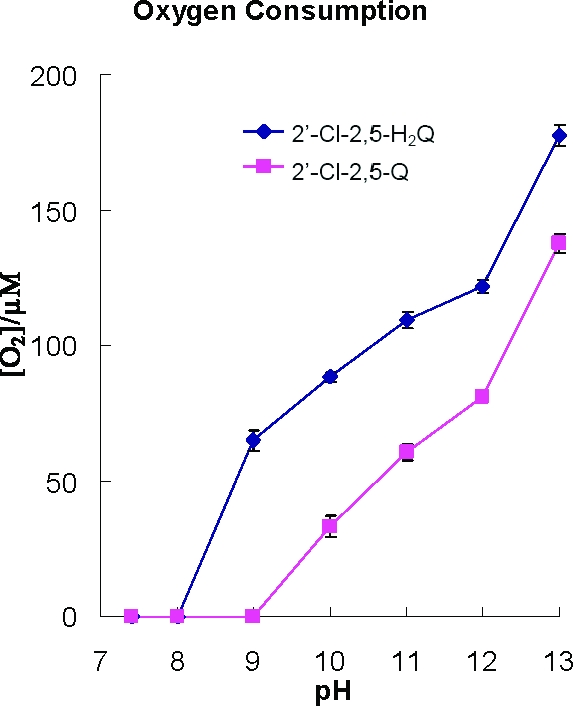
Oxygen consumption of PCB hydroquinone and quinone in different pH solutions. 2′-Cl-2,5-H2Q (100 µM) or 2′-Cl-2,5-Q (100 µM) was introduced into the closed chamber of the YSI Biological Oxygen monitor containing solutions having different pH values. The data points are the means of three determinations; standard errors are in general smaller than the symbols used to present the data.
The rate of consumption of oxygen is dramatically increased upon initiating a pH jump of a near-neutral solution of the oxygenated PCBs that we examined (Figure 7). If reaction 4 is operative, then H2O2 should be formed. We investigated this possible formation of H2O2 using 2′-Cl-2,5-H2Q and 2′-Cl-2,5-Q. Cat disproportionates 2H2O2 into 2H2O and 1O2; thus, it can be used as a tool to examine the stoichiometry of the reduction O2 to H2O2. In near-neutral solution, little or no oxygen was consumed after introduction of 2′-Cl-2,5-H2Q (Table 2, exp 1). However, if the pH is increased to ≈12 by the addition of NaOH, a rapid loss of oxygen occurs (−170 µM) (Table 2, exp 2). If reaction 4 holds, we would have expected to lose only 100 µM O2. These observations are consistent with Figure 7 and the reactions outlined in Scheme 2C. Most surprising was our observation that upon introduction of Cat, little if any oxygen returned, indicating that there was no hydrogen peroxide in the system. (Although high pH inactivates enzymes, this high pH did not inactivate Cat in the short time frame of an experiment; data not shown.)
Table 2. Oxygen Uptake and Changes in [H2O2] upon Oxidation of 2′-Cl-2,5-H2Q or 2′-Cl-2,5-Q by Increasing the pH.
| change in oxygen concentration (µM)a,b |
|||||
|---|---|---|---|---|---|
| additions | exp 1 | exp 2 | exp 3 | exp 4 | exp 5 |
| 2′-Cl-2,5-H2Q (100 µM)c | 0d | 0 | 0 | 0 | 0 |
| H2O2 (100 µM) | e | 0 | 0 | ||
| NaOHf | −170 | −170 | −90 | ||
| H2O2 (100 µM) | 0 | ||||
| Cat (150 U/mL) | 0 | +<5 | +50 | +50 | +30 |
| expected with Catg | 0 | +50 | +50 | +100 | +100 |
| change in oxygen concentration (µM)a,b |
|||||
|---|---|---|---|---|---|
| additions | exp 6 | exp 7 | exp 8 | exp 9 | exp 10 |
| 2′-Cl-2,5-Q (100 µM) | 0 | 0 | 0 | 0 | 0 |
| H2O2 (100 µM) | 0 | 0 | |||
| NaOHf | −70 | −70 | −50 | ||
| H2O2 (100 µM) | 0 | ||||
| Cat (150 U/mL) | 0 | +<5 | +50 | +50 | +5 |
| expected with Catg | 0 | +35 | +50 | +50 | +50 |
Each experiment was done at least three times. Variations between experiments are on the order of 10%.
Each entry represents the change in oxygen concentration after the addition. The additions were made sequentially as shown down column 1.
In PBS, pH 7.4.
The actual loss of oxygen is less than or on the order of the drift of the instrument, which typically is less than 5 µM per 15 min.
A blank cell indicates that there was no addition of this reagent and thus no change in [O2].
The addition of 20 µL of 5 M NaOH into 2 mL of buffer increases the pH of solution to over 12.
Expected return of oxygen assuming that the only important reaction upon introduction of NaOH is reaction 2 when hydroquinone is reactant; if quinone is the reactant, the expected return of oxygen assumes no reaction between the quinone and the H2O2.
To determine if the hydroquinone reacts directly with H2O2, we introduced 100 µM H2O2 into a neutral solution of 100 µM 2′-Cl-2,5-H2Q. After 15 min, all of the H2O2 was still present as the introduction of Cat saw the return of 50 µM O2 (Table 2, exp 3). Thus, there is no rapid direct reaction of hydroquinone with H2O2. In exp 4, we introduced H2O2 after the reactions initiated by the pH jump were complete. The addition of Cat resulted in the return of 50 µM O2, demonstrating that there was no rapid direct reaction of H2O2 with the oxidation products of 2′-Cl-2,5-H2Q. Interestingly, the introduction of H2O2 before the pH jump reduced oxygen consumption (−170 to −90 µM; Table 2, exp 5); Cat returned only 30 µM oxygen, indicating that some of the H2O2 was lost. Experiments 1−5 of Table 2 indicate that H2O2 must be reacting with intermediates in the oxidation reactions of 2′-Cl-2,5-H2Q.
Hydrogen peroxide has been shown to react with benzoquinone (46) and chlorinated benzoquinone (47). Experiments 6−10 of Table 2 examine the possible reaction of H2O2 with a typical PCB-quinone, 2′-Cl-2,5-Q. At near-neutral pH, there was no apparent reaction of H2O2 with 2′-Cl-2,5-Q (exp 8). However, experiments 7 and 10 show a considerable loss of H2O2 upon a pH jump. An explanation for this observation is that in pH 12 solution, some H2O2 will be deprotonated (pK1 for H2O2 to form OOH− is 11.7, (48)). The peroxide anion is a powerful nucleophile, much more reactive than hydroxide anion (47). It will react with PCB quinone, similar to OH−; however, the reaction will likely be much faster (Scheme 2D). Thus, experiments 7 and 10 in Table 2 with 2′-Cl-2,5-Q are consistent with a rapid nucleophilic addition reaction of H2O2, via OOH−, to the quinone.
Conclusions
In this work, we have demonstrated the following:
-
1.
The same semiquinone radical is produced when a PCB-hydroquinone undergoes a one-electron oxidization or the quinone form undergoes a one-electron reduction. This can be accomplished many different ways, including the following:
-
(a)
Xanthine oxidase can reduce quinone PCBs to the corresponding SQ•−.
-
(b)
The heme-containing peroxidases (horseradish and lactoperoxidase) can oxidize hydroquinone PCBs to the corresponding SQ•−.
-
(c)
Tyrosinase acting on PCB ortho-hydroquinones leads to formation of SQ•−.
-
(d)
SQ•− is formed rapidly when hydroquinone-PCBs undergo air oxidation in high pH buffer (≈>pH 8).
-
(e)
Quinone-PCBs in high pH buffer can also form SQ•−.
-
(f)
Mixtures of PCB-quinone and hydroquinone form SQ•− via a comproportionation reaction.
-
(a)
-
2.
The EPR spectra of semiquinone radicals produced from structurally similar PCB hydroquinones and quinones had similar spectral patterns as characterized by hyperfine splittings.
-
3.
The production of superoxide radicals could not be observed using DMPO as a spin trapping agent due to the rapid reaction of superoxide with quinone; however, SOD accelerates the autoxidation of hydroquinone indicating a role for superoxide.
-
4.
At higher pH, hydroxide anion can add to the quinone ring of a PCB-quinone, leading to the formation of a new hydroquinone and rupture of the quinone ring.
-
5.
Using oxygen consumption at higher pH, the autoxidation of both hydroquinone and quinone consumed oxygen—this oxygen consumption could be greater than the 1:1 stoichiometry predicted by reaction 2.
-
6.
H2O2 does not accumulate when a PCB-hydroquinone autoxidizes at high pH; H2O2 appears to react with intermediates formed during the oxidation process.
-
7.
H2O2 accumulates when SOD is present in an autoxidizing PCB-hydroquinone solution at near-neutral pH; without SOD, the reaction is too slow to observe any loss of O2 or accumulation of H2O2.
Acknowledgments
This project was supported by Grant Numbers P42ES013661, P30ES05605, and K25ES012475 (H.-J.L.) from the National Institute of Environmental Health Sciences and R01GM073929 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIMGS or the NIH. The University of Iowa ESR Facility provided invaluable support.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: aH (4), hyperfine splitting constant due to four identical hydrogens; aH3, hyperfine splitting due to the hydrogen on position 3 of the PCB phenyl ring; BQ, benzoquinone; Cat, catalase; DETAPAC, diethylenetriaminepentaacetic acid; DMPO, 5,5-dimethylpyrroline-1-oxide; H2Q, hydroquinone; HRP, horseradish peroxidase; HX, hypoxanthine; LPO, lactoperoxidase; MnSOD, manganese-containing superoxide dismutase; PQ, phenyl-quinone; Q, quinone; QH, quinhydrone; SOD, superoxide dismutase; SQ•−, semiquinone; TYR, tyrosinase; xn′, represents a generic number of chlorines, each at a position n on the secondary ring; XO, xanthine oxidase.
References
- Safe S. H. (1994) Polychlorinated biphenyls (PCBs): Environmental impact, biochemical and toxic responses, and implications for risk assessment. Crit. Rev. Toxicol. 24, 87–149. [DOI] [PubMed] [Google Scholar]
- Silberhorn E. M.; Glauert H. P.; Robertson L. W. (1990) Carcinogenicity of polyhalogenated biphenyls: PCBs and PBBs. Crit. Rev. Toxicol. 20, 440–496. [DOI] [PubMed] [Google Scholar]
- Robertson L. W., and Hansen L. G., Eds. (2001) PCBs: Recent Advances in the Environmental Toxicology and Health Effects, The University Press of Kentucky, Lexington, KY. [Google Scholar]
- Hansen L. G. (1998) Stepping backward to improve assessment of PCB congener toxicities. Environ. Health Perspect. 106, 171–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliano V. J. (1998) Assessing the cancer risk from environmental PCBs. Environ. Health Perspect. 106, 317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson S., and van Esch G. J. (1993) Environmental Health Criteria 140: Polychlorinated Biphenyls and Terphenyls, 2nd ed., World Health Organization, Geneva. Also available at http://www.inchem.org/documents/ehc/ehc/ehc140.htm.
- Kennedy M. W.; Carpentier N. K.; Dymerski P. P.; Adams S. M.; Kaminsky L. S. (1980) Metabolism of monochlorobiphenyls by hepatic microsomal cytochrome P-450. Biochem. Pharmacol. 29, 727–736. [DOI] [PubMed] [Google Scholar]
- Kennedy M. W.; Carpentier N. K.; Dymerski P. P.; Kaminsky L. S. (1981) Metabolism of dichlorobiphenyls by hepatic microsomal cytochrome P-450. Biochem. Pharmacol. 30, 577–588. [DOI] [PubMed] [Google Scholar]
- Kaminsky L. S.; Kennedy M. W.; Adams S. M.; Guengerich F. P. (1981) Metabolism of dichlorobiphenyls by highly purified isozymes of rat liver cytochrome P-450. Biochemistry 20, 7379–7384. [DOI] [PubMed] [Google Scholar]
- McLean M. R.; Bauer U.; Amaro A. R.; Robertson L. W. (1996) Identification of catechol and hydroquinone metabolites of 4-monochlorobiphenyl. Chem. Res. Toxicol. 9, 158–164. [DOI] [PubMed] [Google Scholar]
- Bachur N. R.; Gordon S. L.; Gee M. V. (1978) A general mechanism for microsomal activation of quinone anticancer agents to free radicals. Cancer Res. 38, 1745–1750. [PubMed] [Google Scholar]
- Amaro A. R.; Oakley G. G.; Bauer U.; Spielmann H. P.; Robertson L. W. (1996) Metabolic activation of PCBs to quinones: reactivity toward nitrogen and sulfur nucleophiles and influence of superoxide dismutase. Chem. Res. Toxicol. 9, 623–629. [DOI] [PubMed] [Google Scholar]
- McLean M. R.; Robertson L. W.; Gupta R. C. (1996) Detection of PCB adducts by the 32P-postlabeling technique. Chem. Res. Toxicol. 9, 165–171. [DOI] [PubMed] [Google Scholar]
- Oakley G. G.; Devanaboyina U.; Robertson L. W.; Gupta R. C. (1996) Oxidative DNA damage induced by activation of polychlorinated biphenyls (PCBs): Implications for PCB-induced oxidative stress in breast cancer. Chem. Res. Toxicol. 9, 1285–1292. [DOI] [PubMed] [Google Scholar]
- Oakley G. G.; Robertson L. W.; Gupta R. C. (1996) Analysis of polychlorinated biphenyl-DNA adducts by 32P-postlabeling. Carcinogenesis 17, 109–114. [DOI] [PubMed] [Google Scholar]
- Bolton J. L.; Trush M. A.; Penning T. M.; Dryhurst G.; Monks T. J. (2000) Role of quinones in toxicology. Chem. Res. Toxicol. 13, 135–160. [DOI] [PubMed] [Google Scholar]
- Monks T. J.; Jones D. C. (2002) The metabolism and toxicity of quinones, quinoneimines, quinone methides, and quinone-thioethers. Curr. Drug Metab. 3, 425–438. [DOI] [PubMed] [Google Scholar]
- O’Brien P. J. (1991) Molecular mechanisms of quinone cytotoxicity. Chem.-Biol. Interact. 80, 1–41. [DOI] [PubMed] [Google Scholar]
- Lehmler H.-J.; Robertson L. W. (2001) Synthesis of polychlorinated biphenyls (PCBs) using the Suzuki-coupling. Chemosphere 45, 137–143. [DOI] [PubMed] [Google Scholar]
- Lehmler H.-J.; Robertson L. W. (2001) Synthesis of hydroxylated PCB metabolites with the Suzuki-coupling. Chemosphere 45, 1119–1127. [DOI] [PubMed] [Google Scholar]
- Kania-Korwel I.; Sean P.; Robertson L. W.; Lehmler H.-J. (2004) Synthesis of polychlorinated biphenyls and their metabolites with a modified Suzuki-coupling. Chemosphere 56, 735–744. [DOI] [PubMed] [Google Scholar]
- Duling D. R. (1994) Simulation of multiple isotropic spin-trap EPR spectra. J. Magn. Res. Ser. B 104, 105–110. [DOI] [PubMed] [Google Scholar]
- Bachur N. R.; Gordon S. L.; Gee M. V.; Kon H. (1979) NADPH cytochrome P-450 reductase activation of quinone anticancer agents to free radicals. Proc. Natl. Acad. Sci. U.S.A. 76, 954–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyanagi T.; Yamazaki I. (1969) One-electron transfer reactions in biochemical systems. III. One-electron reduction of quinones by microsomal flavin enzymes. Biochim. Biophys. Acta 172, 370–381. [DOI] [PubMed] [Google Scholar]
- Plancherel D.; Zelewsky A. V. (1982) Proton-triggered complex formation: Radical complexes of o-benzosemiquinone, dopa, dopamine and adrenaline formed by electron transfer reaction from excited tris (2,2′-bipyridy1) ruthenium(II). Helv. Chim. Acta 65, 1929–1940. [Google Scholar]
- Kalyanaraman B.,; Morehouse K. M.; Mason R. P. (1991) An electron paramagnetic resonance study of the interactions between the adriamycin semiquinone, hydrogen peroxide, iron-chelators, and radical scavengers. Arch. Biochem. Biophys. 286, 164–170. [DOI] [PubMed] [Google Scholar]
- Yamazaki I.; Piette L. H. (1965) Electron paramagnetic resonance of undissociated p-benzosemiquinones. J. Am. Chem. Soc. 87, 986–990. [DOI] [PubMed] [Google Scholar]
- Pedersen J. A. (2002) On the application of electron paramagnetic resonance in the study of naturally occurring quinones and quinols. Spectrochim. Acta, Part A 58, 1257–1270. [DOI] [PubMed] [Google Scholar]
- Korytowski W.; Sarna T.; Kalyanaraman B.; Sealy R. C. (1987) Tyrosinase-catalyzed oxidation of dopa and related catechol(amine)s: A kinetic electron spin resonance investigation using spin-stabilization and spin label oximetry. Biochim. Biophys. Acta 924, 383–392. [DOI] [PubMed] [Google Scholar]
- Ferrari R. P.; Laurenti E.; Ghibaudi E. M.; Casella L. (1997) Tyrosinase-catecholic substrates in Vitro model: Kinetic studies on the o-quinone/o-semiquinone radical formation. J. Inorg. Biochem. 68, 61–69. [Google Scholar]
- Venkataraman B.; Fraenkel G. K. (1955) Proton hyperfine interactions in paramagnetic resonance of semiquinones. J. Am. Chem. Soc. 77, 2707–2713. [Google Scholar]
- Ashworth P.; Dixon W. T. (1972) Secondary radicals in the autoxidation of hydroquinones and quinones. J. Chem. Soc., Perkin Trans. 2 1130–1133. [Google Scholar]
- Stone T. J.; Waters W. A. (1965) Aryloxy-radicals. Part IV. Electron spin resonance spectra of some ortho-monobenzosemiquinones and secondary radicals derived therefrom. J. Chem. Soc. 1488–1494. [Google Scholar]
- Ashworth P.; Dixon W. T. (1971) Secondary radicals in the autoxidation of hydroquinones. J. Chem. Soc. D 1150–1152. [Google Scholar]
- Roginsky V.; Barsukova T. (2000) Kinetics of oxidation of hydroquinones by molecular oxygen. Effect of superoxide dismutase. J. Chem. Soc., Perkin Trans. 2 1575–1582. [Google Scholar]
- Pedersen J. A. (1973) Electron spin resonance studies of oxidative processes of quinones and hydroquinones in alkaline solution; formation of primary and secondary semiquinone radicals. J. Chem. Soc., Perkin Trans. 2 424–431. [Google Scholar]
- Ashworth P.; Dixon W. T. (1972) Electron spin resonance spectra of radicals derived from arylhydroquinones. J. Chem. Soc., Perkin Trans. 2 2264–2267. [Google Scholar]
- Venkataraman B.; Segal B. G.; Fraenkel G. K. (1959) Paramagnetic resonance of methyl- and chloro-substituted p-benzosemiquinones. J. Chem. Phys. 30, 1006–1016. [Google Scholar]
- Francesco A. M. D.; Ward T. H.; Butler J. (2004) Diaziridinylbenzoquinones. Methods Enzymol. 382, 174–193. [DOI] [PubMed] [Google Scholar]
- Buettner G. R. (1993) The spin trapping of superoxide and hydroxyl free radicals with DMPO (5,5-dimethylpyrroline-N-oxide): More about iron. Free Radical Res. Commun. 19, Suppl. 1S79−S87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pou S.; Hassett D. J.; Britigan B. E.; Cohen M. S.; Rosen G. M. (1989) Problems associated with spin trapping oxygen-centered free radicals in biological systems. Anal. Biochem. 177, 1–6. [DOI] [PubMed] [Google Scholar]
- Buettner G. R.; Mason R. P. (1990) Spin-trapping methods for detecting oxygen-derived radical formation in vitro and in vivo. Methods Enzymol. 186, 127–133. [DOI] [PubMed] [Google Scholar]
- Roginsky V. A.; Pisarenko L. M.; Bors W.; Michel C. (1999) The kinetics and thermodynamics of quinine−semiquinone−hydroquinone systems under physiological conditions. J. Chem. Soc., Perkin Trans. 2 871–876. [Google Scholar]
- Eyer P. (1991) Effects of superoxide dismutase on the autoxidation of 1,4-hydroquinone. Chem.-Biol. Interact. 80, 159–176. [DOI] [PubMed] [Google Scholar]
- Buettner G. R.; Ng C. F.; Wang W.; Rodgers V. G. J.; Schafer F. Q. (2006) A new paradigm: Manganese superoxide dismutase influences the production of H2O2 in cells and thereby their biological state. Free Radical Biol. Med. 41, 1338–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunmark A. (1989) Formation of electronically excited states during the interaction of benzoquinone with hydrogen peroxide. J. Biolumin. Chemilumine. 4, 219–225. [DOI] [PubMed] [Google Scholar]
- Zhu B. Z.; Kalyanaraman B.; Jiang G. B. (2007) Molecular mechanism for metal-independent production of hydroxyl radicals by hydrogen peroxide and halogenated quinones. Proc. Natl. Acad. Sci. U.S.A. 104, 17575–17578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curci R., and Edwards J. O. (1992) Activation of hydrogen peroxide by organic compounds. In Catalytic Oxidations with Hydrogen Peroxide as Oxidant (Strukul G., Ed.) In the series Catalysis by Metal Complexes, Vol. 9, Chapter 2, Springer-Verlag LLC, New York. [Google Scholar]