

Abstract
Data, both for and against the presence of a mitochondrial nitric-oxide synthase (NOS) isoform, is in the refereed literature. However, irrefutable evidence has not been forthcoming. In light of this controversy, we designed studies to investigate the existence of the putative mitochondrial NOS. Using repeated differential centrifugation followed by Percoll gradient fractionation, ultrapure, never frozen rat liver mitochondria and submitochondrial particles were obtained. Following trypsin digestion and desalting, the mitochondrial samples were analyzed by nano-HPLC-coupled linear ion trap-mass spectrometry. Linear ion trap-mass spectrometry analyses of rat liver mitochondria as well as submitochondrial particles were negative for any peptide from any NOS isoform. However, recombinant neuronal NOS-derived peptides from spiked mitochondrial samples were easily detected, down to 50 fmol on column. The protein calmodulin (CaM), absolutely required for NOS activity, was absent, whereas peptides from CaM-spiked samples were detected. Also, l-[14C]arginine to l-[14C]citrulline conversion assays were negative for NOS activity. Finally, Western blot analyses of rat liver mitochondria, using NOS (neuronal or endothelial) and CaM antibodies, were negative for any NOS isoform or CaM. In conclusion, and in light of our present limits of detection, data from carefully conducted, properly controlled experiments for NOS detection, utilizing three independent yet complementary methodologies, independently as well as collectively, refute the claim that a NOS isoform exists within rat liver mitochondria.
Nitric oxide (NO·)2 is a highly diffusible, hydrophobic, and gaseous free radical (1) that is responsible for autocrine and paracrine signaling activities (2). NO· can readily partition into and through membranes (3–5) to influence biological functions such as blood pressure regulation, platelet aggregation and adhesion, neurotransmission, and cellular defense (4, 6–11). The mechanism by which NO· influences biological functions is by binding to target proteins that contain heme and/or thiol(s). Alternatively, NO· can combine with 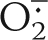 to produce the highly reactive species peroxynitrite.
to produce the highly reactive species peroxynitrite.
Mitochondria are highly compartmentalized, membranous organelles that contain abundant amounts of reactive hemoproteins and thiols (12, 13), to which NO· may bind reversibly (14, 15) or irreversibly (16–18). Mitochondria also generate various amounts of during the process of cellular respiration (19, 20). Studies conducted during the past decade have suggested that NO· can diffuse into mitochondria and cause mitochondrial dysfunction by reversibly inhibiting cytochrome c oxidase (14, 21, 22) and NADH dehydrogenase (23).
In the mid-90s, a putative variant of NOS was proposed to reside within mitochondria. Initially, Kobzik et al. (24) and Hellsten and co-workers (25) observed an apparent endothelial NOS (eNOS) immunoreactivity in skeletal muscle mitochondria. Simultaneously, Bates et al. (26, 27) observed an apparent eNOS histochemical reactivity in inner mitochondrial membrane preparations, isolated from rat liver, brain, heart, skeletal muscle, and kidney. Tatoyan and Giulivi (28), acting on these initial observations, performed experiments in an attempt to confirm the identity of this putative mtNOS. Relying on immunochemical analysis, Tatoyan and Giulivi (28) claimed that inducible NOS (iNOS) was the NOS isoform present in rat liver mitochondria. This same group using mass spectrometry later presented data in support of the putative mtNOS being a variant of nNOS (29). Ghafourifar and Richter (30) had reported previously that the putative mtNOS was calcium-sensitive and constitutive in nature. Since these reports, different groups have reported the presence of each of the three main isoforms of NOS within mitochondria (29, 31, 32). Also, biochemical characterization of the putative mtNOS performed by Giulivi and co-workers (29) revealed certain post-translational modifications (myristoylation and phosphorylation of the protein) that are thought to be unique to eNOS. During the last decade, various reports have supported the presence of at least one of the three main isoforms of NOS residing in mitochondria. However, the more recent reports tend to question this claim (33–36). Because of the contradictory reports regarding the existence of a putative mtNOS, Brookes (33) compiled a critical and thorough review of the literature published up to 2003 dealing with the putative mtNOS. This review brought to light the diverse technical issues involved in the aforementioned studies. Major issues were the degree of purity of mitochondrial preparations (37, 38), shortcomings of measurement methodology (29, 39–41), use of inappropriate, or total lack of, experimental controls and confusing technical practices. Lacza et al. (42) has reviewed the more recent developments in the area of mitochondrial NO· production and discussed some of the shortcomings of certain techniques still being used.
In light of this ongoing controversy regarding the presence or absence of a mtNOS, we designed and carefully conducted properly controlled studies to either confirm or refute the existence of any NOS isoform within mitochondria. Ultrapure rat liver mitochondria were isolated using repeated differential centrifugation followed by Percoll gradient purification. Proteomic analyses were then performed using a nano-HPLC-coupled nanospray LTQ-MS. To avoid the interfering factors that are rampant in NO· trapping assays (43), the NOS-catalyzed conversion of l-[14C]arginine to l-[14C]citrulline was used to probe for NOS activity in mitochondria. Appropriate controls were employed and, for inhibition studies, high concentrations of l-thiocitrulline (TC) (44) were used. Additionally, immunochemical analyses were performed with ultrapure mitochondria using nNOS, eNOS, and CaM antibodies. The problems faced with the commonly used techniques in mtNOS studies are discussed.
EXPERIMENTAL PROCEDURES
Chemicals and Biochemicals
HEPES, l-arginine·HCl, CaCl2·4H2O, bovine brain CaM, (6R)-5,6,7,8-tetrahydro-l-biopterin (BH4), NADPH, l-citrulline, EDTA, EGTA, dithiothreitol, iodoacetamide, sucrose, mannitol, acetone, l-thiocitrulline, N(ω)-nitro-l-arginine (l-NNA), monobasic and dibasic sodium phosphate, SDS, alkaline phosphatase-conjugated anti-rabbit secondary antibody, and horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse secondary antibodies were purchased from Sigma. Dowex 50W-X8 was obtained from Supelco (Bellefonte, PA). Acetonitrile was purchased from Mallinckrodt-Baker, Inc. (Philipsburg, NJ). Trichloroacetic acid was obtained from EMD Chemicals, Inc. (San Diego). Tris and nitrocellulose membranes were purchased from Bio-Rad. SuperSignal West Pico chemiluminescent substrate was purchased from Pierce. Percoll and 2′,5′-ADP-Sepharose 4B were products of GE Healthcare. Complete protease inhibitor mixture tablets were purchased from Roche Applied Science. Primary antibodies for the mitochondrial outer membrane marker, voltage-dependent anion channel (VDAC), were obtained from Affinity Bioreagents (Golden, CO). Primary antibodies for glucose-regulated protein (GRP 75)/mitochondrial heat shock protein 70 (mt hsp70) and CaM were purchased from Abcam, Inc. (Cambridge, MA). nNOS and eNOS primary antibodies were generous gifts from Bettie Sue Masters, University of Texas Health Science Center, San Antonio. The tubulin antibody was a gift from Sukla Roychowdhary, University of Texas, El Paso. All other chemicals and reagents were from common suppliers and were of the highest grade commercially available.
Enzymes
Recombinant nNOS and eNOS, referred to as nNOSr and eNOSr, respectively, were overexpressed in Escherichia coli and purified according to established methodology (45, 46). Superoxide dismutase (SOD) and catalase (CAT) were purchased from Sigma. The specific activities of SOD and CAT were 2,500–7,000 units/mg protein and ≥10,000 units/mg protein, respectively.
Animals
All experimental protocols involving animals were approved by University of Texas, El Paso Institutional Animal Care and Use Committee (IACUC). Male Sprague-Dawley (SD) rats (250–300 g; ∼3 months of age) were obtained from Harlan Sprague-Dawley (Houston, TX) and used in all studies.
Preparation of Mitochondria
Pure rat liver mitochondria were obtained by repeated differential centrifugation followed by Percoll gradient purification. Expertise for the mitochondrial purification procedure was obtained first hand and adopted from the methodology used in the laboratory of James Geddes, University of Kentucky, Lexington. Initially, 2–4 rats were euthanized, and the entire livers were excised and immersed in ice-cold mitochondrial isolation buffer (MIB) containing mannitol (215 mm), sucrose (75 mm), EGTA (1 mm), HEPES/KOH (20 mm), pH 7.2. In addition, a commercially available serine and cysteine protease-inhibiting complete mixture tablet was included. The liver lobes were blotted, washed 2–3 times with fresh MIB, and minced into small pieces with scissors. The resulting minced pieces of liver were washed with MIB to remove blood. Then 6–8 ml of ice-cold MIB was added to the washed and minced tissue. Portions of tissue samples were placed in a glass Dounce homogenizer. The homogenizer was then immersed in ice, and the tissue was gently homogenized with six complete strokes of a somewhat loose-fitting Teflon pestle at 250 rpm, using a variable speed motorized unit (Glas-Col LLC., Terre Haute, IN).
Following homogenization, both differential centrifugation and Percoll gradient fractionation steps were performed using a pre-cooled SM-24 rotor in an RC-5B Sorvall centrifuge at 4 °C. Each sample was centrifuged for 10 min, unless specified otherwise. First, the tissue homogenate (CO) was suspended in ice-cold MIB and centrifuged at 800 × g. After the first 800 × g spin, the white fatty layer covering the supernatant was carefully removed using a lint-free wipe or cotton wool. The 800 × g supernatant (M1) was then collected, and the isolated pellet containing red spots of blood and cellular debris was discarded. M1 was spun at 10,000 × g to obtain a pellet containing mitochondria (M2). M2 was then gently resuspended by homogenization using the same glass Dounce homogenizer as before (four strokes at 250 rpm). The differential centrifugation steps were then repeated with the MIB-resuspended M2 at 800 × g (M3) and 10,000 × g (M4) to obtain a relatively pure mitochondrial preparation. The mitochondrial pellet obtained after the final 10,000 × g spin was resuspended in ice-cold MIB and then centrifuged at the lower speed of 9,000 × g. The pellet (M5) isolated during this step contained highly purified mitochondria.
Mitochondria (M5) were further purified using Percoll gradient centrifugation as described previously (47). Percoll was chosen as the gradient medium because of its chemical inertness and negligible osmolarity (48, 49). Ice-cold MIB without EGTA was used in all steps for preparing the Percoll gradient solutions as well as for the wash steps. Mitochondria were supplemented with an equal volume of 30% Percoll (final concentration, mitochondria (M5) in 15% Percoll). A discontinuous Percoll gradient was used, with the bottom layer containing 40% Percoll, followed by 24% Percoll, and finally by 15% Percoll-containing mitochondria. The density gradient was spun at 30,400 × g, and the band between 24 and 40% Percoll (containing the intact mitochondria) was carefully collected. The broken mitochondria at the bottom of the gradient were discarded. The intact mitochondrial sample was then suspended in MIB without EGTA and centrifuged at 16,700 × g for 15 min. Discarding the supernatant, the resulting loose pellet obtained was resuspended in MIB without EGTA and subsequently centrifuged at 13,000 × g followed by a 10,000 × g spin. The intact mitochondrial pellet obtained after the final 10,000 × g spin was collected and used for experimentation. These intact, pure mitochondria, obtained after repeated differential centrifugation and Percoll gradient fractionation, are referred as “MT” throughout. The protein concentration of the pure mitochondrial sample (MT) was measured using the Bradford protein assay (50) using BSA as a standard.
Isolation of Submitochondrial Particles (SMPs)
Intact MT, were placed in a small glass vial residing within an ice-jacket and sonicated mildly for 20 s, with alternating pulse and pause interval times of 5 s each (amplitude-25%) using an ultrasonic processor (Sonics and Materials, Inc.) equipped with a ⅛-inch diameter probe. For preparation of the positive control, sonication of MT was interrupted after 10 s, and nNOSr (200 fmol) was spiked into the half-sonicated MT sample, and then the spiked samples were sonicated for the remaining 10 s. Submitochondrial particles were subsequently obtained from the experimental as well as the positive control samples by ultracentrifugation at 100,000 × g for 1 h at 4 °C (TLA 100 rotor; Beckman Allegra 64 centrifuge). The supernatant fraction, containing the SMPs, and the solubilized pellet (solubilized using 0.1% octyl β-d-glucopyranoside) were collected for further processing.
Affinity Purification of SMPs
SMPs, the solubilized pellets of MT, as well as the corresponding positive controls, were subjected to 2′,5′-ADP-Sepharose 4B chromatography to enrich samples with NAD(P)H-binding proteins, including NOS, if present. Briefly, the SMPs and the solubilized pellets obtained from purified MT (initial amounts varied depending on the experiment) were incubated with 150 μl of the 2′,5′-ADP-Sepharose 4B resin slurry, equilibrated with ice-cold sodium phosphate buffer (50 mm, pH 7.4), and spun overnight (15 h) at 4 °C. Mini-columns were fabricated in the laboratory using 200-μl pipette tips and the suspension of SMPs that had been preincubated with 2′,5′-ADP-Sepharose 4B beads. The columns were then washed with ice-cold sodium phosphate buffer (5× 100-μl portions). NAD(P)H-dependent proteins bound to the columns were then eluted using 5× 100-μl portions of 5 mm NADPH in Tris (50 mm) containing NaCl (500 mm), pH 7.4.
Sample Processing for Mass Spectrometry
Protein precipitation was performed by treating the affinity-purified eluates of SMP and the corresponding positive controls with 10% trichloroacetic acid for 30 min on ice. Samples were vortexed every 10 min and finally centrifuged at 16,000 × g for 20 min at 4 °C. The trichloroacetic acid-precipitated samples were resuspended in acetone by repeated up-and-down pipetting and centrifuged at 16,000 × g for 20 min at 4 °C. Sample pellets were then dried using a Centri-Vap (Eppendorf).
Dry precipitated proteins were digested as described by Stone and Williams (51). Briefly, dried pellets from acetone washes were solubilized with a solution of 8 m urea, 0.4 m NH4HCO3, and the sulfhydryls were reduced using dithiothreitol (9 mm) for 15 min at 50 °C. The samples were allowed to cool to room temperature and then treated with iodoacetamide (20 mm) for 15 min at room temperature. Subsequently, the iodoacetamide-treated samples were diluted with water to obtain a final concentration of 1 m urea. Finally, the samples were digested overnight with 1 μg of sequencing grade trypsin (Promega) for every 50 μg of mitochondrial protein. Proteolysis was stopped by adding 1 μl of 100% formic acid (Sigma). The resulting samples were desalted using reverse phase ZipTips manufactured with 200-μl micropipette tips and containing POROS 50 R2 resin (Applied Biosystems) (52). Next, strong cation exchange chromatography was performed using POROS 50 HS resin (53), and the samples were fractionated by eluting with increasing concentrations of NaCl (0–500 mm). The eluates were again desalted using reverse phase ZipTips and dried under vacuum centrifugation (Centri-Vap).
Each of the desalted strong cation exchange fractions were solubilized in 30 μl of 0.05% trifluoroacetic acid, and 8 μl were injected onto a trap column (C18, 0.25 μl, OPTI-PAK). Separations were performed using a reverse phase capillary column (Acclaim, 3 μm C18, 75 μm × 25 cm, LC Packings, Dionex) connected to a nano-HPLC system (nano-LC 1D plus, Eksigent). For elution, the mobile phases were as follows: A, 2% acetonitrile, 0.1% formic acid; B, 80% acetonitrile, 0.1% formic acid. A linear gradient from 0 to 40% solvent B over 100 min was used. The eluting peptides were directly introduced into a linear ion trap-mass spectrometer equipped with a nanospray source (LTQ XL, Thermo-Fisher Scientific). MS spectra were collected in centroid mode over the range of 400–1700 m/z, and the five most abundant ions were submitted twice to collision-induced dissociation (35% normalized collision energy), before being dynamically excluded for 120 s. All MS/MS spectra were from peptides of 600–4000 Da, and at least 15 fragments were converted into DTA files using Bioworks version 3.3.1 (Thermo-Fisher Scientific). The DTA files were submitted for data base searching using TurboSequest (54) (available in Bioworks version 3.3.1) and compared against rat NOS sequences (version 3.25) from the International Protein Index. All sequences were submitted in the forward and reverse orientations for calculating the false-positive rate. The data base search parameters included the following: (i) trypsin cleavage in both peptide termini allowing one missed cleavage site; (ii) carbamidomethylation of cysteine residues as a fixed modification; (iii) oxidation of methionine residues as a variable modification; and (iv) 2.0- and 1.0-Da mass tolerance for peptide and fragment, respectively. The following filters in Bioworks were applied as follows: distinct peptides, consensus scores ≥10.2, DCn ≥ 0.1, protein probability ≤1 × 10−3, and Xcorr ≥ 1.5, 2.0 and 2.5 for singly, doubly, and triply charged peptides, respectively. To ensure the quality of the analyses, the false-positive rate was calculated by dividing the number of hits matching the reverse sequences by the total number of identifications.
Assay for NOS Activity
The conversion of l-[14C]arginine to l-[14C]citrulline was used to estimate NOS activity (55). Reaction mixtures consisted of HEPES (50 mm, pH 7.6), NADPH (400 μm), CaCl2, (400 μm), BH4 (5 μm), l-arginine (20 μm, containing 0.5 μCi/ml l-[14C]arginine), and a 1.5-fold molar excess of CaM to nNOS (based on the positive control), in a total volume of 0.25 ml (56). MT (150 μg) were included in all assays, unless mentioned otherwise. A 4-fold excess of NADPH (400 μm), compared with the normal 100 μm assay concentration, was used to ensure that this co-substrate would not become a limiting factor in the reactions. To further confirm that the NADPH concentration was indeed sufficient, an NADPH-regenerating system, consisting of glucose-6-phosphate dehydrogenase (1 unit), glucose 6-phosphate (125 mm), and NADP+ (12.5 mm), was used in some pilot assays. The potent nNOS inhibitor, l-thiocitrulline (800 μm), which is efficacious at this concentration toward any NOS isoform, was preincubated with MT samples for 10 min before initiating the reactions. The NOS inhibitor, l-NNA (400 μm), was also used in some assays. Reactions were run for 10 min at 23 °C. Reaction mixtures were quenched with 133 μl of an ice-cold stop solution containing 1 mm l-citrulline, 10 mm EDTA, and 100 mm HEPES, pH 5.5, and then applied to 2-ml Dowex columns, and l-[14C]citrulline was eluted with 2× 1-ml portions of water. To determine NOS activity, the entire eluates were used to quantify l-[14C]citrulline by liquid scintillation counting. Denatured MTs (dMT), obtained by mild sonication for 5 s (amplitude-25%), followed by incubation at 100 °C for 5 min, were used as negative mitochondrial controls. Overall, controls consisted of complete reaction mix + MIB alone, complete reaction mix + dMT ± nNOSr, complete reaction mix + dMT − NADPH, complete reaction mix + dMT + l-thiocitrulline, and complete reaction mix + l-thiocitrulline alone. Background counts from incubations containing all ingredients except enzyme and/or MT were subtracted from all measurements. The positive controls consisted of MT (150 μg) spiked with 30 nm nNOSr. Data were analyzed using SigmaPlot 9.0 (Systat Software, Inc.) and expressed as the mean ± range of values of experimental data points from two independent experiments, each run in duplicate.
High Performance-Thin Layer Chromatography (HP-TLC) of Amino Acids
Samples from l-[14C]arginine to l-[14C]citrulline conversion assays (either eluted from Dowex columns or not exposed to Dowex) were treated with 80% acetone, and the precipitation was enhanced by a freeze-thaw cycle at −80 °C for 36 h. The precipitated proteins were removed by centrifugation at 16,000 × g for 20 min.
Control experiments were performed to determine loss of radioactivity because of the presence of protein in the MT pellet. This was determined wherein all substrates and cofactors of the l-[14C]arginine to l-[14C]citrulline conversion assay, except l-[14C]arginine, were added to MT, and reactions were quenched with stop solution. l-[14C]Arginine was then added, which was followed by acetone precipitation. Results from control experiments indicated that there was a 0.01% loss of radioactivity because of the presence of the pellet (data not shown).
The supernatants from the 16,000 × g spins, containing the amino acids, were collected separately and dried under a stream of nitrogen. The dried supernatant samples were then dissolved in 100 μl of methanol:water (2:1), and 10 μl of each individual sample was loaded on HP-TLC Silica 60 plates (EMB Chemicals). Ten μl of l-arginine or l-citrulline (2 mg/ml) were used as standards. Butanol:acetic acid:water (60:20:20) was used as the mobile phase. HP-TLC was performed in a glass chamber using standard methodology, and the plates were developed with a 2% ninhydrin solution (in acetone). The developed plates were then dried on a hot plate (medium setting) for 1 min. Afterward, the nonradioactive l-arginine and l-citrulline control bands were identified visually. The dry plates were then exposed to x-ray film (Eastman Kodak Co.) for 108 h (4½ days).
SDS-PAGE and Western Analyses
Immunochemical analyses against MT as well as non-MT markers (GRP 75, VDAC, and tubulin, respectively) were performed wherein 20 μg of rat liver samples (M1, M2, M3, M4, M5, MT, and control tissue homogenate (CO)) were separated by SDS-PAGE (10% polyacrylamide). Initially, samples were placed into 2× loading buffer containing 125 mm Tris-HCl, pH 6.8, 2% SDS, 20% glycerol, 0.2% bromphenol blue, and 0.05% β-mercaptoethanol. Samples were then boiled for 3 min. Electrophoresis was performed using a Bio-Rad Mini Protean II gel apparatus at 100 V for 1.5 h. The separated proteins were then transferred from the gels onto nitrocellulose membranes (30 V) for 15 h, at 4 °C.
Nitrocellulose membranes were then blocked with 5% nonfat dry milk in Tris-buffered saline (TBS: NaCl (0.3 m) and Tris-HCl (40 mm) containing 0.1% Tween 20 (TBST), pH 7.6) for 1 h. After washing for 30 min (three times for 10 min) with TBST, the membranes were incubated overnight at 4 °C with the primary antibodies against either mouse GRP 75 (1:1250), rabbit VDAC (1:1000), or mouse tubulin (1:250). The membranes were washed for 30 min (three times for 10 min) with TBST and incubated with the appropriate HRP-conjugated secondary antibody (anti-mouse (1:10,000 for GRP 75; 1:1,000 for tubulin) or anti-rabbit (1:10,000 for VDAC)) for 1.5 h at 25 °C. Membranes were then washed for 30 min (three times for 10 min) with TBST, incubated with chemiluminescent substrate (Pierce) for 5 min, and developed using Kodak Biomax light film.
Immunochemical analyses for NOS isoforms were performed using rat liver proteins (MT or CO). Positive controls consisted of nNOSr- or eNOSr-spiked MT. Samples (150 μg each) were placed into 2× loading buffer and incubated for 30 min at 37 °C. Samples were loaded in wide lanes (using a 5-well comb), and SDS-PAGE was performed using 7.5% polyacrylamide. Separated proteins were then transferred to nitrocellulose membranes as mentioned previously. The membranes containing transferred proteins were blocked for 20 min with 1% nonfat dry milk plus 1% BSA in TBST. After thoroughly washing the membranes for 15 min (three times for 5 min at 23 °C) with TBST, Western blotting was performed for nNOS and eNOS. Briefly, the membranes were incubated with either rabbit polyclonal nNOS antibody (1:2000) or monoclonal eNOS antibody (1:2000) in TBST for 1 h at 4 °C. Again, the membranes were washed for 15 min (three times for 5 min at 23 °C) with TBST and subsequently incubated for 1 h with the appropriate alkaline phosphatase-conjugated anti-rabbit secondary antibody (1:2000). After washing for 15 min (three times for 5 min at 23 °C) with TBST, the membranes were developed, and proteins were visualized using the 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium substrate (Sigma).
Immunochemical analyses against CaM were performed whereby 150 μg of MT protein or 30 ng of CaM spiked in 150 μg of MT (positive control) were separated by SDS-PAGE (15% polyacrylamide) and transferred to nitrocellulose membranes as mentioned previously. The membranes were then blocked with TBST containing 5% nonfat dry milk and incubated overnight with an anti-rabbit monoclonal antibody against CaM, diluted in TBST containing 5% nonfat dry milk (1:10,000) at 4 °C. After washing for 15 min (three times for 5 min) with TBST, the membranes were incubated with the anti-rabbit HRP-conjugated secondary antibody (1:5000) for 1 h at 23 °C. Membranes were washed for 15 min (three times for 5 min) with TBST, incubated with chemiluminescent substrate (Pierce) for 5 min, and developed using a photo imager (Kodak).
Oxyhemoglobin Capture Assay
Assays containing oxyhemoglobin (8 μm), l-arginine (100 μm), CaCl2 (400 μm), CaM (25 nm), BH4 (10 μm), NADPH (100 μm), and HEPES (50 mm, pH 7.6) were conducted as described previously (57). MT (50 and 500 μg) were subjected to mild sonication two times for 5 s (amplitude 25%) and added to the assay in place of nNOSr. In addition, 50 units each of SOD and CAT were included in all assays to minimize interference from reactive oxygen species, , and H2O2.
RESULTS
Purity of Mitochondrial Preparations
Control tissue homogenate (CO) and fractions obtained during mitochondrial isolation steps (M2, M4, M5, and MT) were subjected to immunochemical analyses for mitochondrial and nonmitochondrial markers to document the purity of the preparation. Western blot analyses probing the mitochondrial matrix marker, GRP 75, indicated sequential enrichment of GRP 75 during purification (Fig. 1A). GRP 75 was neither detected in control tissue homogenate (Fig. 1A) nor in the crude supernatants (M1 or M3; data not shown). The mitochondrial outer membrane marker VDAC was detected in the MT fraction, although it was not detected in control tissue homogenate (Fig. 1B). The nonmitochondrial cytosolic marker tubulin, as a further test of purity, was detected in control whole tissue homogenate, although it was absent in purified MT (Fig. 1C).
FIGURE 1.

Immunochemical analyses of rat liver mitochondrial fractions with antibodies against mitochondrial and nonmitochondrial markers. Proteins collected during mitochondrial isolation and purification steps (M2, 10,000 × g; M4, 10,000 × g; M5, 9,000 × g; MT, intact MT after repeated differential centrifugation and Percoll gradient purification; and CO, control tissue homogenate) were separated by SDS-PAGE and transferred onto nitrocellulose membranes as described (see under “Experimental Procedures”). Western blotting was performed with antibodies against the following: A, mitochondrial matrix marker, GRP 75; B, mitochondrial outer membrane marker, VDAC; or C, cytosol marker, tubulin. The immunocomplexes were visualized by the HRP-conjugate reaction using a chemiluminescence substrate (Pierce). MT lane, run on the same gel as other fractions, was cropped and pasted alongside other fractions after removing the intervening lanes.
Proteomic Analyses of Mitochondria
Initially, purified intact rat liver MT (1.5 mg of protein, Bradford) were trichloroacetic acid-precipitated, acetone-washed, and processed for LTQ-MS analyses after digestion with trypsin as described (see under “Experimental Procedures”). A total of 300 proteins were identified that includes several well established mitochondrial proteins such as components of the respiratory complexes and other mitochondrial markers, including VDAC, NADH-ubiquinone oxidoreductase, and ATP synthase subunits. However, there were no peptide signals in the experimental samples to indicate the presence of any NOS isoform (supplemental 1).
As a next step, SMP were obtained from 2.9 mg of the purified intact MT. SMP and the solubilized pellets obtained from ultracentrifugation were then subjected to affinity chromatography using 2′,5′-ADP-Sepharose 4B, which binds NADPH and NADPH-requiring enzymes. The 2′,5′-ADP-Sepharose 4B eluates of both the SMP as well as the solubilized pellets were processed as described (see under “Experimental Procedures”) and subjected to proteomic analyses. LTQ-MS analyses of the affinity-purified eluates of the SMP led to the identification of 284 proteins with a false-positive rate of ∼2% (supplemental 2). Again, MS analyses of the SMP (supplemental 2) as well as the solubilized pellets (data not shown) failed to provide any evidence for the existence of any NOS-derived peptide(s).
To determine protein loss or recovery, nNOSr was spiked into sequentially purified MT and processed in the exact manner as experimental samples (see under “Experimental Procedures”). To identify the detection threshold for NOS in MT samples, we spiked various amounts of nNOSr (100, 10, 1, and 0.2 pmol) into the purified SMP fractions. Out of the various amounts of nNOSr spiked into SMP, we loaded an aliquot consisting of one-fourth of the total sample. Thus, for the sample spiked with 200 fmol of nNOSr, 50 fmol of nNOSr was loaded in the LC-coupled LTQ-MS. At this level of nNOSr, two nNOSr-derived peptides were easily identified (supplemental 3). We did not attempt to detect much lower amounts of nNOSr in MT samples or determine the limit of detection of the instrument. Furthermore, we did not spike recombinant nNOS in the crude cell extract because we would have lost the nNOSr during purification of the mitochondria.
We examined CaM, the protein binding partner of NOS, and neither CaM nor any known CaM-binding protein could be detected in MT or SMP. Thus, we conclude that NOS (any isoform) is absent from both intact MT as well as SMP, at least at levels that can be detected by the highly sensitive nano-LC-MS/MS methodology.
Assay for NOS Activity
NOS activity was determined by measurement of the conversion of l-[14C]arginine to l-[14C]citrulline (see under “Experimental Procedures”), with intact MT (Fig. 2A) as well as with the SMP fractions (Fig. 2B). The assay containing nNOSr (30 nm) alone, along with all necessary substrates and co-factors, was used as a positive control. The positive control produced a substantial amount of l-[14C]citrulline (Fig. 2, A and B, 1st bar). Addition of the NOS inhibitor, l-thiocitrulline (800 μm) abolished this activity by 98.5% (Fig. 2, A and B, 2nd bar). Initially, in the absence of nNOSr, when intact MT samples were added to the assay, a weak signal of radioactivity was observed (Fig. 2A, 3rd bar). However, this signal could not be inhibited using l-thiocitrulline (Fig. 2A) or l-NNA (data not shown), providing evidence against a NOS-catalyzed reaction. Although l-thiocitrulline is a potent nNOS inhibitor, at 800 μm, it is nonspecific, cross-reacting with all NOS isoforms and resulting in almost complete inhibition of NOS-catalyzed l-[14C]citrulline production.
FIGURE 2.

l-[14C]Arginine to l-[14C]citrulline conversion assay using MT, SMP, and a pure protein, BSA. Measurement of the conversion of l-[14C]arginine to l-[14C]citrulline was performed on MT, SMP, as well as on nNOSr plus BSA. Control values from incubation containing all ingredients except enzyme or MT were subtracted from all data points. Data were expressed as the mean ± range of values, of experimental data points from two independent experiments, each run in duplicate. The positive control was nNOSr (30 nm) in all experiments (A–C, 1st bar). l-[14C]Citrulline production by nNOSr was almost completely inhibited by the NOS inhibitors, thiocitrulline (800 μm; denoted as TC; 2nd bar on A and B) and l-NNA (400 μm; C, 2nd bar). l-[14C]Citrulline production was by the following: A, MT (150 μg; 3rd bar) and MT + TC (150 μg; 4th bar). B, SMP from 150 μg of MT (3rd bar) and SMP + TC (4th bar). C, intact MT of various amounts as follows: 280 μg (3rd bar), 560 μg (4th bar), and 1000 μg (5th bar), and 1000 μg of MT + l-NNA (6th bar). D, nNOSr (30 nm) plus BSA of various amounts as follows: 250 (2nd bar), 500 (3rd bar), and 750 μg (4th bar). Here, values are represented as the percentage change in relation to control radioactivity of nNOSr alone (shown as 1st bar).
Assays conducted with the SMP also showed a weak radioactive signal (Fig. 2B). As with the weak signal observed with intact MT, this signal could not be inhibited by l-thiocitrulline (800 μm) (Fig. 2B, 4th bar). Furthermore, when the solubilized pellet fractions were added to the assay, it did not show any signal (data not shown). When this weak radioactive signal, emanating from intact MT samples, was investigated, it was found that assays performed with increasing amounts of intact MT protein produced a titratable increase in radioactive signal (Fig. 2C). Additionally, this radioactive signal could not be inhibited using l-NNA (400 μm, Fig. 2C). Thus, the radioactive signal obtained with MT or SMP that could not be inhibited by NOS inhibitors indicated that the radioactive signal was probably not a product of an enzymatic reaction catalyzed by NOS. When assays were performed with increasing amounts of another protein, pure BSA (250, 500, and 750 μg) instead of MT, no radioactive signal was apparent in the flow-through (<3%) above control (data not shown). When assays were conducted with pure BSA, increasing amounts of BSA (250, 500, and 750 μg) plus nNOSr (30 nm) slightly increased the radioactive “signal” that was produced when compared with nNOSr in the absence of BSA (Fig. 2D).3 An NADPH-regenerating system was included in some of the assays with MT to ensure that NADPH was not becoming a limiting factor. Again, nNOSr (30 nm) was used as the positive control. Results obtained with MT using the NADPH-regenerating system showed the same weak radioactive signal as samples containing NADPH alone (Fig. 3). When nNOSr was spiked into MT samples, the l-[14C]citrulline signal dramatically increased (82% of the activity of the positive control). Additional control assays using dMT were performed to investigate the reason(s) behind the weak signal that could not be diminished by NOS inhibitors (see under “Experimental Procedures”). nNOSr (30 nm) was used as a positive control, and dMT alone was used as a negative control. nNOSr (30 nm)-catalyzed l-[14C]citrulline production was measured in the presence of increasing amounts of dMT. Following incubation of 150 μg of dMT with nNOSr plus all substrates and cofactors, the corresponding Dowex column eluates displayed a significant increase (63%) in the 14C signal when compared with the nNOSr signal in the absence of MT (Fig. 4, 4th and 5th bars). dMT at concentration >150 μg of protein plus nNOSr caused a protein concentration-dependent increase in the 14C signal (Fig. 4, 6th and 7th bars). Additional control assays were performed with the following: (a) dMT minus NADPH and (b) dMT plus l-thiocitrulline. The Dowex eluates from these control assays were negative for a 14C signal (data not shown). Results from assays containing nNOSr with titrated amounts of dMT indicate that there is an nNOS-independent but protein concentration-dependent increase in the 14C radioactive signal contained in the Dowex column eluates. An important point to note is that this effect occurs even when twice the normal amount of Dowex (2 ml) was used. One ml of Dowex is normally used in our laboratory as well as in the literature. Furthermore, when all the substrates and co-factors, minus nNOSr, MT, NADPH, and BH4 (C-Mix), were incubated with MT, and then eluted from the Dowex columns, once again a titratable increase in 14C signal was observed (Fig. 5, 2nd bar) compared with the C-Mix alone samples (Fig. 5, 1st bar). This signal (Fig. 5, 2nd bar) could not be inhibited by l-thiocitrulline (800 μm, Fig. 5, 3rd bar). Results from Fig. 5 clearly indicate that this signal is observed even in the absence of NADPH and BH4 and is not because of a NOS-catalyzed reaction. Here, the control values from incubations containing all ingredients except enzyme or MT were not subtracted from all data points (Fig. 5, 2nd and 3rd bars). To identify the source of the weak radioactive signal emanating from the MT samples, we probed the conversion assay eluates for amino acids, using HP-TLC (Fig. 6). l-[14C]Arginine to l-[14C]citrulline conversion assay eluates of the respective samples (nNOSr (30 nm; Fig. 6, lane 1); nNOSr + l-thiocitrulline (800 μm; lane 2); MT (150 μg; lane 3); and MT + l-thiocitrulline (lane 4)) and the non-Dowex-treated C-Mix (lane 5) were acetone-precipitated and centrifuged, and the supernatants were subjected to HP-TLC followed by autoradiography (Fig. 6). On the other hand, when C-Mix and the l-[14C]arginine to l-[14C]citrulline conversion assay eluates of the MT samples (Fig. 6, lanes 6 and 7, respectively) were applied to Dowex columns, the eluates were vacuum-centrifuged and then subjected to acetone precipitation (see under “Experimental Procedures”). C-Mix, which contained l-[14C]arginine minus nNOSr, was used as a positive control (Fig. 6, lane 5). In Fig. 6, the nNOSr-containing sample was also used for validation of the assay. Both the reaction substrate (l-[14C]arginine) and the product (l-[14C]citrulline) were present (Fig. 6, lane 1). l-Thiocitrulline was included in the conversion assay as a negative control. Inhibition of nNOSr prevented l-[14C]citrulline production by 98.5% (Fig. 6, lane 2). Results from MT samples indicated no conversion of l-[14C]arginine to l-[14C]citrulline (Fig. 6, lane 3), whereas the migration of authentic standard of L-citrulline was detected by ninhydrin staining. When C-Mix alone was passed through a Dowex column (Fig. 6, lane 6), the positively charged l-[14C]arginine was completely bound to the Dowex resin, and thus no l-[14C]arginine signal was observed in the eluates. Importantly, MT sample eluates that were passed through Dowex (Fig. 6, lane 7) did not contain l-[14C]citrulline. Thus, results obtained from assays measuring the conversion of l-[14C]arginine to l-[14C]citrulline confirmed that the weak radioactive signal associated with MT samples did not represent NOS-catalyzed l-citrulline production. Thus, data from the activity assays conducted in the presence of various amounts of protein validate one another and support the contention that no NOS activity is observed in rat liver mitochondria.
FIGURE 3.

l-[14C]Citrulline production in the presence of NADPH-regenerating system. Measurement of the conversion of l-[14C]arginine to l-[14C]citrulline was performed using an NADPH-regenerating system containing glucose 6-phosphate (125 mm), NADP (12.5 mm), and 1 unit of glucose-6-phosphate dehydrogenase. 1st bar, positive control, nNOSr (30 nm); 2nd bar, MT (150 μg); 3rd bar, MT (150 μg) + nNOSr (30 nm). Values from control incubations containing all ingredients except enzyme were subtracted from all data points. Data were expressed as the mean ± range of values, of experimental data points from two independent experiments, each run in duplicate.
FIGURE 4.

l-[14C]Citrulline production assays containing nNOSr with dMT. Measurement of the conversion of l-[14C]arginine to l-[14C]citrulline was performed on various amounts of dMT in the presence of nNOSr (30 nm). The incubates are as follows: 1st bar, dMT alone; 2nd bar, MIB alone; 3rd bar, nNOSr + intact 150 μg of MT; 4th bar, nNOSr alone; 5th bar, nNOSr + 150 μg of dMT; 6th bar, nNOSr + 280 μg of dMT; 7th bar, nNOSr + 560 μg of dMT. Values from control incubation containing all ingredients except enzyme were subtracted from all data points. Data were expressed as the mean ± range of values, of experimental data points from two independent experiments, each run in duplicate.
FIGURE 5.

Control experiments using l-[14C]citrulline production assays. Data were expressed as the mean ± range of values, of experimental data points from two independent experiments, each run in duplicate. Measurement of the conversion of l-[14C]arginine to l-[14C]citrulline was performed with all substrates and cofactors (l-arginine, CaCl2, CaM, l-[14C]arginine, HEPES) (C-Mix) in the absence of NADPH and BH4. 1st bar, C-Mix; 2nd bar, C-Mix + 150 μg of MT; 3rd bar, C-Mix + 150 μg of MT + 800 μm l-thiocitrulline (denoted as TC). Incubates were eluted with a Dowex column, and scintillation counting was performed.
FIGURE 6.

HP-TLC of the acetone-precipitated supernatants of l-[14C]arginine to l-[14C]citrulline conversion assay eluates. Acetone-precipitated l-[14C]arginine to l-[14C]citrulline conversion assay eluates (lanes 1–7) and the standards, l-arginine and l-citrulline, were loaded on a plate coated with silica gel, and HP-TLC was performed and developed as described (see “Experimental Procedures”). The lane pattern in HP-TLC was as follows: nNOSr, positive control detecting l-[14C]arginine (substrate) and l-[14C]citrulline (product) (lane 1); nNOSr + l-thiocitrulline (denoted as TC), negative control for NOS reaction showing no conversion of l-[14C]arginine to l-[14C]citrulline (lane 2); MT included in the assay, no conversion of l-[14C]arginine to l-[14C]citrulline (lane 3); MT + TC included in the assay, no conversion of l-[14C]arginine to l-[14C]citrulline (lane 4); C-Mix standard without MT or nNOSr, negative control for l-[14C]citrulline (lane 5); C-Mix followed by Dowex chromatography, no l-[14C]arginine, no l-[14C]citrulline, and no unidentified bands (lane 6); MT included in the assay followed by Dowex chromatography, no conversion of l-[14C]arginine to l-[14C]citrulline. The migration of authentic standards of l-arginine and l-citrulline, as detected by ninhydrin staining, is indicated on the right.
Immunochemical Analyses of MT using NOS and CaM Antibodies
Immunochemical analyses were performed with MT using rabbit polyclonal anti-nNOS and rabbit monoclonal anti-eNOS antibodies. SDS-PAGE and gel transfer onto nitrocellulose membranes followed by Western blotting were performed as described (see under “Experimental Procedures”). Western blot analyses of MT (150 μg) using nNOS antibodies were negative for any NOS protein (nNOS/iNOS) (Fig. 7A). Furthermore, no signal for any NOS protein was observed in the control tissue homogenate, confirming the lack of cellular contaminants in these preparations. However, when 30 ng of nNOSr was spiked into control samples containing 150 μg of MT, a clear signal could be detected (Fig. 7A).
FIGURE 7.

Immunochemical analyses of MT using NOS and CaM antibodies. Proteins were separated with SDS-polyacrylamide gel under the conditions described under “Experimental Procedures.” 8 μl of protein prestained standard (Bio-Rad) was loaded (1st lane). Western blots were performed with the following. A, rabbit nNOS antibody. The positive control was 30 ng of NOSr spiked in 150 μg of MT (lane 2). B, rabbit monoclonal eNOS antibody. The positive control was 30 ng of eNOSr spiked in 150 μg of MT (2nd lane). The immunocomplexes of A and B were developed by alkaline phosphatase reaction using 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium substrate. MT lane, run on the same gel as other fractions, was cropped and pasted along with other fractions after removing intervening lanes. C, rabbit CaM antibody. The positive control was 30 ng of CaM spiked in 150 μg of MT (2nd lane). The immunocomplexes were developed by HRP conjugate using a chemiluminescence substrate (Pierce), and MT denotes 150 μg of purified mitochondria (3rd lane).
Western blot analyses, using a highly specific eNOS antibody, also failed to detect the eNOS isoform in MT, whereas the positive control containing 30 ng of eNOSr spiked in 150 μg of mitochondria could be easily detected (Fig. 7B). Also, no clear signal for eNOS was detected in the control tissue homogenate. Although the polyclonal nNOS antibody is 10-fold more specific toward detecting the nNOS isoform, at lower dilutions it cross-reacts with other NOS isoforms, including iNOS.4 In contrast, the monoclonal eNOS antibody is absolutely specific for detecting eNOS (58). Thus, immunochemical analyses using NOS antibodies (with reactivities to nNOS, eNOS, and iNOS) were negative for any NOS isoform in mitochondria. Additionally, Western blot analyses using a rabbit monoclonal antibody to CaM were negative for the presence of CaM in MT. However, the positive control sample containing 30 ng of CaM spiked into a sample of 150 μg of MT could be easily detected (Fig. 7C).
DISCUSSION
This study describes carefully planned and executed studies and presents solid experimental evidence, using several independent but complementary assays, to refute the claims for the presence of any NOS isoform residing in mitochondria. Highly purified rat liver mitochondria were obtained, and to our knowledge we are the first group to have performed repeated differential centrifugation steps as well as a Percoll gradient purification to obtain this level of mitochondrial purity. These ultrapure mitochondria were then used to determine the presence or absence of any NOS isoform residing in or on mitochondria. The Percoll gradient purification procedure was demonstrated by Brown et al. (47) to produce pure mitochondrial preparations devoid of contamination by other organelles. We took painstaking measures to avoid contamination and to obtain pure rat liver mitochondria as follows: for example, use of an ice-jacketed tube during homogenization; avoidance of detergents for washing glassware, centrifuge tubes, and all other containers; removal of the fatty layer floating above the supernatant of the various spins; and avoidance of Mg2+ in MIB to prevent Mg2+ from inhibiting any putative mtNOS activity, as suggested previously (59).
Western blot analyses using mitochondrial and nonmitochondrial markers were used to estimate mitochondrial purity. In addition, results from LTQ-MS analyses revealed that MT contained abundant mitochondrial proteins but only 0.006% cytosolic proteins and zero lysosomal proteins (supplemental 1 and 2).
We focused on designing experiments to confirm or refute the presence of a mtNOS. We began by using a logical stepwise approach, namely the use of nano-HPLC-coupled LTQ-MS utilizing a nanospray source. MS analyses of whole MT did not produce data in support of the presence of any NOS isoform in MT (supplemental 1). To investigate further, we prepared SMP from sonicated whole MT. The SMP and the solubilized pellet were applied to a small column of 2′,5′-ADP-Sepharose 4B affinity resin. This affinity purification step enriched the SMP and the solubilized pellet fractions with NAD(P)H-binding proteins. The eluted proteins were processed and analyzed using LTQ-MS. Even by including this enrichment step, we failed to detect any NOS isoform in the SMP (supplemental 2) or the pellet (data not shown). So that we did not miss anything, samples of the flow-through from both SMP and the solubilized pellet obtained during affinity purification were tested, and LTQ-MS failed to detect the presence of a NOS protein in these flow-through fractions (data not shown). Collectively and without exception, thorough examination of all MT fractions was, in every instance, negative for the presence of any NOS isoform. Importantly, nNOSr, which was spiked into random MT samples and processed in parallel with the other samples, could be detected at absolute levels of ≤50 fmol (supplemental 3). These positive controls confirmed that the chosen analytical methodology, LTQ-MS, possessed more than adequate sensitivity to detect NOS protein, if present.
We then used various other complementary biochemical techniques to test for the presence of NOS in mitochondria. For enzyme activity, l-[14C]citrulline production assays (and not the l-[3H]citrulline production assay) were performed with MT samples. The conversion of l-[14C]arginine to l-[14C]citrulline was used because it provides a more stable signal with little chance of isotope exchange, thus decreasing apparent false-positives because of tritium exchange with water in an aqueous environment. At lower concentrations, l-thiocitrulline is selective for inhibiting nNOS, but at higher concentrations (800 μm), it becomes a nonspecific inhibitor to all NOS isoforms. The background radioactive signal observed with MT samples, which could not be inhibited using l-thiocitrulline (Fig. 2A), was also observed using l-NNA, a widely used and nonspecific NOS inhibitor (data not shown). However, control experiments using dMT (Fig. 4) as well as comparison of radioactive signals of C-Mix, MT, and MT plus l-thiocitrulline (Fig. 5) clearly demonstrated that the radioactive signal was not a NOS-catalyzed product.
Results from HP-TLC of the acetone-precipitated l-[14C]arginine to l-[14C]citrulline conversion assay eluates of MT clearly indicated that there is no NOS-catalyzed l-[14C]citrulline production emanating from MT (Fig. 6, lane 7). However, a fuzzy band was seen at the topmost region of all the HP-TLC lanes that contained MT sample eluates (Fig. 6, lanes 3, 5, and 7), and this signal could not be inhibited by l-thiocitrulline (lane 4). Currently, studies are underway to identify this radioactive signal. Preliminary results using urease indicate that the fuzzy band is because of arginase-produced urea. Thus, results from NOS activity assays showed that the weak radioactive signal observed in the l-[14C]arginine to l-[14C]citrulline conversion assays, which contained MT, did not represent NOS-catalyzed l-citrulline formation. Collectively, our data demonstrate that there is no NOS-catalyzed conversion of l-[14C]arginine to l-[14C]citrulline with MT samples.
Earlier studies separately reported the presence of each NOS isoform within mitochondria (24–30). Several authors found immunoreactivity toward eNOS in the inner mitochondrial membrane (26, 27, 31, 60), although some of those studies were not validated with confirmatory experiments using alternative methods. In some cases, the commercially obtained antibodies have since been shown to be somewhat nonspecific depending on the source. For example, Giulivi and co-workers (29, 61) used iNOS and nNOS antibodies to detect a 130-kDa mitochondrial protein, using protein mass fingerprinting, which they later claimed to be nNOSα. However, Lacza et al. (42) have identified the same 130-kDa protein in nNOSα knock-out mice using the same antibodies from the same source as the Giulivi and co-workers (29, 61). We performed immunochemical analyses by using NOS antibodies that had been well characterized for Western blotting and proven specific (58).5 Unlike the data presentation methods of many groups, which consisted of cropped Western blots, we present the full immunoblot images showing that the nNOS and eNOS antibodies were unable to detect any sign of NOS in mitochondria. On the other hand, the positive controls, nNOSr and eNOSr, when spiked into mitochondrial samples, at levels as low as 30 ng, could be easily detected (Fig. 7, A and B). The eNOS antibody displayed absolute specificity for eNOS alone and no cross-reactivity with the other isoforms (58). At higher dilutions, the nNOS antibody is ≥10-fold more specific toward the nNOS isoform. At lower dilutions, it can cross-react with other isoforms (eNOS and iNOS) as well. Therefore, detection of the three main NOS isoforms was provided for by the nNOS and eNOS antibodies. Also, the protein CaM, required for NOS activity, was not detected by Western analyses of MT (Fig. 7C).
Many of the papers supporting the existence of a mtNOS have used the “oxyhemoglobin conversion to methemoglobin” or “oxymyoglobin conversion to metmyoglobin” assays for measuring NO· production and thus NOS activity. We conducted oxyhemoglobin conversion assays for demonstration and comparison purposes to demonstrate to the reader that this is a useless assay in systems where other free radicals are being produced. The oxymyogobin assay was avoided on purpose, as the extinction coefficient of oxymyoglobin (581–592 nm; 11.6 mm−1 cm−1) (28) is much lower, and thus much less sensitive, than that of oxyhemoglobin measured at 401 nm (59.9 mm−1 cm−1) (57). Because hemoglobin or myoglobin cannot enter the mitochondrial matrix, and because of the considerable background present from the high light absorption and thus cloudiness of mitochondrial suspensions, we used mildly sonicated MT with the oxyhemoglobin capture assay as suggested previously by Ghafourifar (62). Mildly sonicated MTs (50 and 500 μg) along with SOD and CAT (50 units each) were prepared, and the assays were performed as described previously (57) (see under “Experimental Procedures”). Theoretically, this reaction should produce a consistent positive rate in the presence of NOS alone. Results with mildly sonicated MT clearly showed a negative rate for the reaction, which became more negative with increasing amounts of mildly sonicated MT (50 and 500 μg) (Fig. 8). The slope of the hemoglobin capture assay is normally positive when a flux of NO· is present and can go negative when a flux of and/or H2O2 is present (data not shown).3 These results indicate that and/or H2O2, generated by the mildly sonicated MT, interfered with the NO· trapping assays. This effect was evident regardless of any low level NO· fluxes that may have been present. Assays performed using mildly sonicated MT in the absence of SOD and CAT produced even more negative rates than assays conducted in the presence of SOD and CAT (data not shown). When the amounts of SOD and CAT were tripled (150 units each), there was no significant change in the negative slope of the reaction (data not shown). Upon spiking nNOSr (5 nm) into the mildly sonicated MT samples, the slope of the line for the oxyhemoglobin capture assay shifted to the positive direction indicating NO· production (Fig. 8). It should be of note that use of excessive amounts (1000 units/ml) of SOD with the mildly sonicated MT, as suggested previously (59), may totally dysregulate the mitochondrial enzyme systems and may result in interference with protein structure and functions. Any effect observed with such high levels of SOD in techniques such as amperometry would likely be due to dissociated redox-active metal or some nonspecific protein effect. Therefore, by using the oxyhemoglobin capture assay, production of NO· by SMPs could not be confirmed in this laboratory because of interference by oxygen radicals. The inability of SOD and/or catalase to completely scavenge these reactive oxygen species makes this assay useless for detecting NO· production in this model. These data support the results of Tay et al. (36), who also could not detect NO· using the oxymyoglobin capture assay.
FIGURE 8.

Oxyhemoglobin capture assay with mildly sonicated MT. Mildly sonicated MT (50 and 500 μg) along with SOD and CAT (50 units each) were incubated, and the assays were performed as mentioned (57). The rate of absorbance of mildly sonicated MT (50 and 500 μg) was negative in slope. When nNOSr (5 pmol) were spiked, the curves shifted positive.
Electrochemical techniques have been performed by many investigators in attempts to detect NO· production by mitochondria. Results published by Kanai et al. (32) and Schild et al. (63) using mouse heart and rat liver mitochondria, respectively, could not be reproduced by the Lacza et al. (34) or Tay et al. (36). This laboratory also failed to detect NO· production by sonicated MT (150 μg) using a NO· analyzer (Apollo 4000 NO· analyzer). However, the electrode readily detected NO· flux by a positive control consisting of 2.5 nm nNOSr-spiked mitochondria samples or by S-nitrosopenicillamine (250 nm) (data not shown). The fact that we could not detect extremely low levels of NO· production when using the NO·-specific electrode appears to be a sensitivity problem with the method. Furthermore, this method is susceptible to false-positive signals, especially when attempting to detect low levels of NO·, because of the ubiquitous presence of nitrite in buffers and other assay components. However, using the tried-and-true radioactive conversion of l-[14C]arginine to l-[14C]citrulline, our limit of detection for l-citrulline was ≥14.2 pmol/min per 1 pmol of nNOSr, the minimal amount that can produce easily measurable l-citrulline production using our criteria, which was that the signal must be ≥9 times the value of the standard deviation of the control triplicates. Although there are many other proteins in mitochondria, the MS and immunochemical analyses failed to detect the presence of CaM, a protein that is absolutely required for NOS function (supplemental 1 and 2).
NOS in endothelial cells can provide NO· levels at less than nanomolar concentrations over a sustained period of time, and these subnanomolar concentrations do have any effect. The lifetime of NO· at these concentrations is on the order of seconds, thus allowing NO· more than ample opportunity to find, and bind, its specific target guanylate cyclase. However, in contrast to MT, endothelial cells have sensitive and specific targets for NO· to bind and propagate its message and do not support the high concentrations of found in and around mitochondria. High concentrations of dramatically shorten the lifetime of NO· by reacting with NO· to form peroxynitrite, the role of which, in mitochondria, is still unclear. It is without question that sensitive targets do exist in MT, such as aconitase (64). However, the possibility of specific, regulatory, or signal transduction targets for NO· existing in MT is still an open question.
On the other hand, there may be circumstances whereby translocation of NOS into mitochondria could occur, and this possibility cannot be ruled out. However, to our knowledge, at the present time, there is no evidence or precedence for translocation of NOS into mitochondria.
The question of whether or not NOS, and thus NO· production, is localized inside or outside the mitochondria is all about biological significance. NO· produced at, or near, its site of action within mitochondria would not have far to travel to exert its effect(s) and could be easily accepted as playing a role in mitochondrial energy regulation. NO· is freely diffusible across membranes and, for the sake of argument, would not have to be generated by an intra-mitochondrial NOS. However, the biological significance of NO· produced outside the mitochondria becomes an issue of distance if one considers the diffusion-limited reaction rate between NO· and (65). Even though NO· is able to freely diffuse through most, if not all, biological membranes, the issue of NO· being freely diffusible encounters a problem when mitochondria are involved. NO·, diffusing across the plasma membrane or membrane(s) of other cellular organelles, does not encounter the onslaught of as happens when NO· approaches and then diffuses across the outer mitochondrial membrane and into mitochondria. It has been estimated that under certain in vitro conditions, mitochondria can leak up to 2% of the electrons moving down the electron transport chain (66). Therefore, the NO· approaching the mitochondria would encounter a gradient of increasing concentrations of the closer it came to the outer mitochondrial membrane. Thus, regulation of mitochondrial energy production by NO· generated outside the mitochondria would be an unreliable mechanism of regulation upon which the cell could not depend for millisecond-to-millisecond control of energy supplies. Considering carefully conducted studies with aconitase showing that inactivation by either NO· solutions or NO·-producing NONOates under anaerobic conditions is seen for both isoforms (mitochondrial and cytosolic aconitase). This inactivation, which occurs in the presence or absence of substrate, is accompanied by the appearance of a protein-bound dinitrosyl-iron-dithiol complex in the d7 state (64). We contend that the production of by mitochondria may be a protective mechanism that mitochondria use to guard against the possibility of decreased energy production that could occur at the hands of NO· interacting with critical iron and iron/sulfur-containing enzymes of the electron transport chain such as aconitase.
The existence of NOS within mitochondria has been questioned for the past 10 years or more. This situation may be due to the following: 1) differences in the purity of the mitochondrial preparations used among groups; 2) failure to confirm results with complementary techniques; 3) lack of use of appropriate controls; as well as 4) poor reliability and reproducibility of certain techniques as suggested by Brookes (33). Isolation of the purest mitochondria possible is absolutely necessary if groups are to determine with any certainty whether there is a NOS isoform within mitochondria. Although Lacza et al. (34, 35) initially identified eNOS in mitochondria, their subsequent studies conducted with brain, heart, and liver mitochondria from mice, rats, and pigs under stringent conditions did not support their original observations (31). We performed experiments under stringently controlled conditions to isolate, purify, and analyze mitochondria with appropriate controls and markers using a variety of complementary yet independent techniques. Results obtained, using LTQ-MS analyses complemented by other techniques, such as the l-[14C]arginine to l-[14C]citrulline conversion assay as well as immunochemical analyses, collectively and individually refute the claim that any NOS isoform resides within rat liver mitochondria. However, our studies and conclusions pertain only to rat liver. We do not say that our results are all inclusive or that they pertain to every organ, tissue, cell type, or species. The issue of NOS in mitochondria from other organs or other species is still an open issue.
Supplementary Material
Acknowledgment
We thank the Biomedical Analysis Core Facility BBRC/UTEP (supported by National Institutes of Health Grant 5G12RR008124) for full access to the liquid chromatography-MS instruments.
This work was supported, in whole or in part, by National Institutes of Health Grant ES 011982 (to R. T. M.) from NIEHS and by Grant 5G12RR008124 from NCRR to the Border Biomedical Research Center, University of Texas, El Paso.

The on-line version of this article (available at http://www.jbc.org) contains supplemental data 1–3.
R. T. Miller, unpublished observations.
B. S. Masters, personal communication.
P. Martasek and B. S. Masters, personal communication.
- NO·
- nitric oxide
- NOS
- nitric-oxide synthase
- MT
- isolated and purified mitochondria
- dMT
- denatured MT
- superoxide
- mtNOS
- mitochondrial NOS
- eNOS
- endothelial NOS
- iNOS
- inducible NOS
- nNOS
- neuronal NOS
- NOSr
- recombinant NOS
- BH4
- (6R)-5,6,7,8-tetrahydro-l-biopterin
- LTQ-MS
- linear ion trap-mass spectrometry
- CaM
- calmodulin
- HRP
- horseradish peroxidase
- VDAC
- voltage-dependent anion channel
- CAT
- catalase
- BSA
- bovine serum albumin
- HPLC
- high pressure liquid chromatography
- MS
- mass spectrometry
- TC
- l-thiocitrulline
- l-NNA
- nitro-l-arginine
- HP-TLC
- high performance-TLC
- SMP
- submitochondrial particle
- CO
- control.
REFERENCES
- 1.Ignarro L. J. (1989) FASEB J. 3, 31–36 [DOI] [PubMed] [Google Scholar]
- 2.Shin W. S., Sasaki T., Kato M., Hara K., Seko A., Yang W. D., Shimamoto N., Sugimoto T., Toyo-oka T. (1992) J. Biol. Chem. 267, 20377–20382 [PubMed] [Google Scholar]
- 3.Malinski T., Taha Z., Grunfeld S., Patton S., Kapturczak M., Tomboulian P. (1993) Biochem. Biophys. Res. Commun. 193, 1076–1082 [DOI] [PubMed] [Google Scholar]
- 4.Ignarro L. J., Buga G. M., Byrns R. E., Wood K. S., Chaudhuri G. (1988) J. Pharmacol. Exp. Ther. 246, 218–226 [PubMed] [Google Scholar]
- 5.Thomas D. D., Liu X., Kantrow S. P., Lancaster J. R., Jr. (2001) Proc. Natl. Acad. Sci. U. S. A. 98, 355–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Furchgott R. F., Vanhoutte P. M. (1989) FASEB J. 3, 2007–2018 [PubMed] [Google Scholar]
- 7.Förstermann U., Pollock J. S., Schmidt H. H., Heller M., Murad F. (1991) Proc. Natl. Acad. Sci. U. S. A. 88, 1788–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murad F., Forstermann U., Nakane M., Schmidt H., Pollock J., Sheng H., Matsumoto T., Warner T., Mitchell J., Tracey R., et al. ( 1992) Jpn. J. Pharmacol. 58, Suppl. 2, 150– 157 [PubMed] [Google Scholar]
- 9.Pollock J. S., Förstermann U., Mitchell J. A., Warner T. D., Schmidt H. H., Nakane M., Murad F. (1991) Proc. Natl. Acad. Sci. U. S. A. 88, 10480–10484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knowles R. G., Palacios M., Palmer R. M., Moncada S. (1989) Proc. Natl. Acad. Sci. U. S. A. 86, 5159–5162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hibbs J. B., Jr., Taintor R. R., Vavrin Z., Rachlin E. M. (1988) Biochem. Biophys. Res. Commun. 157, 87–94 [DOI] [PubMed] [Google Scholar]
- 12.Radi R., Cassina A., Hodara R. (2002) Biol. Chem. 383, 401–409 [DOI] [PubMed] [Google Scholar]
- 13.Cadenas E., Poderoso J. J., Antunes F., Boveris A. (2000) Free Radic. Res. 33, 747–756 [DOI] [PubMed] [Google Scholar]
- 14.Moncada S. (2000) Verh. K Acad. Geneeskd. Belg. 62, 171–181 [PubMed] [Google Scholar]
- 15.Thomas D. D., Miranda K. M., Colton C. A., Citrin D., Espey M. G., Wink D. A. (2003) Antioxid. Redox. Signal. 5, 307–317 [DOI] [PubMed] [Google Scholar]
- 16.Nathan C. (1992) FASEB J. 6, 3051–3064 [PubMed] [Google Scholar]
- 17.Stuehr D. J., Griffith O. W. (1992) Adv. Enzymol. Relat. Areas Mol. Biol. 65, 287–346 [DOI] [PubMed] [Google Scholar]
- 18.Ebel R. E., O'Keefe D. H., Peterson J. A. (1975) FEBS Lett. 55, 198–201 [DOI] [PubMed] [Google Scholar]
- 19.Dionisi O., Galeotti T., Terranova T., Azzi A. (1975) Biochim. Biophys. Acta 403, 292–300 [DOI] [PubMed] [Google Scholar]
- 20.Boveris A., Cadenas E. (1975) FEBS Lett. 54, 311–314 [DOI] [PubMed] [Google Scholar]
- 21.Cleeter M. W., Cooper J. M., Darley-Usmar V. M., Moncada S., Schapira A. H. (1994) FEBS Lett. 345, 50–54 [DOI] [PubMed] [Google Scholar]
- 22.Brown G. C., Bolaños J. P., Heales S. J., Clark J. B. (1995) Neurosci. Lett. 193, 201–204 [DOI] [PubMed] [Google Scholar]
- 23.Radi R., Rodriguez M., Castro L., Telleri R. (1994) Arch. Biochem. biophys. 308, 89–95 [DOI] [PubMed] [Google Scholar]
- 24.Kobzik L., Stringer B., Balligand J. L., Reid M. B., Stamler J. S. (1995) Biochem. Biophys. Res. Commun. 211, 375–381 [DOI] [PubMed] [Google Scholar]
- 25.Frandsen U., Lopez-Figueroa M., Hellsten Y. (1996) Biochem. Biophys. Res. Commun. 227, 88–93 [DOI] [PubMed] [Google Scholar]
- 26.Bates T. E., Loesch A., Burnstock G., Clark J. B. (1995) Biochem. Biophys. Res. Commun. 213, 896–900 [DOI] [PubMed] [Google Scholar]
- 27.Bates T. E., Loesch A., Burnstock G., Clark J. B. (1996) Biochem. Biophys. Res. Commun. 218, 40–44 [DOI] [PubMed] [Google Scholar]
- 28.Tatoyan A., Giulivi C. (1998) J. Biol. Chem. 273, 11044–11048 [DOI] [PubMed] [Google Scholar]
- 29.Elfering S. L., Sarkela T. M., Giulivi C. (2002) J. Biol. Chem. 277, 38079–38086 [DOI] [PubMed] [Google Scholar]
- 30.Ghafourifar P., Richter C. (1997) FEBS Lett. 418, 291–296 [DOI] [PubMed] [Google Scholar]
- 31.Lacza Z., Puskar M., Figueroa J. P., Zhang J., Rajapakse N., Busija D. W. (2001) Free Radic. Biol. Med. 31, 1609–1615 [DOI] [PubMed] [Google Scholar]
- 32.Kanai A. J., Pearce L. L., Clemens P. R., Birder L. A., VanBibber M. M., Choi S. Y., de Groat W. C., Peterson J. (2001) Proc. Natl. Acad. Sci. U. S. A. 98, 14126–14131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brookes P. S. (2004) Mitochondrion 3, 187–204 [DOI] [PubMed] [Google Scholar]
- 34.Lacza Z., Horn T. F., Snipes J. A., Zhang J., Roychowdhury S., Horváth E. M., Figueroa J. P., Kollai M., Szabó C., Busija D. W. (2004) J. Neurochem. 90, 942–951 [DOI] [PubMed] [Google Scholar]
- 35.Lacza Z., Snipes J. A., Zhang J., Horváth E. M., Figueroa J. P., Szabó C., Busija D. W. (2003) Free Radic. Biol. Med. 35, 1217–1228 [DOI] [PubMed] [Google Scholar]
- 36.Tay Y. M., Lim K. S., Sheu F. S., Jenner A., Whiteman M., Wong K. P., Halliwell B. (2004) Free Radic. Res. 38, 591–599 [DOI] [PubMed] [Google Scholar]
- 37.Kanai A., Epperly M., Pearce L., Birder L., Zeidel M., Meyers S., Greenberger J., de Groat W., Apodaca G., Peterson J. (2004) Am. J. Physiol. Heart Circ. Physiol. 286, H13–21 [DOI] [PubMed] [Google Scholar]
- 38.Giulivi C., Poderoso J. J., Boveris A. (1998) J. Biol. Chem. 273, 11038–11043 [DOI] [PubMed] [Google Scholar]
- 39.Zanella B., Calonghi N., Pagnotta E., Masotti L., Guarnieri C. (2002) Biochem. Biophys. Res. Commun. 290, 1010–1014 [DOI] [PubMed] [Google Scholar]
- 40.López-Figueroa M. O., Caamaño C., Morano M. I., Rønn L. C., Akil H., Watson S. J. (2000) Biochem. Biophys. Res. Commun. 272, 129–133 [DOI] [PubMed] [Google Scholar]
- 41.López-Figueroa M. O., Caamaño C. A., Morano M. I., Akil H., Watson S. J. (2002) Methods Enzymol. 352, 296–303 [DOI] [PubMed] [Google Scholar]
- 42.Lacza Z., Pankotai E., Csordás A., Gero D., Kiss L., Horváth E. M., Kollai M., Busija D. W., Szabó C. (2006) Nitric Oxide 14, 162–168 [DOI] [PubMed] [Google Scholar]
- 43.Schmidt K., Klatt P., Mayer B. (1994) Biochem. J. 301, 645–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Narayanan K., Griffith O. W. (1994) J. Med. Chem. 37, 885–887 [DOI] [PubMed] [Google Scholar]
- 45.Martasek P., Liu Q., Liu J., Roman L. J., Gross S. S., Sessa W. C., Masters B. S. (1996) Biochem. Biophys. Res. Commun. 219, 359–365 [DOI] [PubMed] [Google Scholar]
- 46.Roman L. J., Sheta E. A., Martasek P., Gross S. S., Liu Q., Masters B. S. (1995) Proc. Natl. Acad. Sci. U. S. A. 92, 8428–8432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown M. R., Sullivan P. G., Geddes J. W. (2006) J. Biol. Chem. 281, 11658–11668 [DOI] [PubMed] [Google Scholar]
- 48.Pertoft H., Laurent T. C., Låås T., Kågedal L. (1978) Anal. Biochem. 88, 271–282 [DOI] [PubMed] [Google Scholar]
- 49.Reinhart P. H., Taylor W. M., Bygrave F. L. (1982) Biochem. J. 204, 731–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 51.Stone K. L., Williams K. R. (1996) in The Protein Protocol Handbook (Walker J. M. ed) pp. 415–425, Humana Press Inc.,Totowa, NJ [Google Scholar]
- 52.Jurado J. D., Rael E. D., Lieb C. S., Nakayasu E., Hayes W. K., Bush S. P., Ross J. A. (2007) Toxicon 49, 339–350 [DOI] [PubMed] [Google Scholar]
- 53.Rodrigues M. L., Nakayasu E. S., Oliveira D. L., Nimrichter L., Nosanchuk J. D., Almeida I. C., Casadevall A. (2008) Eukaryot. Cell 7, 58–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eng J. K., McCormack A. L., Yates J. R. (1994) J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 55.Nishimura J. S., Narayanasami R., Miller R. T., Roman L. J., Panda S., Masters B. S. (1999) J. Biol. Chem. 274, 5399–5406 [DOI] [PubMed] [Google Scholar]
- 56.Miller R. T. (2002) Chem. Res. Toxicol. 15, 927–934 [DOI] [PubMed] [Google Scholar]
- 57.Sheta E. A., McMillan K., Masters B. S. (1994) J. Biol. Chem. 269, 15147–15153 [PubMed] [Google Scholar]
- 58.Jáchymová M., Martásek P., Panda S., Roman L. J., Panda M., Shea T. M., Ishimura Y., Kim J. J., Masters B. S. (2005) Proc. Natl. Acad. Sci. U. S. A. 102, 15833–15838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ghafourifar P., Asbury M. L., Joshi S. S., Kincaid E. D. (2005) Methods Enzymol. 396, 424–444 [DOI] [PubMed] [Google Scholar]
- 60.Hotta Y., Otsuka-Murakami H., Fujita M., Nakagawa J., Yajima M., Liu W., Ishikawa N., Kawai N., Masumizu T., Kohno M. (1999) Eur. J. Pharmacol. 380, 37–48 [DOI] [PubMed] [Google Scholar]
- 61.Giulivi C. (2003) Free Radic. Biol. Med. 34, 397–408 [DOI] [PubMed] [Google Scholar]
- 62.Ghafourifar P. (2002) Methods Enzymol. 359, 339–350 [DOI] [PubMed] [Google Scholar]
- 63.Schild L., Reinheckel T., Reiser M., Horn T. F., Wolf G., Augustin W. (2003) FASEB J. 17, 2194–2201 [DOI] [PubMed] [Google Scholar]
- 64.Kennedy M. C., Antholine W. E., Beinert H. (1997) J. Biol. Chem. 272, 20340–20347 [DOI] [PubMed] [Google Scholar]
- 65.Huie R. E., Padmaja S. (1993) Free Radic. Res. Commun. 18, 195–199 [DOI] [PubMed] [Google Scholar]
- 66.Boveris A., Oshino N., Chance B. (1972) Biochem. J. 128, 617–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.