

Abstract
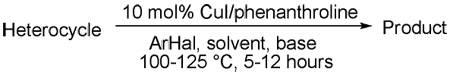
A general method for copper-catalyzed arylation of sp2 C-H bonds with pKa's below 35 has been developed. The method employs aryl halide as the coupling partner, lithium alkoxide or K3PO4 base, and DMF, DMPU, or mixed DMF/xylenes solvent. A variety of electron-rich and electron-poor heterocycles such as azoles, caffeine, thiophenes, benzofuran, pyridine oxides, pyridazine, and pyrimidine can be arylated. Furthermore, electron-poor arenes possessing at least two electron-withdrawing groups on benzene ring can also be arylated. Two arylcopper-phenanthroline complex intermediates were independently synthesized.
1. Introduction
Compounds containing polyaryl moieties are common among natural products, pharmaceuticals, and dyes. As a consequence, regioselective formation of aryl-aryl bonds has attracted substantial interest over the last century.1 The copper-promoted biaryl synthesis was pioneered by Ullmann more than a hundred years ago.2 Until the development of Stille, Suzuki, and Kumada reactions3 in 1970's copper was the only metal widely used for the formation of aryl-aryl bonds. Recently, copper-catalyzed cross-coupling reactions are undergoing resurgence. Efficient methods for carbon-carbon,4 carbon-nitrogen,5 and carbon-oxygen6 bond formation have been demonstrated by using copper complexes. However, copper appears to be underutilized as a catalyst for C-H bond functionalization even though it was the first transition metal shown to promote carbon-hydrogen bond arylation.7 In the last few years palladium-, rhodium-, and ruthenium-catalyzed sp2 C-H bond arylation has undergone explosive growth.8 In contrast, only scattered examples of copper-promoted carbon-hydrogen bond arylation have been described with most reports dating back to 1960's and 1970's.9 Majority of the palladium-, rhodium-, or ruthenium-catalyzed C-H bond functionalization examples involve regioselective arylation of directing-group-containing arenes (Scheme 1A) or electron-rich heterocycles such as azoles or indoles. Several recent reports describe functionalization of arenes possessing no conventional directing groups.10 In the latter case the regioselectivity issues are often unsolved and sometimes only symmetrical arenes can be employed as the C-H coupling component due to the possibility of regioisomer formation (Scheme 1B). Perhaps the only general exception is found in recent elegant work by Fagnou who showed that fluorinated arenes can be regioselectively arylated by aryl halides under palladium catalysis.11 The regioselectivity is imparted by the acidification of the C-H bonds by ortho-fluorine substituents (Scheme 1B, FG = F). Thus, two issues that need to be solved are apparent. First, regioselectivity of arylation is often problematic unless the coupling C-H component contains a directing group. Second, expensive transition metals such as palladium, rhodium, and ruthenium are routinely employed as arylation catalysts. Cheap copper and iron complexes are only rarely used in non-carbene C-H bond functionalization chemistry.12
Scheme 1.

Regioselectivity in C-H Bond Functionalization
We have recently disclosed a method for copper-catalyzed arylation of C-H bonds in electron-poor and electron-rich heterocycles as well as polyfluorobenzenes.13 The reactions proceed by initial deprotonation of a relatively acidic sp2 C-H bond by an alkali metal base (or tBuOCu) followed by transmetallation and coupling with an aryl or vinyl halide (Scheme 2). Even 1,4-difluorobenzene can be arylated, although the efficiency is low, presumably due to insufficient acidity. If pKa of the C-H bond is the major factor determining the arylation efficiency, copper-catalyzed cross-coupling method should be very general.
Scheme 2.

Copper-Catalyzed Arylation
We report here a general method for copper-catalyzed, highly regioselective arylation and alkenylation of electron-rich and electron-poor heterocycles as well as benzenes possessing at least two electron-withdrawing groups. Mechanistic investigations of the arylation process are also described.
2. Results
2.1 Arylation of Electron-Rich Heterocycles
Our initial attempts were directed towards developing optimized conditions for electron-rich heterocycle arylation. We have recently reported a method for copper-catalyzed heterocycle arylation by aryl iodides.13a The best results were obtained by employing lithium t-butoxide base and relatively acidic heterocycle substrates such as oxazoles and thiazoles. For less acidic imidazole and 1,2,4-triazole derivatives a stronger tBuOK base is required and the reaction proceeds by a benzyne-type mechanism.14 Regioisomer mixtures were formed if substituted aryl halides were used in combination with tBuOK base (Scheme 3). Additional issues that had to be considered are as follows. Formation of t-butyl aryl ether by the reaction of t-butoxide bases with aryl iodide was observed, resulting in decreased conversion to the arylation products. Copper catalyst was found to be relatively unstable at the temperature required for the arylation and thus only fast reactions were successful.
Scheme 3.

Benzyne Mechanism
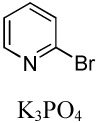
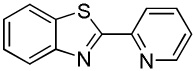
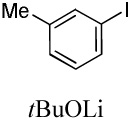
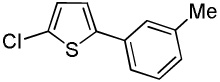
We reasoned that employing a phenanthroline ligand as described by Buchwald and coworkers6a should allow for a more efficient heterocycle arylation by stabilizing the copper catalyst and facilitating the halide displacement step. Replacing tBuOK with a weaker lithium alkoxide or K3PO4 base should shut down the benzyne mechanism thus ensuring arylation regioselectivity. For less reactive substrates employing hindered Et3COLi base instead of tBuOLi should be beneficial by slowing the nucleophilic substitution of aryl iodide while not influencing the arylation rate. We were pleased to discover that addition of phenanthroline ligand allows to use lithium t-butoxide base for less acidic heterocycle arylation avoiding the problems associated with benzyne mechanism. Additionally, the modified reaction conditions allow for the arylation of heterocycles that were not reactive under our previous conditions (Table 1). It is possible to employ K3PO4 base in the arylation of the most acidic heterocycles such as benzothiazole (Entry 1). Caffeine and N-methyl-1,2,4-triazole can be arylated by using tBuOLi base (Entries 2 and 3). Previously, tBuOK was required for the arylation of those substrates.13a For the least acidic heterocycles hindered Et3COLi base is required for optimal results. Arylation of N-methylimidazole (Entry 4), thiophenes (Entries 5, 6, 10, and 11), N-phenylpyrazole (Entry 7), benzofuran and benzothiophene (Entries 8 and 9) can be accomplished in good yields. Reaction of 2-chlorothiophene with 2-iodotoluene afforded only the o-tolylated heterocycle (Entry 10). If benzyne mechanism would be operative, either isomer mixture or m-isomer would be formed. Arylation of 2-chlorothiophene with 3-iodotoluene afforded only the m-tolylated isomer (Entry 11) in contrast with the previous results obtained by employing tBuOK base (Scheme 3). Furans and N-substituted indoles were found to be unreactive under any conditions tried while heterocycles possessing acidic N-H bonds were arylated on the nitrogen as reported by Buchwald.5e The following DMSO pKa's of heterocycle C-H bonds have been reported: N-alkylindoles, about 37; furan, 35; N-methylimidazole, 33.15 It can be concluded that copper-catalyzed electron-rich heterocycle arylation is successful for compounds possessing pKa's below 35.
Table 1.
Electron-rich Heterocycle Arylationa
 | ||||
|---|---|---|---|---|
| entry | heterocycle | aryl halide/base | product | yield, % |
| 1 | 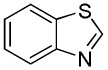 |
 |
 |
89 |
| 2 | 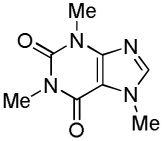 |
C6H5I/tBuOLi | 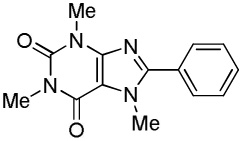 |
85 |
| 3 | 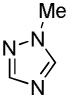 |
C6H5I/tBuOLi | 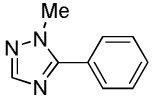 |
88 |
| 4 | 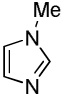 |
C6H5I/Et3COLi | 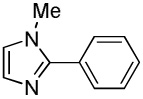 |
82 |
| 5 | 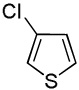 |
C6H5I/Et3COLi | 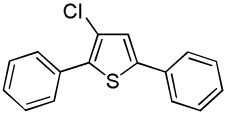 |
87 |
| 6 | 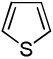 |
C6H5I/Et3COLi | 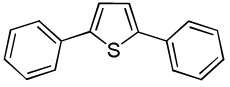 |
85 |
| 7 | 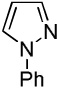 |
C6H5I/Et3COLi | 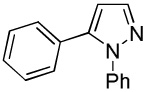 |
52 |
| 8 | 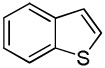 |
C6H5I/Et3COLi | 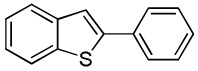 |
86 |
| 9 | 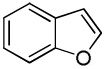 |
C6H5I/Et3COLi | 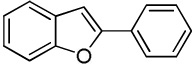 |
60 |
| 10 | 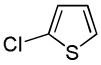 |
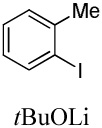 |
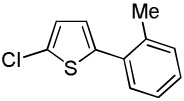 |
89 |
| 11 | 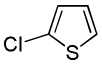 |
 |
 |
91 |
Copper (I) iodide (0.1 mmol), phenanthroline (0.1 mmol), aryl halide (1–3 mmol), heterocycle (1–2 mmol), base (1.7–3 mmol), DMF or DMPU solvent (0.5–0.6 mL). Yields are isolated yields. See the Supporting Information for details.
2.2 Arylation of Electron-Poor Heterocycles
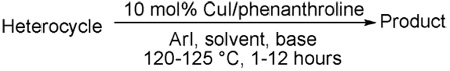
We have previously reported one example of copper-catalyzed electron-poor heterocycle arylation.13a If the mechanistic considerations presented in Scheme 2 are correct, arylation of electron-poor heterocycles with C-H bond DMSO pKa's below 35 should be feasible. Gratifyingly, conditions developed for electron-rich heterocycle arylation worked well also in this case (Table 2). While most pyridines are not reactive, more acidic pyridine oxides can be arylated by using either tBuOLi or K3PO4 base (Entries 1–5). 2-Iodopyridine is incompatible with alkoxides due to the formation of 2-t-butoxypyridine under the reaction conditions and K3PO4 base has to be used (Entry 2). 2-Methylpyridine oxide is also reactive, but the yield is diminished compared to other substrates, presumably due to acidic benzylic protons decreasing effective concentration of the arylcopper intermediate. 2-Phenylpyridine oxide is efficiently arylated by substituted aryl iodides and the products are obtained in excellent yield (Entries 4 and 5). More interestingly, pyridazine can be arylated in a good yield (Entry 6). A four-step synthesis of 4-phenylpyridazine has been reported.16 In contrast, direct arylation methodology allows to synthesize this compound in a single step from commercially available starting materials. Pyrimidine is arylated in a low yield, presumably due to insufficient acidity (pKa = 37;15 Entry 7). As expected, the most acidic positions in pyrimidine and pyridazine are arylated.15a Cyanidine and 1,2,3-triazine decompose under the reaction conditions.
Table 2.
Electron-poor Heterocycle Arylationa
 | ||||
|---|---|---|---|---|
| entry | heterocycle | aryl halide/base | product | yield, % |
| 1b |  |
C6H5I/tBuOLi | 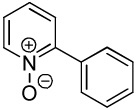 |
58 |
| 2 | 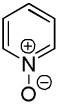 |
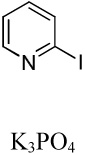 |
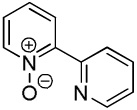 |
41 |
| 3 | 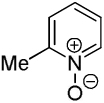 |
C6H5I/tBuOLi | 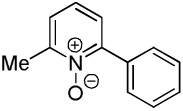 |
43 |
| 4 |  |
 |
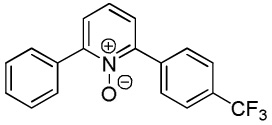 |
80 |
| 5 | 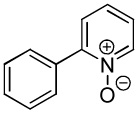 |
 |
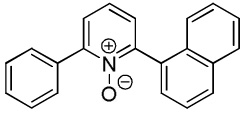 |
91 |
| 6 | 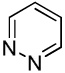 |
C6H5I/Et3COLi | 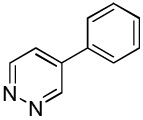 |
60 |
| 7 | 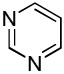 |
C6H5I/Et3COLi | 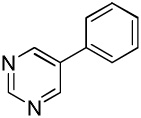 |
31 |
Copper (I) iodide (0.1 mmol), phenanthroline (0.1 mmol), aryl halide (1–2 mmol), heterocycle (1–2 mmol), base (1.7–2 mmol), DMF or DMPU solvent (0.5–0.6 mL). Yields are isolated yields. See the Supporting Information for details.
2,6-Diphenylpyridine oxide also isolated (20%).
2.3 Arylation of Electron-Poor Benzenes
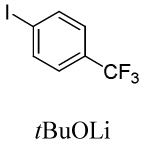
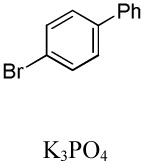
We have recently disclosed preliminary results showing that polyfluorobenzene derivatives can be arylated and alkenylated under copper catalysis.13b Both aryl iodide and bromide reagents can be employed. The reactivity parallels the acidity of C-H bonds, with the most acidic C-H bonds, those flanked by two C-F bonds, arylated most efficiently. The arylation of C-H bonds that are not flanked by two C-F bonds was inefficient and only 10% yield was obtained in the reaction of 4-iodotoluene with 1,2,3,4-tetrafluorobenzene. Since introduction of electron-withdrawing substituents in aromatic ring is expected to decrease the pKa of C-H bonds, we reasoned that arylation of a variety of other electron-deficient arenes should be possible. The improved conditions for arylation of electron-rich heterocycles were successfully applied to the arylation of electron-deficient arenes (Table 3). Pentafluorobenzene and tetrafluoroarenes can be arylated by aryl iodides (Entries 2 and 6) as well as aryl bromides (Entries 1, 7, 8). Even some heteroaryl chlorides can be used (Entries 3 and 4) although for 2-pyridyl chloride 150° C reaction temperature is required. Alkenylation is also possible (Entries 5 and 9). Potassium phosphate can be used as a base if arene contains more than two fluorine substituents or two fluorine substituents and an additional electron-withdrawing group (Entry 11). For 1,4-difluorobenzene arylation, hindered Et3COLi base is required. Previously we were unable to efficiently arylate such compounds by using tBuOLi base due to formation of tBuOAr byproduct. Penta- and tetrachlorobenzenes can be phenylated in excellent yield (Entries 12 and 13). Less acidic 1,3-dichlorobenzene is regioselectively phenylated in an acceptable 43% yield by employing Et3COLi base (Entry 14). If tBuOLi base was used, arylation product was isolated in only 18% yield. 1,3-Dinitrobenzene and 3-nitrobenzonitrile are also reactive affording the arylation products in moderate yields (Entries 15 and 16). The latter two arenes are slowly decomposed by the base and thus only the most reactive aryl iodides can be used. High arylation yield and absence of cyclized products for Entry 6 suggests that SRN1 mechanism is unlikely.17 Recent data obtained by Hartwig and coworkers argue against intermediacy of aryl radicals for copper-catalyzed C-N bond formation reactions.18 The following limitations have been observed. If aryl bromides are used in combination with lithium alkoxide bases, low yields of arylation products are obtained. Low conversions (<5%) are obtained in arylation of fluorobenzene, nitrobenzene, and α,α,α-trifluorotoluene.
Table 3.
Arylation of Electron-poor Arenesa
 | ||||
|---|---|---|---|---|
| entry | arene | aryl halide/base | product | yield, % |
| 1b | C6F5H | 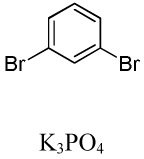 |
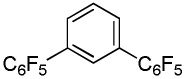 |
51 |
| 2b | C6F5H | 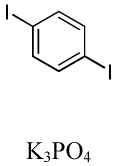 |
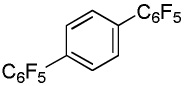 |
73 |
| 3 | C6F5H | 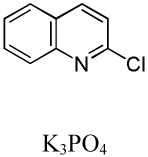 |
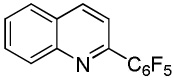 |
85 |
| 4 | C6F5H | 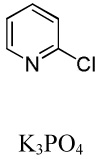 |
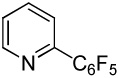 |
41 |
| 5 | C6F5H | 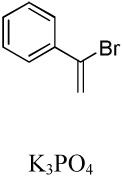 |
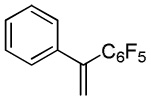 |
81 |
| 6 | C6F5H | 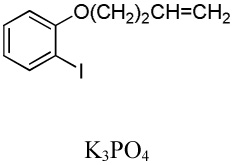 |
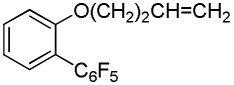 |
89 |
| 7 | 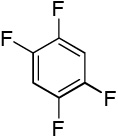 |
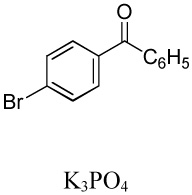 |
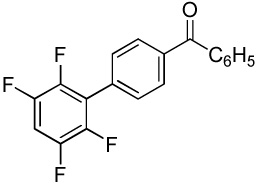 |
52 |
| 8 | 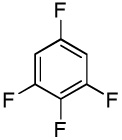 |
 |
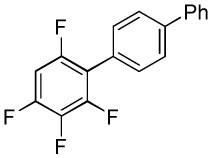 |
70 |
| 9 | 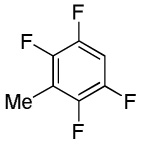 |
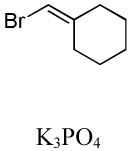 |
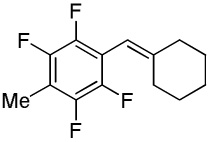 |
95 |
| 10 | 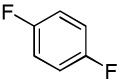 |
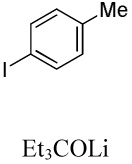 |
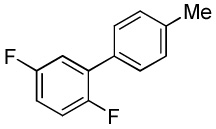 |
54 |
| 11 | 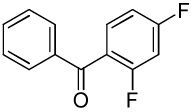 |
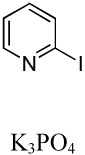 |
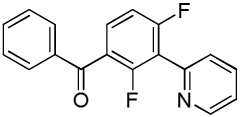 |
68 |
| 12 | C6C15H | C6H5I/tBuOLi | 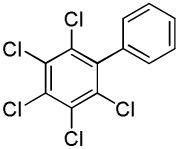 |
91 |
| 13 | 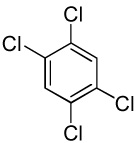 |
C6H5I/tBuOLi | 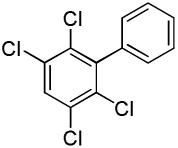 |
74 |
| 14 | 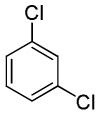 |
C6H5I/Et3COLi | 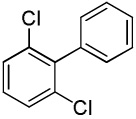 |
43 |
| 18c | ||||
| 15 | 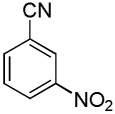 |
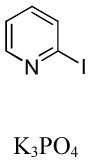 |
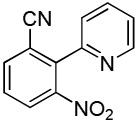 |
51 |
| 16 | 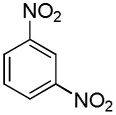 |
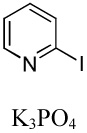 |
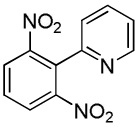 |
72 |
Copper (I) iodide (0.1 mmol), phenanthroline (0.1 mmol), halide (1–2 mmol), arene (1–3 mmol), base (1.7–4 mmol), DMF, DMPU, or DMF/xylenes solvent (0.5–0.8 mL). Yields are isolated yields. See the Supporting Information for details.
Copper (I) iodide (0.15 mmol), phenanthroline (0.15 mmol), halide (1 mmol), arene (3 mmol), base (4 mmol).
tBuOLi base.
2.4 Mechanistic Considerations
As shown in Scheme 2, the arylation reaction can be divided into three parts: metallation, transmetallation with copper halide, and reaction of arylcopper with haloarene. Metallation and reaction of arylcopper with haloarene steps will be discussed in more detail.
2.4.1. Metallation Step
One can expect that metallation step may be facilitated by coordination of copper species to Lewis-basic heteroatoms of the substrate. However, base-promoted H/D exchange in polyfluoroarenes, electron-rich, and electron-poor heterocycles occurs with the same efficiency both in the presence or absence of CuI (Scheme 4). Consequently, the acidity of substrate determines the position and efficiency of metallation even though the substrates belong to different classes of compounds and some of them possess heteroatoms capable of coordinating transition metals. Copper t-butoxide is a competent metallating reagent under the arylation conditions (Scheme 4B) complicating the mechanistic situation. The lifetime of aryllithium and arylpotassium species must be short since significant amounts of benzyne-derived products are not formed from polyfluoro- or polychloroarenes under catalytic or H/D exchange conditions.
Scheme 4.

H/D Exchange Experiments.
The rate of metallation/demetallation relative to subsequent reaction steps also has been considered (Scheme 5). If benzothiophene is arylated under the usual reaction conditions but with added tBuOD, incorporation of deuterium in the unreacted starting material is observed. The protonation of aryllithium and/or arylcopper intermediates by relatively weak t-butanol acid is competitive with the arylation step.
Scheme 5.

Deuteration under Reaction Conditions.
The ease of electron-deficient arene metallation demonstrated in this work may have other synthetic implications since strong alkyllithium bases and cryogenic conditions are not required. For substrates possessing DMSO pKa's below 27 even K3PO4 base is an efficient metallating agent.
2.4.2. Arylcopper Reaction with Haloarene Step
Several competition experiments were undertaken to determine relative reactivities of aryl iodides and arenes. The intermediate arylcopper species were identified by NMR as well as independently synthesized.
2.4.2.1. Relative Reactivities
Competition between arylation of pentafluorobenzene and tetrafluorobenzene by 4-iodotoluene results in preferential functionalization of pentafluorobenzene (Scheme 6). This result may be explained by the higher concentration of arylmetal intermediate for the more acidic pentafluorobenzene.
Scheme 6.

Competition Between Pentafluorobenzene and Tetrafluorobenzene
The reactivity of electron-rich and electron-poor aryl halides was compared by reacting a mixture of 4-trifluoromethylhalobenzene and 4-halotoluene with pentafluorobenzene (Scheme 7). A 4/1 product ratio was observed favoring trifluoromethylphenylation for both Hal = I and Br. Thus, electron-deficient aryl halides are more reactive as reported earlier.9b
Scheme 7.

Comparison of Aryl Halide Reactivity
2.4.2.2. Arylcopper Intermediates
We independently synthesized one of the presumed arylation intermediates, pentafluorophenylcopper-phenanthroline complex 1 (Scheme 8). It exists as a moisture-and temperature-sensitive dark orange solid that is either insoluble or poorly soluble in most common organic solvents. The connectivity was verified by X-ray crystallography; however, it was not possible to fully refine the structure due to twinning of the crystals. The reaction of copper iodide, potassium phosphate, pentafluorobenzene, and phenanthroline in DMF under the conditions of the catalytic process affords complex 1 as determined by 19F NMR of the crude reaction mixture. The complex reacts with aryl iodides producing cross-coupled biaryls. An analogous 4-methoxy-2,3,5,6-tetrafluorophenylcopper-phenanthroline complex 2 was prepared as dark rust-colored crystals by reacting tBuOCu with 2,3,5,6-tetrafluoroanisole followed by addition of phenanthroline ligand. The complex is stable in solid state under inert atmosphere at −20 °C; however, slow decomposition is observed in CH2Cl2 solution. It is sparingly soluble in most organic solvents and can be recrystallized from dichloromethane at −30 °C. The structure of 2 was verified by single-crystal X-ray diffraction analysis. The ORTEP diagram of 2 is shown in Figure 1. The complex exists as a dimer in solid state with a Cu-Cu distance of 2.5570(6) Å that is shorter than the van der Waals radii sum of 2.80 Å signifying a Cu-Cu bonding interaction.19 Copper assumes a distorted tetrahedral geometry with C(13)-Cu-N(1) angle of 135.68(9)°. As expected, phenanthroline complexes to Cu in a bidentate fashion with Cu-N(1) distance of 2.0720(18) Å and Cu-N(2) distance of 2.0949(19) Å. The copper-C(aryl) bond length is 1.932(2) Å, which is slightly shortened compared to the corresponding Cu-C distance in tetrameric pentafluorophenylcopper (1.962(2) to 2.007(2) Å).20 However, Cu-C distance in pentafluorophenylcopper-pyridine complex is shorter at 1.8913(17) Å.21 No arylcopper-phenanthroline complexes appear to have been crystalographically characterized. Isomeric tolylcopper-phenanthroline complexes are known.22
Scheme 8.

Polyfluorophenylcopper-Phenanthroline Complexes
Figure 1.

ORTEP view of 2. Selected interatomic distances (Å) and angles (deg): Cu-C(13) = 1.932(2), Cu-N(1) = 2.0720(18), Cu-N(2) = 2.0949(19), Cu-Cu = 2.5770(6), C(13)-Cu-N(1) = 135.68(9), C(13)-Cu-N(2) = 132.69(8).
3. Summary
A general method for copper-catalyzed arylation of sp2 C-H bonds possessing DMSO pKa's below 35 has been developed. The choice of base is dependent on the acidity of the C-H bond to be arylated. For comparatively acidic C-H bonds with pKa below 27 K3PO4 base may be employed. If the substrates are less acidic (pKa 27–35), a stronger lithium alkoxide base is required. A variety of electron-rich and electron-poor heterocycles such as azoles, caffeine, thiophenes, benzofuran, pyridine oxides, pyridazine, and pyrimidine can be arylated. Furthermore, electron-poor arenes possessing at least two electron-withdrawing groups on benzene ring can also be arylated. Unusual regioselectivity has been achieved allowing the arylation of the most hindered position. This method supplements the well-known C-H activation/borylation methodology23 where functionalization usually occurs at the least hindered position. Additionally, the copper-catalyzed arylation methodology is complementary to existing lithiation/boronation/cross-coupling methods and in some cases may offer advantages with regards to the number of synthetic steps and functional group tolerance.24
4. Experimental Section
General procedure for coupling reactions
Outside the glovebox a 1-dram vial equipped with a magnetic stir bar was charged with haloarene, phenanthroline (10 mol%), substrate, and solvent (DMF or a 1/1 mixture of DMF and xylenes). If anhydrous DMPU was used, the reaction was set up inside the glovebox. The vial was flushed with argon, capped and placed inside a glovebox. To this mixture was added CuI (10 mol %) and base (1.7–4.0 equiv). The sealed vial was taken out of the glovebox, stirred at room temperature for 5 min and placed in a preheated oil bath. After the completion of the reaction, the mixture was cooled to room temperature and diluted with ethyl acetate (50 mL). The resulting solution was washed with brine (15 mL), dried over anhydrous MgSO4, and concentrated under vacuum to a volume of about 1 mL. The mixture containing the product was subjected to column chromatography on silica gel (hexanes followed by appropriate solvent to elute the products). After concentrating the fractions containing the product, the residue was dried under reduced pressure to yield pure product.
Supplementary Material
Detailed experimental procedures, characterization data for new compounds, and X-ray crystallography data for 2. This material is available free of charge via the Internet at http://pubs.acs.org.
ACKNOWLEDGMENTS
We thank the Welch Foundation (Grant No. E-1571), National Institute of General Medical Sciences (Grant No. R01GM077635), A. P. Sloan Foundation, Camille and Henry Dreyfus Foundation, and Norman Hackerman Advanced Research Program for supporting this research. We thank Dr. James Korp for collecting and solving the X-ray structure of 2, and Dr. Ashok Krishnaswami (JEOL USA, Peabody MA) for acquiring a 13C spectrum of 1.
REFERENCES
- 1.Hassan J, Sévignon M, Gozzi C, Schulz E, Lemaire M. Chem. Rev. 2002;102:1359. doi: 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]
- 2.Ullmann F, Bielecki J. Chem. Ber. 1901;34:2174. [Google Scholar]
- 3.Reviews: Suzuki A. Chem. Commun. 2005:4759. doi: 10.1039/b507375h. Nicolaou KC, Bulger PG, Sarlah D. Angew. Chem. Int. Ed. 2005;44:4442. doi: 10.1002/anie.200500368. Miura M. Angew. Chem. Int. Ed. 2004;43:2201. doi: 10.1002/anie.200301753. Stanforth SP. Tetrahedron. 1998;54:263.
- 4.(a) Allred GD, Liebeskind LS. J. Am. Chem. Soc. 1996;118:2748. [Google Scholar]; (b) Thathagar MB, Beckers J, Rothenberg G. J. Am. Chem. Soc. 2002;124:11858. doi: 10.1021/ja027716+. [DOI] [PubMed] [Google Scholar]; (c) Ma D, Liu F. Chem. Commun. 2004:1934. doi: 10.1039/b407090a. [DOI] [PubMed] [Google Scholar]; (d) Kamata K, Yamaguchi S, Kotani M, Yamaguchi K, Mizuno N. Angew. Chem. Int. Ed. 2008;47:2407. doi: 10.1002/anie.200705126. [DOI] [PubMed] [Google Scholar]; (e) Usui S, Hashimoto Y, Morey JV, Wheatley AEH, Uchiyama M. J. Am. Chem. Soc. 2007;129:15102. doi: 10.1021/ja074669i. [DOI] [PubMed] [Google Scholar]
- 5. Klapars A, Antilla JC, Huang X, Buchwald SL. J. Am. Chem. Soc. 2001;123:7727. doi: 10.1021/ja016226z. Campbell MJ, Johnson JS. Org. Lett. 2007;9:1521. doi: 10.1021/ol0702829. Hamada T, Ye X, Stahl SS. J. Am. Chem. Soc. 2008;130:833. doi: 10.1021/ja077406x. Bolshan Y, Batey RA. Angew. Chem. Int. Ed. 2008;47:2109. doi: 10.1002/anie.200704711. Antilla JC, Baskin JM, Barder TE, Buchwald SL. J. Org. Chem. 2004;69:5578. doi: 10.1021/jo049658b. del Amo V, Dubbaka SR, Krasovskiy A, Knochel P. Angew. Chem. Int Ed. 2006;45:7838. doi: 10.1002/anie.200603089. Shafir A, Lichtor PA, Buchwald SL. J. Am. Chem. Soc. 2007;129:3490. doi: 10.1021/ja068926f. Munro-Leighton C, Blue ED, Gunnoe TB. J. Am. Chem. Soc. 2006;128:1446. doi: 10.1021/ja057622a. Taillefer M, Xia N, Ouali A. Angew. Chem. Int. Ed. 2007;46:934. doi: 10.1002/anie.200603173. A review about copper catalysis in C-heteroatom bond formation: Ley SV, Thomas AW. Angew. Chem. Int. Ed. 2003;42:5400. doi: 10.1002/anie.200300594.
- 6.(a) Altman RA, Shafir A, Choi A, Lichtor PA, Buchwald SL. J. Org. Chem. 2008;73:284. doi: 10.1021/jo702024p. [DOI] [PubMed] [Google Scholar]; (b) Cai Q, Zou B, Ma D. Angew. Chem. Int. Ed. 2006;45:1276. doi: 10.1002/anie.200503538. [DOI] [PubMed] [Google Scholar]
- 7.(a) Steinkopf W, Leitsmann R, Hofmann KH. Liebigs Ann. Chem. 1941;546:180. [Google Scholar]; (b) Sease JW, Zechmeister L. J. Am. Chem. Soc. 1947;69:270. [Google Scholar]
- 8.Reviews: Dick AR, Sanford MS. Tetrahedron. 2006;62:2439. Kakiuchi F, Chatani N. Adv. Synth. Catal. 2003;345:1077. Seregin IV, Gevorgyan V. Chem. Soc. Rev. 2007;36:1173. doi: 10.1039/b606984n. Dyker G. Angew. Chem. Int. Ed. 1999;38:1698. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1698::AID-ANIE1698>3.0.CO;2-6. Ritleng V, Sirlin C, Pfeffer M. Chem. Rev. 2002;102:1731. doi: 10.1021/cr0104330. Alberico D, Scott ME, Lautens M. Chem. Rev. 2007;107:174. doi: 10.1021/cr0509760. Ackermann L. Synlett. 2007:507. Campeau L-C, Fagnou K. Chem. Commun. 2006:1253. doi: 10.1039/b515481m. Yu J-Q, Giri R, Chen X. Org. Biomol. Chem. 2006;4:4041. doi: 10.1039/b611094k. Lewis JC, Bergman RG, Ellman JA. Acc. Chem. Res. 2008;41:1013. doi: 10.1021/ar800042p.
- 9.(a) Björklund C, Nilsson M. Acta Chem. Scand. 1968;22:2338. [Google Scholar]; (b) Ljusberg H, Wahren R. Acta Chem. Scand. 1973;27:2717. [Google Scholar]; (c) Nilsson M. Tetrahedron Lett. 1966;7:679. [Google Scholar]; (d) Forrest J. J. Chem. Soc. 1960:574. [Google Scholar]
- 10.(a) Fujita K-i, Nonogawa M, Yamaguchi R. Chem. Commun. 2004:1926. doi: 10.1039/b407116f. [DOI] [PubMed] [Google Scholar]; (b) Fuchita Y, Oka H, Okamura M. Inorg. Chim. Acta. 1992;194:213. [Google Scholar]; (c) Tani M, Sakaguchi S, Ishii Y. J. Org. Chem. 2004;69:1221. doi: 10.1021/jo035568f. [DOI] [PubMed] [Google Scholar]; (d) Jintoku T, Fujiwara Y, Kawata I, Kawauchi T, Taniguchi H. J. Organomet. Chem. 1990;385:297. [Google Scholar]; (e) Ackerman LJ, Sadighi JP, Kurtz DM, Labinger JA, Bercaw JE. Organometallics. 2003;22:3884. [Google Scholar]; (f) Proch S, Kempe R. Angew. Chem. Int. Ed. 2007;46:3135. doi: 10.1002/anie.200604988. [DOI] [PubMed] [Google Scholar]; (g) Hull KL, Sanford MS. J. Am. Chem. Soc. 2007;129:11904. doi: 10.1021/ja074395z. [DOI] [PubMed] [Google Scholar]; (h) Brasche G, Garcia-Fortanet J, Buchwald SL. Org. Lett. 2008;10:2207. doi: 10.1021/ol800619c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Dwight TA, Rue NR, Charyk D, Josselyn R, DeBoef B. Org. Lett. 2007;9:3137. doi: 10.1021/ol071308z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Stuart DR, Fagnou K. Science. 2007;316:1172. doi: 10.1126/science.1141956. [DOI] [PubMed] [Google Scholar]
- 11.(a) Lafrance M, Rowley CN, Woo TK, Fagnou K. J. Am. Chem. Soc. 2006;128:8754. doi: 10.1021/ja062509l. [DOI] [PubMed] [Google Scholar]; (b) Lafrance M, Shore D, Fagnou K. Org. Lett. 2006;8:5097. doi: 10.1021/ol0619967. [DOI] [PubMed] [Google Scholar]
- 12.(a) Brasche G, Buchwald SL. Angew. Chem. Int. Ed. 2008;47:1932. doi: 10.1002/anie.200705420. [DOI] [PubMed] [Google Scholar]; (b) Norinder J, Matsumoto A, Yoshikai N, Nakamura E. J. Am. Chem. Soc. 2008;130:5858. doi: 10.1021/ja800818b. [DOI] [PubMed] [Google Scholar]; (c) Zhang Y, Li C-J. Angew. Chem. Int. Ed. 2006;45:1949. doi: 10.1002/anie.200503255. [DOI] [PubMed] [Google Scholar]; (d) Chen X, Hao X-S, Goodhue CE, Yu J-Q. J. Am. Chem. Soc. 2006;128:6790. doi: 10.1021/ja061715q. [DOI] [PubMed] [Google Scholar]; (e) Uemura T, Imoto S, Chatani N. Chem. Lett. 2006;35:842. [Google Scholar]; (f) Li Z, Cao L, Li C-J. Angew. Chem. Int. Ed. 2007;46:6505. doi: 10.1002/anie.200701782. [DOI] [PubMed] [Google Scholar]; (g) Phipps RJ, Grimster NP, Gaunt MJ. J. Am. Chem. Soc. 2008;130:8172. doi: 10.1021/ja801767s. [DOI] [PubMed] [Google Scholar]
- 13.(a) Do H-Q, Daugulis O. J. Am. Chem. Soc. 2007;129:12404. doi: 10.1021/ja075802+. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Do H-Q, Daugulis O. J. Am. Chem. Soc. 2008;130:1128. doi: 10.1021/ja077862l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Pellissier H, Santelli M. Tetrahedron. 2003;59:701. [Google Scholar]; (b) Liu Z, Larock RC. J. Org. Chem. 2007;72:223. doi: 10.1021/jo0619534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Shen K, Fu Y, Li J-N, Liu L, Guo Q-X. Tetrahedron. 2007;63:1568. [Google Scholar]; (b) Bordwell FG. Acc. Chem. Res. 1988;21:456. [Google Scholar]
- 16.Helm MD, Moore JE, Plant A, Harrity JPA. Angew. Chem. Int. Ed. 2005;44:3889. doi: 10.1002/anie.200500288. [DOI] [PubMed] [Google Scholar]
- 17.(a) Beckwith ALJ, Gara WB. J. Chem. Soc. Perkin Trans. 2. 1975;7:795. [Google Scholar]; (b) Branchi B, Galli C, Gentili P. Eur. J. Org. Chem. 2002:2844. [Google Scholar]
- 18.Tye JW, Weng Z, Johns AM, Incarvito CD, Hartwig JF. J. Am. Chem. Soc. 2008;130:9971. doi: 10.1021/ja076668w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Bondi A. J. Phys. Chem. 1964;68:441. [Google Scholar]; (b) Carvajal MA, Alvarez S, Novoa JJ. Chem. Eur. J. 2004;10:2117. doi: 10.1002/chem.200305249. [DOI] [PubMed] [Google Scholar]
- 20.Sundararaman A, Lalancette RA, Zakharov LN, Rheingold AL, Jäkle F. Organometallics. 2003;22:3526. [Google Scholar]
- 21.Sundararaman A, Zakharov LN, Rheingold AL, Jäkle F. Chem. Commun. 2005:1708. doi: 10.1039/b417532h. [DOI] [PubMed] [Google Scholar]
- 22.Camus A, Marsich N. J. Organomet. Chem. 1970;21:249. [Google Scholar]
- 23.(a) Ishiyama T, Takagi J, Ishida K, Miyaura N, Anastasi NR, Hartwig JF. J. Am. Chem. Soc. 2002;124:390. doi: 10.1021/ja0173019. [DOI] [PubMed] [Google Scholar]; (b) Cho J-Y, Tse MK, Holmes D, Maleczka RE, Jr, Smith MR., III Science. 2002;295:305. doi: 10.1126/science.1067074. [DOI] [PubMed] [Google Scholar]
- 24.(a) Anctil EJ-G, Snieckus V. J. Organomet. Chem. 2002;653:150. [Google Scholar]; (b) Snieckus V. Chem. Rev. 1990;90:879. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed experimental procedures, characterization data for new compounds, and X-ray crystallography data for 2. This material is available free of charge via the Internet at http://pubs.acs.org.