

Abstract
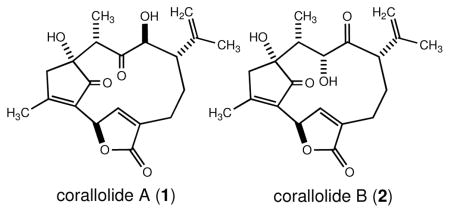
Corallolides A (1) and B (2) are naturally occurring diterpenes isolated from the Caribbean gorgonian octocoral Pseudopterogorgia bipinnata collected near Providencia Island, Colombia. Their tricyclic structures are based on a uniquely substituted bicyclo[9.2.1]tetradecane ring system that was established through detailed spectroscopic analysis. Compounds 1 and 2 were shown to exhibit anti-parasitic and anti-tuberculosis activity, respectively.
For nearly four decades, investigations into the natural products chemistry of Caribbean gorgonian octocorals of the genus Pseudopterogorgia have yielded a plethora of diterpenoids of diverse molecular architectures that endow them with remarkable biological activities.1 The gorgonian species P. bipinnata, P. kallos, P. acerosa, P. americana, and P. elisabethae are particularly noteworthy as they alone appear to account for the production of the vast majority of these fascinating natural products.2 Recently, the groups of Mulzer, Trauner, and Kerr have highlighted the ever increasing number of diterpenoid skeletal classes produced by these chemically prolific Pseudopterogorgia species. Furthermore, many of the metabolites engendered by these animals are highly likely to experience subsequent rearrangement to yield even more perplexing structural variants.3 The chemical investigation of these five octocoral species has led to important pharmacological advances which in turn have spurred novel synthetic methodology and inspired many chemists and biochemists to speculate on the plausible biogenetic relationships among the diverse skeletal families of diterpenes that are routinely co-isolated from them.
As part of our continuing interest in the natural products chemistry of Caribbean gorgonian octocorals and their potential utilization in the development of novel anti-infective and anti-cancer drugs, we have previously reported on several pseudopterane lactones from a Southwestern Caribbean specimen of P. bipinnata.4 From the same specimen, we now report the isolation and structure elucidation of two isomeric diterpenes which feature a uniquely substituted bicyclo[9.2.1]tetradecane ring system. These compounds, which have been trivially named corallolides A (1) and B (2), feature a twelve-membered macrocycle that appears to be linked biosynthetically to the pseudopterane class of diterpenes through additional C–C bond formation leading to an intriguing bridged ring system. Their molecular structures were established by analysis of 1D and 2D NMR, IR, UV, and high-resolution mass spectral data. Biological screening of metabolites 1 and 2 revealed that they are bestowed with anti-tubercular and anti-parasitic activity.
A detailed scheme for the extraction of the dry gorgonian specimen (0.11 kg) has been provided as Supporting Information. The crude CHCl3 extract (4.1 g) was fractionated over silica gel (150 g) with a 99:1 mixture of CHCl3/MeOH to give 22 fractions, denoted I–XXII. Fractions IX and X were combined (126 mg) and chromatographed over silica gel (7 g) using 1% acetone in CHCl3 leading to 7 secondary fractions (A–G). Further purification of sub-fraction D (37 mg) by normal-phase HPLC afforded pure corallolide A (1) (4.5 mg, 4.1 × 10−3% yield) along with corallolide B (2) (5.0 mg, 4.5 × 10−3% yield) (dry gorgonian weight basis).
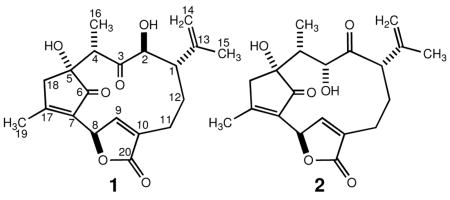
The molecular formula of corallolide A (1)5 was assigned as C20H24O6 on the basis of high-resolution mass measurement of the [M + Na]+ ion at m/z 383.1483 and overall NMR information indicating the presence of nine degrees of unsaturation in the molecule. The IR data for 1 indicated the presence of hydroxyl (3467 cm−1), carbonyl (1756 and 1706 cm−1) and olefin (1628 cm−1) bands. The carbonyl bands at 1756 and 1706 cm−1 showed comparable intensity suggesting the presence in 1 of a conjugated γ-lactone and a 2-cyclopentenone ring, respectively. The latter contention was supported by strong UV absorptions at λmax 214 nm (ε 14 200) and 235 nm (ε 12 300). Although 1 was soluble in CDCl3, the 1H and 13C NMR signals in this solvent were broad and poorly dispersed. Consequently, 1D and 2D NMR data were gathered in CD3OD. The 13C NMR spectrum (75 MHz, CD3OD) of 1 showed 20 resolved resonances (Table 1). Six olefinic [δ 174.0 (C), 153.8 (CH), 146.1 (C), 137.0 (C), 132.6 (C), 114.7 (CH2)] and three carbonyl [δ 212.9 (C), 208.0 (C), 174.8 (C)] resonances in the 13C NMR spectrum accounted for six sites of unsaturation. Therefore, the remaining sites of unsaturation required by the molecular formula had to be explained by the presence of three rings. HMQC and DEPT-135 data showed that 22 of the 24 hydrogen atoms were attached to carbons (3 × CH3, 4 × CH2, 5 × CH); therefore, compound 1 had to have 2 OH groups.
Table 1.
1H NMR (300 MHz), 13C NMR (75 MHz), 1H—1H COSY, NOESY, and HMBC spectral data for corallolide A (1)a
| position | δH, mult, intgt (J, Hz) | δC (mult)b | 1H–1H COSY | NOESY | HMBCc |
|---|---|---|---|---|---|
| 1 | 2.04, br m, 1H | 47.4 (CH) | H2, H12αβ | H4, H14αβ | H2, H14αβ, H3-15 |
| 2 | 3.95, d, 1H (4.8) | 78.4 (CH) | H1 | H9, H12α | |
| 3 | 212.9 (C) | H2, H4, H3-16 | |||
| 4 | 2.68, q, 1H (6.8) | 48.6 (CH) | H3-16 | H1, H14αβ, H3-16, H18β | H3-16, H18β |
| 5 | 83.6 (C) | H4, H3-16, H18β | |||
| 6 | 208.0 (C) | H18β | |||
| 7 | 137.0 (C) | H18β, H3-19 | |||
| 8 | 5.93, br s, 1H | 77.1 (CH) | H9, H18α, H3-19 | H9, H3-19 | H9 |
| 9 | 8.20, br s, 1H | 153.8 (CH) | H8 | H2, H8, H11α, H12α | |
| 10 | 132.6 (C) | H9 | |||
| 11αβ | 2.53, br m, 2H | 22.5 (CH2) | H12αβ | H9 | |
| 12α | 1.71, br m, 1H | 27.4 (CH2) | H1, H11αβ, H12β | H2, H9 | H1 |
| 12β | 2.00, br m, 1H | H1, H11αβ, H12α | |||
| 13 | 146.1 (C) | H2, H3-15 | |||
| 14α | 4.73, br s, 1H | 114.7 (CH2) | H14β, H3-15 | H4, H14β, H3-15 | H3-15 |
| 14β | 4.75, br s, 1H | H14α, H3-15 | H1, H4, H14α | ||
| 15 | 1.60, br s, 3H | 21.1 (CH3) | H14αβ | H14αβ | H14αβ |
| 16 | 1.16, d, 3H (6.8) | 13.2 (CH3) | H4 | H4, H18β | H4 |
| 17 | 174.0 (C) | H18αβ, H3-19 | |||
| 18α | 2.60, br d, 1H (17.4) | 46.7 (CH2) | H8, H18β, H3-19 | H18β | H3-19 |
| 18β | 2.81, br d, 1H (17.4) | H18α | H4, H3-16, H18α | ||
| 19 | 2.29, br s, 3H | 19.1 (CH3) | H8, H18α | H8 | |
| 20 | 174.8 (C) | H9, H11αβ |
Spectra were recorded in CD3OD at 25 °C. Chemical shift values are in parts per million relative to the residual CH3OH (3.30 ppm) or CD3OD (49.0 ppm) signals. Assignments were aided by 2D NMR experiments, spin-splitting patterns, number of attached protons, and chemical shift values.
13C NMR multiplicities were obtained from a DEPT-135 experiment.
Protons correlated to carbon resonances in the 13C column. Parameters were optimized for 2,3JCH = 6 and 8 Hz.
Four partial structures (A–D) were deduced from extensive analyses of the 2D NMR data of 1 including COSY, NOESY, HMQC, and HMBC spectra in CD3OD (Figure 1). The COSY, NOESY, and HMBC correlations for corallolide A (1) are presented in Table 1.
Figure 1.
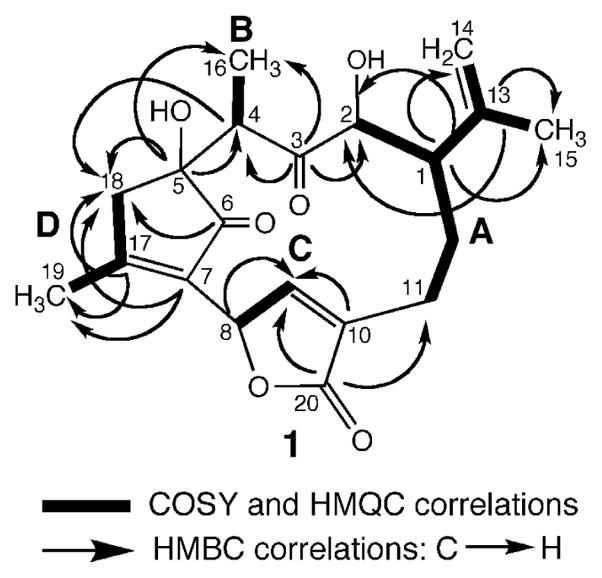
Partial structures for corallolide A (1) generated from 1H–1H COSY, HMQC, and HMBC spectral data.
Connectivities from C11 to C2 in substructure A, including additional connectivities to an isopropylene group attached to C1, were inferred from the 1H–1H COSY cross-peaks (Table 1). HMBC correlations of C1 (δC 47.4) with H2 (δH 3.95), H2-14 (δH 4.73 and 4.75), and H3-15 (δH 1.60), and long-range couplings of C13 (δC 146.1) with H2 and H3-15 allowed assignment of the structure for unit A. Partial structure B, an isolated –CH–CH3 spin system (C4 and C16), was clearly revealed by the 1H NMR, COSY, and HMQC spectra. The chemical shifts of H4 (δH 2.68) and C4 (δC 48.6) (as well as the multiplicities and coupling constants of all the protons) allowed us to further link units A and B through C3 (δC 212.9). This was supported by complementary HMBC correlations between C3 and protons H2, H4, and H3-16.
The presence of a butenolide moiety in the molecule (unit C) was deduced from the COSY and HMBC spectra, the proton and carbon chemical shifts at positions C8–C10 and C20, together with IR and UV data (vide supra). Long-range couplings between H9 (δH 8.20) and C8, C10, and C20 were consistent with partial structure C. Furthermore, substructures A and C were confidently linked by the 3JCH correlations between H2-11 (δH 2.53) and C20 (δC 174.8). Applying these combined NMR methods resulted in the unambiguous assignment of all the protons and carbons in units A–C.
For partial structure D (C5–C7 and C17–C19), connectivities from C17 through C19 were clearly revealed by the 1H–1H COSY and HMQC spectra. It was, however, difficult to obtain unambiguous evidence for connecting H2-18 (an AB system at δH 2.60 and 2.81) to other spin systems. Notwithstanding, the connectivity of unit D was completed by careful analysis of the proton and carbon chemical shifts at positions C5, C6, C7, C17, C18 and C19, and by HMBC correlations for H18β/C5/C6/C7, H18αβ/C17, and H3-19/C7/C17/C18. Thus, unit D was formulated as a 2,3,5,5-tetrasubstituted 2-cyclopentenone moiety, a contention that was in full agreement with the IR and UV spectra recorded for 1. The pivotal 13C NMR resonances at δC 208.0 (C), 174.0 (C), and 137.0 (C) were thus ascribed, respectively, to C6, C17, and C7 of the 2-cyclopentenone ring system. Confirmation of the substitution pattern of the latter system as well as its connectivity to the other ring systems found in 1 came from additional HMBC and TOCSY correlation data. Critically, HMBC cross-peaks between the hydroxyl-bearing quaternary carbon C5 (δC 83.6) and protons H4, H3-16, and H18β allowed the attachment of units B and D through C4 and C5, respectively. At this point, the link between units D and C through C7 and C8, allowed the complete planar structure for 1 to be assigned. Weak but diagnostic homoallylic couplings (5J) between the protons of C18 and C19 with lactone methine H8 confirmed the latter connectivity.6 Interestingly, such linkage, which renders H8 bisallylic (δH 5.93), imposes upon the geometry of 1 in such a manner that H9, which lies close to the C=O bond of unit D, cuts into the deshielding cone of the induced magnetic field causing H9 to be unusually deshielded (δH 8.20). These analyses confidently established the gross structure shown for 1.
The relative configurations (1S*,2S*,4S*,5R*,8R*) for the five stereocenters about the tricyclic framework of corallolide A were assigned primarily on the basis of 1H NMR coupling constants and NOESY data supported by distance calculations (McSpartan’04 Program). As 3JH1-H2 was only 4.8 Hz, a refutable trans-relationship between these protons was proposed.4 This assignment, however, was firmly established by the strong NOESY correlations of H1 (β-orientation in planar conformation) and H4, plus the conspicuous absence of a NOE between H2 and H4 (calcd inter-proton distance = 3.2 Å), thus placing the isopropenyl side chain at C1 and H3-16 in the same α-plane. Synchronous NOESY cross-peaks between H2 and pivotal protons H9 and H12α further supported these assignments. After calculating the conformer distribution of each possible stereomeric structure of 1 at C5 and C8, the coincident NOESY correlations of H2/H9, H4/H18β, H3-16/H18β, and H9/H12α were consistent only with structure 1 (Figure 2cf Supporting Information). Only in this stereoisomer, which also possessed the lowest calculated energy (312 kJmol−1), the intramolecular distances for the latter protons were calculated to be within 2.6–2.0 Å.
The structural characterization of corallolide B (2) was carried out in an analogous manner.7 The HRESIMS and 13C NMR data for 2 were consistent with a molecular formula of C20H24O6. A side-by-side comparison between the NMR spectra of isomers 1 and 2 rapidly pinpointed their structural similarities. Nevertheless, some major differences between these compounds were observed in the 13C NMR spectra: the signals ascribed to C1–C4 and C16 shifted from δC 47.4 (CH), 78.4 (CH), 212.9 (C), 48.6 (CH), and 13.2 (CH3) in corallolide A (1) to 50.5 (CH), 209.8 (C), 73.1 (CH), 41.6 (CH), and 7.1 (CH3) in 2, respectively. Placement in corallolide B of a carbonyl at C2 and a hydroxyl (in the α-orientation) at C3 would account for these spectral differences. Consideration of 1H and 13C NMR data as well as COSY, NOESY, and HMBC data allowed the complete structure to be assigned as 2 and led to the unambiguous assignment of all the protons and carbons as listed in Table 2. The NOEs and the small coupling constant between H3 and H4 (JH3/H4 < 1 Hz), which require a dihedral angle close to 90° (calcd θ = −88.2°), correlated with the lowest energy conformer of a model representing the relative stereochemistry shown in structure 2.
Table 2.
1H NMR (300 MHz), 13C NMR (75 MHz), 1H—1H COSY, NOESY, and HMBC spectral data for corallolide B (2)a
| position | δH, mult, intgt (J, Hz) | δC (mult)b | 1H–1H COSY | NOESY | HMBCc |
|---|---|---|---|---|---|
| 1 | 2.74, br d, 1H (10.4) | 50.5 (CH) | H12β | H3, H4, H14αβ | H14αβ, H3-15 |
| 2 | 209.8 (C) | H1, H3 | |||
| 3 | 4.44, s, 1H | 73.1 (CH) | H1, H4, H9, H12α | H3-16 | |
| 4 | 1.78, br q (6.9) | 41.6 (CH) | H3-16 | H1, H3, H3-16 | H3, H3-16 |
| 5 | 84.3 (C) | H3, H3-16 | |||
| 6 | 208.1 (C) | H18α | |||
| 7 | 135.7 (C) | H18α, H3-19 | |||
| 8 | 5.89, br s, 1H | 76.1 (CH) | H9, H18αβ, H3-19 | H9 | H9 |
| 9 | 8.09, br s, 1H | 150.5 (CH) | H8 | H3, H8, H12αβ | |
| 10 | 133.6 (C) | H9, H11αβ | |||
| 11αβ | 2.42, br m, 2H | 20.8 (CH2) | H12αβ | H1, H12α | |
| 12αβ | 1.54, br m, 1H; 2.17, br m, 1H | 31.6 (CH2) | H1, H11αβ | H3, H9 | H1 |
| 13 | 138.2 (C) | H1, H3-15 | |||
| 14αβ | 5.08, br s, 1H; 4.80, br s, 1H | 115.3 (CH2) | H3-15 | H1, H3-15 | H1, H3-15 |
| 15 | 1.70, br s, 3H | 21.8 (CH3) | H14αβ | H14α | H1, H14αβ |
| 16 | 0.88, d, 3H (6.9) | 7.1 (CH3) | H4 | H4, H18α | H3, H4 |
| 17 | 169.1 (C) | H18α, H3-19 | |||
| 18αβ | 2.64, br d, 1H (17.3); 2.77, br d, 1H (17.3) | 44.3 (CH2) | H8 | H3-16 | H3-19 |
| 19 | 2.33, br s, 3H | 19.4 (CH3) | H8 | ||
| 20 | 174.0 (C)d | H9 |
Spectra were recorded in CDCl3 at 25 °C. Chemical shift values are in parts per million relative to the residual CHCl3 (7.26 ppm) or CDCl3 (77.0 ppm) signals. Assignments were aided by 2D NMR experiments, spin-splitting patterns, number of attached protons, and chemical shift values.
13C NMR multiplicities were obtained from a DEPT-135 experiment.
Protons correlated to carbon resonances in the 13C column. Parameters were optimized for 2,3JCH = 6 and 8 Hz.
Due to its low intensity the chemical shift value of this peak was carefully estimated from HMBC experiments.
The co-ocurrence of compounds 1 and 2 with various pseudopterane lactones4 within P. bipinnata raises the possibility that the corallolides represent a further modification of an existing metabolite, thus suggesting the biogenetic pathway shown in Scheme 1. We suggest the semisystematic name “corallolane” to define this new carbon skeleton and a numbering system that preserves the C1–C20 numbering of the pseudopterane skeleton.8
In our primary in vitro anti-tuberculosis assay (at 128 μg/mL), compound 2 inhibited growth of Mycobacterium tuberculosis by 95%, but was inactive against the malaria parasite Plasmodium falciparum. Compound 1, however, displayed strong antimalarial activity (IC50 = 10 μg/mL).
Supplementary Material
Acknowledgments
Financial support from the NIH-SCORE/RISE Program (Grant No. S06GM08102) is gratefully acknowledged. We thank Dr. Dirk Trauner (CIPSM) for his valuable assistance in preparing Scheme 1B and Dr. Juan A. Sánchez for collecting and identifying the biological material.
Footnotes
Supporting Information Available: Figure 2, Schemes 1A and 1B, copies of NMR spectra of 1 and 2, and experimental details for the extraction of P. bipinnata and the computational methods. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Fenical W. J Nat Prod. 1987;50:1001–1008. doi: 10.1021/np50054a001. [DOI] [PubMed] [Google Scholar]; (b) Rodríguez AD. Tetrahedron. 1995;51:4571–4618. doi: 10.1016/0040-4020(95)00216-U. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blunt JW, Copp BR, Hu WP, Munro MHG, Northcote PT, Prinsep MR. Nat Prod Rep. 2009;26:170–244. doi: 10.1039/b805113p. and previous articles in this series. [DOI] [PubMed] [Google Scholar]
- 3.(a) Heckrodt TJ, Mulzer J. Top Curr Chem. 2005;244:1–41. [Google Scholar]; (b) Roethle PA, Trauner D. Nat Prod Rep. 2008;25:298–317. doi: 10.1039/b705660p. [DOI] [PubMed] [Google Scholar]; (c) Berrue F, Kerr RG. Nat Prod Rep. 2009;26:681–710. doi: 10.1039/b821918b. [DOI] [PubMed] [Google Scholar]
- 4.(a) Marrero J, Ospina CA, Rodríguez AD, Baran P, Zhao H, Franzblau SG, Ortega-Barria E. Tetrahedron. 2006;62:6998–7008. [Google Scholar]; (b) Ospina CA, Rodríguez AD, Zhao H, Raptis RG. Tetrahedron Lett. 2007;48:7520–7523. [Google Scholar]
- 5.Corallolide A (1): colorless oil; [α]20D–12 (c 0.5, CHCl3); IR (neat) νmax 3467, 3098, 3077, 2941, 1756, 1706, 1628, 1442, 1379, 1332, 1111, 1068, 1039, 752 cm−1; UV (MeOH) λmax 214 (ε 14 200), 235 (ε 12 300), 278 (ε 1900) nm; HRESIMS m/z [M + Na]+ 383.1483 (calcd for C20H24O6Na, 383.1471).
- 6.In some alkenes coupling can occur between the C–H σ bonds on either side of the double bond. This type of coupling is generally very small or even nonexistent in most molecules, but it sometimes appears in NMR spectra; see: Jackman LM, Sternhell S. Applications of Nuclear Magnetic Resonance Spectroscopy in Organic Chemistry. 2. Pergamon Press; New York: 1969. pp. 316–328.
- 7.Corallolide B (2): colorless oil; [α]20D–98 (c 0.5, CHCl3); IR (neat) νmax 3458, 3084, 2929, 1756, 1707, 1629, 1442, 1379, 1336, 1204, 1070, 758 cm−1; UV (MeOH) λmax 208 (ε 8100), 234 (ε 6400), 279 (ε 900) nm; HRESIMS m/z [M + Na]+ 383.1483 (calcd for C20H24O6Na, 383.1471).
- 8.Erythrolide K, a diterpene isolated from the gorgonian coral Erythropodium caribaeorum, is based on a bicyclo[9.2.1]tetradecane ring system. Notwithstanding, the alkylation/substitution pattern about the corallolane skeleton is unprecedented in Nature; see: Banjoo D, Maxwell AR, Mootoo BS, Lough AJ, McLean S, Reynolds WF. Tetrahedron Lett. 1998;39:1469–1472.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.