

Abstract
Streptokinase (SK) conformationally activates the central zymogen of the fibrinolytic system, plasminogen (Pg). The SK·Pg* catalytic complex binds Pg as a specific substrate and cleaves it into plasmin (Pm), which binds SK to form the SK·Pm complex that propagates Pm generation. Catalytic complex formation is dependent on lysine-binding site (LBS) interactions between a Pg/Pm kringle and the SK COOH-terminal Lys414. Pg substrate recognition is also LBS-dependent, but the kringle and SK structural element(s) responsible have not been identified. SK mutants lacking Lys414 with Ala substitutions of charged residues in the SK β-domain 250-loop were evaluated in kinetic studies that resolved conformational and proteolytic Pg activation. Activation of [Lys]Pg and mini-Pg (containing only kringle 5 of Pg) by SK with Ala substitutions of Arg253, Lys256, and Lys257 showed decreases in the bimolecular rate constant for Pm generation, with nearly total inhibition for the SK Lys256/Lys257 double mutant. Binding of bovine Pg (BPg) to the SK·Pm complex containing fluorescently labeled Pm demonstrated LBS-dependent assembly of a SK·labeled Pm·BPg ternary complex, whereas BPg did not bind to the complex containing the SK Lys256/Lys257 mutant. BPg was activated by SK·Pm with a Km indistinguishable from the KD for BPg binding to form the ternary complex, whereas the SK Lys256/Lys257 mutant did not support BPg activation. We conclude that SK residues Arg253, Lys256, and Lys257 mediate Pg substrate recognition through kringle 5 of the [Lys]Pg and mini-Pg substrates. A molecular model of the SK·kringle 5 complex identifies the putative interactions involved in LBS-dependent Pg substrate recognition.
Streptokinase (SK)6 activates the human fibrinolytic system by activating plasminogen (Pg) through a unique mechanism that is responsible for the use of SK as a thrombolytic drug and its role as a key pathogenicity factor in Group A streptococcal infection (1, 2). The crystal structure of SK bound to the catalytic domain of plasmin (μPm) shows that SK consists of three β-grasp, tightly folded domains, α, β, and γ, linked by flexible segments (3). In solution, SK is highly flexible and behaves hydrodynamically like three beads on a string (4). When bound to μPm, SK assumes a highly ordered structure resembling a three-sided crater surrounding the catalytic site that provides an exosite(s) for binding the catalytic domain of Pg as a substrate (3, 5). In the first step of the SK-mediated Pg activation pathway, SK binds the catalytic domain of the Pg zymogen in a rapid equilibrium process and inserts its NH2-terminal Ile1 residue into the NH2-terminal binding cleft of Pg, activating the catalytic site nonproteolytically (6–10). Although structural proof is lacking, SK Ile1 presumably forms a critical salt bridge with Asp740(194) (plasminogen numbering; chymotrypsinogen numbering is in parentheses) that initiates conformational activation of the substrate binding site and oxyanion hole required for proteolytic activity (6, 8–10). The activated SK·Pg* complex binds a second molecule of Pg as a specific substrate and cleaves it at Arg561(15)-Val562(16) to form the fibrin-degrading proteinase, plasmin (Pm) (10–14). Proteolytic generation of Pm is propagated by formation of a high affinity SK·Pm complex that converts the remaining free Pg into Pm (5, 11).
[Glu]Pg, the full-length form of Pg circulating in blood, consists of an NH2-terminal PAN (Pg/Apple/Nematode (15, 16)) module, followed by five kringle domains (K1–K5), and the trypsin-like serine proteinase catalytic domain (17). Formation of the SK·Pg* and SK·Pm catalytic complexes and Pg substrate binding are inhibited by the lysine analog, 6-aminohexanoic acid (6-AHA), which binds to lysine-binding sites (LBS) located primarily in kringles K1, K4, and K5 of Pg and Pm (10, 11, 18–23). Cleavage of the Lys77-Lys78 peptide bond in [Glu]Pg by Pm releases the PAN module and generates the truncated form, [Lys]Pg. Formation of [Lys]Pg is accompanied by a conformational change of [Glu]Pg from a compact, closed α-conformation to a partially extended β-conformation with expression of higher affinity LBS for 6-AHA (24, 25). The fourth kringle module mediates a second conformational change, from the β-conformation to the extended γ-conformation (25).
Binding of SK to [Glu]Pg is independent of LBS, with a dissociation constant of 100–150 nm, whereas formation of SK·[Lys]Pg is LBS-dependent with a 13–20-fold higher affinity that is reduced to that of [Glu]Pg by saturating concentrations of 6-AHA (10, 21). Activation of the catalytic domain in [Lys]Pm increases affinity for SK about 830-fold, which is reduced 11–20-fold by 6-AHA (5, 21). Interaction of the COOH-terminal Lys414 residue of SK with a Pg/Pm kringle domain is responsible for the LBS-dependent enhancement of the affinity of SK·[Lys]Pg* and SK·Pm catalytic complex formation (22). Recent rapid reaction kinetic studies of the SK·Pm binding pathway demonstrated that interaction of Lys414 with a Pm kringle enhances formation of an initial rapid equilibrium SK·Pm encounter complex, succeeded by two sequential, tightening conformational changes, to achieve an overall dissociation constant of ∼12 pm (26). The Pg/Pm kringle domain responsible for the enhancement of SK·Pg* and SK·Pm complex formation is not known. Productive interaction of Pg as a substrate of the SK·Pg*/Pm complexes is also greatly inhibited by saturating 6-AHA (11). Kinetic and equilibrium binding studies of SK-mediated Pm formation resolved the conformational activation process from the coupled proteolytic generation of Pm (10, 11). The kinetic approach demonstrated that Lys414 deletion reduced the affinity of formation of the SK·Pg* catalytic complex specifically, whereas the subsequent LBS-dependent proteolytic formation of Pm was unaffected, indicating that Pg substrate recognition is mediated by a structurally distinct region of SK and an unknown kringle (22).
Previous structure-function studies have yielded diverse interpretations and conclusions regarding the structural basis of LBS-dependent Pg substrate recognition (23, 27–34). Each of the three domains of SK has been implicated in this regard (29, 30, 35, 36), and binding of two Pg molecules to the residue 1–59 sequence of the α-domain has been reported (36). In particular, segments 16–36, 41–48, 48–59, and 88–97 of the SK α-domain have been concluded to play a role in Pg substrate recognition (32, 33, 37, 38). For several SK mutants, a complex mixture of functional effects on their binding to [Glu]Pg and its conformational and proteolytic activation has been reported (28, 31, 33). Some of these effects may result from the inherent flexibility of SK when bound to Pg or Pm (39), and others may be due to the use of kinetic approaches that do not clearly discriminate between conformational and proteolytic activation.
Some observations implicate a protruding hairpin loop called the 250-loop (residues Ala251–Ile264) in the SK β-domain in Pg substrate recognition (27, 28, 31, 34). This loop is disordered in the structure of the SK·μPm complex but is ordered in the structure of the isolated β-domain (3, 40). Deletion of the 250-loop, Ala substitution of Lys256 and Lys257 at the apex of the loop, and substitution of multiple residues near and within the loop resulted in disparate effects on Km and kcat for [Glu]Pg activation (27, 28, 31). The conclusions of these studies were that Lys256 and Lys257 are involved in SK binding and conformational activation of [Glu]Pg in addition to proteolytic processing of Pg as a substrate. Some of these studies are problematic because the natural NH2-terminal Ile1 residue necessary for conformational activation is preceded either by an additional methionine (27, 31) or maltose-binding protein (28) in the recombinant SK species used.
Because of the diverse conclusions regarding the functional properties of the 250-loop mutations and the possibility of other potential Pg substrate binding sites, the present studies were undertaken to resolve the function of residues in the 250-loop in LBS-dependent Pg substrate recognition by the SK·Pg* complex. The kringle domain of Pg involved in Pg substrate recognition has not been clearly identified but has been suggested to be K5 (27) on the basis that the isolated β-domain bound Pg (30) and K5 (29) in an LBS-dependent manner. Given the general specificity of Pg kringles for COOH-terminal Lys residues and zwitterionic ligands, such as 6-AHA, and the internal sequence of the 250-loop, it appeared possible that a pseudolysine motif on SK was involved. In the binding of a 30-residue peptide from plasminogen binding Group A streptococcal M-like protein (PAM), VEK-30, to K2 of Pg, Castellino and co-workers (41, 42) showed by crystallography and mutagenesis that residues with cationic (Arg and His) and anionic side chains (Glu) arranged spatially on a helix constituted a pseudolysine structure similar to 6-AHA that binds specifically to the LBS of K2. Additional evidence for pseudolysine structures in Pg binding comes from studies of α-enolase from Streptococcus pneumoniae, which has a 9-residue internal binding site for Pg containing essential basic (two Lys residues) and acidic (Asp and Glu residues) located on a surface loop (43, 44).
To determine whether a similar SK structure is involved in [Lys]Pg substrate recognition, anionic and cationic residues in the 250-loop were substituted with Ala and characterized in kinetic studies using methods that resolve conformational and proteolytic activation. Studies with [Lys]Pg and mini-Pg, which contains only K5 and the catalytic domain, showed that Arg253, Lys256, and Lys257 facilitate LBS-dependent substrate recognition through interactions with K5. The absence of evidence for a pseudolysine structure in the 250-loop is compatible with the established atypical specificity of K5 for cationic ligands, such as benzamidine, Nα-acetyl-Lys-methyl ester, 6-aminohexane, and 5-aminopentane, in addition to zwitterionic ligands (19, 45–47). The studies resolve for the first time the structural features of SK that mediate the LBS-dependent interactions that enhance affinity of SK·Pg* and SK·Pm catalytic complex formation and those that facilitate binding of Pg as a substrate of these complexes.
EXPERIMENTAL PROCEDURES
Protein Purification and Characterization
Human [Glu]Pg carbohydrate form 2 ([Glu]Pg2) was purified from plasma by published procedures (10, 18, 21, 48–50). Activation of 10 μm [Glu]Pg2 to [Lys]Pm2 with 90 units/ml urokinase (Calbiochem) was performed in 10 mm MES, 10 mm HEPES, 0.15 m NaCl, 20 mm 6-AHA, and 1 mg/ml polyethylene glycol (PEG) 8000 at pH 7.4 and 25 °C (5). Native SK (Diapharma) was obtained as outdated therapeutic material and purified as described (18). Bovine Pg (BPg) was purchased from Hematologic Technologies. Mini-Pg was purified from a limited digestion of [Glu]Pg2 with porcine pancreatic elastase (Sigma) by a modification of previous procedures (50, 51). [Glu]Pg2 (30 mg) was diluted to a final concentration of 30 μm in 12 ml of 50 mm HEPES, 125 mm NaCl, 1 mm EDTA, pH 7.4. Elastase and bovine pancreatic trypsin inhibitor (Sigma) were added to final concentrations of 6 μm and 0.1 mg/ml, respectively. Digestion was performed at 25 °C for 60 min before inactivation of elastase by the addition of 120 μm N-(methoxysuccinyl)-Ala-Ala-Pro-Val-CH2Cl (Calbiochem) and further incubation for 45 min. d-Phe-Phe-Arg-CH2Cl (FFR-CH2Cl) and d-Phe-Pro-Arg-CH2Cl (FPR-CH2Cl) were added to final concentrations of 1 μm and phenylmethylsufonyl fluoride to 1 mm. The digest was concentrated to ∼10 mg/ml by YM-10 ultrafiltration and dialyzed at 4 °C against 8 liters of lysine-Sepharose equilibration buffer (150 mm NaH2PO4, 3 mm EDTA, 0.1 μm FFR-CH2Cl and FPR-CH2Cl, 0.02% NaN3, pH 7.4). Mini-Pg appeared in the flow-through of a lysine-Sepharose column (30 ml) at 22 °C, with subsequent elution of kringles 1–4 with 3 mm 6-AHA. Mini-Pg was dialyzed at 4 °C against 8 liters of 20 mm MES, 25 mm NaCl, 0.02% NaN3, pH 6.0, and chromatographed on Resource S (1 ml) (GE Healthcare) in the above buffer. Mini-Pg was pooled from the major peak eluted at ∼110 mm NaCl from the column developed with a 50-ml gradient up to 110 mm NaCl in the equilibration buffer. Purified mini-Pg was dialyzed against three changes of 4 liters of 50 mm HEPES, 125 mm NaCl, pH 7.4.
Cloning, Expression, and Purification of Wild-type SK and SK Mutants
Wild-type SK (wtSK) and SKΔK414 constructs were cloned into the pET-30b(+) vector as fusion proteins with a tobacco etch virus proteinase cleavage site, as described previously (22). Deletion of the Arg253–Leu260 segment and Ala substitutions for Glu249, Arg253, Lys256, Lys257, Glu262, and Glu263 in the β-domain of SK were performed on the SKΔK414 background. Deletion of Arg253–Leu260 was performed by QuikChange (Stratagene) mutagenesis with the sense strand primer 5′-CGGGAACAAGCTTATAATGAAGAAATAAACAACACTGACCTG-3′ and antisense primer 5′-CAGGTCAGTGTTGTTTATTTCTTCATTATAAGCTTGTTCCCG-3′. Confirmed mutants were transformed in Escherichia coli Rosetta 2 (DE3) pLysS cells, and the fusion proteins were expressed and purified by Ni2+-iminodiacetic acid-Sepharose chromatography, cleaved with tobacco etch virus protease, and subjected to chromatography on Ni2+-iminodiacetic acid-Sepharose to remove tobacco etch virus protease and noncleaved protein, as described previously (22). When necessary to remove minor impurities, SK mutants were chromatographed on Q-Sepharose (21 ml) equilibrated with 50 mm HEPES, 50 mm NaCl, 1 mm EDTA, pH 7.4, and eluted with a 400-ml gradient of 50–500 mm NaCl in the equilibration buffer.
Met-SK(K256A/K257A)
The construct for this mutant was cloned into the pET-3 vector and expressed in BL21 (DE3) pLysS cells (Novagen). The cells were grown to an optical density of 0.5 at 600 nm and induced by 0.8 mm isopropyl-β-d-thiogalactopyranoside for 4 h at 37 °C. Lysozyme (Sigma) was added to a final concentration of 1 mg/ml, and cells were lysed by three cycles of freeze-thawing followed by sonication to reduce DNA-associated viscosity. The lysate was centrifuged at 43,000 × g for 30 min, and the supernatant was loaded onto a Pm-SulfoLink column (16 ml) (18, 48) and washed with 0.1 m HEPES, 0.5 m NaCl, 1 mm EDTA, 0.02% NaN3, pH 7.4, to remove impurities. Bound Met-SK(K256A/K257A) was eluted with 0.1 m HEPES, 0.5 m NaCl, 3 m NaSCN, 1 mm EDTA, 0.02% NaN3, pH 7.4. Additional purification was done by fast protein liquid chromatography on a 1-ml Resource Q (GE Healthcare) column equilibrated with 50 mm HEPES, 50 mm NaCl, pH 7.4. Met-SK(K256A/K257A) was eluted with a 50–1000 mm NaCl gradient in the equilibration buffer.
Active Site Labeling of Pm
The active site specific inhibitor, Nα-[(acetylthio)acetyl]-(d-Phe)-Pro-Arg-CH2Cl, prepared by published procedures (52–54), was incubated at a 10–15-fold molar excess with 10 μm [Lys]Pm2 in 0.1 m HEPES, 0.3 m NaCl, 1 mm EDTA, 10 mm 6-AHA, 1 mg/ml PEG, pH 7.0, at 25 °C for 60 min. Inactivation was monitored from the initial rate of VLK-pNA hydrolysis until complete (<0.1% active). Excess inhibitor was removed by chromatography on a 27-ml column of Sephadex G25 equilibrated with the above pH 7.0 buffer. Na-[(acetylthio)acetyl]-(d-Phe)-Pro-Arg-Pm was labeled with tetramethylrhodamine-5-iodoacetamide (Molecular Probes) in 10-fold molar excess initiated by the subsequent addition of 1 m NH2OH to a final concentration of 0.1 m and incubation in the dark at 25 °C for 1 h. Excess probe was removed by chromatography on Sephadex G25 (39 ml) equilibrated with 50 mm HEPES, 125 mm NaCl, 5 mm 6-AHA, 1 mm EDTA, pH 7.4, and dialysis against the same buffer. Probe incorporation and protein concentration were calculated from the absorbance at 280 nm and the probe absorbance in 6 m guanidine buffer as described (52), with an absorption coefficient of 90,527 m−1 cm−1 at 557 nm and an absorbance ratio A280 nm/A557 nm of 0.215. Probe incorporation was 0.9 mol of probe/mol of Pm.
Proteins were homogeneous by SDS gel electrophoresis. All proteins were dialyzed against 50 mm HEPES, 125 mm NaCl, pH 7.4, quick frozen in a dry ice/2-propanol bath and stored at −70 °C, except Pm, for which the dialysis buffer contained 5 mm 6-AHA. Protein concentrations were determined by absorbance at 280 nm using the following absorption coefficients ((mg/ml)−1cm−1) and molecular masses (Da): [Glu]Pg, 1.69 and 92,000; [Lys]Pg, 1.69 and 84,000; [Lys]Pm, 1.9 and 84,000 (50, 55, 56); bovine Pg, 1.7 and 88,000 (57); mini-Pg, 1.6 and 38,000 (51); SK and SK mutants, 0.81 and 47,000 (58, 59). The active Pm concentration was determined from the initial rate of hydrolysis of 200 μm VLK-pNA at 405 nm and 25 °C, using the kinetic constants determined previously (5).
Plasminogen Activation Kinetics
Continuous assays of conformational and proteolytic activation of [Lys]Pg by SK species were performed as described previously (10, 11, 22). Parabolic progress curves of hydrolysis of 200 μm VLK-pNA were obtained in the presence of 15 nm [Lys]Pg as a function of SK concentration.
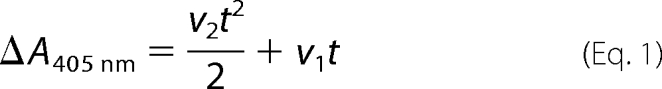 |
The first order term in the second order polynomial (Equation 1) fit to the progress curves represents the initial rate (v1) of VLK-pNA hydrolysis by the conformationally activated SK·Pg* complex, and the rate of the activity increase (v2) obtained from the second order term reflects the initial rate of Pm generation. The kinetics were analyzed based on the simplified mechanism (Reactions 1 and 2), which assumes a negligible contribution from Pm generated by the SK·Pm complex (10, 11).
 |
KA and KA′ represent the dissociation constants for the SK·Pg* and SK·Pm complexes, respectively, and kPg is the bimolecular rate constant (kcat/Km) for proteolytic generation of Pm by SK·Pg*. Under the conditions of the experiments, where the concentration of free [Lys]Pg ([Pg]free) was much less than Km (270 nm) for formation of the productive ternary SK·Pg*·Pg complex, generation of Pm can be represented as a bimolecular reaction (11). Under initial velocity conditions, the product is assumed to be SK·Pm, because this complex is formed with a dissociation constant, KA′, of 12 pm, much lower than [SK]o and [Pg]free (5). When [SK]o is >[Pg]o, all of the product is SK·Pm, whereas when [SK]o is <[Pg]o, all of the SK binds to Pm as the reaction progresses. v1 is given by Equation 4, where kcat and Km are the kinetic constants for VLK-pNA hydrolysis by the SK·Pg* complex (10).
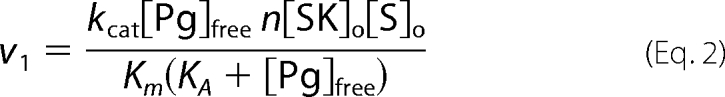 |
For analysis of v1 as a function of SK species concentration, Km was fixed at its determined value of 3000 μm (10), and KA and kcat were fitted parameters. The stoichiometric factor (n) was also fit for tight binding of wild-type and native SK and otherwise fixed at 1. v2 is given by Equation 3, where KI is the empirically determined competitive inhibition constant (330 ± 80 μm) of the bimolecular generation of Pm by the chromogenic substrate (11). [Pg]free is given by the quadratic Equation 4.
 |
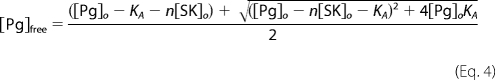 |
For analysis of v2, KI, and KA′ were fixed at their previously determined values (5, 11), and kPg and KA were the fitted parameters.
Kinetics of BPg Activation by the SK·Pm Complex
The SK·Pm complex was formed by incubation of 0.1 nm Pm with 1.2 nm native SK or Met-SK(K256A/K257A) for 12 min before initiation of the reactions by the addition of 200 μm VLK-pNA and BPg. The rate of Pm formation as a function of BPg concentration, obtained from the parabolic progress curves as described above, was fit by the Michaelis-Menten equation to obtain kcat and Km.
Fluorescence Equilibrium Binding
Fluorescence was measured with an SLM 8100 spectrofluorometer in the ratio mode, using PEG 20,000-coated acrylic cuvettes. Experiments were performed in 50 mm HEPES, 125 mm NaCl, 1 mm EDTA, 1 mg/ml PEG 8000, 1 mg/ml bovine serum albumin, 10 μm FFR-CH2Cl in the presence or absence of 40 mm 6-AHA, as previously described (5, 10, 11, 21, 22, 39). [TMR]FPR-Pm (35 nm) titrations were performed with an excitation wavelength of 556 nm (8 nm band pass) and emission wavelength of 576 nm (16 nm band pass). Photon counting for 2.1 nm [TMR]FPR-Pm titrations was performed at the same wavelengths (16 nm band pass). All titrations were corrected for dilution (<10%) and for background using a buffer blank. BPg titrations of complexes of [TMR]FPR-Pm with native SK and Met-SK(K256A/K257A) in the absence and presence of 6-AHA were also corrected for small (≤1%/μm) nonspecific fluorescence changes measured in titrations of [TMR]FPR-Pm in the absence of SK. Fluorescence changes (Fobs − Fo)/Fo = ΔF/Fo, were measured as a function of total SK species concentration and fit by the quadratic binding equation, with the maximum fluorescence change ((Fmax − Fo)/Fo = ΔFmax/Fo), dissociation constant (KD), and stoichiometric factor (n) the fitted parameters for tight binding. For weaker binding, n was fixed at 1. Nonlinear least-squares fitting was performed with SCIENTIST (MicroMath). Error estimates represent the 95% confidence interval.
Molecular Modeling
X-ray coordinates for the isolated SK β-domain (Protein Data Bank entry 1QQR)7 were superimposed on the structure of the SK·μPm complex (1BML) (3). The 250-loop was reconstructed from the superimposed structure of the free β-domain, and the resulting model was minimized. The coordinates for human Pg K5 were taken from PDB entry 5HPG (47). Docking of the kringle module to the SK·μPm model with a restored 250-loop was performed with ZDOCK (60). Structure figures were generated with PyMOL (68).
RESULTS
To evaluate the possibility that the 250-loop of the SK β-domain contained a pseudolysine motif, basic (Arg253, Lys256, and Lys257) and acidic residues (Glu249, Glu262, and Glu263) were substituted with Ala. The mutants were made on the backbone of SKΔK414 to eliminate the interaction of Lys414 with a Pg kringle that enhances formation of the SK·Pg* and SK·Pm catalytic complexes (22). Purified SK mutants were evaluated in kinetic studies of [Lys]Pg and mini-Pg activation measured in continuous assays collected as a function of SK concentration. Fitting of the parabolic rate equation (Equation 1) to the progress curves yielded v1 from the first order term as described under “Experimental Procedures.” Analysis of the hyperbolic dependence of v1 on SK concentration gave the dissociation constant for the conformationally activated SK·Pg* complex (KA) and the specificity constant (kcat/Km) for chromogenic substrate hydrolysis by the complex (Equation 2). The rate of Pm formation was obtained from the second order term (v2), the SK dependence of which increases at approximate concentration ratios <0.5[SK]o/[[Lys]Pg]o and approaches zero at concentration ratios >0.5[SK]o/[[Lys]Pg]o (Equation 3). This is due to Pg acting both as the catalyst in the SK·Pg* complex and as the substrate of this complex, resulting in inhibition of the rate at SK concentrations higher than the optimum due to depletion of free Pg (11). Analysis of v2 yielded a second estimate of KA because of the coupling of the conformational and proteolytic activation reactions and the bimolecular rate constant for proteolytic generation of Pm (kPg) (11). Values of KA derived from v2 are typically smaller and less well determined than those from v1 because of the contribution of the SK·Pm catalytic complex to Pm generation, which is not included in the simplified mechanism (Reactions 1 and 2) (11). Only values of KA from v1 are reported for this reason.
Kinetics of [Lys]Pg Activation by SK and SK Mutants
Results of the kinetic analysis for [Lys]Pg activation by native SK and wtSK and those for SK mutants that showed significant decreases in kPg, including the loop deletion mutant SKΔ(R253-L260), as well as SK(R253A), SK(K256A), SK(K257A), and SK(K256A/K257A) are shown in Fig. 1. The kinetic parameters for all of the mutants are listed in Table 1. The kinetic studies were done in the absence and presence of 10 mm 6-AHA to identify LBS-dependent reactions (Fig. 1 and Table 1). wtSK and native SK gave the same results, with KA (v1) values of 2 ± 2 and 1.4 ± 0.8 nm, respectively, that were weakened 18- and 28-fold, respectively, by 6-AHA. kPg in the absence of 6-AHA was reduced to 1–3% of the wild-type and native SK values in the presence of the lysine analog. There is some bias in KA (v1) when reactions are performed at Pg concentrations greater than 5–10 nm, as done in these experiments (15 nm [Lys]Pg), which results in slightly lower KA values. The dissociation constant for SK·Pg* measured from equilibrium binding studies is 6 ± 2 nm in the absence of 6-AHA and 115 ± 32 nm in 10 mm 6-AHA (10), whereas the kinetic values were ∼2 and ∼40 nm, both ∼3-fold lower (Table 1). Repeating the kinetic analysis of wtSK and native SK at 5 nm [Lys]Pg gave KA(v1) values of 5 ± 1 and 5 ± 7 nm, respectively (not shown). This presumably also reflects a limitation of the simplified mechanism. On the basis that these differences were fairly small for such tight binding, all of the experiments were conducted at 15 nm [Lys]Pg to maintain internal consistency. Reliable values of kPg from v2 could not be obtained in the presence of 10 mm 6-AHA or for SKΔ(R253-L260) or SK(K256A/K257A), because these were essentially zero, reflecting 1–3% residual Pg activator activity relative to wtSK (Fig. 1 and Table 1).
FIGURE 1.

Kinetics of [Lys]Pg activation by SK and SK mutants. A and B, v1 as a function of the total SK or SK mutant concentration ([SK]o) and v2 as a function of log total SK or SK mutant concentration ([SK]o), respectively, for wtSK in the absence (●) and presence of 10 mm 6-AHA (○), native SK in the absence (▴) and presence of 10 mm 6-AHA (▵), and SKΔ414 in the absence (■) and presence of 10 mm 6-AHA (□). C and D, v1 and v2, respectively, as in A for SKΔ(R253-L260) in the absence (●) and presence of 10 mm 6-AHA (○) and SK(R253A) in the absence (▴) and presence of 10 mm 6-AHA (▵). E and F, v1 and v2, respectively, as in A for SK(K256A) in the absence (●) and presence of 10 mm 6-AHA (○), SK(K257A) in the absence (▴) and presence of 10 mm 6-AHA (▵), and SK(K256A/K257A) in the absence (■) and presence of 10 mm 6-AHA (□). Solid lines, nonlinear least-squares fits to the data with Equations 2 and 4 for v1 and Equations 3 and 4 for v2 with the parameters listed in Table 1. Kinetic data were collected and analyzed as described under ”Experimental Procedures.“
TABLE 1.
Kinetic parameters determined for conformational and proteolytic [Lys]Pg and mini-Pg activation
Values of KA(v1) and kcat/Km(v1) (for VLK-pNA hydrolysis by the SK·Pg* complexes) derived from analysis of v1, and kPg(v2) from analysis of v2 are listed for wtSK, native SK, SKΔK414, the indicated SK deletion mutant, and Ala substitution mutants. All of the mutants were made on the background of SKΔK414. The parameters determined for [Lys]Pg and mini-Pg activation are listed for reactions in the absence of 6-AHA (No 6-AHA) and presence of 10 mm 6-AHA (10 mm 6-AHA). Experimental error represents ±2 S.D. NM, not measurable. Kinetics experiments were performed, and the progress curves were analyzed as described under “Experimental Procedures.”
| No 6-AHA |
10 mm 6-AHA |
||||
|---|---|---|---|---|---|
| KA (v1) | kcat/Km (v1) | kPg (v2) | KA (v1) | kcat/Km (v1) | |
| nm | nm−1s−1 | nm−1s−1 | nm | nm−1s−1 | |
| [Lys]Pg | |||||
| Wild-type SK | 2 ± 2 | 12.6 ± 0.3 | 780 ± 20 | 39 ± 6 | 12.5 ± 0.5 |
| Native SK | 1.4 ± 0.8 | 12.8 ± 0.4 | 880 ± 60 | 40 ± 5 | 12.2 ± 0.3 |
| SKΔK414 | 22 ± 7 | 13 ± 1 | 920 ± 60 | 38 ± 6 | 12.2 ± 0.5 |
| SKΔK414Δ(R253-L260) | 22 ± 5 | 13.6 ± 0.4 | NM | 54 ± 13 | 11.8 ± 0.4 |
| SKΔK414(E249A) | 21 ± 8 | 12.2 ± 0.9 | 1030 ± 90 | 58 ± 15 | 11.5 ± 0.8 |
| SKΔK414(R253A) | 27 ± 7 | 12.7 ± 0.7 | 370 ± 30 | 27 ± 7 | 11.3 ± 0.6 |
| SKΔK414(K256A) | 29 ± 5 | 12.3 ± 0.5 | 180 ± 10 | 39 ± 4 | 12.0 ± 0.3 |
| SKΔK414(K257A) | 32 ± 7 | 12.2 ± 0.6 | 390 ± 20 | 53 ± 7 | 11.5 ± 0.4 |
| SKΔK414(K256A/K257A) | 28 ± 5 | 13.8 ± 0.5 | NM | 54 ± 6 | 12.9 ± 0.4 |
| SKΔK414(E262A) | 22 ± 8 | 17.9 ± 1.5 | 1570 ± 70 | 37 ± 7 | 16.8 ± 0.9 |
| SKΔK414(E262A/E263A) | 23 ± 11 | 13.7 ± 1.3 | 1370 ± 50 | 58 ± 6 | 12.9 ± 0.3 |
| Mini-Pg | |||||
| Wild-type SK | 40 ± 13 | 16 ± 1 | 1110 ± 80 | 38 ± 4 | 14.5 ± 0.4 |
| Native SK | 52 ± 21 | 16 ± 1 | 1220 ± 160 | 47 ± 3 | 13.8 ± 0.3 |
| SKΔK414 | 36 ± 12 | 15 ± 1 | 1070 ± 30 | 33 ± 2 | 14.6 ± 0.2 |
| SKΔK414Δ(R253-L260) | 42 ± 4 | 13.8 ± 0.4 | NM | 38 ± 7 | 12.4 ± 0.6 |
| SKΔK414(E249A) | 50 ± 19 | 13 ± 1 | 960 ± 40 | 55 ± 26 | 10 ± 1 |
| SKΔK414(R253A) | 29 ± 7 | 13.1 ± 0.7 | 280 ± 10 | 27 ± 3 | 14.1 ± 0.3 |
| SKΔK414(K256A) | 24 ± 5 | 15.3 ± 0.8 | 160 ± 10 | 32 ± 5 | 16.3 ± 0.7 |
| SKΔK414(K257A) | 45 ± 17 | 15 ± 1 | 260 ± 10 | 47 ± 10 | 14.9 ± 0.9 |
| SKΔK414(K256A/K257A) | 39 ± 9 | 16 ± 1 | NM | 36 ± 5 | 15.2 ± 0.6 |
| SKΔK414(E262A) | 27 ± 9 | 15 ± 2 | 1570 ± 50 | 37 ± 5 | 14.7 ± 0.7 |
| SKΔK414(E262A/E263A) | 30 ± 10 | 13 ± 1 | 1030 ± 40 | 32 ± 4 | 14.6 ± 0.4 |
[Lys]Pg activation by SKΔK414 was characterized by an 11-fold decrease in affinity of SK·Pg* formation in the absence of 6-AHA that was reduced only 1.7-fold in the presence of 6-AHA (Fig. 1A), whereas kPg was indistinguishable from that of wild-type and native SK and nearly totally inhibited by 6-AHA (Fig. 1B and Table 1). These results were consistent with previous equilibrium binding and kinetic experiments (10, 11, 22). SKΔ(R253-L260) and the substitution mutants SK(R253A), SK(K256A), SK(K257A), and SK(K256A/K257A) showed no significant difference from SKΔK414 in KA(v1), with a mean and range of 24 ± 13 nm, indicating no significant effect of the mutations on SK·Pg* formation. In contrast, in the absence of 6-AHA, certain SK mutants showed decreases in kPg compared with wtSK of 2.1-fold (R253A), 4.3-fold (K256A), and 2.0-fold (K257A), whereas kPg for SKΔ(R253-L260) and SK(K256A/K257A) was below the limit of detection (Fig. 1, D and F). The rates of Pm formation were inhibited totally by 10 mm 6-AHA (Fig. 1, B, D, and F, and Table 1). Substitution of acidic residues in SK(E249A), SK(E262A), and SK(E262A/E263A) produced no significant effects on the dissociation constant and substrate hydrolysis specificity constant for the SK·Pg* complex, except for SK(E262A), where the substrate specificity constant was 1.4-fold higher relative to SKΔK414 (Table 1). kPg was increased by substitution of the acidic residues: 1.3-fold SK(E249A), 2.0-fold SK(E262A), and 1.8-fold SK(E262A/E263A).
Kinetics of Mini-Pg Activation by Native SK, wtSK, and SK Mutants
The ability of the same SK species to activate mini-Pg, which lacks kringles 1–4, was also evaluated in kinetic studies of mini-Pg activation (Fig. 2 and Table 1). KA(v1) was indistinguishable from that of wtSK, with a mean and range of 38 ± 14 nm for native, wild type, and all of the SK mutants, and was affected ≤1.4-fold by 6-AHA (Fig. 2, A, C, and E). The observation that the affinity of SK·mini-Pg* was independent of Lys414 deletion, and 6-AHA demonstrated that Lys414 does not interact with K5, the only kringle present in mini-Pg, to enhance SK·mini-Pg* catalytic complex formation (Fig. 2A). The specificity constant for chromogenic substrate hydrolysis by wtSK·mini-Pg* of 16 ± 1 nm−1 s−1 was similar to that of wild-type and native SK·[Lys]Pg* of 12.6 ± 0.3 and 12.8 ± 0.4 nm−1 s−1, respectively, and was also independent of Lys414 and 6-AHA.
FIGURE 2.

Kinetics of mini-Pg activation by SK and SK mutants. A and B, v1 as a function of the total SK or SK mutant concentration ([SK]o) and v2 as a function of log total SK or SK mutant concentration ([SK]o), respectively, for wtSK in the absence (●) and presence of 10 mm 6-AHA (○), native SK in the absence (▴) and presence of 10 mm 6-AHA (▵), and SKΔ414 in the absence (■) and presence of 10 mm 6-AHA (□). C and D, v1 and v2, respectively, as in A for SKΔ(R253-L260) in the absence (●) and presence of 10 mm 6-AHA (○) and SK(R253A) in the absence (▴) and presence of 10 mm 6-AHA (▵). E and F, v1 and v2, respectively, as in A for SK(K256A) in the absence (●) and presence of 10 mm 6-AHA (○), SK(K257A) in the absence (▴) and presence of 10 mm 6-AHA (▵), and SK(K256A/K257A) in the absence (■) and presence of 10 mm 6-AHA (□). Solid lines represent the nonlinear least-squares fits to the data with Equations 2 and 4 for v1 and Equations 3 and 4 for v2 with the parameters listed in Table 1. Kinetic data were collected and analyzed as described under ”Experimental Procedures.“
In contrast, β-domain substitutions and 250-loop deletion in SK greatly affected recognition of mini-Pg as a substrate. The value of kPg for mini-Pg decreased 4.0-fold for the SK mutant R253A, 6.9-fold for K256A, and 4.3-fold for K257A and was essentially abolished for SKΔ(R253-L260) and SK(K256A/K257A). The value of kPg was affected no more than 1.4-fold for the other mutants (Fig. 2, B, D, and F, and Table 1). SK(E262A) exhibited the largest decrease in KA(v1) of 1.5-fold and the largest increase in kPg of 1.4-fold (Table 1). Similar to the results with [Lys]Pg, kPg for wild-type SK, native SK, and the SK mutants was fully inhibited (1–3% activity) by 6-AHA (Fig. 2, B, D, and F). The results demonstrated that substrate recognition of mini-Pg by the SK·mini-Pg* catalytic complex was facilitated by interaction of residues Arg253, Lys256, and Lys257 through K5 of mini-Pg.
Recognition of BPg as a Substrate of the SK·Pm Complex
Equilibrium binding and kinetic studies of BPg were performed to evaluate the role of Lys256 and Lys257 in substrate binding to the SK·Pm complex. BPg binds SK weakly in the catalytic mode and is not thought to be conformationally activated by SK but acts as a specific substrate of the SK·human Pm catalytic complex, allowing a clear distinction between conformational activation and substrate recognition (12, 13, 61).8 For these experiments, we used a full-length SK construct (i.e. with the COOH-terminal Lys414) containing a Met residue preceding Ile1. Blockage of the natural NH2 terminus of SK prevents conformational activation of Pg but is inconsequential in studies of the SK·Pm complex, where the NH2 terminus of Pm is inserted, as shown in the SK·μPm structure (3). Pm was labeled specifically at the active site with tetramethylrhodamine-5-iodoacetamide linked by Nα-[(acetylthio)acetyl]-d-Phe-Pro-Arg-CH2Cl ([TMR]FPR-Pm).
Native SK or full-length Met-SK(K256A/K257A) binding to [TMR]FPR-Pm resulted in 48 ± 1 and 51 ± 2% fluorescence enhancements, signaling binding with unit stoichiometries and indistinguishable KD values of 290 ± 100 and 130 ± 80 pm (Fig. 3A and Table 2). These values were 24- and 11-fold weaker than determined previously for native Pm (12 pm) (5) and Pm labeled at the active site with fluorescein (11–19 pm) (5, 21, 22). The presence of 40 mm 6-AHA reduced the affinity of native SK and Met-SK(K256A/K257A) 5- and 10-fold, respectively (Fig. 3A and Table 2). The loop deletion mutant, SKΔ(R253-L260), also lacking Lys414, bound to [TMR]FPR-Pm with a KD of 1.2 ± 0.2 nm, essentially the same as Met-SK(K256A/K257A) in the presence of 6-AHA (KD = 1.3 nm), suggesting that there were no major global structural changes in SK accompanying deletion of the loop (Table 2).
FIGURE 3.

Ternary complex formation among SK or Met-SK(K256A/K257A), [TMR]FPR-Pm, and BPg and kinetics of BPg activation by SK·Pm. A, fractional change in fluorescence intensity (ΔF/Fo) as a function of total native SK concentration (●) or total Met-SK(K256A/K257A) concentration ([SK]o) (○) in the absence of 6-AHA. Titrations with native SK (▴) or Met-SK(K256A/K257A) (▵) in the presence of 40 mm 6-AHA. Solid lines represent the nonlinear least-squares fits of the quadratic binding equation with the parameters given under ”Results.“ Fluorescence titrations were performed and analyzed as described under ”Experimental Procedures.“ B, fractional change in fluorescence intensity (ΔF/Fo) as a function of total BPg concentration ([BPg]o) for 35 nm [TMR]FPR-Pm·native SK complex formed in the presence of 98 nm SK in the absence of 6-AHA (●) and in the presence of 230 nm SK and 40 mm 6-AHA (○). Titrations of the Met-SK(K256A/K257A)·[TMR]FPR-Pm complex formed in the presence of 98 nm Met-SK(K256A/K257A) in the absence of 6-AHA (▴) and in the presence of 230 nm Met-SK(K256A/K257A) and 40 mm 6-AHA (▵). Solid lines, least-squares fit of the quadratic binding equation to the [TMR]FPR-Pm·native SK titration (●) with the parameters given under ”Results“ and the fit of the [TMR]FPR-Pm·native SK titration in the presence of 40 mm 6-AHA (○), assuming the same maximum fluorescence change and a KD value of 31 μm. Fluorescence titrations were performed and analyzed as described under ”Experimental Procedures.“ C, initial velocity (v2) of BPg activation by 0.1 nm native SK·Pm complex as a function of total BPg concentration ([BPg]o) in the absence (●) and presence of 40 mm 6-AHA (○) and initial rates of BPg activation by 0.09 nm Met-SK(K256A/K257A)·Pm complex in the absence of 6-AHA (▴). The solid line represents the fit by the Michaelis-Menten equation with the parameters given under ”Results.“ Initial velocities of BPg activation were measured and analyzed as described under ”Experimental Procedures.“
TABLE 2.
Equilibrium binding and kinetic parameters for BPg substrate mode interactions with the SK·Pm complex
The dissociation constant (KD), maximum fluorescence change (ΔFmax/Fo), and stoichiometric factor (n) determined from titrations of [TMR]FPR-Pm ([TMR]-Pm) in the absence (No 6-AHA) and presence of 40 mm 6-AHA (40 mm 6-AHA) are listed for the indicated ligand-acceptor interactions. Km, kcat, and kcat/Km from kinetics experiments of BPg activation by the indicated substrate-enzyme pairs are listed for reactions in the absence and presence of 40 mm 6-AHA. Experimental error represents ±2 S.D. NM, not measurable. Fluorescence titrations and kinetics experiments were performed and analyzed as described under “Experimental Procedures.”
| Binding interaction | No 6-AHA |
40 mm 6-AHA |
||||
|---|---|---|---|---|---|---|
| KD | ΔFmax/Fo | n | KD | ΔFmax/Fo | n | |
| nm | % | mol/mol | nm | % | mol/mol | |
| SK–[TMR]-Pm | 0.29 ± 0.10 | 48 ± 1 | 0.92 ± 0.11 | 1.4 ± 0.2 | 60 ± 1 | |
| Met-SK(K256/257A)–[TMR]-Pm | 0.13 ± 0.08 | 51 ± 2 | 0.98 ± 0.11 | 1.3 ± 0.2 | 59 ± 2 | |
| SKΔK414Δ(R253-L260)–[TMR]-Pm | 1.2 ± 0.2 | 56 ± 2 | ||||
| BPg–(SK·[TMR]-Pm) | 990 ± 130 | 114 ± 5 | ∼31,000a | |||
| BPg–(Met-SK(K256/257A)·[TMR]-Pm) | NM | NM | ||||
| Kinetics | Km | kcat | kcat/Km | Km | kcat | kcat/Km |
|---|---|---|---|---|---|---|
| nm | s−2 | nm−1 s−2 | nm | s−2 | nm−1 s−2 | |
| BPg–SK·Pm | 1120 ± 140 | 2.4 ± 0.1 | 0.0021 | NM | ||
| BPg–(Met-SK(K256A/K257A)·Pm) | NM | NM |
a Estimated KD from the linear dependence of the fluorescence change on BPg concentration in the presence of 40 mm 6-AHA (Fig. 3B) assuming ΔFmax/Fo of 60%.
Titrations of the fluorescence of [TMR]FPR-Pm complexes formed in the presence of saturating SK with BPg resulted in an additional 114 ± 5% fluorescence enhancement with a dissociation constant of 990 ± 130 nm (Fig. 3B). By contrast, BPg produced only a ∼3% maximum fluorescence change in the Met-SK(K256A/K257A)·[TMR]FPR-Pm complex over the same concentration range, indicating greatly reduced affinity of the mutant for BPg (Fig. 3B). Steady-state kinetics of BPg activation by the SK·Pm complex gave a Km of 1120 ± 140 nm and kcat of 2.4 ± 0.1 s−2 (Fig. 3C and Table 2). The Km was indistinguishable from the KD for BPg binding in the fluorescence experiments. Titrations of SK·[TMR]FPR-Pm complex with BPg in the presence of 40 mm 6-AHA reduced the fluorescence change to a small linear increase (Fig. 3B). Assuming the same fluorescence maximum gave an estimate of ∼30-fold decreased affinity (Table 2). Likewise, no significant rate of BPg activation was observed with the Met-SK(K256A/K257A)·Pm complex (Fig. 3C). The rate of BPg activation was reduced to an unmeasurable level by 40 mm 6-AHA (Fig. 3C). The absence of a fluorescence change in BPg titrations of the Met-SK(K256A/K257A)·[TMR]FPR-Pm complex was also unaffected by 6-AHA (Fig. 3B). These results indicated that BPg substrate binding was facilitated by the same LBS-dependent interactions with the principal residues identified kinetically and was independent of the weaker affinity of the fluorescently labeled Pm when it was saturated with SK.
DISCUSSION
The results of kinetics and equilibrium binding studies support the conclusion that residues Arg253, Lys256, and Lys257 in the SK β-domain facilitate LBS-dependent Pg substrate recognition through interaction with kringle 5 of the substrate. The increases in [Lys]Pg processing to Pm accompanying alanine substitution of acidic residues in and near the 250-loop argues against a pseudolysine structure in the 250-loop. Application of kinetic approaches that resolve Pg conformational activation and proteolytic generation of Pm made it possible to demonstrate a specific function of residues in the 250-loop in Pg substrate recognition, which was independent of conformational Pg activation. This is apparently not in agreement with the results of some previous studies indicating that both conformational and proteolytic activation are affected by mutations in the 250-loop (28, 31). The results also extend previous work by Sahni and co-workers (27, 31, 34), from which it was concluded that the 250-loop was involved in Pg substrate interactions by the SK·Pm complex. Our studies and those of others have also shown that two molecules bind to SK in LBS-dependent interactions through different sites to assemble the product-forming SK·Pg*/Pm·Pg ternary complexes (10, 11, 13, 35). The results obtained here with mini-Pg activation are the first to identify kringle 5 as the site of LBS-dependent interactions with SK residues Arg253, Lys256, and Lys257 in the SK 250-loop that facilitate Pg substrate recognition.
Binding of the COOH-terminal Lys414 residue of SK is responsible for LBS-dependent enhanced affinity of formation of the SK·Pg* and SK·Pm catalytic complexes (22). Recent rapid reaction kinetic studies of the SK·Pm binding pathway and the effects of 6-AHA and benzamidine on the elementary steps suggested that Lys414 probably binds to K4 (26). The results of the present studies with mini-Pg show that Lys414 does not bind to K5 in this substrate. Remarkably, mini-Pg had a KA(v1) of 36 ± 12 nm with SKΔK414, whereas [Lys]Pg had only a 1.6-fold higher affinity of 22 ± 7 nm, and the specificity constants for chromogenic substrate hydrolysis by SKΔK414·[Lys]Pg* and SKΔK414·mini-Pg* were similar at 12.6 ± 0.3 and 16 ± 1 nm−1 s−1, respectively. It appears that in the absence of Lys414, kringles 1–4 only have a small effect on conformational activation of Pg, indicating that the affinity and activation of the Pg catalytic domain, when unassisted by Lys414, is dominated by catalytic domain interactions. This can also be appreciated by the similar affinities and kcat/Km values of substrate hydrolysis for conformational activation of [Lys]Pg and mini-Pg with wild-type and native SK, compared with SKΔK414 when LBS interactions are blocked by 6-AHA (Table 1).
K5 binds cationic ligands such as benzamidine, Nα-acetyl-Lys-methyl ester, 6-aminohexane, and 5-aminopentane, in addition to zwitterionic lysine analogs (19, 20, 45–47). The specificity of K5 might be explained in principle by the absence of basic residues that interact with the anionic groups of lysine analogs, the so-called cationic center residues Arg117 and/or Arg153 in K1 (62), Arg234 in K2 (63), and Lys392 and/or Arg426 in K4 (64). By contrast, the topologically equivalent residues in human Pg kringle 5 possess aliphatic/aromatic side chains (Ile496, Phe497, and in particular Leu532) (47). This specificity is compatible with the absence of a pseudolysine structure in the 250-loop of SK. However, we stress that most of the information regarding LBS specificity has been derived from studies with low molecular weight compounds, which might not accurately reproduce the binding mode of kringle-interacting macromolecules. For instance, binding of an internal lysine residue to the LBS of human K4 has been reported (65) (Protein Data Bank code 1PML). Although the physiological significance of the trimer of kringles trapped in this crystal structure has not been clarified, it highlights the possibility of “noncanonical” binding to a kringle module, even in those specific for small zwitterionic compounds. In the structures of the internal PAM peptide, VEK-30, bound to either a triple mutant of human K2 (41) or to recombinant human angiostatin (Pg kringles K1–K3 (42)), both anionic and cationic centers of the kringle engage in electrostatic interactions with basic (Arg101 and His102) and acidic ligand residues (Glu104; PAM numbering system). However, a recent study identified two residues that do not belong to the “canonical” LBS of K2 as responsible for the specific binding of VEK-30 to human but not murine Pg (Arg220 and Leu222 in the human protein) (66). These observations, among others, prompt further structural and functional investigations of kringle interactions with macromolecular ligands.
The SK β-domain, and in particular its 250-loop, is one of the most variable regions in SK from different strains (67) (Fig. S1). In one of the deposited crystal structures of the β-domain (1QQR),7 the sequence of the 250-loop corresponds to the protein used in the current investigation. Interestingly, this loop adopts essentially the same conformation in all four crystallographically independent molecules, with Arg253 forming a hydrogen-bonded salt bridge with Glu262. In the other reported structure of the SK β-domain (1C4P) (40), Arg253 is replaced by a Glu residue found in a different streptococcus species (40) (Fig. S1). In this case, the 250-loop presents slightly different conformations in all four independent molecules, with side chains of Glu253 and Glu262 rotated away to minimize electrostatic repulsion. In all of these eight independent molecules, Glu262 is oriented with its carboxylic acid located at least ∼9 Å from the ϵ-amino group of Lys257 and between ∼10 and 14 Å from the ϵ-amino group of Lys256, whereas the carboxylic acid of Glu263 points in the opposite direction. Although the high flexibility of the 250-loop could theoretically allow the lysine residues to approach the carboxylates from Glu262/Glu263, it is important to notice that the overall extended (hairpin-like) conformation of this loop is maintained in all independent copies of the SK β-domain, which is stabilized by a large number of mostly main chain to main chain hydrogen bonds. These interactions extend to the very tip of the 250-loop, with, for example, the Asn255 carboxamide accepting hydrogen bonds from the backbone nitrogen atoms of residue 257, 258, and/or 259. Accordingly, alanine substitution of both Glu residues produced a modest 1.8-fold increase in kPg, indicating that they do not participate in a pseudolysine structure. The increase in kPg produced by mutating these acidic residues to alanine suggests that the negative charge of these residues inhibits the Lys256 and Lys257 interaction with substrate Pg or alters the conformation of the 250-loop in a favorable way for Pm generation.
It should be appreciated that we have focused on identifying the SK structure responsible for the LBS dependence of Pg substrate recognition. The results do not rule out roles for other structural elements of SK in Pg recognition and processing to Pm. Notably, some studies proposed that a region in the SK α-domain (residues 45–70) that is disordered in the SK·μPm crystal structure is important for Pg substrate recognition (32, 33, 36). Although the present results show a nearly total dependence of the bimolecular rate constant for Pm formation on Lys256 and Lys257 and 6-AHA, how much of this is due to increases in Km or decreases in kcat remains to be determined.
The results of equilibrium binding studies with BPg as a substrate indicated that loss of binding affinity contributes significantly to the loss of activity in Pm generation by the SK·Pg/Pm complexes associated with Lys256 and Lys257 substitution. The use of [TMR]FPR-Pm in these experiments allowed both SK binding and formation of a catalytically inactive, SK·[TMR]FPR-Pm·BPg ternary complex to be quantitated in the absence of proteolysis. Ternary complex formation was inhibited roughly 30-fold by 6-AHA, demonstrating the involvement of LBS interactions. The demonstration that BPg substrate binding was undetectable for the Met-SK(K256A/K257A)·[TMR]FPR-Pm complex shows the importance of Lys256 and Lys257 in this interaction. Importantly, these results were quantitatively paralleled by the kinetics of BPg activation catalyzed by the SK·Pm complex. The Km and KD for BPg activation and binding were indistinguishable; BPg activation was not supported by Met-SK(K256A/K257A) and was weakened similarly by 6-AHA. The close correspondence between KD and Km indicates that although labeling the active site of Pm reduced its affinity for SK and Met-SK(K256A/K257A) binding, there were no significant effects on BPg binding as a substrate at saturating concentrations of SK.
The critical role played by the triplet of basic residues Arg253, Lys256, and Lys257 in Pg substrate recognition and processing can be rationalized on the basis of a model of the human Pg K5 domain docked to the SK·μPm structure with an intact 250-loop (Fig. 4, A and B; see above). This unique docking solution allows simultaneous interaction of all three basic residues with the kringle module. In this model, both nitrogen atoms of the Arg253 guanidinium group are within hydrogen bond distance of the Tyr525 hydroxyl oxygen, and the Arg side chain also engages in close van der Waals contacts with Ile496 and Leu532. Lys256 occupies the LBS via strong interactions with the Phe497 aromatic ring and with its ammonium group in position to form either a salt bridge to Asp516 or a hydrogen bond with the Asn502 carboxamide oxygen. Finally, Lys257 is within hydrogen bonding distance of the Glu500 carboxylate (Fig. 4B). These interactions would help in positioning of the presumably rigidly assembled K5-catalytic domain tandem into the active site of SK-bound Pg*/Pm, in such a manner that the disulfide-constrained activation loop of substrate Pg inserts into the proteinase active site cleft. It is noteworthy that Lys257 is the only residue within the 253–265 stretch that is strictly conserved among SK from different streptococcal species (Fig. S1). It is not possible to assess whether SK variants with nonconservative variations at positions 253 (Glu, Gln, or Gly) and/or 256 (Pro, Ser, or Asn) would bind in similar conformations to the Pg K5 domain or rely on other surface-exposed loops for substrate recognition. However, it is important to appreciate that over half of the SK sequences deposited to date contain the Lys256/Lys257 pair (74 of 138, or 53.6%), whereas position 253 among “Lys257 only variants” is usually occupied by a second lysine residue (44 isolates; 31.9%). Thus, it would seem that different combinations of basic residues along the tip of the 250-loop are suitable for Pg K5 recognition. In conclusion, the combined kinetic, equilibrium binding, and molecular modeling results support the conclusion that Pg substrate binding and proteolytic generation of Pm are facilitated primarily by noncanonical interactions between SK residues Arg253, Lys256, and Lys257 and K5 of the substrate.
FIGURE 4.

Putative model of plasminogen K5 recognition by SK β-domain. A, overall assembly of the ternary K5·SK·μPm complex predicted by FTDOCK. The SK moiety is shown as a solid surface model colored according to its electrostatic surface potential (blue, positive; red, negative), and the bound μPm is represented as a yellow ribbon. SK domains are indicated. The kringle domain of the incoming Pg substrate is given as a green ribbon. B, close-up of interactions between SK 250-loop and human Pg kringle 5 module. For simplicity, only residues of SK 250-loop and neighboring K5 residues are included in this plot. Hydrogen bonds are shown as green dotted lines. Molecular modeling was performed as described under ”Experimental Procedures.“
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health, NHLBI, Grant RO1 HL056181 (to P. E. B.) and by Spanish Ministerio de Ciencia e Innovación (MICINN) Grant SAF2007-64140 (to P. F.-P.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
G. Spraggon, X. C. Zhang, C. P. Ponting, V. F. Fox, C. Phillips, R. A. G. Smith, E. Y. Jones, C. M. Dobson, and D. I. Stuart, unpublished results.
P. E. Bock, unpublished results.
- SK
- streptokinase
- wtSK
- recombinant wild-type SK
- SKΔK414
- SK lacking the COOH-terminal Lys414 residue
- SK(R253-L260)
- SKΔK414 with residues Arg253 to Leu260 deleted
- SK(E249A)
- SKΔK414 with Glu249 substituted with Ala
- SK(R253A)
- SKΔK414 with Arg253 substituted with Ala
- SK(K256A)
- SKΔK414 with Lys256 substituted with Ala
- SK(K257A)
- SKΔK414 with Lys257 substituted with Ala
- SK(K256A/K257A)
- SKΔK414 with Lys256 and Lys257 substituted with Ala
- SK(E262A/E263A)
- SKΔK414 with Glu262 and Glu263 substituted with Ala
- Pg
- plasminogen
- [Glu]Pg
- intact native plasminogen
- [Lys]Pg
- native Pg lacking the NH2-terminal 77 residues
- Pm
- [Lys]plasmin
- μPm
- micro-Pm; Pm catalytic domain
- BPg
- bovine Pg
- FFR-CH2Cl
- d-Phe-Phe-Arg-CH2Cl
- FPR-CH2Cl
- d-Phe-Pro-Arg-CH2Cl
- 6-AHA
- 6-aminohexanoic acid
- PAM
- plasminogen-binding Group A streptococcal M-like protein
- Pg*
- nonproteolytically activated form of the plasminogen zymogen
- LBS
- lysine-binding site(s)
- PEG
- polyethylene glycol
- pNA
- para-nitroaniline
- MES
- 4-morpholineethanesulfonic acid; [TMR]FPR-Pm, Pm inactivated with Nα-[(acetylthio)acetyl]-d-Phe-Pro-Arg-CH2Cl and labeled with tetramethylrhodamine-5-iodoacetamide.
REFERENCES
- 1.Sun H., Ringdahl U., Homeister J. W., Fay W. P., Engleberg N. C., Yang A. Y., Rozek L. S., Wang X., Sjöbring U., Ginsburg D. (2004) Science 305, 1283–1286 [DOI] [PubMed] [Google Scholar]
- 2.Collen D., Lijnen H. R. (1991) Blood 78, 3114–3124 [PubMed] [Google Scholar]
- 3.Wang X., Lin X., Loy J. A., Tang J., Zhang X. C. (1998) Science 281, 1662–1665 [DOI] [PubMed] [Google Scholar]
- 4.Damaschun G., Damaschun H., Gast K., Gerlach D., Misselwitz R., Welfle H., Zirwer D. (1992) Eur. Biophys. J. 20, 355–361 [DOI] [PubMed] [Google Scholar]
- 5.Boxrud P. D., Fay W. P., Bock P. E. (2000) J. Biol. Chem. 275, 14579–14589 [DOI] [PubMed] [Google Scholar]
- 6.Boxrud P. D., Verhamme I. M., Fay W. P., Bock P. E. (2001) J. Biol. Chem. 276, 26084–26089 [DOI] [PubMed] [Google Scholar]
- 7.Schick L. A., Castellino F. J. (1974) Biochem. Biophys. Res. Commun. 57, 47–54 [DOI] [PubMed] [Google Scholar]
- 8.Wang S., Reed G. L., Hedstrom L. (1999) Biochemistry 38, 5232–5240 [DOI] [PubMed] [Google Scholar]
- 9.Wang S., Reed G. L., Hedstrom L. (2000) Eur. J. Biochem. 267, 3994–4001 [DOI] [PubMed] [Google Scholar]
- 10.Boxrud P. D., Verhamme I. M., Bock P. E. (2004) J. Biol. Chem. 279, 36633–36641 [DOI] [PubMed] [Google Scholar]
- 11.Boxrud P. D., Bock P. E. (2004) J. Biol. Chem. 279, 36642–36649 [DOI] [PubMed] [Google Scholar]
- 12.Wohl R. C., Summaria L., Arzadon L., Robbins K. C. (1978) J. Biol. Chem. 253, 1402–1407 [PubMed] [Google Scholar]
- 13.Davidson D. J., Higgins D. L., Castellino F. J. (1990) Biochemistry 29, 3585–3590 [DOI] [PubMed] [Google Scholar]
- 14.Bajaj A. P., Castellino F. J. (1977) J. Biol. Chem. 252, 492–498 [PubMed] [Google Scholar]
- 15.Brown P. J., Mulvey D., Potts J. R., Tomley F. M., Campbell I. D. (2003) J. Struct. Funct. Genomics 4, 227–234 [DOI] [PubMed] [Google Scholar]
- 16.Tordai H., Bányai L., Patthy L. (1999) FEBS Lett. 461, 63–67 [DOI] [PubMed] [Google Scholar]
- 17.Ponting C. P., Marshall J. M., Cederholm-Williams S. A. (1992) Blood Coagul. Fibrinolysis 3, 605–614 [PubMed] [Google Scholar]
- 18.Bock P. E., Day D. E., Verhamme I. M., Bernardo M. M., Olson S. T., Shore J. D. (1996) J. Biol. Chem. 271, 1072–1080 [DOI] [PubMed] [Google Scholar]
- 19.Marti D. N., Hu C. K., An S. S., von Haller P., Schaller J., Llinás M. (1997) Biochemistry 36, 11591–11604 [DOI] [PubMed] [Google Scholar]
- 20.Castellino F. J., McCance S. G. (1997) Ciba Found. Symp. 212, 46–60 [DOI] [PubMed] [Google Scholar]
- 21.Boxrud P. D., Bock P. E. (2000) Biochemistry 39, 13974–13981 [DOI] [PubMed] [Google Scholar]
- 22.Panizzi P., Boxrud P. D., Verhamme I. M., Bock P. E. (2006) J. Biol. Chem. 281, 26774–26778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin L. F., Houng A., Reed G. L. (2000) Biochemistry 39, 4740–4745 [DOI] [PubMed] [Google Scholar]
- 24.Ponting C. P., Holland S. K., Cederholm-Williams S. A., Marshall J. M., Brown A. J., Spraggon G., Blake C. C. (1992) Biochim. Biophys. Acta 1159, 155–161 [DOI] [PubMed] [Google Scholar]
- 25.Marshall J. M., Brown A. J., Ponting C. P. (1994) Biochemistry 33, 3599–3606 [DOI] [PubMed] [Google Scholar]
- 26.Verhamme I. M., Bock P. E. (2008) J. Biol. Chem. 283, 26137–26147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dhar J., Pande A. H., Sundram V., Nanda J. S., Mande S. C., Sahni G. (2002) J. Biol. Chem. 277, 13257–13267 [DOI] [PubMed] [Google Scholar]
- 28.Lin L. F., Oeun S., Houng A., Reed G. L. (1996) Biochemistry 35, 16879–16885 [DOI] [PubMed] [Google Scholar]
- 29.Loy J. A., Lin X., Schenone M., Castellino F. J., Zhang X. C., Tang J. (2001) Biochemistry 40, 14686–14695 [DOI] [PubMed] [Google Scholar]
- 30.Conejero-Lara F., Parrado J., Azuaga A. I., Dobson C. M., Ponting C. P. (1998) Protein Sci. 7, 2190–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chaudhary A., Vasudha S., Rajagopal K., Komath S. S., Garg N., Yadav M., Mande S. C., Sahni G. (1999) Protein Sci. 8, 2791–2805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim D. M., Lee S. J., Kim I. C., Kim S. T., Byun S. M. (2000) Thromb. Res. 99, 93–98 [DOI] [PubMed] [Google Scholar]
- 33.Wakeham N., Terzyan S., Zhai P., Loy J. A., Tang J., Zhang X. C. (2002) Protein Eng. 15, 753–761 [DOI] [PubMed] [Google Scholar]
- 34.Nihalani D., Raghava G. P., Sahni G. (1997) Protein Sci. 6, 1284–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Young K. C., Shi G. Y., Wu D. H., Chang L. C., Chang B. I., Ou C. P., Wu H. L. (1998) J. Biol. Chem. 273, 3110–3116 [DOI] [PubMed] [Google Scholar]
- 36.Liu L., Sazonova I. Y., Turner R. B., Chowdhry S. A., Tsai J., Houng A. K., Reed G. L. (2000) J. Biol. Chem. 275, 37686–37691 [DOI] [PubMed] [Google Scholar]
- 37.Nihalani D., Kumar R., Rajagopal K., Sahni G. (1998) Protein Sci. 7, 637–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yadav S., Datt M., Singh B., Sahni G. (2008) Biochim. Biophys. Acta 1784, 1310–1318 [DOI] [PubMed] [Google Scholar]
- 39.Bean R. R., Verhamme I. M., Bock P. E. (2005) J. Biol. Chem. 280, 7504–7510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang X., Tang J., Hunter B., Zhang X. C. (1999) FEBS Lett. 459, 85–89 [DOI] [PubMed] [Google Scholar]
- 41.Rios-Steiner J. L., Schenone M., Mochalkin I., Tulinsky A., Castellino F. J. (2001) J. Mol. Biol. 308, 705–719 [DOI] [PubMed] [Google Scholar]
- 42.Cnudde S. E., Prorok M., Castellino F. J., Geiger J. H. (2006) Biochemistry 45, 11052–11060 [DOI] [PubMed] [Google Scholar]
- 43.Bergmann S., Wild D., Diekmann O., Frank R., Bracht D., Chhatwal G. S., Hammerschmidt S. (2003) Mol. Microbiol. 49, 411–423 [DOI] [PubMed] [Google Scholar]
- 44.Ehinger S., Schubert W. D., Bergmann S., Hammerschmidt S., Heinz D. W. (2004) J. Mol. Biol. 343, 997–1005 [DOI] [PubMed] [Google Scholar]
- 45.Christensen U. (1984) Biochem. J. 223, 413–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thewes T., Constantine K., Byeon I. J., Llinas M. (1990) J. Biol. Chem. 265, 3906–3915 [PubMed] [Google Scholar]
- 47.Chang Y., Mochalkin I., McCance S. G., Cheng B., Tulinsky A., Castellino F. J. (1998) Biochemistry 37, 3258–3271 [DOI] [PubMed] [Google Scholar]
- 48.Dharmawardana K. R., Bock P. E. (1998) Biochemistry 37, 13143–13152 [DOI] [PubMed] [Google Scholar]
- 49.Nieuwenhuizen W., Traas D. W. (1989) Thromb. Haemost. 61, 208–210 [PubMed] [Google Scholar]
- 50.Castellino F. J., Powell J. R. (1981) Methods Enzymol. 80, 365–378 [DOI] [PubMed] [Google Scholar]
- 51.Sottrup-Jensen L., Claeys H., Zajdel M., Petersen T. E., Magnusson S. (1978) in Progress in Chemical Fibrinolysis and Thrombolysis (Davidson J. F., Rowan R. M., Samama M. M., Desnoyers P. C. eds) pp. 191–209, Raven Press, New York [Google Scholar]
- 52.Bock P. E. (1992) J. Biol. Chem. 267, 14963–14973 [PubMed] [Google Scholar]
- 53.Bock P. E. (1992) J. Biol. Chem. 267, 14974–14981 [PubMed] [Google Scholar]
- 54.Bock P. E. (1993) Methods Enzymol. 222, 478–503 [DOI] [PubMed] [Google Scholar]
- 55.Sjöholm I. (1973) Eur. J. Biochem. 39, 471–479 [DOI] [PubMed] [Google Scholar]
- 56.Violand B. N., Castellino F. J. (1976) J. Biol. Chem. 251, 3906–3912 [PubMed] [Google Scholar]
- 57.Schaller J., Moser P. W., Dannegger-Müller G. A., Rösselet S. J., Kämpfer U., Rickli E. E. (1985) Eur. J. Biochem. 149, 267–278 [DOI] [PubMed] [Google Scholar]
- 58.Jackson K. W., Tang J. (1982) Biochemistry 21, 6620–6625 [DOI] [PubMed] [Google Scholar]
- 59.Pace C. N., Vajdos F., Fee L., Grimsley G., Gray T. (1995) Protein Sci. 4, 2411–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen R., Li L., Weng Z. (2003) Proteins 52, 80–87 [DOI] [PubMed] [Google Scholar]
- 61.Wohl R. C., Sinio L., Summaria L., Robbins K. C. (1983) Biochim. Biophys. Acta 745, 20–31 [DOI] [PubMed] [Google Scholar]
- 62.Mathews I. I., Vanderhoff-Hanaver P., Castellino F. J., Tulinsky A. (1996) Biochemistry 35, 2567–2576 [DOI] [PubMed] [Google Scholar]
- 63.Marti D. N., Schaller J., Llinás M. (1999) Biochemistry 38, 15741–15755 [DOI] [PubMed] [Google Scholar]
- 64.Wu T. P., Padmanabhan K., Tulinsky A., Mulichak A. M. (1991) Biochemistry 30, 10589–10594 [DOI] [PubMed] [Google Scholar]
- 65.Padmanabhan K., Wu T. P., Ravichandran K. G., Tulinsky A. (1994) Protein Sci. 3, 898–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fu Q., Figuera-Losada M., Ploplis V. A., Cnudde S., Geiger J. H., Prorok M., Castellino F. J. (2008) J. Biol. Chem. 283, 1580–1587 [DOI] [PubMed] [Google Scholar]
- 67.Kalia A., Bessen D. E. (2004) J. Bacteriol. 186, 110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.