

Abstract
Thyroid hormone receptors (TRs) are hormone-regulated transcription factors that play multiple roles in vertebrate endocrinology and development. TRs are expressed as a series of distinct receptor isoforms that mediate different biological functions. The TRβ2 isoform is expressed primarily in the hypothalamus, pituitary, cochlea, and retina, and displays an enhanced response to hormone agonist relative to the other TR isoforms. We report here that the unusual transcriptional properties of TRβ2 parallel the ability of this isoform to bind p160 coactivators cooperatively through multiple contact surfaces; the more broadly expressed TRβ1 isoform, in contrast, utilizes a single contact mechanism. Intriguingly, the PAS-B domain in the p160 N terminus plays a previously unanticipated role in permitting TRβ2 to recruit coactivator at limiting triiodothyronine concentrations. The PAS-B sequences also play an important role in coactivator binding by estrogen receptor-α. We propose that the PAS-B domain of the p160 coactivators is an important modulator of coactivator recruitment for a specific subset of nuclear receptors, permitting stronger transcriptional activation at lower hormone concentrations than would otherwise occur, and allowing isoform-specific mRNA splicing to customize the hormone response in different tissues.
Thyroid hormone receptors (TRs)4 are members of a larger family of nuclear receptors that play multiple roles in vertebrate development, differentiation, and homeostasis (1–3). Nuclear receptors function as hormone-regulated transcription factors that bind to specific target DNA sequences and either repress or activate expression of adjacent genes by recruiting accessory proteins, denoted corepressors and coactivators (3–14). TRs are encoded by two distinct genetic loci, α and β, each of which is also subject to alternative mRNA splicing to generate a series of interrelated receptor isoforms (12, 14–19) (Fig. 1A). The TRα1 and TRβ1 isoforms are expressed in a wide variety of tissues, whereas the TRβ2 isoform is found primarily in the pituitary, hypothalamus, the auditory hair cells in the inner ear, and the cone cells of the retina (1, 14, 16, 18–21). The different TR isoforms play distinct roles in endocrine physiology (14, 17, 21–24). The TRβ2 isoform in the hypothalamus and pituitary plays a particularly crucial role in a negative feedback regulatory loop by which increases in circulating T3/T4 thyroid hormone levels result in suppression of thyroid releasing hormone and thyroid stimulating hormone synthesis, thereby restoring proper endocrine homeostasis (19, 20, 22, 25–29).
FIGURE 1.

Schematic representations of TR isoforms and p160 coactivators. A, thyroid hormone receptor isoforms. DNA-binding and hormone-binding domains are indicated. Different shadings indicate regions of sequence divergence among the different isoforms. Codon numbering is presented for the TRβ2 isoform. B, coactivators. The locations of the motifs described in the text are indicated. Filled squares above each coactivator indicate an LDELL/LLEQL region known to interact with CREB-binding protein and with certain PAS-B domains; circular dots indicate additional LXXLL sequences of unknown function. Codons are numbered from the N terminus. SRC1a subdomains employed in the GST pulldown assays are shown as horizontal bars.
Notably the transcriptional properties of TRβ2 differ from those of the otherwise closely related TRβ1 isoform. The TRβ1 isoform represses classical target genes in the absence of hormone, and only becomes a transcriptional activator on binding to hormone agonist such as T3 (30–34). TRβ2 in contrast does not repress, but instead modestly activates target genes even in the absence of hormone, and displays an enhanced ability to activate target genes compared with TRβ1 over a wide range of T3 concentrations (30–35). We have suggested that the enhanced T3 response of TRβ2 permits tissues that preferentially express this isoform to respond to lower concentrations of hormone than do tissues that exclusively express TRα1 or TRβ1 (34).
We wished to more fully understand the molecular basis behind the unusual transcriptional properties of the TRβ2 isoform. We report here that the enhanced transcriptional response of TRβ2 is closely paralleled by the enhanced ability of this isoform to bind to the p160 family of coactivators: SRC1 (also known as CoA-1), GRIP1 (also known as SRC2 or CoA-2), and activator of thyroid hormone receptor (ACTR) (also known as SRC3, or CoA-3). TRβ2, unlike TRβ1, binds to the p160 proteins in the absence of T3 due to a hormone-independent interaction between the TRβ2 N terminus and a glutamine-rich (Gln-rich) region in the C-terminal domain of these coactivators. However, the enhanced ability of TRβ2 to recruit p160 coactivators at limiting T3 concentration can be observed even in the absence of the Gln-rich domain, a phenomenon that requires the PAS-B domain within the p160 N-terminal region. The PAS-B domain itself does not bind detectably to any TR isoform tested, but instead greatly elevates the ability of the LXXLL motifs in the same coactivator to bind to TRβ2 under low T3 conditions. Notably the PAS-B domain is also required for estrogen receptor (ER)-α to efficiently bind p160 coactivator in response to estradiol (E2), but plays little or no role in p160 coactivator recruitment by TRβ1, TRβ0, or farnesoid X receptor. We propose that the TRβ2 isoform possesses an enhanced transcriptional response to T3 due to its ability to preferentially recruit p160 coactivators through a synergistic array of protein-protein interactions not available to TRβ1. We further propose that the PAS-B domain of the p160s plays an important, and previously undetected role in stabilizing coactivator recruitment by a subset of nuclear receptors and their isoforms.
EXPERIMENTAL PROCEDURES
Molecular Clones
The pSG5 expression vectors containing avian or human TRβ0, TRα1, TRβ1, or TRβ2, and the DR4 M-pTK-luciferase reporter construct were previously described (32, 34, 36–39). Full-length or subdomains of the SRC1, GRIP1, ACTR, or DRIP205/TRAP220 coactivators were either obtained in, or generated by subcloning into a pSG5 or pCR3.1 expression vector backbone. Glutathione S-transferase (GST)-coactivator and GST-receptor fusions were created in pGEX vectors by using standard recombinant DNA methodologies (32, 34, 36, 37). Mutations were created by the QuikChange Site-directed Mutagenesis Kit using the protocol recommended by the manufacturer (Stratagene, La Jolla, CA). All mutations were subsequently confirmed by sequence analysis.
Transient Transfections
Transfections using CV-1 cells were performed by the Effectene protocol as recommended by the manufacturer (Qiagen, Valencia, CA), using 10 ng of pSG5-TR expression vector, 100 ng of reporter plasmid, 60 ng of pCH110 as an internal transfection control, and sufficient pUC18 to bring the total DNA concentration to 200 ng (34). After a 24-h incubation, the transfection medium was replaced with fresh, hormone-depleted medium, and either ethanol carrier alone, or 3,3′,5-triiodo-l-thyronine (T3) (Sigma) was added. The cells were incubated for an additional 24 h, harvested, and lysed in 100 μl of Triton Lysis Buffer (0.2% Triton X-100, 91 mm K2HPO4, and 9.2 mm KH2PO4). Luciferase and β-galactosidase activities were measured as reported (34). Protein expression levels were analyzed by immunoblot (34).
Protein-Protein Interaction Assays
GST pulldown assays were adapted to a microplate format that enhanced the reproducibility and sensitivity of the methodology (40). Briefly, GST-coactivator fusion proteins were synthesized in Escherichia coli strain BL-21 transformed by the corresponding pGEX vector. The bacteria were lysed, and the GST fusion proteins were recovered and purified by binding to a glutathione-agarose matrix. The pSG5-TR plasmids were synthesized as 35S-radiolabeled proteins in vitro using a TnT Quick kit (Promega Corp., Madison WI). Each radiolabeled protein (typically 2–5 μl of TnT reaction product per assay) was incubated at 4 °C with the immobilized GST fusion protein of interest (∼10–20 ng immobilized to 5 μl of agarose matrix per reaction) in a total volume of 100 μl of Binding Buffer A (40). The binding reactions were carried out in 96-well multiscreen filter plates (Millipore, Bedford, MA); any given comparison of TRβ2 to TRβ1 was performed in parallel in the same plate. After a 2-h incubation with rocking at 4 °C, the filter wells were washed 3 times with 200 μl of ice-cold wash buffer (40), and any radiolabeled proteins remaining bound to the immobilized GST fusion proteins were subsequently eluted with 50 μl of 20 mm glutathione in 100 mm Tris-HCl, pH 7.8. The eluted proteins were resolved by SDS-PAGE and were visualized and quantified using a PhosphorImager/STORM system (GE Healthcare) and the GraphPad Prism 4 statistical/plotting package (La Jolla, CA). Results were reproducible over different days and with different preparations of protein.
Coimmunoprecipitations were performed by introducing 200 ng of pSG5-SRC1a and 200 ng of either pSG5-HA-tagged TRβ1 or pSG5-HA-tagged TRβ2 into HeLa cells by the Effectin-mediated transient transfection protocol and modification as described in Ref. 33. Approximately 1.2 × 105 cells were used per assay. After 47 h, 100 nm T3 was added, or not, and the cells were incubated for an additional 1 h. The cells were lysed and the coactivator was immunoprecipitated with anti-SRC1 antibodies (Affinity BioReagents, PA1–840). The immunoprecipitates were analyzed by SDS-PAGE and immunoblotted using antisera directed against hormone-binding domain sequences shared by TRβ1 and β2 (Affinity BioReagents, MA1–215). The bands were visualized and quantified by Flurochem8900 Imager (Alpha Innotech, San Leandro, CA).
Protease Resistance Assay
All steps, unless otherwise noted, were performed on ice. 35S-Radiolabeled SRC1-(1–781) or SRC1-(595–781) was synthesized using the TnT system and bound to the immobilized GST-TRβ1 or -β2 in Binding Buffer A in the presence of 625 nm T3, as described above. The protein complexes were washed 3 times in 50 mm Tris-Cl, pH 7.5, containing 625 nm T3, and were resuspended in 25 μl of the same buffer for each protease concentration employed. Twenty-five microliters of serially diluted protease (either Endoproteinase Glu-c “V8,” or elastase, Sigma) were added to each sample, and the samples were incubated 15 min at 20 °C with rocking (41). The reactions were terminated with 25 μl of concentrated SDS-PAGE sample buffer, boiled 5 min, resolved by SDS-PAGE, and the proteolytic degradation products were visualized and quantified by phosphorimager analysis.
RESULTS
TRβ2 Displays an Enhanced Transcriptional Response to T3 Hormone Compared with the Other TR Isoforms Due to Its Unique N-terminal Domain
To compare the transcriptional properties of the different human TR isoforms (Fig. 1), we expressed each isoform in CV-1 cells together with a luciferase reporter containing a cognate direct repeat (DR)-4 response element (34). CV-1 cells lack endogenous TRs, and there was virtually no effect of T3 on the DR4-luciferase reporter in the absence of ectopic TR expression (see Ref. 34, and data not shown). As anticipated, ectopic introduction of the human TRβ1 isoform into these cells repressed luciferase expression below basal level in the absence of T3, and induced luciferase expression in the presence of T3 (Fig. 2A). In contrast, introduction of the human TRβ2 isoform into the CV-1 cells did not repress the DR4-luciferase reporter, but instead induced a modest activation of luciferase expression even in the absence of hormone (Fig. 2A). Significantly, TRβ2 also displayed enhanced transcriptional activity in the presence of T3, with TRβ2 inducing higher levels of luciferase activity than did TRβ1 at all subsaturating T3 concentrations (Fig. 2A). Stated reciprocally, human TRβ2 induced an equal reporter gene activation as did human TRβ1, but at a ∼5-fold lower T3 concentration: half-maximal stimulation for TRβ2 was ∼0.2 nm T3 compared with ∼1.0 nm T3 for TRβ1. TRβ1 and β2 were expressed at nearly equal levels in the transfected cells, and the enhanced transcriptional activation properties of TRβ2 over TRβ1 were also observed in Chinese hamster ovary and 293 cells, and over a range of TR expression vector inputs (see Ref. 34, and data not shown). The third major mammalian TR isoform, TRα1, closely resembled TRβ1 in our transfection assay, repressing in the absence of hormone and activating only in response to T3 (Fig. 2A).
FIGURE 2.

Enhanced reporter gene activation by TRβ2. An expression construct for each TR isoform was introduced into CV-1 cells by lipofection, together with a DR4-luciferase reporter and a pCH100-lacZ internal control. After 24 h in hormone-depleted medium, the cells exposed to T3 as indicated on the bottom of each panel, were incubated for an additional 24 h, harvested, and the relative luciferase levels (absolute luciferase/β-galactosidase units) were calculated. Fold induction equals the relative luciferase levels observed in the presence of each receptor compared with that observed for an empty expression vector control. The data represent the mean ± S.D. (error bars) of two or more independent experiments; error bars smaller than data point symbols may not be visible. The EC50 values for TRβ2 differed from that of TRβ0, TRβ1, TRβ2, and ΔN-TRβ2 at a p value < 0.001. A, human-derived TR isoforms. B, avian (Gallus/chicken)-derived TR isoforms.
The TRβ1 and TRβ2 isoforms differ only in their N-terminal domains, which are derived from different exons (Fig. 1A). We therefore examined two truncated forms of TRβ to determine if these N-terminal domains contributed positively or negatively to TR function. TRβ0 is a naturally occurring isoform found in birds, reptiles, and amphibians and encodes a receptor with a highly truncated N-terminal domain (42, 43) (Fig. 1A). TRβ0 is believed to represent an ortholog of mammalian TRβ1, and avian TRβ0 displayed transcriptional properties nearly identical to those of human TRβ1 when introduced into the CV-1 cells, whereas avian TRβ2 mimicked the properties of human TRβ2 (Fig. 2B). A human TRβ2 bearing an artificial N-terminal truncation (denoted TRβ1ΔN; Fig. 2A, dashed curve) yielded results similar to avian TRβ0 (Fig. 2A). We conclude that the TRβ2 N-terminal sequences contribute in a positive manner, and are essential for the enhanced T3 transcriptional response of this isoform in diverse vertebrates.
The Enhanced Transcriptional Response of the TRβ2 Isoform Parallels an Enhanced Ability to Bind to p160 Coactivators in Vitro and in Vivo
TRβ1 and TRβ2 bind T3 and release SMRT and N-CoR corepressors in a comparable fashion, suggesting these properties are not the basis for the enhanced transcriptional properties of TRβ2 (34). We therefore focused on the interactions of TRβ1 and TRβ2 with coactivators. We first tested the ability of a full-length p160 coactivator, SRC1a, to bind to either TRβ1 or TRβ2. TRβ2 displayed an elevated ability to bind to SRC1a in a GST pulldown assay compared with TRβ1, and this was observed both in the absence of hormone and over a range of T3 concentrations (Fig. 3, A, and quantified in B). Mutational disruption of all of the LXXLL motifs in SRC1a prevented hormone-dependent binding to either TRβ1 or TRβ2, although this LXXAA SRC1 mutant retained a modest, hormone independent binding to TRβ2 that was not observed for TRβ1 (data not shown). Analogous results were observed for the other two members of the p160 family: GRIP1 and ACTR (Fig. 3, C and D). The enhanced ability of TRβ2 versus TRβ1 to bind SRC1a at limiting T3 could also be observed in vivo using a co-immunoprecipitation method (Fig. 3E). In contrast to the p160 coactivators, binding of DRIP205, a distinct receptor coactivator (44, 45), was indistinguishable between TRβ1 and TRβ2 (Fig. 3F).
FIGURE 3.

Enhanced p160 coactivator recruitment by TRβ2. A, preferential binding of SRC1a by TRβ2 compared with TRβ1. Full-length, 35S-radiolabeled SRC1a was incubated with immobilized, GST fusions of full-length human TRβ1 or TRβ2 in the presence of increasing [T3] as indicated. The resulting coactivator-TR complexes were characterized by SDS-PAGE and phosphorimager analysis. A representative phosphorimager scan is presented. B, preferential binding of SRC1a by TRβ2 compared with TRβ1 quantification. A series of GST pulldowns were performed as in panel A and quantified by phosphorimager analysis. The coactivator bound to each receptor is expressed as a percent of input. Data represent the mean ± S.D. (error bars) of at least two independent experiments. C, preferential binding of GRIP1 by TRβ2 compared with TRβ1. Protocol was as in panel B. D, preferential binding of ACTR by TRβ2 compared with TRβ1. Protocol was as in panel B. E, preferential co-immunoprecipitation (IP) of TRβ2 by SRC1a. SRC1a and either HA-tagged TRβ1 or HA-tagged TRβ2 were introduced into HeLa cells by lipofection of the corresponding expression vectors. After 48 h, 100 nm T3 was added, or not, and the cells were incubated for 1 h more. The cells were lysed and the coactivator was immunoprecipitated with anti-SRC1 antibodies. The immunoprecipitates were analyzed by SDS-PAGE and immunoblot using anti-TRβ antisera. F, equal binding of DRIP205 by TRβ2 and TRβ1. Protocol was as in panel B. G, equalization of the transcriptional properties of TRβ1 and TRβ2 by ectopic expression of SRC1a. An expression construct for either TRβ1 or TRβ2 was transfected into CV-1 cells together with the DR4-luciferase reporter, the pCH100-lacZ internal control, and increasing amounts of an expression vector for SRC1a. The cells were incubated −T3 or +T3 and were analyzed for relative luciferase expression as described in the legend to Fig. 2. H, failure to equalize the transcriptional properties of TRβ1 and TRβ2 by ectopic expression of DRIP205. An expression construct for each TR isoform was introduced into CV-1 cells by lipofection, together with a DR4-luciferase reporter, a pCH100-lacZ construct, and increasing amounts of an expression vector for DRIP205. The cells were incubated and analyzed for relative luciferase expression as described in the legend to Fig. 2. Symbols in each panel indicate statistical confidence that TRβ2 differs in EC50 or Bmax from TRβ1 as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001; #, curves are not statistically distinguishable.
Our results suggested that an enhanced affinity for p160 coactivators was at least one mechanism underlying the enhanced transcriptional activity of TRβ2 relative to TRβ1. If true, sufficient p160 expression in cells might overcome this difference in coactivator affinity between TRβ1 and TRβ2; consistent with this concept, increasing ectopic expression of SRC1a in CV-1 cells first elevated, then equalized the transcriptional activity of TRβ1 to that of TRβ2 (Fig. 3G). This equalization of TRβ1 and TRβ2 activity was not observed using a p205 DRIP coactivator, which enhanced both TRβ1 and TRβ2 activity in parallel, consistent with the equal affinities for this coactivator seen in vitro (Fig. 3H).
Enhanced p160 Binding by TRβ2 Reflects Both Hormone-independent and Hormone-dependent Interactions with the Coactivator
An enhanced ability of TRβ2 to bind to coactivator compared with that of TRβ1 was also observed when an SRC-1a-(595–1441) construct containing only the central LXXLL motifs plus the Gln-rich region, and was observed both in the absence and presence of T3 (Fig. 4A, and schematic in Fig. 1B). SRC1a contains a fourth LXXLL receptor-interaction motif at its extreme C terminus; an SRC-1a construct limited to the Gln-rich domain and this C-terminal LXXLL also displayed preferred binding to TRβ2 compared with TRβ1 (Fig. 4B). SRC1a constructs containing the Gln-rich domain, but lacking all LXXLLs, bound to the TRβ2 independent of T3 status and failed to bind to TRβ1 (Fig. 4C). Conversely, SRC1a constructs restricted to the central LXXLL motifs, or to the C-terminal LXXLL, but lacking the Gln-rich region, demonstrated little or no binding to either TRβ2 or TRβ1 minus T3, and exhibited near equal binding to both isoforms plus T3 (Fig. 4D, and data not shown). These results indicate that the Gln-rich coactivator domain is necessary and sufficient for hormone-independent SRC1a binding by TRβ2, and contributes to the elevated ability of TRβ2 to bind SRC1a in the presence of T3; the latter, however, also requires the presence of at least one LXXLL receptor-interaction motif in the coactivator. Similar results were observed with GRIP1 (data not shown).
FIGURE 4.

Identification of three domains on the p160 coactivators required for preferential recruitment by TRβ2. A, preferential TRβ2 binding to an SRC1a-(595–1441) construct containing the central LXXLL motifs, the Gln-rich domain, and the C-terminal LXXLL motif. Protocol was as described in the legend to Fig. 3B. B, preferential TRβ2 binding to an SRC1a-(891–1441) construct containing the central LXXLL motifs, the Gln-rich domain, and the C-terminal LXXLL motif. Protocol was as described in the legend to Fig. 3B, except a GST-SRC1-(891–1441) construct was incubated with 35S-radiolabeled full-length TRβ1 or TRβ2, as indicated. Percent bound is presented, normalized to TRβ2 levels. C, preferential, hormone-independent TRβ2 binding to an SRC1a-(891–1441) construct containing the Gln-rich domain but lacking the C-terminal LXXLL motif. Protocol was as in panel B, except the C-terminal LXXLL in the GST-SRC1-(891–1441) construct was mutated to LXXAA. D, equal TRβ2 and TRβ1 binding to an SRC1a-(566–891) construct limited to the central LXXLL motifs. Protocol was as in panel B. E, preferential TRβ2 binding to an SRC1a-(1–781) construct containing the N-terminal and central LXXLL domains, but lacking the Gln-rich region. Protocol was as in panel A. F, preferential TRβ2 binding to a GRIP1-(1–776) construct containing the N-terminal and central LXXLL domains, but lacking the Gln-rich region. Protocol was as in panel A. Statistical confidence symbols are as described in the legend to Fig. 3.
The N-terminal Domain of SRC1 Selectively Enhances TRβ2 Binding Even in the Absence of the Gln-rich Domain
We made an unexpected observation when expanding these studies to an SRC1-(1–781) construct lacking the Gln-rich region, but retaining the SRC1 N-terminal and central LXXLL domains (Fig. 1B). Although the loss of the Gln-rich domain virtually eliminated the ability of the SRC1-(1–781) construct to bind to TRβ2 in the total absence of T3, this construct still retained a strong preference for binding to TRβ2 compared with TRβ1 at low and intermediate T3 levels (Fig. 4E). The N-terminal domain of SRC1 was required for this phenomenon, given SRC1 constructs limited to the LXXLL motifs bound nearly equally to TRβ1 and TRβ2 (Figs. 4D and 5). Similar results were obtained with GRIP1 (Fig. 4F).
FIGURE 5.

Positive contribution of the N-terminal domain of TRβ2 to recruitment of SRC1a. 35S-Radiolabeled versions of SRC1 (codons 1–781) and SRC1 (codons 595–781) were mixed together and incubated with each immobilized GST-TRβ construct over a range of T3 concentrations, as described in the legend to Fig. 4E. The SRC1-(1–781) and SRC1-(595–781) coactivator proteins bound by each receptor were eluted, resolved from one another by SDS-PAGE, quantified, and are presented as the percent of their input values. A, strong preference of TRβ2 for SRC1 constructs retaining the N-terminal domain. B, minor preference of TRβ1 for SRC1 constructs retaining the N-terminal domain. C, lack of preference of ΔN-TRβ2 for SRC1 constructs retaining the N-terminal domain. Statistical confidence symbols are as described in the legend to Fig. 3.
To better define this phenomenon by an internally controlled protocol, we mixed an 35S-labeled SRC1-(1–781) construct (containing both the N-terminal and LXXLL domains, Fig. 1B) and an 35S-labeled SRC1-(595–781) construct (containing only the LXXLL domain, Fig. 1B) together and assayed their ability to competitively bind to GST-TRβ1, GST-TRβ2, or a GST-ΔN-TR construct (Fig. 5). The TRβ2 N terminus greatly enhanced the preference of this receptor for SRC1-(1–781) versus SRC1-(595–781) under limiting hormone (Fig. 5A), the TRβ1 N terminus had a detectable, but much less of an effect (Fig. 5B), and a TRβ mutant lacking the N-terminal domain displayed no preference (Fig. 5C).
The Enhanced Binding of TRβ2 by SRC1a Maps to a PAS-B Domain within the p160 Coactivator
The N terminus of the p160 coactivators contains a basic helix-loop-helix domain, a PAS-A and a PAS-B domain (13, 46–48) (Fig. 1B). To map the specific domain responsible for the enhanced interaction with TRβ2 under limiting T3, we tested a series of N-terminal deletions (Fig. 1B) of the SRC1-(1–781) construct in the GST pulldown assay. Deletion of the basic helix-loop-helix domain had no observable effect on either TRβ1 or TRβ2 binding (Fig. 6A). Further truncation of the SRC1 N terminus, resulting in loss of the PAS-A domain, increased the overall binding of the SRC1 coactivator to both TRβ1 and TRβ2, but nonetheless, preserved the TRβ2 > TRβ1 binding phenotype in response to limiting T3 (Fig. 6, B and C). Larger N-terminal truncations that deleted the PAS-B domain of SRC1 virtually eliminated the preferential binding of TRβ2 compared with TRβ1 (Fig. 6, D–F). Internal deletions restricted exclusively to PAS-A or to PAS-B mimicked the results seen with the sequential N-terminal deletions (Fig. 6, G versus H). Although the N terminus of SRC-1 contains two “cryptic” LXXLL sequences (Fig. 1B), these are not known to bind nuclear receptors and had no effect on the interaction of the coactivator with either TR isoform (Fig. 6). We conclude that the PAS-B domain, conserved in all p160 coactivators, is responsible for the preferential ability of TRβ2 to bind this coregulator in the absence of the Gln-rich domain.
FIGURE 6.

Requirement for the coactivator PAS-B domain in preferential recruitment by TRβ2. The ability of GST-TRβ1 and GST-TRβ2 to bind to various radiolabeled SRC1 constructs was assayed as described in the legend to Fig. 4E. A, TR binding of SRC1-(83–781). B, TR binding of SRC1-(199–781). C, TR binding of SRC1-(263–781). D, TR binding of SRC1-(318–781). E, TR binding of SRC1-(381–781). F, TR binding of SRC1-(487–781). G, TR binding of SRC1-(1–781) bearing a PAS-A domain deletion. H, TR binding of SRC1-(1–781) bearing a PAS-B domain deletion. Statistical confidence symbols are as described in the legend to Fig. 3.
It was conceivable that the PAS-B motif in SRC1 functioned through a direct interaction with TRβ2. However, an SRC1-(1–338) construct containing this motif failed to interact detectably with either TRβ2 or TRβ1 in a GST pulldown assay (data not shown). The PAS-B domain of SRC1 is known to interact with an LDELL/LLEQL motif region in the SRC1 C-terminal region (Fig. 1B, box), potentially generating intra- or intermolecular SRC1-SRC1 interactions (49); however, this region is absent from the SRC1-(1–781) construct used in our studies. Consistent with prior studies (49), we also failed to detect any interaction between the SRC1-(1–338) N-terminal domain and the central LXXLL motifs that are retained in the SRC1-(1–781) fragment (data not shown). Our evidence therefore suggests that the PAS-B SRC1a domain enhances TRβ2 binding under limiting T3 concentrations through an indirect effect.
The PAS-B Domain Enhances the Ability of TRβ2 to Bind to LXXLL Motif Pairings Not Recognized by TRβ1
We next tested if the PAS-B motif worked by enhancing in some fashion the affinity of TRβ2 for the central LXXLL coactivator motifs known to mediate agonist-dependent nuclear receptor binding. We employed GRIP1 for these experiments due to the prior availability of the appropriate mutations. It has been proposed that receptor dimers interact with two LXXLL motifs within one p160 coactivator (50–53). TRs reportedly prefer LXXLL-2 and LXXLL-3 (54–57), and consistent with this observation, mutation of LXXLL-1 to an LXXAA sequence had little or no effect on the ability of full-length GRIP1 to bind to either TRβ1 or TRβ2 (Fig. 7A). In contrast, inactivation of either LXXLL-2 or LXXLL-3 by mutagenesis virtually eliminated the ability of GRIP1 to bind to TRβ1, but preserved substantial binding to TRβ2 (Fig. 7, B and C). These results suggest that TRβ2 is capable of recognizing a different combination of LXXLL motifs than TRβ1.
FIGURE 7.

Broadened specificity of TRβ2 for coactivator LXXLL motifs not recognized by TRβ1. The ability of GST-TRβ1 and GST-TRβ2 to bind to the 35S-radiolabeled GRIP1 constructs indicated was assayed as described in the legend to Fig. 4E. A, TR binding of full-length GRIP1 in which LXXLL-1 was mutated to LXXAA. B, TR binding of full-length GRIP1 in which LXXLL-2 was mutated to LXXAA. C, TR binding of full-length GRIP1 in which LXXLL-3 was mutated to LXXAA. D, TR binding of GRIP1 in which LXXLL-2 was mutated to LXXAA and bearing an N-terminal deletion. E, TR binding of GRIP1 in which LXXLL-2 was mutated to LXXAA and bearing a deletion of the Q-rich domain. Statistical confidence symbols are as described in the legend to Fig. 3.
Deletion of the GRIP1 PAS-B region strongly reduced the ability of TRβ2 to bind to the LXXLL-2 mutant in response to T3 (Fig. 7D). Interestingly, removal of the GRIP1 C-terminal Gln-rich domain also interfered with the ability of TRβ2 to recognize the LXXLL-2 mutant (Fig. 7E). These results indicate that the N- and C-terminal regions of the coactivator operate together to stabilize TRβ2 binding to LXXLL motifs that are not recognized by TRβ1.
Partial Protease Degradation Suggests That SRC1a Can Assume a Different Conformation Once Bound to TRβ2 Compared with That When Bound to TRβ1
Our results suggest that TRβ2 interacts with p160 coactivators differently than does TRβ1. If so, this different mode of interaction might impose a different conformation on the coactivator. To examine this question, we bound radiolabeled SRC1a to either GST-TRβ1 or GST-TRβ2 under high T3 concentrations and probed the coactivator conformation by use of a limited protease digestion, with the expectation that differences in coactivator conformation would manifest as differences in protease susceptibility (58). There were consistent differences in the elastase and V8 protease degradation patterns of the same SRC1a preparation when bound to TRβ2 than when bound to TRβ1 (Fig. 8A and data not shown). Different protease protection patterns were also observed using an SRC1-(1–781) construct (Fig. 8B), but not with an SRC-(595–781) construct (data not shown). These results are consistent with the SRC1a coactivator assuming a different conformation when bound to TRβ2 than when bound to TRβ1.
FIGURE 8.
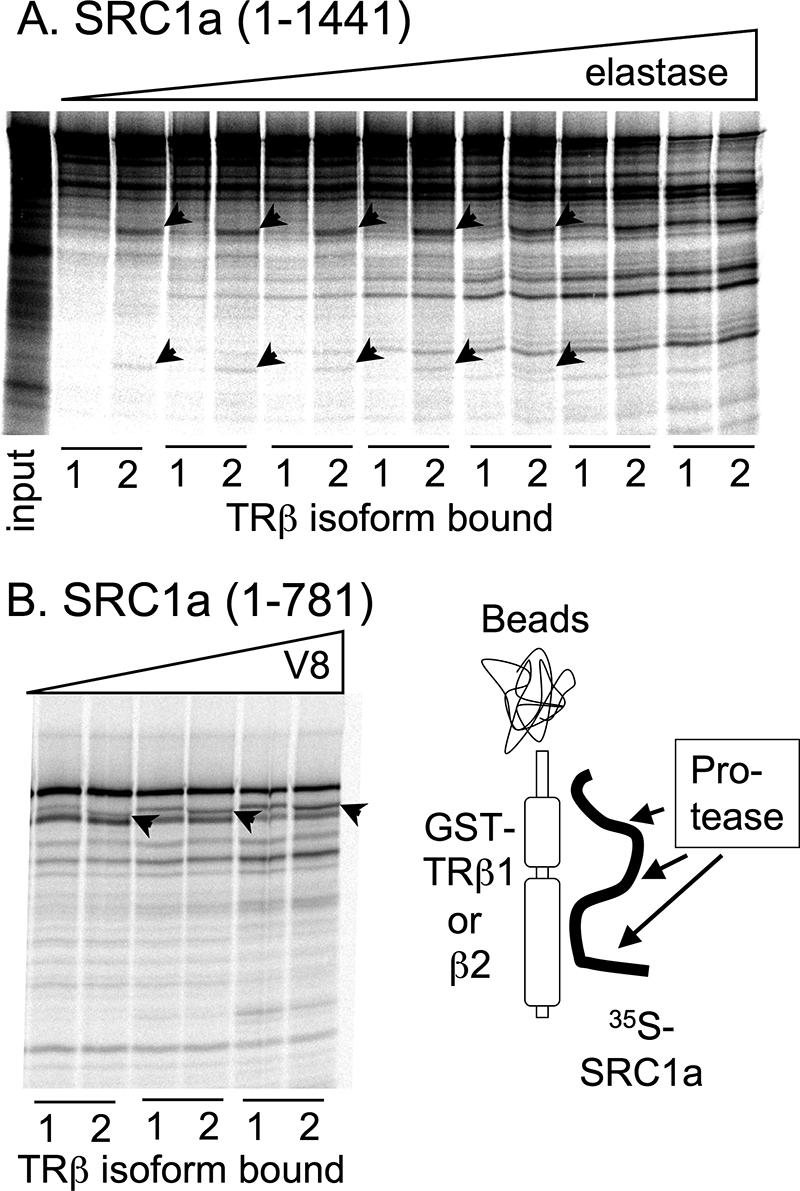
Alterations in protease susceptibility of SRC1a when bound to TRβ2 versus TRβ1. The 35S- radiolabeled SRC1 constructs indicated were bound to either GST-TRβ1 or GST-TRβ2 in the presence of 625 nm T3. The complexes were washed and incubated with increasing amounts of either elastase or V8 protease (represented schematically), and the resulting coactivator peptides were analyzed by SDS-PAGE and phosphorimager visualization. A, elastase degradation of full-length SRC1a bound to GST-TRβ1 or GST-TRβ2. Arrowheads indicate radiolabeled SRC1 fragments generated from this coactivator when bound to GST-TRβ2 but not when bound to GST-TRβ1. Leftmost lane, input. B, V8 degradation of SRC1-(1–781) bound to GST-TRβ1 or GST-TRβ2. Arrowheads indicate radiolabeled SRC1 fragments that differ in size when this coactivator was bound to TRβ2 versus TRβ1.
The N-terminal Domain of SRC1 Also Contributes to Efficient p160 Coactivator Binding by Estrogen Receptor-α
To determine if the PAS-B sequences contribute to p160 binding not only for specific TR splice forms, but also for other classes of nuclear receptors, we examined the comparative ability of the SRC1-(1–781) and SRC1-(595–781) constructs to bind to ERα. Notably the SRC1 construct containing the N-terminal domain bound much more strongly to ERα in response to estradiol than did the SRC1 construct lacking the N-terminal domain (Fig. 9A), whereas both SRC1 constructs bind equally well to the ΔN-TRβ construct (e.g. Fig. 5). The enhanced binding of the SRC-(1–781) construct to ERα required the PAS-B domain of the coactivator (Fig. 9B) and was not observed using a ERα construct lacking the receptor N-terminal domain, or a full-length FXR construct (data not shown). We conclude that the PAS-B domain of SRC1a enhances the ability of the p160 coactivators to recruit a subset of nuclear receptors in response to hormone agonist.
FIGURE 9.

Requirement for the coactivator PAS-B domain for efficient recruitment by ERα. 35S-Radiolabeled versions of SRC1 (codons 1–781) and SRC1 (codons 595–781) were mixed together and incubated with immobilized GST-ERα in the presence of increasing E2, using the same general protocol as described in the legend to Fig. 4E. The SRC1-(1–781) and SRC1-(595–781) coactivator proteins bound by the GST-ERα were eluted, resolved from one another by SDS-PAGE, quantified, and are presented as the percent of their input values. A, stabilization of SRC1 recruitment by the coactivator N-terminal domain. B, mapping of the stabilizing function to the SRC1 PAS-B domain. Statistical confidence symbols are as described in the legend to Fig. 3.
DISCUSSION
Wild-type TRβ2 displays enhanced transcriptional activation properties not seen with other TR isoforms
Unlike the TRβ1 isoform, the unliganded TRβ2 isoform fails to repress transcription, and instead activates transcription even in the absence of hormone (33). TRβ2 also displays an elevated ability to activate positive response elements (and to repress negative response elements) in response to hormone compared with TRβ1 (29–32, 34, 35). The divergent transcriptional properties of these two isoforms are not due to differences in their levels of expression or different affinities for T3 but instead appear to reflect inherent differences in the ability of these two different isoforms to recruit certain coactivators (34).
In this report, we confirm that this enhanced transcriptional responsiveness is unique to TRβ2, and that TRα1 closely parallels the TRβ1 response. Given the preferential expression of TRβ2 in the hypothalamus and pituitary, where it plays an important role in negative feedback regulation of circulating thyroid hormone levels, we have suggested that this enhanced responsiveness of TRβ2 to T3 can help it sense and suppress surges of thyroid hormone before the less responsive, but more widely expressed TRα1, TRβ0, and TRβ1 isoforms do so, thereby avoiding peripheral thyrotoxicity (34). Consistent with this proposal, TR mutations that selectively impair the enhanced T3 response of TRβ2 result in Pituitary Resistance to Thyroid Hormone Syndrome, an endocrine disease characterized by central resistance but peripheral thyrotoxicity (34). The role the enhanced transcriptional response of TRβ2 might play in the other cell types in which this isoform is expressed, such as the retina and the inner ear, remains to be established.
The Unique N Terminus of TRβ2 Is Essential for the Enhanced Transcriptional Activation Properties of This Isoform
TRβ2 and TRβ1 are identical in sequence with the exception of their N-terminal domains, which are encoded by different exons in the two different isoforms. Significantly, the avian TRβ0 isoform, which possesses a severely truncated N-terminal domain, more closely resembles mammalian TRβ1 in its transcriptional properties, and is readily distinguished from mammalian or avian TRβ2. The same is true of an artificial, N-terminal truncation mutation of mammalian TRβ2. We conclude that the N terminus of TRβ2 contributes positively to the enhanced T3 response seen for this isoform.
The TRβ2 N-terminal Domain, Unlike That of TRβ1, Interacts with and Helps Recruit p160 Coactivators in a Hormone-independent Manner
In common with other nuclear receptors, the hormone-binding domains of TRα1, TRβ0, TRβ1, and TRβ2 interact with the LXXLL motifs found in many coactivators, and this interaction requires hormone agonist. Interestingly, the N-terminal domain of TRβ2, but not of TRβ1 or TRα1, can also interact in a hormone-independent manner with a Gln-rich region located near the C terminus of all three p160 coactivators (30, 32). This coactivator interaction likely contributes to the in vivo ability of TRβ2 to activate target gene expression in the absence of T3 (32). Furthermore, we report here that in the presence of T3 these TRβ2 N-terminal domain/coactivator contacts cooperate with the LXXLL coactivator contacts mediated by the hormone-binding domain of the receptor, yielding stronger p160 recruitment by TRβ2 than by TRβ1 under limiting T3 concentrations.
Both in the CV-1 cells used here, and in gene knock-out studies, SRC1a appears to play a particularly important role in defining the transcriptional activity of TRβ2 (59, 60). However, our current study does not exclude the possibility that coactivators outside of the p160 family also contribute to the unique hormone responsiveness of this isoform. CREB-binding protein, pCIP, and NRC, for example, display an enhanced interaction with TRβ2 in a phenomenon that requires the TRβ2 N-terminal domain, although probably by a mechanism different in its details from that of the p160 coactivators (30, 31). It appears likely that the relative transcriptional properties of TRβ2 and TRβ1 will differ in different cell types, on different target genes, and perhaps in different species (31) depending on which coactivators predominate in each context.
A PAS-B Motif in the SRC1 Unexpectedly Modulates Isoform-specific Nuclear Receptor Binding, Strongly Enhancing the p160 Interaction with TRβ2 under Limiting T3 Conditions
The Gln-rich region alone was insufficient to account for the enhanced ability of TRβ2 to recognize the p160 coactivators. Instead, enhanced recruitment of SRC1 by TRβ2 under limiting T3 was also observed for coactivators containing only the LXXLL motifs and an N-terminal domain PAS-B motif. The PAS-B domain not only increased the ability of TRβ2 to bind to p160 coactivator under limiting T3, but also permitted TRβ2 to bind to GRIP1 and SRC1 derivatives in which LXXLL-2 or -3 motifs, crucial for TRβ1 binding, had been disrupted by mutation.
No direct interaction of the PAS-B domain with any portion of TRβ2 was detected. Instead, our results suggest that the PAS-B domain functions indirectly, most likely by enhancing the ability of LXXLLs in the center of the p160 coactivators to bind to TRβ2. We explored several means by which this might occur. The PAS-B domain is known to interact with a subset of LXXLL motifs, including an LDELL/LLEQL region within the C-terminal region of the full-length p160 coactivators (49, 61). However, these motifs are absent from the SRC1-(1–781) construct, and therefore cannot contribute to the PAS-B phenotype we report here. Alternatively, it was possible that, despite the absence of the necessary flanking basic amino acids, the central, receptor-interaction LXXLL motifs retained in our SRC1-(1–781) constructs might interact with the PAS-B domain; however, no such contacts were observed in our hands or by other researchers (49).
Although we have been unable to define the specific mechanisms of action of the PAS-B and TRβ2 N-terminal domains in stabilizing the LXXLL/receptor interaction under limiting T3 conditions, we strongly suspect that these domains are acting through global conformational effects on their respective proteins. In support of this conjecture, SRC1 appears to assume a different conformation (based on protease susceptibility) when bound to TRβ2 than when bound to TRβ1. Conversely, TR with the β2 N-terminal domain appears to assume a different conformation state (by similar protease probes) than does TRβ1 (data not shown). We suggest that the SRC1 and TRβ2 N termini confer conformations on their respective proteins that permit and help stabilize higher affinity interactions between them. Unfortunately little is currently known about the secondary or tertiary structure of these N-terminal nuclear receptor domains, which are believed to be relatively disordered in solution and to assume an induced-fit conformation only in response to contacts with other proteins (62, 63).
The PAS-B Domain Also Contributes to the Ability of the p160 Coactivators to Bind to Other Nuclear Receptors, Including ERα
In common with the TRβ2 N-terminal region, the N-terminal domains of estrogen, glucocorticoid, and androgen receptors interact with the Gln-rich region of the p160 coactivators (64–67). We report here an additional parallel between TRβ2 and these steroid receptors, in that the PAS-B domain also contributes to the ability of SRC1 to bind to ERα, and this further requires the presence of the ERα N terminus. Further experiments will be required to define how wide a role the PAS-B domain plays in nuclear receptor function.
PAS domains serve as chemical and environmental sensors in a number of proteins in which they are found (68). For example, a recent report reveals that the PAS-B domain of hypoxia inducible factor 2α forms a large internal cavity, and that artificial compounds that dock to this cavity can modulate the interaction of hypoxia inducible factor 2 with its transcription factor partner, ARNT (69). If the PAS-B domain of the p160 coactivators can similarly bind and be regulated by small molecules, a cross-talk may exist by which the recruitment of the p160s to nuclear receptors can be controlled not just by receptor ligands, but also by coactivator ligands. These hypothetical coactivator ligands may be endocrine hormones, cellular metabolites, or yet some other regulatory molecule.
Acknowledgments
We thank Dr. Leonard Freedman for the generous gift of the DRIP 205 molecular clone, Dr. Wei Wan for the experiments that served as a foundation for this work, and Liming Liu for superb technical support and assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant R01DK53528 from the NIDDK.
- TR
- thyroid hormone receptor
- ER
- estrogen receptor
- CREB
- cAMP-response element-binding protein
- E2
- estradiol
- GST
- glutathione S-transferase
- HA
- hemagglutinin
- T3
- 3,3′,5-triiodo-l-thyronine
- DR
- direct repeat
- T4
- thyroxine
- ACTR
- activator of thyroid hormone receptor.
REFERENCES
- 1.Flamant F., Baxter J. D., Forrest D., Refetoff S., Samuels H., Scanlan T. S., Vennström B., Samarut J. (2006) Pharmacol. Rev. 58, 705–711 [DOI] [PubMed] [Google Scholar]
- 2.Larsen P. R., Davies T. F., Schlumberger M.-J., Hay I. D. (2003) in Williams Textbook of Endocrinology (Larsen P. R., Kronenberg H. M., Melmed S., Polonsky K. S. eds) pp. 331–373, Saunders, Philadelphia [Google Scholar]
- 3.Yen P. M. (2001) Physiol. Rev. 81, 1097–1142 [DOI] [PubMed] [Google Scholar]
- 4.Apriletti J. W., Ribeiro R. C., Wagner R. L., Feng W., Webb P., Kushner P. J., West B. L., Nilsson S., Scanlan T. S., Fletterick R. J., Baxter J. D. (1998) Clin. Exp. Pharmacol. Physiol. Suppl. 25, S2–11 [DOI] [PubMed] [Google Scholar]
- 5.Aranda A., Pascual A. (2001) Physiol. Rev. 81, 1269–1304 [DOI] [PubMed] [Google Scholar]
- 6.Bassett J. H., Harvey C. B., Williams G. R. (2003) Mol. Cell. Endocrinol. 213, 1–11 [DOI] [PubMed] [Google Scholar]
- 7.Cheng S. Y. (2000) Rev. Endocr. Metab. Disord. 1, 9–18 [DOI] [PubMed] [Google Scholar]
- 8.Lee J. W., Lee Y. C., Na S. Y., Jung D. J., Lee S. K. (2001) Cell Mol. Life Sci. 58, 289–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perissi V., Aggarwal A., Glass C. K., Rose D. W., Rosenfeld M. G. (2004) Cell 116, 511–526 [DOI] [PubMed] [Google Scholar]
- 10.Privalsky M. L. (2004) Annu. Rev. Physiol. 66, 315–360 [DOI] [PubMed] [Google Scholar]
- 11.Robyr D., Wolffe A. P., Wahli W. (2000) Mol. Endocrinol. 14, 329–347 [DOI] [PubMed] [Google Scholar]
- 12.Tsai C. C., Fondell J. D. (2004) Vitam. Horm. 68, 93–122 [DOI] [PubMed] [Google Scholar]
- 13.Xu J., O'Malley B. W. (2002) Rev. Endocr. Metab. Disord. 3, 185–192 [DOI] [PubMed] [Google Scholar]
- 14.Zhang J., Lazar M. A. (2000) Annu. Rev. Physiol. 62, 439–466 [DOI] [PubMed] [Google Scholar]
- 15.Brent G. A. (2000) Rev. Endocr. Metab. Disord. 1, 27–33 [DOI] [PubMed] [Google Scholar]
- 16.Cheng S. Y. (2005) Steroids 70, 450–454 [DOI] [PubMed] [Google Scholar]
- 17.Flamant F., Samarut J. (2003) Trends Endocrinol. Metab. 14, 85–90 [DOI] [PubMed] [Google Scholar]
- 18.Murata Y. (1998) Nagoya J. Med. Sci. 61, 103–115 [PubMed] [Google Scholar]
- 19.Wondisford F. E. (2003) J. Investig. Med. 51, 215–220 [DOI] [PubMed] [Google Scholar]
- 20.Abel E. D., Boers M. E., Pazos-Moura C., Moura E., Kaulbach H., Zakaria M., Lowell B., Radovick S., Liberman M. C., Wondisford F. (1999) J. Clin. Invest. 104, 291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forrest D., Vennström B. (2000) Thyroid 10, 41–52 [DOI] [PubMed] [Google Scholar]
- 22.Abel E. D., Ahima R. S., Boers M. E., Elmquist J. K., Wondisford F. E. (2001) J. Clin. Invest. 107, 1017–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhat M. K., Dace A., Cheng S. Y. (1999) Thyroid 9, 411–418 [DOI] [PubMed] [Google Scholar]
- 24.Gauthier K., Chassande O., Plateroti M., Roux J. P., Legrand C., Pain B., Rousset B., Weiss R., Trouillas J., Samarut J. (1999) EMBO J. 18, 623–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abel E. D., Kaulbach H. C., Campos-Barros A., Ahima R. S., Boers M. E., Hashimoto K., Forrest D., Wondisford F. E. (1999) J. Clin. Invest. 103, 271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abel E. D., Moura E. G., Ahima R. S., Campos-Barros A., Pazos-Moura C. C., Boers M. E., Kaulbach H. C., Forrest D., Wondisford F. E. (2003) Mol. Endocrinol. 17, 1767–1776 [DOI] [PubMed] [Google Scholar]
- 27.Forrest D., Hanebuth E., Smeyne R. J., Everds N., Stewart C. L., Wehner J. M., Curran T. (1996) EMBO J. 15, 3006–3015 [PMC free article] [PubMed] [Google Scholar]
- 28.Ng L., Forrest D., Haugen B. R., Wood W. M., Curran T. (1995) Mol. Endocrinol. 9, 1202–1213 [DOI] [PubMed] [Google Scholar]
- 29.Safer J. D., Langlois M. F., Cohen R., Monden T., John-Hope D., Madura J., Hollenberg A. N., Wondisford F. E. (1997) Mol. Endocrinol. 11, 16–26 [DOI] [PubMed] [Google Scholar]
- 30.Oberste-Berghaus C., Zanger K., Hashimoto K., Cohen R. N., Hollenberg A. N., Wondisford F. E. (2000) J. Biol. Chem. 275, 1787–1792 [DOI] [PubMed] [Google Scholar]
- 31.Tian H., Mahajan M. A., Wong C. T., Habeos I., Samuels H. H. (2006) Mol. Endocrinol. 20, 2036–2051 [DOI] [PubMed] [Google Scholar]
- 32.Yang Z., Privalsky M. L. (2001) Mol. Endocrinol. 15, 1170–1185 [DOI] [PubMed] [Google Scholar]
- 33.Yang Z., Hong S. H., Privalsky M. L. (1999) J. Biol. Chem. 274, 37131–37138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wan W., Farboud B., Privalsky M. L. (2005) Mol. Endocrinol. 19, 1529–1542 [DOI] [PubMed] [Google Scholar]
- 35.Sjöberg M., Vennström B. (1995) Mol. Cell. Biol. 15, 4718–4726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee S., Privalsky M. L. (2005) Mol. Endocrinol. 19, 863–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mengeling B. J., Pan F., Privalsky M. L. (2005) Mol. Endocrinol. 19, 35–51 [DOI] [PubMed] [Google Scholar]
- 38.Tzagarakis-Foster C., Privalsky M. L. (1998) J. Biol. Chem. 273, 10926–10932 [DOI] [PubMed] [Google Scholar]
- 39.Vivanco Ruiz M. M., Bugge T. H., Hirschmann P., Stunnenberg H. G. (1991) EMBO J. 10, 3829–3838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goodson M. L., Farboud B., Privalsky M. L. (2007) Nucl. Recept. Signal 5, e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Farboud B., Hauksdottir H., Wu Y., Privalsky M. L. (2003) Mol. Cell. Biol. 23, 2844–2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forrest D., Sjöberg M., Vennström B. (1990) EMBO J. 9, 1519–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sjöberg M., Vennström B., Forrest D. (1992) Development 114, 39–47 [DOI] [PubMed] [Google Scholar]
- 44.Ito M., Roeder R. G. (2001) Trends Endocrinol. Metab. 12, 127–134 [DOI] [PubMed] [Google Scholar]
- 45.Rachez C., Gamble M., Chang C. P., Atkins G. B., Lazar M. A., Freedman L. P. (2000) Mol. Cell. Biol. 20, 2718–2726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leo C., Chen J. D. (2000) Gene 245, 1–11 [DOI] [PubMed] [Google Scholar]
- 47.Ma H., Hong H., Huang S. M., Irvine R. A., Webb P., Kushner P. J., Coetzee G. A., Stallcup M. R. (1999) Mol. Cell. Biol. 19, 6164–6173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu J., Li Q. (2003) Mol. Endocrinol. 17, 1681–1692 [DOI] [PubMed] [Google Scholar]
- 49.Lodrini M., Münz T., Coudevylle N., Griesinger C., Becker S., Pfitzner E. (2008) Nucleic Acids Res. 36, 1847–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McInerney E. M., Rose D. W., Flynn S. E., Westin S., Mullen T. M., Krones A., Inostroza J., Torchia J., Nolte R. T., Assa-Munt N., Milburn M. V., Glass C. K., Rosenfeld M. G. (1998) Genes Dev. 12, 3357–3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nolte R. T., Wisely G. B., Westin S., Cobb J. E., Lambert M. H., Kurokawa R., Rosenfeld M. G., Willson T. M., Glass C. K., Milburn M. V. (1998) Nature 395, 137–143 [DOI] [PubMed] [Google Scholar]
- 52.Schmidt S., Baniahmad A., Eggert M., Schneider S., Renkawitz R. (1998) Nucleic Acids Res. 26, 1191–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Westin S., Kurokawa R., Nolte R. T., Wisely G. B., McInerney E. M., Rose D. W., Milburn M. V., Rosenfeld M. G., Glass C. K. (1998) Nature 395, 199–202 [DOI] [PubMed] [Google Scholar]
- 54.Darimont B. D., Wagner R. L., Apriletti J. W., Stallcup M. R., Kushner P. J., Baxter J. D., Fletterick R. J., Yamamoto K. R. (1998) Genes Dev. 12, 3343–3356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ding X. F., Anderson C. M., Ma H., Hong H., Uht R. M., Kushner P. J., Stallcup M. R. (1998) Mol. Endocrinol. 12, 302–313 [DOI] [PubMed] [Google Scholar]
- 56.Leers J., Treuter E., Gustafsson J. A. (1998) Mol. Cell. Biol. 18, 6001–6013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Northrop J. P., Nguyen D., Piplani S., Olivan S. E., Kwan S. T., Go N. F., Hart C. P., Schatz P. J. (2000) Mol. Endocrinol. 14, 605–622 [DOI] [PubMed] [Google Scholar]
- 58.Lin B. C., Hong S. H., Krig S., Yoh S. M., Privalsky M. L. (1997) Mol. Cell. Biol. 17, 6131–6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kamiya Y., Zhang X. Y., Ying H., Kato Y., Willingham M. C., Xu J., O'Malley B. W., Cheng S. Y. (2003) Endocrinology 144, 4144–4153 [DOI] [PubMed] [Google Scholar]
- 60.Takeuchi Y., Murata Y., Sadow P., Hayashi Y., Seo H., Xu J., O'Malley B. W., Weiss R. E., Refetoff S. (2002) Endocrinology 143, 1346–1352 [DOI] [PubMed] [Google Scholar]
- 61.Plevin M. J., Mills M. M., Ikura M. (2005) Trends Biochem. Sci. 30, 66–69 [DOI] [PubMed] [Google Scholar]
- 62.Wärnmark A., Treuter E., Wright A. P., Gustafsson J. A. (2003) Mol. Endocrinol. 17, 1901–1909 [DOI] [PubMed] [Google Scholar]
- 63.Chandra V., Huang P., Hamuro Y., Raghuram S., Wang Y., Burris T. P., Rastinejad F. (2008) Nature 456, 350–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alen P., Claessens F., Verhoeven G., Rombauts W., Peeters B. (1999) Mol. Cell. Biol. 19, 6085–6097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bevan C. L., Hoare S., Claessens F., Heery D. M., Parker M. G. (1999) Mol. Cell. Biol. 19, 8383–8392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Christiaens V., Bevan C. L., Callewaert L., Haelens A., Verrijdt G., Rombauts W., Claessens F. (2002) J. Biol. Chem. 277, 49230–49237 [DOI] [PubMed] [Google Scholar]
- 67.Hong H., Darimont B. D., Ma H., Yang L., Yamamoto K. R., Stallcup M. R. (1999) J. Biol. Chem. 274, 3496–3502 [DOI] [PubMed] [Google Scholar]
- 68.Gu Y. Z., Hogenesch J. B., Bradfield C. A. (2000) Annu. Rev. Pharmacol. Toxicol. 40, 519–561 [DOI] [PubMed] [Google Scholar]
- 69.Scheuermann T. H., Tomchick D. R., Machius M., Guo Y., Bruick R. K., Gardner K. H. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 450–455 [DOI] [PMC free article] [PubMed] [Google Scholar]