

Summary
Background
Identifying therapeutic drugs that block the release or effects of T-helper type 2 (Th2) cytokines after allergen exposure is an important goal for the treatment of allergic inflammatory diseases including asthma. We recently showed, using a murine model of allergic airway inflammation, that poly(ADP-ribose) polymerase (PARP) plays an important role in the pathogenesis of asthma-related lung inflammation. PARP inhibition, by single injection of a novel inhibitor, thieno[2,3-c]isoquinolin-5-one (TIQ-A), before ovalbumin (OVA) challenge, prevented airway eosinophilia in C57BL/6 mice with concomitant suppression of Th2 cytokine production and mucus secretion.
Objective
To evaluate the efficacy of the drug when it is given after OVA challenge for its possible therapeutic potential.
Methods
This study was conducted using a murine model of allergic airway inflammation.
Results
A single injection of TIQ-A (6 mg/kg) one or 6 h post-allergen challenge conferred similar reduction in OVA challenge-induced eosinophilia. More significantly, post-allergen challenge administration of the drug exerted even better suppression on the production of IL-4, IL-5, IL-13, and IgE and prevented airway hyperresponsiveness to inhaled-methacholine. The significant decrease in IL-13 was accompanied by a complete absence of airways mucus production indicating a potential protection against allergen-induced airway remodelling.
Conclusion
The coincidence of the inflammation trigger and the time of drug administration appear to be important for the drug’s more pronounced protection. The observed time window for efficacy, 1 or 6 h after allergen challenge may be of great clinical interest. These findings may provide a novel therapeutic strategy for the treatment of allergic airway inflammation, including asthma.
Keywords: airway hyperresponsiveness, allergy, cytokines, eosinophils, inflammation, interleukin, lung, mucus, PARP, therapeutic potential
Introduction
Asthma is, in part, a T-helper type 2 (Th2) lymphocytemediated inflammatory airway disease characterized by pulmonary eosinophilia, increased mucus production by goblet cells, and airway hyperresonsiveness (AHR) [1-3]. The presence of high levels of allergen-specific IgE is a reflection of an aberrant Th2 immune response [4, 5]. In recent years, an accumulation of data from mice and humans has identified Th2 cytokines IL-4, IL-5, and IL-13, as major contributors to allergy and asthma. Such studies suggest that blocking the release or effects of these cytokines may have important therapeutic potential. Significant areas of asthma therapy research and development are emerging, which include humanized monoclonal antibodies (mAbs) targeting IgE, IL-4, IL-5, TNF, or IL-13 as well as cytokine and chemokine receptors [6]. Furthermore, removal of cytokines or chemokines has been attempted using soluble receptors or small molecule receptor antagonists [6-8]. Additionally, drugs have been designed that block the signal transduction pathways that become activated when cytokines or chemokines interact with their receptors [6, 9]. Overall, these strategies showed very promising results in experimental animal models of asthma but exhibited serious limitations when applied to treat the disease in humans both in terms of efficacy and cost [8]. These limitations and failed clinical trials clearly argue for additional studies to discover new strategies for the treatment of the disease.
Poly(ADP-ribose) polymerase (PARP)-1 plays an important role in tissue injury associated with oxidative stress and inflammation. In several pathological situations that involve oxidative DNA damage, excessive activation of PARP depletes cellular stores of both NAD and its precursor ATP. This leads to irreversible cytotoxicity and can cause cell death [10, 11]. In addition to its effects through NAD metabolism on cell and tissue homeostasis, PARP is thought to participate in inflammation by regulating, directly or indirectly, the expression of several inflammatory factors including adhesion molecules, TNF, ILs, and inducible nitric oxide synthase [12].
Airway eosinophilia is a consistent characteristic feature of allergic inflammation and asthma [13]. Eosinophil release lipid mediators, cytokines, and cytotoxic proteins [6, 13]. Despite the fact that the number of eosinophils is associated with asthma severity in human patients, the actual role of these leucocytes in the pathophysiology of the disease remains elusive. Recently, Lee et al. [14] and Humbles et al. [15] reported, using transgenic animals deficient in eosinophils, that eosinophilia is an integral trait of experimental allergic asthma. However, these two studies reported contradictory findings on the role of eosinophils on AHR.
We recently showed that PARP play a role in eosinophil recruitment into lungs in our experimental model of allergic airway inflammation [16]. Administration of the novel PARP inhibitor thieno[2,3-c]isoquinolin-5-one (TIQ-A) before allergen challenge resulted in suppression of Th2 cytokines, including IL-4, IL-5, and IL-13, and suppression of the ensuing eosinophil infiltration and associated mucus production in lung airways [16]. Our results led us to examine the effect of TIQ-A on the eosinophil recruitment when it is administered after the allergen challenge. This is a clinically more relevant approach for the treatment of inflammation-associated asthma. Our results show that a single injection of TIQ-A one or 6 h after allergen challenge significantly suppress the production of Th2 cytokines, the consequent infiltration of eosinophils and mucus production, and AHR. Taken together, our results show a strong therapeutic potential of TIQ-A in blocking important traits that result from allergen exposure.
Materials and methods
Animals, protocols for sensitization and challenge
Mice were bred in a specific-pathogen free facility at LSUHSC, New Orleans, LA, and allowed unlimited access to sterilized chow and water. Maintenance, experimental protocols, and procedures were all approved by the LSUHSC Animal Care & Use Committee. C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA).
Six-week-old C57BL/6 mice (n = at least six) were sensitized with intraperitoneally (i.p.) injections of 100 μg Grade V chicken ovalbumin (OVA) (Sigma-Aldrich, St Louis MO, USA) mixed with 2 mg aluminum hydroxide in saline once a week for two consecutive weeks. The mice were then challenged with aerosolized OVA (3% OVA in saline) a week after the second sensitization in a Plexiglas chamber for 30 min. The OVA aerosol was generated by a Bennett nebulizer (DeVilbiss, Somerset, PA, USA). Control groups were not sensitized or challenged. Additional groups of mice received 6 mg/kg TIQ-A (i.p.) 2 h prior, one post, or 6 h post OVA challenge. The mice used in each experiment were of the same litter or the same family. Mice were then left to recover and killed 24–48 h later for bronchio-alveolar lavage (BAL) or lung fixations and processing for histological analysis.
Organ recovery, staining, and T-helper type 2 cytokine and immunoglobulin E assessments
Animals were killed by CO2 asphyxiation and lungs were fixed with formalin for histological analysis or subjected to BAL. A final volume of 5 mL saline was used for BAL to assess inflammatory cells (48 h post OVA-challenge) or 1 mL to assess cytokines or IgE (24 h post OVA-challenge). Formalin fixed lungs were sectioned and subjected to hematoxylin and eosin (H&E), Periodic Acid-Schiff (PAS) staining using standard protocols. Histological mucin index was assessed essentially as described [17]. Collected BAL fluids were subjected to cyto-spin and stained with H&E for the assessment of inflammatory cells. The cytokine assessment was conducted using the Bio-Rad Bioplex System for mouse Th2 cytokines namely IL-4, IL-5 and IL-13 according to the manufacturer instructions and specifications. OVA-specific IgE was quantified by a sandwich ELISA (Serotec, Raleigh, NC, USA) essentially as described [16].
Assessment of pulmonary function
Lung resistance to increasing doses of methacholine (MeCh, Sigma, St Louis, MO, USA; 0, 25, and 50 mg/mL in isotonic saline) was assessed 24 h after OVA challenge using the forced oscillation technique as previously described [18]. Anesthetized animals were mechanically ventilated with a tidal volume of 10 mL/kg and a frequency of 2.5 Hz using a computer-controlled piston ventilator (FlexiVent, SCIREQ; Montreal, Canada). Just before data collection, the volume history of the respiratory system was standardized by inflating the lungs to total lung capacity. Resistance data were collected using single compartment model and plotted after normalization to baseline (i.e. resistance at 0 mg/mL MeCh). Data are plotted as means ± SEM.
Data analysis
All data are expressed as means ± SD of values from at least six mice per group unless stated otherwise. PRISM software (GraphPad, San Diego, CA, USA) was used to analyze the differences between experimental groups by one way ANOVA followed by Dunnett’s multiple comparison test.
Results
Comparison of the effects of thieno [2, 3-c] isoquinolin-5-one administration before vs. after ovalbumin challenge
OVA sensitization and challenge induced a clear and marked perivascular and peribronchial infiltration by eosinophils in lungs of wild-type mice (Figs 1a and b). Administration of the PARP inhibitor TIQ-A before OVA-challenge significantly suppressed eosinophil recruitment (Figs 1a and b), which is consistent with our previous report [16].
Fig. 1.

Effect of PARP inhibition by TIQ-A on eosinophil recruitment following OVA sensitization and challenge. Mice received an i.p. injection of TIQ-A (6 mg/kg) or vehicle alone 2 h prior, 1 h post, or 6 h post-OVA challenge; mice were killed 48 h later. Lungs were either fixed with formalin or subjected to BAL. (a) Lung sections were stained with H&E and analyzed by light microscopy; bars: 4 μm. (b) BAL fluids were collected and centrifuged; cells were then differentially stained, and eosinophils were counted. Data are expressed as percent eosinophils from total cell number collected by BAL. Data are means ± SD of values from at least six mice per group. *Difference from challenged mice without TIQ-A treatment, P<0.01; #difference from TIQ-A-treated mice before OVA-challenge, P<0.01. PARP, poly(ADP-ribose) polymerase; TIQ-A, thieno[2,3-c]isoquinolin-5-one; BAL, bronchio-alveolar lavage; H&E, hematoxylin and eosin; OVA, ovalbumin; i.p., intraperitoneally.
We tested the efficacy of TIQ-A in controlling inflammation when administered after allergen (OVA) challenge. As shown in Fig. 1a, infiltration of inflammatory cells into the airways of OVA-challenged mice was greatly reduced in animals that received a single TIQ-A i.p. injection either 1 or 6 h after challenge. An assessment of the number of eosinophils in BAL fluids showed that post-challenge TIQ-A administration exerted a similar pronounced reduction in eosinophil recruitment compared with administration of the drug before challenge (Fig. 1b). These results not only show that PARP plays a critical role in allergen-induced lung inflammation but also presents PARP inhibition as a potential therapeutic strategy for avoiding the manifestation of eosinophilia after allergen exposure.
Post-challenge thieno[2,3-c]isoquinolin-5-one administration severely suppress T-helper type 2 cytokine and immunoglobulin E production after ovalbumin challenge
The expression of Th2-type cytokines and production of IgE are critical factors in promoting allergen-induced lung inflammation. Cytokine expression is crucial for the generation and recruitment of eosinophils into the lung upon allergen exposure [19, 20]. Accordingly, we compared the effects of PARP inhibition post vs. prior OVA challenge on the expression of the Th2 cytokines IL-4, IL-5, IL-13. Cytokine production was measured from BAL fluids 24 h after challenge. As expected, OVA exposure induced a robust production of each of these cytokines (Figs 2a—c). TIQ-A administration post-challenge suppressed production of Th2 cytokines even more than TIQ-A administration before OVA challenge (Figs 2a—c). These results not only demonstrate that PARP inhibition exerts its anti-inflammatory effect by reducing the production of Th2 cytokines but also show that post-challenge TIQ-A administration is more efficacious in blocking such an allergen-induced airway inflammatory response.
Fig. 2.

Effect of PARP inhibition by TIQ-A on Th2 cytokine production following OVA sensitization and challenge. Mice were sensitized to and challenged with OVA or left unchallenged. Different experimental groups received prior or post-challenge administration of TIQ-A as described in Fig. 1. Mice were killed 24 h later, after which they were subjected to BAL. BAL fluids were collected and centrifuged. The supernatant was used for cytokine assessment using the Bio-Rad Bioplex system (a—c) or IgE (d) levels by ELISA. Data are given as means ± SD of values obtained from at least six mice per group. *Difference from challenged mice without TIQ-A treatment, P<0.01; #difference from TIQ-A-treated mice before OVA-challenged, P<0.01; ?difference is not statistically significant. PARP, poly(ADP-ribose) polymerase; TIQ-A, thieno[2,3-c]isoquinolin-5-one; BAL, bronchio-alveolar lavage; Th2, T-helper type 2; OVA, ovalbumin.
We next examined whether the marked reduction in eosinophil recruitment into airways of OVA-challenged mice conferred by PARP inhibition was associated with a reduction in IgE production. Levels of OVA-specific IgE were measured in BAL fluid samples 24 h post-challenge. Figure 2d shows that TIQ-A caused a moderate reduction in the levels of OVA-specific IgE when administered before OVA challenge but almost completely blocked IgE production when administered after OVA challenge. These results are consistent with the effect of TIQ-A administration on IL-4.
Post-challenge thieno[2,3-c]isoquinolin-5-one administration completely stopped mucus production in airways of ovalbumin-challenged mice
Mucus hypersecretion is a consistent trait of allergic inflammatory response, especially in fatal episodes of asthma [21]. A number of reports have provided evidence that support a critical role for IL-13 in mucus hypersecretion during asthma [17]. Strategies that block IL-13 production, such as administration of the soluble IL-13Ra2-Ig, or through IL-13 gene deletion in mice, prevent airway production of mucus [6]. Given the severe effect of PARP inhibition post allergen-challenge on IL-13 production, we wanted to determine its effect on mucus hypersecretion in our experimental murine model. Lung sections from different experimental mouse groups were stained with PAS to detect mucus-secreting goblet cells.
While lungs of OVA-challenged wild-type mice exhibited substantial hyper-production of mucus, as evidenced by the pronounced red staining of goblet cells, the lungs of OVA-challenged wild-type mice that received TIQ-A either 1–6 h after challenge exhibited a complete absence of PAS-positive goblet cells (Fig. 3a). Quantitative assessment of mucus production expressed as a histological mucin index show that while treatment with TIQ-A before OVA challenge significantly reduced mucus production, post-challenge treatment with the drug completely blocked such mucus production (Fig. 3b).
Fig. 3.

Effect of PARP inhibition by TIQ-A on mucus hypersecretion by goblet cells in airways of OVA-sensitized and challenged mice. C57BL/6 mice were sensitized to OVA and subjected to a challenge with aerosolized OVA or left unsensitized and unchallenged. Mice received an i.p. injection of TIQ-A or vehicle alone 2 h prior, 1 h post, or 6 h post-OVA challenge. Mice were killed 48 h later. Lungs were fixed with formalin. Lung sections were stained with PAS, after which histological analysis was conducted by light microscopy; bars: 4 μm; arrows indicate PAS positive goblet cells. (b) The extent of mucus production (histological mucin index) was assessed using Image-Pro Plus software. Data are means ± SD of values from at least six mice per group. Difference from challenged mice without TIQ-A treatment, P<0.01; #difference from TIQ-A-treated mice before OVA-challenged, P<0.01. PARP, poly(ADP-ribose) polymerase; TIQ-A, thieno[2,3-c]isoquinolin-5-one; BAL, bronchio-alveolar lavage; H&E, hematoxylin and eosin; OVA, ovalbumin; i.p., intraperitoneally.
Post-challenge thieno[2,3-c]isoquinolin-5-one administration prevents methacholine-induced airway hyperrresponsiveness in ovalbumin-challenged mice
We next assessed whether the protective effects of PARP inhibition by TIQ-A against eosinophilia and induction of the Th2 response coincided with prevention of AHR, a major hallmark of human asthma. Lung resistance to increasing doses of MeCh was assessed using the forced oscillation technique (see ‘Materials and methods’). Figure 4 shows that administration of TIQ-A post-OVA challenge almost completely blocked AHR to inhaled MeCh at a dose of 50 mg/mL 24 h after OVA exposure.
Fig. 4.
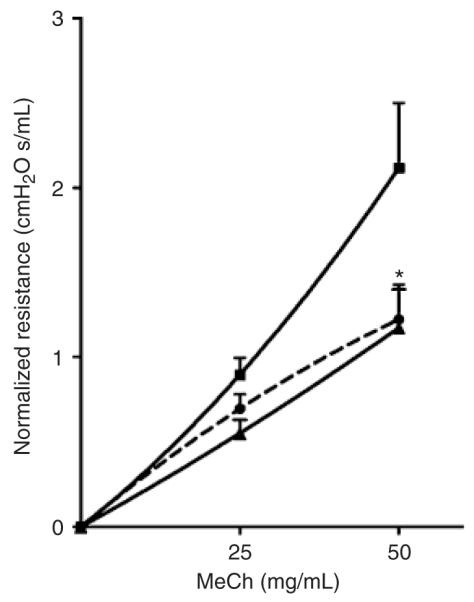
Effect of PARP inhibition by TIQ-A on airway hyperreactivity in OVA-sensitized and challenged mice. C57BL/6 mice were sensitized to OVA and subjected to a challenge with aerosolized OVA (∎). A group of mice received an i.p. injection of TIQ-A (▴) or vehicle alone (●)after OVA exposure. Lung resistance to increasing doses of MeCh was assessed using the forced oscillation technique. Lung resistance is plotted as a function of increasing doses of inhaled MeCh, using single compartment model and expressed as mean ± SEM. n = 5 per group. *Difference from challenged mice without TIQ-A treatment, P < 0.01. PARP, poly(ADP-ribose) polymerase; TIQ-A, thieno[2,3-c]isoquinolin-5-one; BAL, bronchio-alveolar lavage; MeCh, methacholine; OVA, ovalbumin; i.p. intraperitoneally.
Taken together, our results strongly suggest a great clinical potential for PARP inhibition in blocking the inflammatory phenotype and AHR associated with allergen exposure such as that observed in asthma.
Discussion
Asthma is a complex disease that involves a variety of cell and non-cell components [1, 2, 22]. Antigen exposure initiates this disease in sensitive subjects and results in an array of events leading to the generation of CD4+ Th2 cells, IgE production, recruitment of eosinophils into the AHR, hyperplasia of goblet cells and the ensuing mucus production [20, 23]. CD4+ Th2 cells play a pivotal role in orchestrating the allergic response, including effects on the production of key Th2 cytokines necessary for the build up of eosinophils in the lungs as well as activation of B lymphocytes to generate IgE [20].
It is now widely accepted that airway inflammation is the key factor underlying the pathogenesis of asthma [1, 22]. While corticosteroids remain the most important anti-inflammatory treatment for asthma, they are rather nonspecific in their actions [6, 7]. Their use also raises concerns over side-effects and compliance issues, particularly in children and adolescents [7]. Thus, there is much effort underway to develop novel specific and safer therapies for the treatment of asthma. Inhibitors of cytokines figure prominently in these novel therapeutic approaches [6, 9].
As Th2 lymphocytes play a key role in orchestrating the eosinophilic inflammatory response in asthma, blocking the release or effects of these cytokines may have therapeutic potential. Numerous studies suggest that blocking individual Th2 cytokines have shown some promise, but not complete success, in the treatment of asthma [6, 8]. Inhibition of IL-5 with mAb has been effective in reducing circulating and airway eosinophils, but, disappointingly, does not appear to be beneficial to functional responses [8, 24]. IL-4 blocking antibodies inhibit allergen-induced AHR, goblet cell metaplasia and pulmonary eosinophilia in a murine model [6]. Inhibition of IL-4, despite promising early results in asthma, has been disappointing at the clinical level [25]. A blocking antibody to IL-13 is very effective in reversing chronic allergic inflammation, indicating the potential for IL-13 blockage in the treatment of chronic asthma [7]. However, Perkins et al. [26] have recently shown that IL-4 can induce all of the acute changes in airway inflammation that have been associated with IL-13, including BAL fluid eosinophilia, AHR, and goblet cell hyperplasia, in the absence of IL-13 signaling.
Taken together, these finding suggest that attempts to block specific cytokines may not be adequate for blocking chronic inflammation in asthma, as so many cytokines are involved and there is considerable redundancy of effects. Development of drugs that have a more general effect on cytokine synthesis may be more successful. Accordingly, PARP inhibition by TIQ-A following allergen exposure plays a critical role in the amelioration of the pathogenic process of allergic-induced lung inflammation in mice. TIQ-A down-regulates the production of Th2 cytokines IL-4, IL-5, and IL-13 and IgE in a concerted manner as well as AHR as measured in response to inhaled MeCh. Tissue eosinophilia [27] and expression of eotaxin-1 [28], a critical chemokine for eosinophil recruitment to the airways after allergen exposure, are important variables in animal models of asthma. However, the effect of PARP inhibition on such variables remains to be determined.
We have reported that PARP inhibition before allergen exposure protects against infiltration of eosinophils and mucus production into lung airways by suppressing Th2 cytokine production [16]. However, our current findings show an equally potent action of TIQ-A, a novel PARP inhibitor, against the allergen-induced airway eosinophilia and mucus production, when it is given after the allergen exposure. The effects of TIQ-A administration on mucus production is consistent with its effect on IL-13 and the other cytokines. Further, we show for the first time that PARP inhibition almost completely prevents AHR. Because most asthma symptoms are associated with accidental exposure to known or unknown allergens, it becomes crucially relevant to determine whether promising new drugs with preventive outcomes, would also harbor a therapeutic potential.
Our findings suggest that timing of drug administration is crucial for its maximal beneficial action. The reduction in eosinophilia was accompanied by more pronounced suppression of IL-4, IL-5, IL-13, and IgE and an absence of mucus production by airway goblet cells. These effects were also accompanied with the prevention of AHR, a major feature of human asthma. Importantly, the marked protection observed as late as 6 h after allergen exposure enhances the clinical potential of the drug considerably. These findings broaden the current perspectives in our understanding of the functions of PARP in an allergen-induced airway and present new insight into the immunopharmacological effects of PARP inhibition in blocking critical inflammatory traits associated with asthma.
Acknowledgements
We thank Sangeetha Kandan for maintaining our mouse colony and for her technical assistance. We also thank Drs Alberto Martinez and Cooper Woods and the Ochsner Clinic Foundation (Jefferson, LA, USA) for providing us with laboratory space to pursue our research following Hurricane Katrina.
Funding Sources: This work was supported in part by grants HL072889 and 1P20RR18766 (overall P. I. Dr D. Kapusta) from the National Institute of Health to H. Boulares.
Footnotes
Disclosures: none
References
- 1.Fireman P. Understanding asthma pathophysiology. Allergy Asthma Proc. 2003;24:79–83. [PubMed] [Google Scholar]
- 2.Maddox L, Schwartz DA. The pathophysiology of asthma. Annu Rev Med. 2002;53:477–98. doi: 10.1146/annurev.med.53.082901.103921. [DOI] [PubMed] [Google Scholar]
- 3.Shen HH, Ochkur SI, McGarry MP, et al. A causative relationship exists between eosinophils and the development of allergic pulmonary pathologies in the mouse. J Immunol. 2003;170:3296–305. doi: 10.4049/jimmunol.170.6.3296. [DOI] [PubMed] [Google Scholar]
- 4.Prescott SL. New concepts of cytokines in asthma: is the Th2/Th1 paradigm out the window? J Paediatr Child Health. 2003;39:575–9. doi: 10.1046/j.1440-1754.2003.00237.x. [DOI] [PubMed] [Google Scholar]
- 5.Poole JA, Matangkasombut P, Rosenwasser LJ. Targeting the IgE molecule in allergic and asthmatic diseases: review of the IgE molecule and clinical efficacy. J Allergy Clin Immunol. 2005;115:S376–85. doi: 10.1016/j.jaci.2004.12.1124. [DOI] [PubMed] [Google Scholar]
- 6.Barnes PJ. New therapies for asthma. Trends Mol Med. 2006;12:515–20. doi: 10.1016/j.molmed.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Walsh GM. Targeting airway inflammation: novel therapies for the treatment of asthma. Curr Med Chem. 2006;13:3105–11. doi: 10.2174/092986706778521779. [DOI] [PubMed] [Google Scholar]
- 8.Finegold I. Allergen immunotherapy: present and future. Allergy Asthma Proc. 2007;28:44–9. doi: 10.2500/aap.2007.28.2971. [DOI] [PubMed] [Google Scholar]
- 9.Barnes PJ. Transcription factors in airway diseases. Lab Invest. 2006;86:867–72. doi: 10.1038/labinvest.3700456. [DOI] [PubMed] [Google Scholar]
- 10.Chiarugi A, Meli E, Calvani M, et al. Novel isoquinolinone-derived inhibitors of poly(ADP-ribose) polymerase-1: pharmacological characterization and neuroprotective effects in an in vitro model of cerebral ischemia. J Pharmacol Exp Ther. 2003;305:943–9. doi: 10.1124/jpet.103.048934. [DOI] [PubMed] [Google Scholar]
- 11.Nicoletti VG, Stella AM. Role of PARP under stress conditions: cell death or protection? Neurochem Res. 2003;28:187–94. doi: 10.1023/a:1022316914492. [DOI] [PubMed] [Google Scholar]
- 12.Kraus WL, Lis JT. PARP goes transcription. Cell. 2003;113:677–83. doi: 10.1016/s0092-8674(03)00433-1. [DOI] [PubMed] [Google Scholar]
- 13.Rosenberg HF, Phipps S, Foster PS. Eosinophil trafficking in allergy and asthma. J Allergy Clin Immunol. 2007;119:1303–10. doi: 10.1016/j.jaci.2007.03.048. [DOI] [PubMed] [Google Scholar]
- 14.Lee JJ, Dimina D, Macias MP, et al. Defining a link with asthma in mice congenitally deficient in eosinophils. Science. 2004;305:1773–6. doi: 10.1126/science.1099472. [DOI] [PubMed] [Google Scholar]
- 15.Humbles AA, Lloyd CM, McMillan SJ, et al. A critical role for eosinophils in allergic airways remodeling. Science. 2004;305:1776–9. doi: 10.1126/science.1100283. [DOI] [PubMed] [Google Scholar]
- 16.Oumouna M, Datta R, Oumouna-Benachour K, et al. PARP-1 inhibition prevents eosinophil recruitment by modulating Th2 cytokines in a murine model of allergic airway inflammation: a potential specific effect on IL-5. J Immunol. 2006;177:6489–96. doi: 10.4049/jimmunol.177.9.6489. [DOI] [PubMed] [Google Scholar]
- 17.Whittaker L, Niu N, Temann UA, et al. Interleukin-13 mediates a fundamental pathway for airway epithelial mucus induced by CD4 T cells and interleukin-9. Am J Respir Cell Mol Biol. 2002;27:593–602. doi: 10.1165/rcmb.4838. [DOI] [PubMed] [Google Scholar]
- 18.You D, Becnel D, Wang K, Ripple M, Daly M, Cormier SA. Exposure of neonates to respiratory syncytial virus is critical in determining subsequent airway response in adults. Respir Res. 2006;7:107. doi: 10.1186/1465-9921-7-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lampinen M, Carlson M, Hakansson LD, Venge P. Cytokine-regulated accumulation of eosinophils in inflammatory disease. Allergy. 2004;59:793–805. doi: 10.1111/j.1398-9995.2004.00469.x. [DOI] [PubMed] [Google Scholar]
- 20.Ngoc PL, Gold DR, Tzianabos AO, Weiss ST, Celedon JC. Cytokines, allergy, and asthma. Curr Opin Allergy Clin Immunol. 2005;5:161–6. doi: 10.1097/01.all.0000162309.97480.45. [DOI] [PubMed] [Google Scholar]
- 21.Morcillo EJ, Cortijo J. Mucus and MUC in asthma. Curr Opin Pulm Med. 2006;12:1–6. doi: 10.1097/01.mcp.0000198064.27586.37. [DOI] [PubMed] [Google Scholar]
- 22.Elias JA, Lee CG, Zheng T, Ma B, Homer RJ, Zhu Z. New insights into the pathogenesis of asthma. J Clin Invest. 2003;111:291–7. doi: 10.1172/JCI17748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hogg JC. The pathology of asthma. Apmis. 1997;105:735–45. doi: 10.1111/j.1699-0463.1997.tb05079.x. [DOI] [PubMed] [Google Scholar]
- 24.Leckie MJ. Anti-interleukin-5 monoclonal antibodies: preclinical and clinical evidence in asthma models. Am J Respir Med. 2003;2:245–59. doi: 10.1007/BF03256653. [DOI] [PubMed] [Google Scholar]
- 25.Steinke JW. Anti-interleukin-4 therapy. Immunol Allergy Clin North Am. 2004;24:599–614. doi: 10.1016/j.iac.2004.06.008. vi. [DOI] [PubMed] [Google Scholar]
- 26.Perkins C, Wills-Karp M, Finkelman FD. IL-4 induces IL-13-independent allergic airway inflammation. J Allergy Clin Immunol. 2006;118:410–9. doi: 10.1016/j.jaci.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 27.Kumar RK, Herbert C, Yang M, Koskinen AM, McKenzie AN, Foster PS. Role of interleukin-13 in eosinophil accumulation and airway remodelling in a mouse model of chronic asthma. Clin Exp Allergy. 2002;32:1104–11. doi: 10.1046/j.1365-2222.2002.01420.x. [DOI] [PubMed] [Google Scholar]
- 28.Foster PS, Mould AW, Yang M, et al. Elemental signals regulating eosinophil accumulation in the lung. Immunol Rev. 2001;179:173–81. doi: 10.1034/j.1600-065x.2001.790117.x. [DOI] [PubMed] [Google Scholar]