

Abstract
What could be a better way to study virus trafficking than ‘miniaturizing oneself’ and ‘taking a ride with the virus particle’ on its journey into the cell? Single-virus tracking in living cells potentially provides us with the means to visualize the virus journey. This approach allows us to follow the fate of individual virus particles and monitor dynamic interactions between viruses and cellular structures, revealing previously unobservable infection steps. The entry, trafficking and egress mechanisms of various animal viruses have been elucidated using this method. The combination of single-virus trafficking with systems approaches and state-of-the-art imaging technologies should prove exciting in the future.
Introduction
Viruses live brief but eventful lives. During its short life cycle, a virus interacts with various cellular structures, taking advantage of different cellular environments to optimize the chances of replication. The infection starts with the virus binding to specific receptors or attachment factors on the cell surface1. Although some viruses release their genomes directly into the cell by breaking the plasma membrane barrier, most viruses enter cells by endocytosis2,3. After internalization, virus particles are sequestered in endocytic organelles until the proper conditions are met for viral-genome release. Two main strategies are used for genome release: enveloped viruses fuse with a cell membrane, thereby exposing the genome or capsid to the cytosol, whereas non-enveloped viruses partially disrupt cellular membranes to release viral RNA or DNA. After entry, the viral genome is transported to specific sites in the cytoplasm or nucleus for replication and expression. The newly synthesized viral proteins and genetic material are then transported to specific sites for assembly into progeny viruses. Although virus assembly can occur at the plasma membrane, many viruses begin the assembly process in intracellular organelles, such as the endoplasmic reticulum or multivesicular bodies. Assembled virions exit by exocytosis or after lysis of the host cell. Mature viruses can subsequently infect new target cells.
It is evident that viral infections are complex processes that include many steps and interactions with different cellular structures. Not only are these interactions dynamic, but often the cellular structures are themselves dynamic. Furthermore, the same virus might infect cells by several different routes and most viral entry events might be futile. Therefore, studying viral infection mechanisms should benefit from an experimental approach that allows researchers to follow the fate of individual virus particles, to probe dynamic interactions between viruses and cellular machinery, and to dissect the steps of the infection process so that the mechanisms underlying each step can be determined. Single-virus tracking in living cells offers precisely this opportunity.
Single-virus tracking in live cells
Single-virus tracking is a real-time imaging technique that uses fluorescence microscopy to monitor individual virus particles or viral components in live cells. To successfully track a single virus particle, the virus and cellular structures of interest must be fluorescently labelled, the microscope must be sufficiently powerful for the detection of single viruses or viral components in real time, and the acquired image series must be analysed to extract useful information.
Technical aspects of single-virus imaging
The first step towards single-virus tracking in live cells is labeling the viral components and relevant cellular structures. A crucial requirement is that both viruses and cellular structures need to be labelled with a sufficient number of fluorophors for detection at the single particle level without inhibiting viral infectivity and cell functions. The external components of a virus, such as the capsids of a non-enveloped virus or the membrane of an enveloped virus, can be readily labelled with fluorescent dyes (BOX 1). Tracking virions that are labelled in this manner can provide important insights into the transport or uncoating mechanisms of viruses (FIG. 1a; Supplementary information S1 (Movie)). Internal components, such as the capsid or genome of an enveloped virus, are typically inaccessible for labelling after the completion of viral assembly and purification. The development of fluorescent proteins (FPs)4 solved this technical problem. The key feature of FPs is that they are genetically encoded and therefore allow specific viral components to be labelled during viral replication and prior to assembly. Moreover, FPs can also be used to label endogenous cellular proteins, which facilitates the study of virus–cell interactions (FIG. 1b; Supplementary information S2 (movie)).
Box 1 | Strategies for labelling various viral components
Two general strategies exist for labelling viruses: the fusion of a target viral protein with a fluorescent protein (FP), or direct chemical labelling with small dye molecules. The palette of FPs has recently expanded to include brighter and more photostable FPs for a broad range of colours7,80. In addition, pH sensitive and photo-switchable FPs have also been developed4. To fuse an FP with a viral structure, the DNA sequence of the FP has to be inserted into the open reading frame of the target viral protein. Labelling occurs during virus replication in host cells. In order to generate a virus that is visible at the single particle level with a high signal-to-noise ratio, attachment of multiple FPs per virus is necessary. Therefore viral components that exist in multiple copies per virion, such as capsid or tegument proteins, are useful labeling targets (see figure).
In contrast to FPs, chemical labels (CLs) can be attached to viral structures at different time-points during the viral life cycle. An increasing number of CLs with different characteristics can be either covalently attached or non-covalently associated with the target protein8. For example, capsids of purified non-enveloped viruses can be covalently labelled with amino-reactive dyes (for example, cynine or Alexa dyes)6,18,42. The outer membrane of purified enveloped viruses can be labeled by the incorporation of lipophilic dyes for example, DiD, DiI and analogues5. Metabolic labelling can be used to label viruses during viral replication. For instance, cell-permeable biarsenical dyes can specifically bind to viral proteins that contain tetracystein peptide sequences9,57. Labelling of viral genomes can be done by infecting cells in the presence of fluorescently labeled nucleotides or nucleic acid-binding dyes (B.B. et al., unpublished data). Antibodies are typically not useful for virus labelling in tracking experiments in live cells because they can block the function of viral proteins after binding.
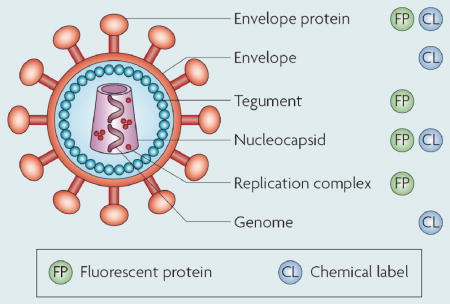
Figure 1. Time-lapse images of influenza viruses in live cells.

a | Stacked, time-lapse images of influenza viruses in living cells, revealing actin and microtubule-dependent transport. The virus is labelled with the lipophilic dye, DiD. The sudden colour change from blue/pink to yellow/white indicates a dramatic increase in the fluorescence signal of DiD, indicating the fusion of the virus with an endosome. A movie showing the time course of one of these virus particles can be found in the Supplementary Information S1 (movie). b | Simultaneous images of a DiD-labelled virus (upper panels and red in lower panels) and fluorescent protein-labelled clathrin (middle panels and green in lower panels) in a cell show the internalization of the virus by a clathrin-coated vesicle. The centres of dotted circles in the middle panels indicate the virus positions. Overlay of green and red signals appears yellow. The time (in seconds) after viral attachment and different stages of viral entry are shown below the images. A movie showing the time course of this virus particle is shown in Supplementary Information S2 (movie). Part (a) reproduced with permission from REF.5 © (2003) National Academy of Sciences, USA. Part (b) reproduced with permission from Nature Structural and Molecular Biology REF.13 © (2004) Macmillan Publishers Ltd.
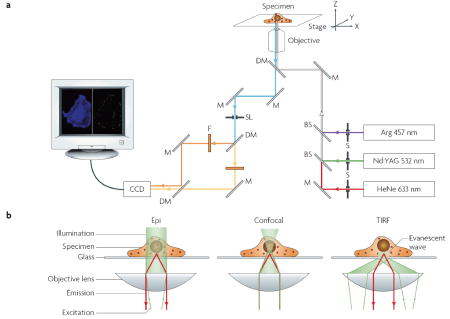
Labelled viruses are visualized in live cells using a fluorescence microscopy set-up. Three imaging geometries are used most often. Among these, epifluorescence microscopy is the simplest to set-up (BOX 2). This method also has the largest imaging depth and is often the method of choice when long-range viral trafficking or transport is being studied. However, owing to the background autofluorescence (noise) of cells, epidetection is inadequate for imaging virus particles that contain only a few fluorescent molecules. To reduce the background noise, confocal detection, or excitation by total internal reflection is used (BOX 2). Confocal microscopy requires a rapid scanning scheme (laser scanning confocal) or multiplexed detection (spinning disc confocal) to acquire images of living cells (BOX 2). By using confocal imaging it is possible to acquire 3-dimensional images, but this advantage is mitigated by a relatively severe signal loss. Total internal reflection fluorescence (TIRF) microscopy is a wide-field imaging technique that has minimum signal loss. However, the excitation depth of an evanescent wave generated by total internal reflection is only 100–200 nm, limiting the application to events that occur close to the cell surface. Therefore TIRF imaging is typically used to monitor the early events of viral entry. In addition to these background-reduction techniques, the development of charge-coupled device (CCD) cameras with high quantum yields (60–80%) and rapid frame rates (1–500 ms) was also an important technological advance that benefited single-virus-imaging technology.
Box 2 | Fluorescence microscopes for single-virus imaging
A typical single-virus tracking setup includes an inverted microscope with a temperature controlled stage, several lasers, optics and a sensitive detector. In the figure, part (a) shows the microscope setup. Various laser lines, such as Argon ion (Arg; 457 nm, 488 nm and 514 nm), Krypton ion (647 nm; not shown), helium-neon (HeNe; 543 nm, 594 nm and 633 nm) and Nd:YAG (532 nm) lasers, can function as light sources to excite different fluorophors. Multicolour imaging is enabled by a combination of several different laser lines with specific excitation and detection optics that allow different coloured excitation to be combined at the sample, and different coloured fluorescence emissions to be separately detected. In the figure, part (b) shows imaging geometries. In the epi-fluorescence geometry (left panel), a collimated light beam illuminates a large sample area (~ 100 μm in linear dimension)81. Fluorescence emission from the sample is collected by a high numerical aperture objective and detected by a CCD camera. The advantages of this scheme are the low signal loss, rapid wide-field detection and large excitation depth, which allow single particles to be tracked in a large sample volume. The disadvantage is its poor rejection of the fluorescence background signal from the cell. Confocal microscopy (middle panel) uses a focused light beam and spatial filtering techniques (pinholes in excitation and detection paths) to eliminate out-of-focus background fluorescence, but at the cost of signal reduction82. For the observation of fast dynamics in live cells, a spinning disk confocal setup is often preferred over the relatively slow scanning confocal microscope, which relies on rotating mirrors to scan a focused laser beam in the imaging plane and a point detector, such as avalanche photodiode or photomultiplier tubes, for signal acquisition81. The spinning disk confocal microscope relies on a pair of rapidly rotating discs, one with an array of pinholes and the other with correspondingly aligned micro-lenses. Thereby the excitation light is subdivided into thousands of beams, which simultaneously scan the entire field at a rate of >1000 frames per second. This scheme effectively creates a wide-field image detected by a CCD camera. When combined with Z-direction scanning of the sample stage, both confocal schemes allow the construction of 3-dimensional images. In a TIRF microscope (right panel), the incident light strikes the interface between two optical media of different refractive indices (for example, a glass substrate and a cell) at a sufficiently large angle to induce total reflection83. As a result, an evanescent excitation field is generated, extending only a few hundred nanometers into the second medium. This wide-field imaging geometry offers the best rejection of background signal, but can only be used to detect events occurring close to the adhering surface of the cell. In order to simultaneously track a large number viruses, a camera chip with a high number of pixels is needed, which allows a large field of view. BS, beam splitter; CCD, charge-coupled device; DM, dichroic mirrors; F, filter; M, mirrors; SL, optical slits to control image size; S, shutters; TIRF, total internal reflection fluorescence.
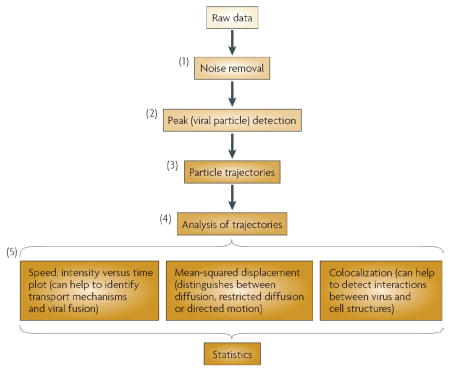
Finally, live-cell imaging is a technique with a high information content. Each frame of an image contains megabytes of data and each viral infection movie typically comprises hundreds to thousands of frames. Recently, developments in automated image analysis have enabled us to convert these movies into large statistical ensembles of viral trajectories that show viral transport mechanisms and virus–cell interactions. We briefly describe the image-analysis principles that are crucial for single-virus tracking in BOX 3.
Box 3 | Image analysis
Single particle-tracking algorithms have made it possible to capture the detailed dynamics of viral entry and trafficking mechanisms. The algorithms convert the raw stack of fluorescence images into a set of trajectories that specify the coordinates and fluorescence intensity of each particle as a function of time. The analysis typically begins with processing each image frame with a spatial filter to remove noise (step 1 in the figure) and is followed by automated identification of isolated fluorescent peaks that correspond to virus particles (step 2)13. To identify peaks, filtered images are scanned for local maxima above a threshold, giving an estimate of the coordinates of the particles84. Trajectories can be established by connecting particles from one frame to the next using nearest-neighbour association and the motion history of individual particles (step 3)13,57,85. Furthermore, a split-and-merge mechanism allows the continued tracking of particles even when they merge or separate into smaller entities86. The overlap in fluorescence emission between two or more coloured channels can be used to probe the assembly or disassembly of different viral components or the interactions between viral and cellular structures. Analysis of the spatial and temporal trajectories of viruses allows us to identify the crucial events in infection pathways (such as viral fusion), to analyse the transport mechanisms of viruses and to detect interactions between virus particles and cellular structures (step 4). Other analyses are also possible at this stage. The automated analysis of particle trajectories is typically followed by a manual check for ambiguities and errors. A large number of trajectories need to be analysed to generate a statistically sound description of viral movement and virus–cell interaction (step 5). The variety of viral behaviours should be observed in multiple independent experiments and ideally in different cell types.
Timeline | Key developments for single-virus tracking.
Challenges in single-virus tracking
Virus and cell labeling
Viruses are tiny — the smallest viruses have a diameter of only 20–30 nm, limiting the number of fluorescent probes that can be attached to a virus particle without causing self-quenching effects between dye molecules or affecting viral infectivity. For example, whereas hundreds to thousands of dye molecules can be attached to a relatively large influenza virion without affecting its infectivity5, only 30–50 dye molecules can be attached to a polio capsid owing to its small size (B.B. et al., unpublished data). In extreme examples, such as the adeno-associated virus, attaching more than a few dye molecules to the capsid renders the virus non-infectious6. This limit can be even more stringent for the labelling of internal viral components, such as the genome. Because cells also fluoresce, it can be difficult for microscopists to distinguish between the signal from a single-virus particle and the background fluorescence of a cell. As the cell auto-fluorescence background levels tend to be weaker at longer excitation wavelengths, it is helpful to use fluorescent probes with longer wavelengths for virus labelling. The red fluorescent dyes, such as Cy5, Alexa 647 and DiD (excitation maximum ~ 650 nm) and red fluorescent proteins, such as mCherry (excitation maximum ~ 590 nm7) are often good choices for virus labelling. The situation becomes more troublesome when studying viral assembly, because fluorescent viral proteins are usually overexpressed in an infected cell, which produces a considerable amount of background signal and makes it difficult to identify assembling virions. In addition, the relatively bulky size of fluorescent proteins might interfere with viral structures and functions, especially when attached to the internal components of tightly packed viruses. One advance that might help to solve this problem is the development of small molecules that specifically bind to genetically encoded peptide sequences8,9.
Inorganic nanoparticles, such as semiconductor quantum dots, are also promising for use as probes10,11. Quantum dots are much brighter and more robust than fluorescent dyes and proteins, but their bulky size places severe limits on their application. Research is underway to reduce the size of quantum dots (M. Bawendi, A. Ting and S. Weiss, personal communication) and to synthesize other small inorganic probes12.
In order to study the interaction of viral particles with cellular structures, it is often helpful to label cellular proteins with FPs. Although this can be accomplished by fusing the FP gene with the gene of interest and expressing the fusion protein in host cells, this approach can be fraught with problems. It is sometimes challenging to obtain an optimum level of fusion-protein expression that allows the detection of individual cellular structures yet does not perturb cellular processes. Production of fluorescently tagged proteins needs to be accompanied by a proof-of-function study. For example, when clathrin-coated pits are marked with FP-tagged clathrin, the uptake of well characterized cargos that are known to enter cells by clathrin-dependent endocytosis, such as transferrin and low-density lipoprotein (LDL), should be similar to the uptake behaviour that is observed in wild-type cells. Even when such conditions are met, endogenous unlabelled proteins might still complicate quantitative analysis and leave open the question of whether all relevant cellular structures are indeed fluorescently labelled. Functional and morphological control experiments are necessary to remove such doubt. Again, using clathrin-coated pits as an example, we and others have shown that when FP-tagged clathrin-light chain A (CLa) was expressed in cells in the presence of endogenous CLa, more than 96% of clathrin-coated pits visualized by immunofluorescence or with fluorescently labelled transferrin co-localize with FP-marked structures13,14, showing that nearly all clathrin-coated pits are detected by the FP signal.
Large particle-to-pfu ratio
Many viruses have high particle-to-pfu (plaque forming unit) ratios; that is, hundreds to thousands of virus particles are necessary to infect a single cell. How can one be sure of tracking an infectious (as opposed to a non-infectious) virus particle? It is advisable to simultaneously track the maximum possible number of virus particles that are entering one cell and separate tracked particles into groups that have similar behaviours. Among these viruses, it should be possible to focus on the group of virus particles that are most likely to cause a productive infection. For example, fusion between viral and cellular membranes allows the virus to release its contents into the cell and is therefore a crucial infection step for enveloped viruses. So monitoring the fusion event of enveloped viruses, by fluorescence dequenching5, enables the researcher to selectively focus on a subgroup of viruses that will have a productive infection. For non-enveloped viruses, viral genome release can be monitored as a marker of a productive infection. This can be done by separately labelling the viral capsids and genome with different fluorescent probes (B.B. et al., unpublished results) as a marker for successful viral entry. In most cases, single-virus-tracking experiments cannot cover a full cycle of infection. In order to confirm that the tracked and analysed viral behaviours are indicative of those of the infectious virus particles, a combination of single-virus tracking with virological assays, such as a plaque assay, is necessary. For example, when inhibitors (drugs or siRNA) of viral entry are monitored using both single-particle tracking and viral titer assays, complete correlation between the two assays should give confidence that the single-particle tracking experiments are monitoring entry pathways that are relevant to infection.
Challenges associated with image instrumentation and analysis
In common with other imaging techniques, other challenges for single-virus tracking arise from the limited spatial and time resolution of fluorescence imaging and the need to analyse a rapidly increasing amount of image data, both discussed later.
Despite the challenges described above, recent advances in optics, device physics, chemistry and molecular biology have enabled us to track individual virus particles in living cells and to probe dynamic interactions between viruses and cellular machinery in real time (TIMELINE). In the following sections, we will review advances made in viral entry, transport and egress due to single-virus tracking in live cells.
Viral entry
Viruses exploit specific receptors on the cell surface to identify and infect target cells. However, these receptors are often rare or distributed in regions that are not readily accessible to incoming virions. Therefore, many viruses first bind to relatively non-specific attachment factors, such as carbohydrates, and migrate along the cell surface to locate specific receptor(s)1 (FIG. 2). Specific virus–receptor interactions activate signaling cascades, guide the virus into endocytic pathways and/or trigger conformational changes in the virus envelope or capsid proteins for genome release1 (FIG. 2). Single-virus tracking offers an ideal method for the visualization of virus movement. Multicolour live-cell imaging that allows tracking of single-virus particles together with fluorescent-protein labelled cellular structures provides a powerful tool for studying viral entry mechanisms. Using this approach, even transient interactions between viruses and cellular proteins can be detected. Multicolour labelling of distinct virus components also allows viral disassembly during entry to be monitored. Such dynamic information is crucial for a more complete understanding of viral entry mechanisms.
Figure 2. Viral entry and transport.

Viruses attach to the plasma membrane, surf on the cell surface or along the filopodia (1–3), and bind to specific receptors before entering the cell. Viruses can directly fuse with the plasma membrane (2). They also hijack endocytic pathways, including clathrin- dependent (1), caveolin-dependent (3) or clathrin- and caveolin-independent (4) pathways for internalization. After internalization and transport through the actin matrix, vesicles that contain virus are transported by dynein or dynactin along microtubules towards the microtubule organizing centre (MTOC). This might include trafficking of viruses through endosomes, caveosomes or the endoplasmic reticulum, prior to the release of the virus into the cytoplasm. Capsids can also be transported by dynein or dynactin along microtubules. From the MTOC, capsids can be transported by kinesin towards the replication site of the nucleus (5). Some viruses release their genetic material into the cytosol whereas others transport their genomes into the nucleus. The key shows how the different components have been labelled previously. The inset panel shows the caveolin-mediated endocytosis of Simian virus 40 (SV40). The arrowheads indicate SV40-containing caveolae co-localized with actin tails. The dye-labelled SV40 particles are shown in red and the fluorescent protein-labelled caveolin and actin are in purple and green, respectively. Scale bar represents 3μm. Inset panel reproduced with permission from REF.21 © (2002) American Association for the Advancement of Science.
The trajectories of single influenza viruses in live cells revealed actin-dependent movement of the virus before the particles were taken up by endocytosis5,13. Several viruses, such as Murine Leukaemia virus, Avian Leukosis virus, HIV and Vesicular Stomatitis virus (VSV) use the cortical actin cytoskeleton, together with myosin II, for directed movement along microvilli or filopodia towards entry ‘hot spots’ at the cell body15. Murine Polyoma virus-like particles have distinct modes of movement on the cell surface: an initial phase of free diffusion is followed by actin-dependent, confined movement in small domains16. A virus-imaging experiment shows that, in order to reach specific receptors located at tight junctions, Coxsackieviruses first binds to the attachment factor DAF (decay-accelerating factor), which through Abl kinase activates a Rac-dependent actin rearrangement that allows the virus particle to move towards the tight junction17. Together, these imaging experiments have substantially improved our understanding of viral entry and revealed an early stage of entry that had been largely overlooked previously: viral attachment is followed by organized transport on the cell surface, often mediated by actin, probably to locate specific receptors or special active zones that are efficient in virus uptake.
Multicolour live-cell imaging provides additional information on the interactions between virus and cellular structures, further elucidating viral-entry mechanisms. Using this approach, single Simian virus 40 (SV40) particles were simultaneously tracked with GFP-tagged caveolae. These experiments revealed the dynamic SV40 entry process and a new endocytic organelle, the caveosome18,19,20 (FIG. 2). Most SV40 co-localizes with caveolae, activating tyrosin kinases and inducing rearrangement of actin stress fibres to form actin tails on virus-loaded caveolae21 (FIG. 2). After a dynamin-dependent pinching process, the SV40-loaded caveolae leaves the plasma membrane and enters the caveosome before finally reaching the ER through microtubule-dependent transport. Polyoma virus and Echovirus 1 also enter cells through a similar pathway to SV40 (REFS 22–24).
Influenza virus uses a distinct entry mechanism13,25,26. As directly visualized in a virus tracking experiment, Influenza viruses can simultaneously use two pathways to enter cells13 (FIG. 2): most virus particles are internalized through clathrin-mediated endocytosis by promoting the de novo formation of clathrin-coated pits (CCPs) at the viral binding site; the remaining virus particles enter cells through a clathrin- and caveolin-independent pathway. After entry by these pathways the virus particles have similar post-endocytic trafficking behaviour, which leads to viral fusion with similar efficiencies. As obligatory parasites that exploit constitutive cellular functions for their own replication, viruses are also natural probes for understanding cell biology. Experiments tracking fluorescent Rab proteins together with single Influenza particles (as well as transferrin, LDL and EGF (epidermal growth factor)) in live cells reveal two distinct populations of early endosomes27: a dynamic population that matures rapidly to form late endosomes, and a relatively static population that matures much more slowly. Different cargos are differentially targeted to the two endosomal populations. Influenza viruses are preferentially delivered, by microtubule-dependent transport, to the dynamic early endosomes27. Viral fusion typically occurs in the midst of the endosomal maturation process with both early and late endosomal markers present on the endosome — an intermediate state of endosomal maturation that has only recently been discovered27–29.
Other viruses have also been observed to co-localize with CCPs in live cells, including reovirus and adenovirus30,31. In common with Influenza, reovirus enters by the de novo formation of CCPs30. Experiments tracking Reoviruses, transferrin, LDL and labelled CCPs revealed a random initiation behaviour of coated pits, which are only stabilized after cargo binding30. For HIV, although direct fusion with the plasma membrane is the main pathway to deliver the capsid into the cytoplasm, a smaller fraction of viruses also infect cells by clathrin-mediated endocytosis32. Besides clathrin- or caveolin-mediated endocytosis, various viruses, including Polyoma virus and Herpes Simplex virus (HSV)33–35, exploit other clathrin- and caveolin-independent entry pathways. As shown by these examples, the ability to monitor time-dependent behaviour of individual virus particles and to probe dynamic interactions between viral and cellular structures has substantially enriched our understanding of viral-entry mechanisms at the molecular level. These studies have not only revealed previously unknown virus–cell interactions that are crucial for infection, but have also shed new light on fundamental cellular mechanisms and pathways. Emerging single-virus tracking experiments have also begun to study fusion mechanisms of enveloped viruses in live cells36–38. Compared with enveloped viruses, the uncoating mechanisms used by non-enveloped viruses are less well understood, but we expect future single-virus experiments to contribute to this important area of virus research.
Viral transport
After uncoating, the viral contents need to be transported to proper sites for replication. Objects larger than 20 nm (or significantly heavier than 500 kDa) cannot freely diffuse through the crowded cytoplasm39. Instead, molecular motors shuttle on cytoskeletal tracks to actively transport cargo. Kinesin and dynein motors move on microtubule tracks, whereas myosin motors interact with actin filaments40,41. The polymerization of actin can also propel a cargo particle in and out of the cell40. All of these transport systems have been successfully hijacked by viruses.
As is evident in single-virus trajectories, many viruses are transported by minus and plus end-directed motor proteins along microtubules towards the microtubule organizing centre (MTOC), either by directly interacting with molecular motors or through inclusion in a motor-interacting vesicle (FIG. 2). This was observed for Reovirus, Adenovirus, HSV, influenza virus and HIV5,42–45. Viral transport is often highly regulated as with, for example, HSV in neurons. After fusion of incoming virions with the plasma membrane, HSV capsids are transported towards the minus end of microtubules by dynein and its cofactor dynactin43. In the axons of sensory neurons, incoming HSV capsids are moved by retrograde transport, whereas newly assembled capsids undergo bidirectional and salutatory motions, indicating a modulation of the plus-end directed motility46. This modulation involves HSV tegument proteins47–51, the removal of which precedes the retrograde transport of the incoming capsids to the cell body, whereas the addition is coupled to anterograde transport of progeny capsids to the distal axon52,53. Important signalling events might occur during viral transport. During its dynein-dependent motion along microtubules, Adenoviruses transiently activate distinct signalling cascades, such as cAMP dependent protein kinase A, p38/mitogen activated protein kinase (MAPK) and the downstream MAPK activated protein kinase 2, to balance nuclear targeting and enhance infection42,54.
The mechanisms by which capsids are transported to the nuclear pore complex for the import of viral genomes are less well known. Capsids might be transported by kinesin from the MTOC towards the nuclear pore complex and nuclear import factors might be involved in the unloading of capsids from microtubules that are proximal to the nucleus55 (FIG. 2). The direct tracking of an incoming viral genome in live cells has been challenging due to difficulties encountered in generating infectious virions harbouring labelled viral DNA or RNA. Tracking microinjected viral ribonucleoprotein (vRNP) particles from Influenza showed diffusion as the predominant mechanism for the transport of vRNPs both towards the nuclear pore complexes and inside the nucleus56. In a recent study, the HIV genome-associated integrase was labelled, and automated 3-dimensional particle tracking revealed that HIV-1 complexes underwent both microtubule- and actin-dependent movements toward the nucleus and more diffuse movements once inside the nucleus57. Remarkably, reverse transcription of the HIV genome seems to begin while the virus is being transported to MTOC44.
Viral assembly and egress
A successful viral infection is ultimately marked by the assembly and release of progeny virus particles. After genome replication and protein synthesis, the subviral components are transported to the assembly site and progeny virions leave the cell by directly budding from the plasma membrane, controlled exocytosis or lysis of the cell (FIG. 3). Labelling viral structural components with fluorescent proteins allows the assembly of individual virions to be monitored, and shows the location and kinetics of assembly as well as the exit mechanisms of matured viruses.
Figure 3. Viral assembly and egress.

Viral genomes are packaged into capsids and are transported along microtubules (1). Viral membrane proteins are translated at the endoplasmic reticulum membrane (2) and transported along microtubules to the Golgi apparatus, where capsids can bud into an envelope (3). Viruses also bud into the multivesicular bodies (MVB) (4). Complete virions inside transport vesicles (5), or subviral particles (6), are transported by kinesin on microtubules towards the plasma membrane, and exit the cell by exocytosis (7) or budding (8) at the plasma membrane. During egress, the actin cortex might propel viruses towards neighbouring cells through a dynamic actin tail (9). The key shows how the different components have been labelled previously. The inset image shows actin-tail formation during the egress of vaccinia virus. Actin (green)and the phosphorylated viral membrane protein A36R (red) were detected by immunofluorescence. MTOC, microtubule organization centre. Scale bar represents 10 μm. Inset image reproduced with permission from REF.65 © (2004) American Association for the Advancement of Science.
A series of imaging experiments by time-lapse microscopy revealed the exit mode of Vaccinia virus40. Intracellular enveloped Vaccinia virus particles move from their perinuclear assembly site to the plasma membrane along microtubules58–63. These particles fuse with the plasma membrane, but remain attached as cell-associated enveloped virus (CEV)64. The envelope protein B5R activates the Src family kinases, which phosphorylate another viral protein A36R, which in turn recruits the actin polymerization machinery, forming a characteristic actin tail beneath the CEV to enhance cell-to-cell spread of the virus65 (FIG. 3). Interestingly, the phosphorylation of A36R also triggers the release of kinesin from the virus65, indicating that the viral envelope protein coordinates the transition from microtubule-based to actin-based transport, which is crucial for the egress of the virus. The actin-tail formation can also be independently initiated by the Abl-family kinase, which induces the release of CEV from the cell66. It was recently discovered that African swine fever virus also shows a microtubule-dependent movement towards the plasma membrane, followed by actin polymerization that propels the virus away from the cell67,68.
Compared with imaging the entry and transport of single virions, which typically involves labelling incoming viruses and therefore does not suffer from strong background cell fluorescence, it is more challenging to probe the assembly and egress of viruses at the single particle level. The background fluorescence from newly synthesized viral components makes it extremely difficult to monitor early assembly steps. Determining the distribution of viral proteins in living cells is currently more tractable. For example, HIV-1 Gag is initially diffuse in the cytosol and then accumulates in perinuclear clusters, after which it transiently passes through multivesicular bodies before moving to the plasma membrane69. The Bluetongue virus core protein VP3 has highly distinct cellular distributions in the presence and absence of other viral proteins (VP7 and NS2) that are related to assembly70. After initial stages of assembly which accumulate sufficient proteins to form a virion, the signal might become sufficiently large to allow viral egress to be tracked at the single-virion level. Experiments with labelled Pseudorabies virus tegument and capsid proteins demonstrate viral assembly in the cell body before entering the axon of cultured neurons71. The association of human Cytomegalovirus tegument and capsid proteins occurs inside nuclear inclusions72. As brighter probes become available and new imaging methods continue to improve background suppression, we expect viral assembly and egress to become as fertile a ground for single-particle tracking experiments as viral entry and transport.
Future perspectives
In this article we have reviewed technical developments that have enabled single-virus imaging in live cells and new insights into viral infection derived from these experiments. Viral infection is complex: viruses can exploit a number of different pathways to infect cells, each pathway consists of many steps and each step features specific interactions between viral and cellular constituents. This level of complexity demands investigation of viral infection at a systems biology level and such research is now taking place. A tour-de-force, high-throughput siRNA screen that compared the involvement of the human kinome (the collection of genes in the human genome that encode kinases) in clathrin-mediated and caveolinmediated endocytosis revealed distinct but overlapping sets of kinases that regulate VSV and SV40 entry73: 92 kinases regulate VSV entry, 80 regulate SV40 entry and 36 kinases are involved in both. Remarkably, most of the overlapping kinases have opposite effects on VSV and SV40. Extending this approach to other viruses and to a much larger set of genes potentially related to viral infection will provide a systematic analysis of viral infection at the molecular level.
It will be extremely fruitful to combine this systemsbiology approach with the ability of single-virus tracking to dissect multiple infection pathways and multiple infection steps, such that the mechanism of action can be clearly identified for a large set of host genes that are involved in viral infection. This requires the development of automated acquisition of high-resolution live-cell images that are compatible with high-throughput screening, and algorithms that would allow the analysis of the resulting fast-growing data sets.
Many of the viral infection steps involve the action of only a few viral and cellular molecules. To track these steps in live cells, the development of brighter and smaller probes that allow single-molecule detection in live cells is needed. Furthermore, molecular interactions typically occur at the nanometer scale, beyond the resolving power of conventional optical microscopy. Ultra-structural information is typically obtained by electron microscopy, which is unfortunately not compatible with live-cell imaging. Recently, a number of nanometer-resolution optical imaging methods have been reported, including stimulated emission depletion (STED) microscopy74, stochastic optical reconstruction microscopy (STORM)75 photoactivation light microscopy (PALM)76 and structured illumination microscopy (SIM)77. Although substantial improvement in imaging speed is needed before cellular processes can be imaged using these methods in real time, the accomplishment of this goal will definitely open up the investigation of virus–cell interactions.
Finally, it is important to note that cultured cells are simplified model systems. A complete understanding of viral infection would greatly benefit from in vivo experiments. It is therefore particularly exciting to track virus particles in live tissues and animals to see how viruses break host defence barriers to reach target cells for infection and how viral infection is transmitted from cell to cell. With the rapid development of in vivo imaging methods, such as non-linear optical microscopy78,79 and endoscopy, this dream might be realized in the not-too-distant future.
Supplementary Material
Acknowledgments
We thank J. C. Vaughan for his critical reading of the manuscript and other members of the Zhuang laboratory for helpful discussions. We apologize to colleagues whose work might not have been cited due to space constraints. This work is supported in part by the National Institutes of Health. X. Z. is a Howard Hughes Medical Institute investigator.
References
- 1.Marsh M, Helenius A. Virus entry: open sesame. Cell. 2006;124:729–740. doi: 10.1016/j.cell.2006.02.007. A comprehensive review of the main mechanisms of viral entry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pelkmans L, Helenius A. Insider information: what viruses tell us about endocytosis. Curr Opin Cell Biol. 2003;15:414–422. doi: 10.1016/s0955-0674(03)00081-4. [DOI] [PubMed] [Google Scholar]
- 3.Conner SD, Schmid SL. Regulated portals of entry into cells. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 4.Giepmans BN, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. A review of the uses of fluorescent probes for studying proteins and protein–protein interaction in cells. [DOI] [PubMed] [Google Scholar]
- 5.Lakadamyali M, Rust MJ, Babcock HP, Zhuang X. Visualizing infection of individual influenza viruses. Proc Natl Acad Sci USA. 2003;100:9280–9285. doi: 10.1073/pnas.0832269100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seisenberger G, et al. Real-time single-molecule imaging of the infection pathway of an adenoassociated virus. Science. 2001;294:1929–1932. doi: 10.1126/science.1064103. [DOI] [PubMed] [Google Scholar]
- 7.Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nature Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 8.Marks KM, Nolan GP. Chemical labeling strategies for cell biology. Nature Methods. 2006;3:591–596. doi: 10.1038/nmeth906. [DOI] [PubMed] [Google Scholar]
- 9.Griffin BA, Adams SR, Tsien RY. Specific covalent labeling of recombinant protein molecules inside live cells. Science. 1998;281:269–272. doi: 10.1126/science.281.5374.269. [DOI] [PubMed] [Google Scholar]
- 10.Michalet X, et al. Quantum dots for live cells, in vivo imaging, and diagnostics. Science. 2005;307:538–544. doi: 10.1126/science.1104274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dixit SK, et al. Quantum dot encapsulation in viral capsids. Nano Lett. 2006;6:1993–1999. doi: 10.1021/nl061165u. [DOI] [PubMed] [Google Scholar]
- 12.Zheng J, Dickson RM. Individual water-soluble dendrimer-encapsulated silver nanodot fluorescence. J Am Chem Soc. 2002;124:13982–13983. doi: 10.1021/ja028282l. [DOI] [PubMed] [Google Scholar]
- 13.Rust MJ, Lakadamyali M, Zhang F, Zhuang X. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nature Struct Mol Biol. 2004;11:567–573. doi: 10.1038/nsmb769. By real-time imaging of individual virus particles in live cells, this paper reveals that influenza viruses can exploit two endocytic pathways in parallel and enter cells either by de novo formation of clathrincoated pits or by a clathrin- and caveolinindependent pathway. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaidarov I, Santini F, Warren RA, Keen JH. Spatial control of coated-pit dynamics in living cells. Nature Cell Biol. 1999;1:1–7. doi: 10.1038/8971. [DOI] [PubMed] [Google Scholar]
- 15.Lehmann MJ, Sherer NM, Marks CB, Pypaert M, Mothes W. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J Cell Biol. 2005;170:317–325. doi: 10.1083/jcb.200503059. A vivid demonstration of actin-dependent virus surfing along the filopodia of polarized epithelial cells towards the viral entry site. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ewers H, et al. Singleparticle tracking of murine polyoma virus-like particles on live cells and artificial membranes. Proc Natl Acad Sci USA. 2005;102:15110–15115. doi: 10.1073/pnas.0504407102. Single-virus tracking revealed distinct types of lateral movement of polyoma virus-like particles on the cell surface. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coyne CB, Bergelson JM. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell. 2006;124:119–131. doi: 10.1016/j.cell.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 18.Pelkmans L, Kartenbeck J, Helenius A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nature Cell Biol. 2001;3:473–483. doi: 10.1038/35074539. An early single-virus tracking work that demonstrated the caveolin-mediated entry mechanism for Simian virus 40 and the presence of a new intracellular organelle, the caveosome. [DOI] [PubMed] [Google Scholar]
- 19.Pelkmans L, Burli T, Zerial M, Helenius A. Caveolin-stabilized membrane domains as multifunctional transport and sorting devices in endocytic membrane traffic. Cell. 2004;118:767–780. doi: 10.1016/j.cell.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Tagawa A, et al. Assembly and trafficking of caveolar domains in the cell: caveolae as stable, cargo-triggered, vesicular transporters. J Cell Biol. 2005;170:769–779. doi: 10.1083/jcb.200506103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pelkmans L, Puntener D, Helenius A. Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science. 2002;296:535–539. doi: 10.1126/science.1069784. [DOI] [PubMed] [Google Scholar]
- 22.Elphick GF, et al. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science. 2004;306:1380–1383. doi: 10.1126/science.1103492. [DOI] [PubMed] [Google Scholar]
- 23.Pietiainen V, et al. Echovirus 1 endocytosis into caveosomes requires lipid rafts, dynamin II, and signaling events. Mol Biol Cell. 2004;15:4911–4925. doi: 10.1091/mbc.E04-01-0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Upla P, et al. Clustering induces a lateral redistribution of α 2 β 1 integrin from membrane rafts to caveolae and subsequent protein kinase C-dependent internalization. Mol Biol Cell. 2004;15:625–636. doi: 10.1091/mbc.E03-08-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matlin KS, Reggio H, Helenius A, Simons K. Infectious entry pathway of influenza virus in a canine kidney-cell line. J Cell Biol. 1981;91:601–613. doi: 10.1083/jcb.91.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sieczkarski SB, Whittaker GR. Influenza virus can enter and infect cells in the absence of clathrinmediated endocytosis. J Virol. 2002;76:10455–10464. doi: 10.1128/JVI.76.20.10455-10464.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lakadamyali M, Rust MJ, Zhuang X. Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell. 2006;124:997–1009. doi: 10.1016/j.cell.2005.12.038. Real-time tracking of single-virus particles and other endocytic ligands in live cells enabled the discovery of a new endocytic sorting mechanism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell. 2005;122:735–749. doi: 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 29.Vonderheit A, Helenius A. Rab7 associates with early endosomes to mediate sorting and transport of Semliki forest virus to late endosomes. PLoS Biol. 2005;3:e233. doi: 10.1371/journal.pbio.0030233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ehrlich M, et al. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell. 2004;118:591–605. doi: 10.1016/j.cell.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 31.Meier O, et al. Adenovirus triggers macropinocytosis and endosomal leakage together with its clathrinmediated uptake. J Cell Biol. 2002;158:1119–1131. doi: 10.1083/jcb.200112067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daecke J, Fackler OT, Dittmar MT, Krausslich HG. Involvement of clathrin-mediated endocytosis in human immunodeficiency virus type 1 entry. J Virol. 2005;79:1581–1594. doi: 10.1128/JVI.79.3.1581-1594.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Damm EM, et al. Clathrin- and caveolin-1-independent endocytosis: entry of simian virus 40 into cells devoid of caveolae. J Cell Biol. 2005;168:477–488. doi: 10.1083/jcb.200407113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liebl D, et al. Mouse polyomavirus enters early endosomes, requires their acidic pH for productive infection, and meets transferrin cargo in Rab11-positive endosomes. J Virol. 2006;80:4610–4622. doi: 10.1128/JVI.80.9.4610-4622.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nicola AV, Straus SE. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J Virol. 2004;78:7508–7517. doi: 10.1128/JVI.78.14.7508-7517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Melikyan GB, Barnard RJ, Abrahamyan LG, Mothes W, Young JA. Imaging individual retroviral fusion events: from hemifusion to pore formation and growth. Proc Natl Acad Sci USA. 2005;102:8728–8733. doi: 10.1073/pnas.0501864102. Real-time imaging of the fusion processes between individual virus particles and cell membranes revealed semi-fusion and small fusion pores as fusion intermediate states. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Markosyan RM, Cohen FS, Melikyan GB. Time-resolved imaging of HIV-1 Env-mediated lipid and content mixing between a single virion and cell membrane. Mol Biol Cell. 2005;16:5502–5513. doi: 10.1091/mbc.E05-06-0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Le Blanc I, et al. Endosome-to-cytosol transport of viral nucleocapsids. Nature Cell Biol. 2005;7:653–664. doi: 10.1038/ncb1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luby-Phelps K. Cytoarchitecture and physical properties of cytoplasm: volume, viscosity, diffusion, intracellular surface area. Int Rev Cytol. 2000;192:189–221. doi: 10.1016/s0074-7696(08)60527-6. [DOI] [PubMed] [Google Scholar]
- 40.Greber UF, Way M. A superhighway to virus infection. Cell. 2006;124:741–754. doi: 10.1016/j.cell.2006.02.018. A comprehensive review of the intracellular transport mechanisms used by viruses. [DOI] [PubMed] [Google Scholar]
- 41.Radtke K, Dohner K, Sodeik B. Viral interactions with the cytoskeleton: a hitchhiker’s guide to the cell. Cell Microbiol. 2006;8:387–400. doi: 10.1111/j.1462-5822.2005.00679.x. [DOI] [PubMed] [Google Scholar]
- 42.Suomalainen M, et al. Microtubule-dependent plus and minus end-directed motilities are competing processes for nuclear targeting of adenovirus. J Cell Biol. 1999;144:657–672. doi: 10.1083/jcb.144.4.657. An early single-virus tracking study that revealed the active-transport mechanisms of adenoviruses in cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dohner K, et al. Function of dynein and dynactin in herpes simplex virus capsid transport. Mol Biol Cell. 2002;13:2795–2809. doi: 10.1091/mbc.01-07-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McDonald D, et al. Visualization of the intracellular behavior of HIV in living cells. J Cell Biol. 2002;159:441–452. doi: 10.1083/jcb.200203150. A single-virus tracking study that revealed the transport mechanisms of HIV-1 virus at various stages of the virus life cycle. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Georgi A, Mottola-hartshorn C, Warner W, Fields B, Chen LB. Detection of individual fluorescently labelled reovirions in living cells. Proc Natl Acad Sci USA. 1990;87:6579–6583. doi: 10.1073/pnas.87.17.6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith GA, Pomeranz L, Gross SP, Enquist LW. Local modulation of plus-end transport targets herpesvirus entry and egress in sensory axons. Proc Natl Acad Sci USA. 2004;101:16034–16039. doi: 10.1073/pnas.0404686101. This time-lapse imaging study of herpes simplex viruses in live cells that elucidated distinct microtubule-dependent transport mechanisms for incoming and progeny HSV virus capsids. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Elliott G, O’Hare P. Live-cell analysis of a green fluorescent protein-tagged herpes simplex virus infection. J Virol. 1999;73:4110–4119. doi: 10.1128/jvi.73.5.4110-4119.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Antinone SE, et al. The Herpesvirus capsid surface protein, VP26, and the majority of the tegument proteins are dispensable for capsid transport toward the nucleus. J Virol. 2006;80:5494–5498. doi: 10.1128/JVI.00026-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee GE, Murray JW, Wolkoff AW, Wilson DW. Reconstitution of herpes simplex virus microtubuledependent trafficking in vitro. J Virol. 2006;80:4264–4275. doi: 10.1128/JVI.80.9.4264-4275.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolfstein A, et al. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic. 2006;7:227–237. doi: 10.1111/j.1600-0854.2005.00379.x. [DOI] [PubMed] [Google Scholar]
- 51.Dohner K, Radtke K, Schmidt S, Sodeik B. Eclipse phase of herpes simplex virus type 1 infection: Efficient dynein-mediated capsid transport without the small capsid protein VP26. J Virol. 2006;80:8211–8224. doi: 10.1128/JVI.02528-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luxton GW, et al. Targeting of herpesvirus capsids transport in axons is coupled to association with specific sets of tegument proteins. Proc Natl Acad Sci USA. 2005;102:5832–5837. doi: 10.1073/pnas.0500803102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luxton GW, Lee JI, Haverlock-Moyns S, Schober JM, Smith GA. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsids transport. J Virol. 2006;80:201–209. doi: 10.1128/JVI.80.1.201-209.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suomalainen M, Nakano MY, Boucke K, Keller S, Greber UF. Adenovirus-activated PKA and p38/MAPK pathways boost microtubule-mediated nuclear targeting of virus. EMBO J. 2001;20:1310–1319. doi: 10.1093/emboj/20.6.1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Strunze S, Trotman LC, Boucke K, Greber UF. Nuclear targeting of adenovirus type 2 requires CRM1-mediated nuclear export. Mol Biol Cell. 2005;16:2999–3009. doi: 10.1091/mbc.E05-02-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Babcock HP, Chen C, Zhuang X. Using singleparticle tracking to study nuclear trafficking of viral genes. Biophys J. 2004;87:2749–2758. doi: 10.1529/biophysj.104.042234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arhel N, et al. Quantitative four-dimensional tracking of cytoplasmic and nuclear HIV-1 complexes. Nature Methods. 2006;3:817–824. doi: 10.1038/nmeth928. This paper demonstrated the use of advanced 3D tracking methods for the study of viral motion in live cells. [DOI] [PubMed] [Google Scholar]
- 58.Ward BM, Moss B. Vaccinia virus intracellular movement is associated with microtubules and independent of actin tails. J Virol. 2001;75:11651–11663. doi: 10.1128/JVI.75.23.11651-11663.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hollinshead M, et al. Vaccinia virus utilizes microtubules for movement to the cell surface. J Cell Biol. 2001;154:389–402. doi: 10.1083/jcb.200104124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rietdorf J, et al. Kinesin-dependent movement on microtubules precedes actin-based motility of Vaccinia virus. Nature Cell Biol. 2001;3:992–1000. doi: 10.1038/ncb1101-992. [DOI] [PubMed] [Google Scholar]
- 61.Ward BM, Moss B. Visualization of intracellular movement of vaccinia virus virions containing a green fluorescent protein-B5R membrane protein chimera. J Virol. 2001;75:4802–4813. doi: 10.1128/JVI.75.10.4802-4813.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Herrero-Martinez E, Roberts KL, Hollinshead M, Smith GL. Vaccinia virus intracellular enveloped virions move to the cell periphery on microtubules in the absence of the A36R protein. J Gen Virol. 2005;86:2961–2968. doi: 10.1099/vir.0.81260-0. [DOI] [PubMed] [Google Scholar]
- 63.Ward BM. Visualization and characterization of the intracellular movement of vaccinia virus intracellular mature virions. J Virol. 2005;79:4755–4763. doi: 10.1128/JVI.79.8.4755-4763.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith GL, Murphy BJ, Law M. Vaccinia virus motility. Annu Rev Microbiol. 2003;57:323–342. doi: 10.1146/annurev.micro.57.030502.091037. [DOI] [PubMed] [Google Scholar]
- 65.Newsome TP, Scaplehorn N, Way M. SRC mediates a switch from microtubule- to actin-based motility of vaccinia virus. Science. 2004;306:124–129. doi: 10.1126/science.1101509. An elegant analysis of the molecular mechanisms used by vaccinia virus to regulate switching between microtubule-dependent and actindependent modes of transport within the cell. [DOI] [PubMed] [Google Scholar]
- 66.Newsome TP, Weisswange I, Frischknecht F, Way M. Abl collaborates with Src family kinases to stimulate actin-based motility of vaccinia virus. Cell Microbiol. 2006;8:233–241. doi: 10.1111/j.1462-5822.2005.00613.x. [DOI] [PubMed] [Google Scholar]
- 67.Jouvenet N, Monaghan P, Way M, Wileman T. Transport of African swine fever virus from assembly sites to the plasma membrane is dependent on microtubules and conventional kinesin. J Virol. 2004;78:7990–8001. doi: 10.1128/JVI.78.15.7990-8001.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jouvenet N, et al. African swine fever virus induces filopodia-like projections at the plasma membrane. Cell Microbiol. 2006 doi: 10.1111/j.1462-5822.2006.00750.x. [DOI] [PubMed] [Google Scholar]
- 69.Perlman M, Resh MD. Identification of an intracellular trafficking and assembly pathway for HIV-1 gag. Traffic. 2006;7:731–745. doi: 10.1111/j.1398-9219.2006.00428.x. [DOI] [PubMed] [Google Scholar]
- 70.Kar AK, Iwatani N, Roy P. Assembly and intracellular localization of the bluetongue virus core protein VP3. J Virol. 2005;79:11487–11495. doi: 10.1128/JVI.79.17.11487-11495.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.del Rio T, Ch’ng TH, Flood EA, Gross SP, Enquist LW. Heterogeneity of a fluorescent tegument component in single pseudorabies virus virions and enveloped axonal assemblies. J Virol. 2005;79:3903–3919. doi: 10.1128/JVI.79.7.3903-3919.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sampaio KL, Cavignac Y, Stierhof YD, Sinzger C. Human cytomegalovirus labeled with green fluorescent protein for live analysis of intracellular particle movements. J Virol. 2005;79:2754–2767. doi: 10.1128/JVI.79.5.2754-2767.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pelkmans L, et al. Genome-wide analysis of human kinases in clathrin- and caveolae/raft-mediated endocytosis. Nature. 2005;436:78–86. doi: 10.1038/nature03571. The first systems-biology study of viral entry mechanisms by high-throughput siRNA screening. [DOI] [PubMed] [Google Scholar]
- 74.Hell SW. Toward fluorescence nanoscopy. Nature Biotech. 2003;21:1347–1355. doi: 10.1038/nbt895. [DOI] [PubMed] [Google Scholar]
- 75.Rust M, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nature Methods. 2006 doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Betzig E, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 77.Gustafsson MGL. Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution. Proc Natl Acad Sci USA. 2005;102:13081–13086. doi: 10.1073/pnas.0406877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zipfel WR, Williams RM, Webb WW. Nonlinear magic: multiphoton microscopy in the biosciences. Nature Biotech. 2003;21:1368–1376. doi: 10.1038/nbt899. [DOI] [PubMed] [Google Scholar]
- 79.Evans CL, et al. Chemical imaging of tissue in vivo with videl-rate coherent anti-Stokes Raman scattering microscopy. Proc Natl Acad Sci USA. 2005;102:16807–16812. doi: 10.1073/pnas.0508282102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chudakov DM, Lukyanov S, Lukyanov KA. Fluorescent proteins as a toolkit for in vivo imaging. Trends Biotechnol. 2005;23:605–613. doi: 10.1016/j.tibtech.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 81.Stephens DJ, Allan VJ. Light microscopy techniques for live cell imaging. Science. 2003;300:82–86. doi: 10.1126/science.1082160. [DOI] [PubMed] [Google Scholar]
- 82.Amos WB, White JG. How the confocal laser scanning microscope entered biological research. Biol Cell. 2003;95:335–342. doi: 10.1016/s0248-4900(03)00078-9. [DOI] [PubMed] [Google Scholar]
- 83.Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Traffic. 2001;2:764–774. doi: 10.1034/j.1600-0854.2001.21104.x. [DOI] [PubMed] [Google Scholar]
- 84.Olivo-Marin JC. Extraction of spots in biological images using multiscale products. Pattern Recognition. 2002;35:1989–1996. [Google Scholar]
- 85.Sbalzarini IF, Koumoutsakos P. Feature point tracking and trajectory analysis for video imaging in cell biology. J Struct Biol. 2005;151:182–195. doi: 10.1016/j.jsb.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 86.Genovesio A, Olivo-Marin JC. Split and merge data association filter for dense multi-target tracking. IEEE ICP. 2004;4:677–680. [Google Scholar]
- 87.Helenius A, Kartenbeck J, Simons K, Fries E. On the entry of Semliki forest virus into BHK-21 cells. J Cell Biol. 1980;84:404–420. doi: 10.1083/jcb.84.2.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Barak LS, Webb WW. Diffusion of low density lipoprotein-receptor complex on human fibroblasts. J Cell Biol. 1982;95:846–852. doi: 10.1083/jcb.95.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.De Brabander M, Geuens G, Nuydens R, Moeremans M, De Mey J. Probing microtubuledependent intracellular motility with nanometer particle video ultramicroscopy (nanovid ultramicroscopy) Cytobios. 1985;43:273–283. [PubMed] [Google Scholar]
- 90.Gelles J, Schnapp BJ, Sheetz MP. Tracking kinesin-driven movements with nanometre-scale precision. Nature. 1988;331:450–453. doi: 10.1038/331450a0. [DOI] [PubMed] [Google Scholar]
- 91.Inoue S. Imaging of unresolved objects, superresolution, and precision of distance measurement with video microscopy. Methods Cell Biol. 1989;30:85–112. doi: 10.1016/s0091-679x(08)60976-0. [DOI] [PubMed] [Google Scholar]
- 92.Qian H, Sheetz MP, Elson EL. Single particle tracking. Analysis of diffusion and flow in twodimensional systems. Biophys J. 1991;60:910–921. doi: 10.1016/S0006-3495(91)82125-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Saxton MJ. Single-particle tracking: models of directed transport. Biophys J. 1994;67:2110–2119. doi: 10.1016/S0006-3495(94)80694-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ghosh RN, Webb WW. Automated detection and tracking of individual and clustered cell surface low density lipoprotein receptor molecules. Biophys J. 1994;66:1301–1318. doi: 10.1016/S0006-3495(94)80939-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bachi T. Direct observation of the budding and fusion of an enveloped virus by video microscopy of viable cells. J Cell Biol. 1988;107:1689–1695. doi: 10.1083/jcb.107.5.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lowy RJ, Sarkar DP, Chen Y, Blumenthal R. Observation of single influenza virus-cell fusion and measurement by fluorescence video microscopy. Proc Natl Acad Sci USA. 1990;87:1850–1854. doi: 10.1073/pnas.87.5.1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Georgi A, Mottola-Hartshorn C, Warner A, Fields B, Chen LB. Detection of individual fluorescently labeled reovirions in living cells. Proc Natl Acad Sci USA. 1990;87:6579–6583. doi: 10.1073/pnas.87.17.6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Anderson CM, Georgiou GN, Morrison IE, Stevenson GV, Cherry RJ. Tracking of cell surface receptors by fluorescence digital imaging microscopy using a charge-coupled device camera. Low-density lipoprotein and influenza virus receptor mobility at 4 degrees C. J Cell Sci. 1992;101:415–425. doi: 10.1242/jcs.101.2.415. [DOI] [PubMed] [Google Scholar]
- 99.Prasher DC, Eckenrode VK, Ward WW, Prendergast FG, Cormier MJ. Primary structure of the Aequorea victoria green-fluorescent protein. Gene. 1992;111:229–233. doi: 10.1016/0378-1119(92)90691-h. [DOI] [PubMed] [Google Scholar]
- 100.Inouye S, Tsuji FI. Aequorea green fluorescent protein. Expression of the gene and fluorescence characteristics of the recombinant protein. FEBS Lett. 1994;341:277–280. doi: 10.1016/0014-5793(94)80472-9. [DOI] [PubMed] [Google Scholar]
- 101.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 102.Heim R, Prasher DC, Tsien RY. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc Natl Acad Sci USA. 1994;91:12501–12504. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cudmore S, Cossart P, Griffiths G, Way M. Actinbased motility of vaccinia virus. Nature. 1995;378:636–638. doi: 10.1038/378636a0. [DOI] [PubMed] [Google Scholar]
- 104.Funatsu T, Harada Y, Tokunaga M, Saito K, Yanagida T. Imaging of single fluorescent molecules and individual ATP turnovers by single myosin molecules in aqueous solution. Nature. 1995;374:555–559. doi: 10.1038/374555a0. [DOI] [PubMed] [Google Scholar]
- 105.Lewis JD, et al. Viral nanoparticles as tools for intravital vascular imaging. Nature Med. 2006;12:354–360. doi: 10.1038/nm1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Genovesio A, et al. Multiple particle tracking in 3-D+t microscopy: method and application to the tracking of endocytosed quantum dots. IEEE TIP. 2006;15:1062–1070. doi: 10.1109/tip.2006.872323. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.