

Abstract
Postnatal bone marrow houses mesenchymal progenitor cells that are osteoblast precursors. These cells have established therapeutic potential 1 but they are difficult to maintain and expand in vitro, presumably because little is known about the mechanisms controlling their fate decisions. To investigate the potential role of Notch signaling in osteoblastogenesis, we used conditional alleles to genetically remove components of the Notch signaling system during skeletal development. We find that Notch disruption in the limb skeletogenic mesenchyme markedly enhanced trabecular bone mass in adolescent mice. Notably, mesenchymal progenitors were virtually depleted in the bone marrow of the high-bone-mass animals. As a result, these animals developed severe osteopenia as they aged. Moreover, Notch appeared to inhibit osteoblast differentiation through Hes/Hey proteins that diminished Runx2 transcriptional activity via physical interaction. These results support a model wherein Notch signaling in bone marrow normally acts to maintain a pool of mesenchymal progenitors by suppressing osteoblast differentiation. Thus, mesechymal progenitors may be expanded in vitro by activating Notch, whereas bone formation in vivo may be enhanced by transiently suppressing this pathway.
Notch signaling mediates communication between neighboring cells to control cell fate decisions during embryogenesis 2 and in postnatal life 3. In the canonical Notch pathway, the single-pass transmembrane cell surface Notch receptors (Notch1-4 in mammals) undergo two sequential proteolytic cleavages upon binding of ligands (Jagged1, 2 and Delta-like 1, 3, 4 in mammals) presented on a neighboring cell surface 4. As a result, the Notch intracellular domain (NICD) is released from the plasma membrane and translocates to the nucleus where it interacts with a transcription factor of the CSL family (RBP-Jκ/CBF-1 in mammals) to activate transcription of target genes 5. The transmembrane domain cleavage and release of NICD is mediated by the γ-secretase complex which in mammals contains either presenilin 1 (PS1) or 2 (PS2) as the catalytic subunit 6; removal of both PS1 and PS2 completely abolishes NICD production 7,8.
Clear genetic evidence demonstrating a direct role for Notch signaling in the skeleton is lacking. Mouse mutants lacking individual receptors 9-12, most ligands 13-17, or PS2 8 either died in utero prior to overt skeletogenesis, or exhibited no overt skeletal phenotype. Although mice lacking either Delta-like 318 or PS119,20 exhibited defects in the axial skeleton, a direct effect on skeletal development was difficult to discern due to the early deficiency in somite segmentation and maintenance. Finally, mice lacking PS1 and PS2 in the limb mesenchyme missed certain phalanges, but it was not clear whether the phenotype was a direct consequence of losing Notch signaling in skeletogenic cells 21.
Removal of γ-secretase in limb mesenchyme affects long bones
To address this gap in our knowledge, we systematically analyzed the skeleton of mice in which PS1 and PS2 were removed using Prx1Cre (Prx1Cre; PS1c/c; PS2n/n, hereafter PPS mutants, c: floxed allele, n: null allele) 21,22. In these mice, all Notch signaling were abolished selectively from the early mesenchyme of the calvaria and the limb primordium. The PPS mice were born at the Mendelian ratio and appeared normal except for variable digit anomalies 21, but all died between 9 and 10 weeks of age. The reasons for premature death are not clear at present, but the animals exhibited severe ulcerative dermatitis in the ventral cervical area, possibly reflecting excessive inflammatory response.
Morphological analyses of the skeleton revealed a dramatic phenotype in mutant mice at 8 weeks of age. X-ray radiography indicated that PPS mutant mice not only had shorter long bones when compared with littermate controls (PS2n/n, no obvious phenotype), but also exhibited a marked increase in radiodensity within the bone marrow cavity (Fig. 1a). 3-D reconstruction of the tibia using micro computed-tomography (μCT) confirmed that a massive accumulation of bone (asterisk) occluded much of the presumptive marrow cavity in the mutant (Fig. 1b). In addition, the radiolucent growth plate cartilage was greatly elongated in the mutant. Histology of longitudinal sections through the medial portion of the tibia further demonstrated that PPS mutants contained excessive cancellous bone encapsulating a dramatic “wedge shaped” extension of the growth plate cartilage (Fig. 1c,d). Examination at a higher magnification indicated that the cartilage extension was chiefly due to accumulation of hypertrophic chondrocytes (Fig. 1f–h), whereas the length of the non-hypertrophic region appeared unaltered (Fig. 1f, “NH”). In the most severe case, the mutant diaphysis appeared to be entirely filled with cancellous bone, with no recognizable marrow cavity or clear demarcation of cortical bone (Fig. 1e). Similarly, the secondary ossification center of the PPS mutant also contained excessive bone and no obvious marrow (Fig. 1f, “2°”). Interestingly, the calvarium, also targeted by Prx1Cre, was not affected in the PPS mutant, a result confirmed by both X-ray radiography (data not shown) and histology (Supplementary Data Fig. S1). Thus, removal of PS1 and PS2 results in expansion of the hypertrophic cartilage and an excess of trabecular bone specifically in endochondral bones of 8-week-old animals.
Figure 1.
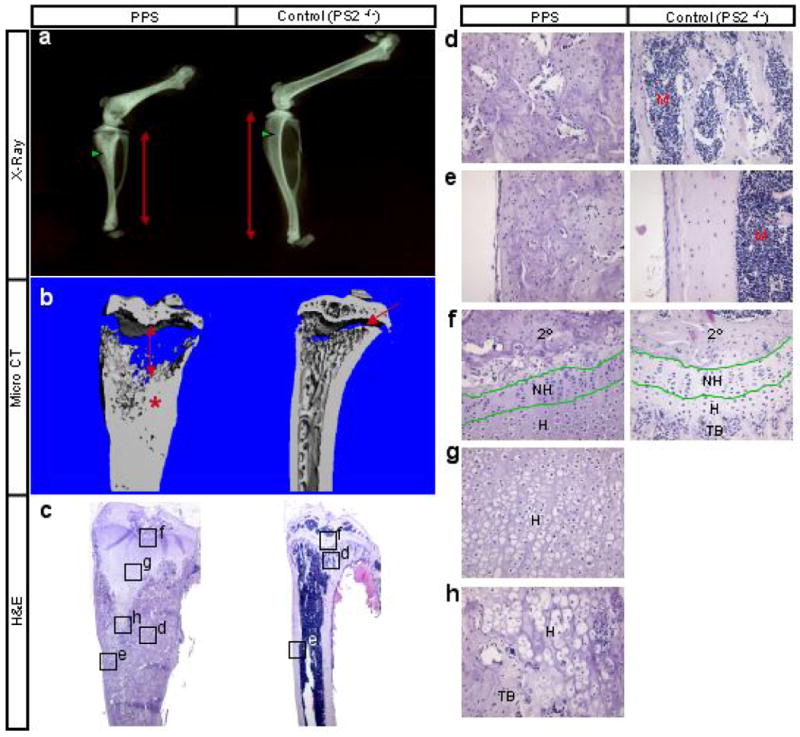
Skeletal phenotype of PPS mice at 8 weeks of age. (a) X-ray radiographs of hindlimbs. Red double-headed arrows denote length of tibia. Green arrowheads point to trabecular bone region. (b) Medial, longitudinal section through 3-D reconstruction of the tibia by μCT. Double-headed arrow: expanded growth plate; asterisk: excessive bone; arrow: normal growth plate. (c) H&E staining of medial longitudinal sections through the tibia. (d-h) Higher magnification of boxed areas in c. 2°: secondary ossification center; NH: nonhypertrophic region; H: hypertrophic region; TB: trabecular bone; M: marrow.
Deletion of Notch1 and Notch2 resembles removal of γ-secretase
The skeletal phenotype caused by removal of γ-secretase prompted us to determine whether abrogation of Notch signaling was solely responsible. To this end, we used the same strategy to remove Notch1 (N1) and Notch2 (N2) from the early limb mesenchyme. Mice of the genotype Prx1Cre; N1n/c; N2c/c (hereafter PNN mutants) were born at the Mendelian ratio and exhibited a skeletal phenotype remarkably similar to that of PPS mutants. In particular, X-ray radiography revealed that at 8 weeks of age the PNN mutants had notably shorter long bones, and markedly higher radiodensity within the trabecular bone region, when compared to wild type (N1c/c; N2c/c) littermates (Fig. 2a). μCT analyses of the tibia confirmed both elongation of the radiolucent growth plate cartilage, and increase of bone mass within the marrow cavity of PNN mutants (Fig. 2b). Finally, histology of the tibia revealed excessive trabecular bone encasing a “wedge-shaped” extension of the growth plate cartilage, and a notable reduction of the marrow space in both primary and secondary ossification centers of PNN mutants (Fig. 2c). As seen in PPS mice, cartilage elongation in PNN animals was due to expansion of the hypertrophic zone, with no apparent increase in the non-hypertrophic region (Fig. 2d). Interestingly, the increase in bone mass appeared to be limited to the trabecular region; although the organization of the cortical bone appeared to be altered, no significant changes in cortical thickness were observed between mutant and wild type littermates at this stage (Fig. 2d). Overall, PNN mutants exhibit a postnatal skeletal phenotype qualitatively identical to that of PPS mutants.
Figure 2.
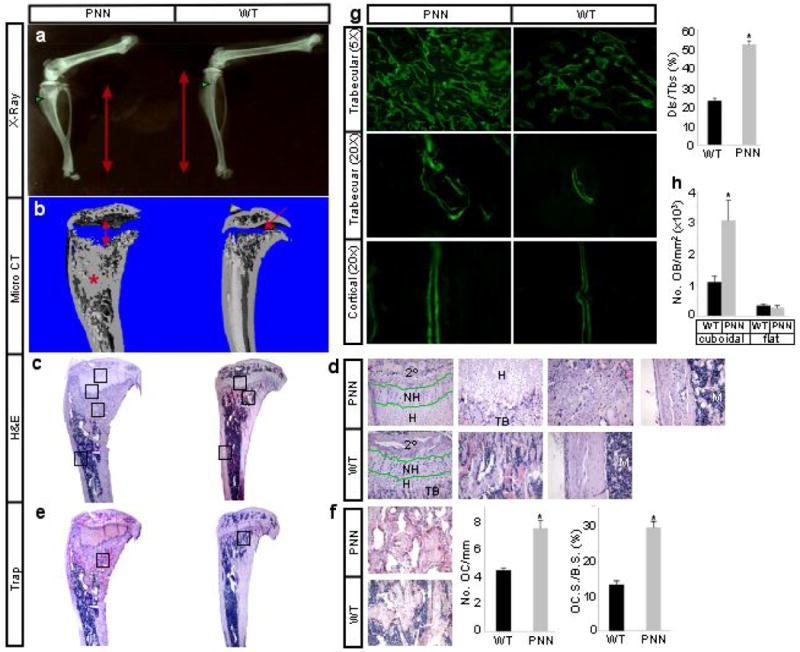
Skeletal phenotype of PNN mice at 8 weeks of age. (a) X-ray radiographs of hindlimbs. Red double-headed arrows denote length of tibia. Green arrowheads point to trabecular bone region. (b) Medial longitudinal section through 3-D reconstruction of the tibia by μCT. Double-headed arrow: expanded growth plate; asterisk: excessive bone; arrow: normal growth plate. (c) H&E staining of medial longitudinal sections through the tibia. (d) Higher magnification of boxed areas in c from top to bottom shown in left-to-right order. 2°: secondary ossification center; NH: nonhypertrophic region; H: hypertrophic region; TB: trabecular bone; M: marrow. (e) TRAP staining on medial longitudinal sections through the tibia. Osteoclasts stain red. (f) Higher magnification of boxed areas in e, number of osteoclasts normalized to trabecular bone perimeter (No. OC/mm), and osteoclast surface normalized to bone surface (OC.S./B.S.). (g) Area micrographs from longitudinal sections of tibia after calcein double labeling, and percentage of calcein double-labeled bone surface over total bone surface (Dls/Tbs) in trabecular bone region. (h) Number of osteoblasts per trabecular bone area on sections. Cuboidal and flat osteoblasts were scored separately. *: p<0.05, n=3.
The resemblance between the PPS and the PNN mice indicated that loss of Notch signaling was responsible for the bone phenotypes in both settings, and prompted us to analyze the PNN mice in further detail. We first explored the cellular basis for the high-bone-mass phenotype. Staining for tartrate resistant acid phosphatase (Trap) activity on tibia sections of mice at 8 weeks of age revealed that the mutants in fact had more osteoclasts per bone surface area than wild type littermates (Fig. 2e–f). The percentage of bone surfaces eroded by osteoclasts also increased in the PNN mice (Fig. 2f). On the other hand, calcein double-labeling experiments indicated that whereas the mineral apposition rate (MAR) as indicated by the distance between the double labels was not significantly altered in either the trabecular or the cortical bone, PNN mutants exhibited a marked increase in the number of double-labeled surfaces within the trabecular bone (Fig. 2g). Indeed, cuboidal (active) osteoblasts normalized to areas of trabecular bone on sections were greatly increased in the mutant animals, although the number of flat (inactive) osteoblasts did not change (Fig. 2h), and the osteoblast density (osteoblast number normalized to trabecular bone perimeter) was not altered (data not shown). Thus, the increase of bone mass in PNN animals was not due to defects in osteoclasts or upregulation of osteoblast activity, but rather to an increase in the total number of active osteoblasts.
Notch signaling regulates embryonic skeletal development
We next determined whether Notch removal affects the embryonic skeleton. At E18.5, histology revealed that both PPS (Supplementary Data Fig. S2) and PNN embryos (Fig. 3a) contained elongated hypertrophic cartilage, and excessive osteoblasts within the marrow region. Interestingly, embryos with one Notch1 allele remaining (Prx1Cre; N1c/+; N2c/c) showed similar phenotypes to PNN mutants, but those with one Notch2 allele remaining (Prx1Cre; N1c/c; N2c/+) had a relatively normal bone marrow and only a slight elongation of the hypertrophic zone (Fig. 3a), indicating that Notch2 is likely the predominant regulator in the endochondral skeleton. The increased osteoblast population in both PPS (Supplementary Data Fig. S2) and PNN embryos expressed high levels of bone sialoprotein (Ibsp) (Fig. 3b), as well as type I collagen (Col1a1) and alkaline phosphatase (Akp2) (data not shown). Interestingly, the number of osteoclasts expressing Trap (Acp5) also increased within the trabecular region of PNN mutants (Fig. 3b). The excessive trabecular osteoblasts in PNN mutants did not reflect changes in cell proliferation, as BrdU labeling experiments revealed a ∼20% reduction in the labeling index when mutant trabecular osteoblasts were compared to the wild type (n=3, p=0.05). Similarly, immunohistochemistry with an antibody specific for activated caspase 3 detected no difference in apoptosis of trabecular osteoblasts between mutant and wild type animals (data not shown). Thus, genetic disruption of Notch signaling from the limb mesenchyme results in elongation of the hypertrophic cartilage, and overproduction of trabecular osteoblasts, in embryonic long bones.
Figure 3.
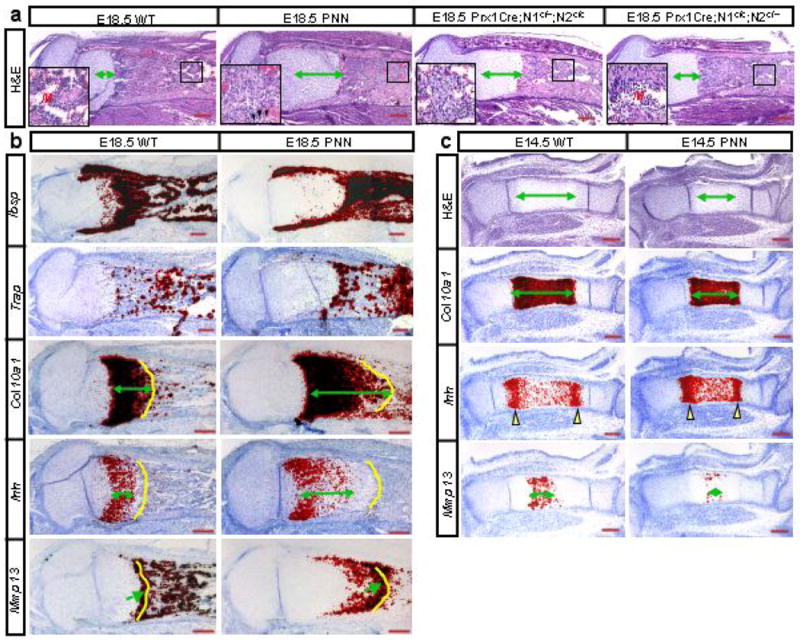
Molecular and histological analyses in PNN and wild type (WT) embryos. (a) H&E staining of medial longitudinal sections through the tibia of E18.5 embryos with various combinations of Notch1 (N1) and Notch2 (N2) alleles. Insets: higher magnification of boxed regions: M: marrow; arrowheads: occasional myeloid cells. Double-headed arrows: hypertrophic zone. (b) In situ hybridization on adjacent longitudinal sections through the tibia from E18.5 wild type (WT) versus PNN littermates. Double-headed arrows denote length of expression domain; yellow contours demarcate chondro-osseous junction; arrows indicate last low of hypertrophic chondrocytes. (c) H&E staining and in situ hybridization of adjacent medial longitudinal sections through the tibia of E14.5 littermates. Double-headed arrows: lengths of hypertrophic zones or expression domains. Yellow arrowheads denote major expression domains of Ihh.
To further characterize the cartilage defect, we performed molecular analyses on sections of embryonic tibia. At E18.5, the hypertrophic zone expressing type X collagen (Col10a1) was significantly longer in PPS (Supplementary Data Fig. S2) or PNN mutants than in wild type littermates (Fig. 3b). Indian hedgehog (Ihh), normally restricted to the prehypertrophic/early hypertrophic region at this stage, persisted at a lower level throughout much of the mutant hypertrophic zone (Fig. 3b). Matrix metallopeptidase 13 (Mmp13), normally expressed at high levels in a single row of terminal hypertrophic chondrocytes before they were removed, was activated in the mutant mouse at a lower level in many rows of hypertrophic cells before eventually reaching the higher level (Fig. 3b). The delayed progression towards terminal hypertrophy (Mmp13-positive) may have contributed to the overall lengthening of the hypertrophic zone. In addition, in vivo BrdU labeling experiments revealed that the proliferation rate of growth plate chondrocytes was reduced by ∼25% in PNN mutants versus wild type littermates (n=3, p=0.004). At E14.5, both histology and the expression profile of Col10a1 indicated that the hypertrophic region was significantly reduced in PNN mutants (Fig. 3c). The two major Ihh-expressing domains were less well separated, and the number of terminal hypotrophic chondrocytes expressing Mmp13 was markedly reduced in PNN mutants (Fig. 3c). Thus, Notch signaling in the growth plate positively controls the proliferation of nonhypertrophic chondrocytes, the onset of chondrocyte hypertrophy and also subsequent progression toward terminal hypertrophy.
Notch regulation of bone marrow mesenchymal progenitors
The results so far indicate that Notch removal enhances osteoblast differentiation from precursors. We reasoned that unchecked differentiation might lead to a deficit in bone marrow mesenchymal progenitors (including both mesenchymal stem cells and the more committed cell types such as osteoblast progenitors). Because these cells are found in the clonogenic subset of adherent bone marrow stromal cells (BMSCs), known as colony-forming unit-fibroblasts (CFU-Fs) 1,23,24, we cultured BMSCs to assay for the frequency of CFU-Fs in the bone marrow of 8-week-old animals. Remarkably, although a similar number of total marrow cells was retrieved from the PNN mutant and the wild type bones, the mutant marrow contained hardly any “type I” (see Methods) CFU-Fs (0-1 per 106 marrow cells), whereas wild type samples typically contained ∼300 per 106 marrow cells (Fig. 4a). In addition, the mutant samples formed significantly fewer (∼5 per 106 marrow cells) and smaller (fewer cells within each cluster, data not shown) “type II” CFU-Fs than the wild type (∼20 per 106 marrow cells). Thus, removal of Notch signaling from the limb mesenchyme severely reduced the number of bone marrow mesenchymal progenitors in adolescent mice.
Figure 4.
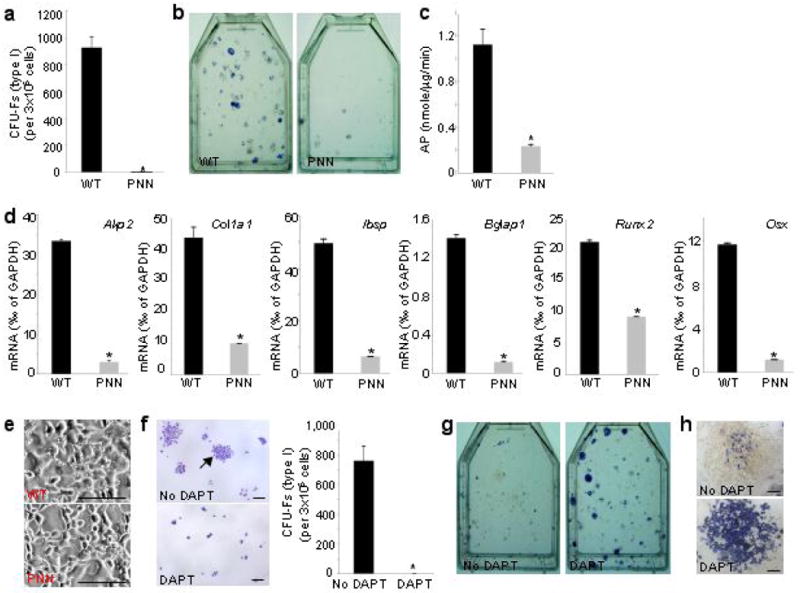
Notch regulation of bone marrow mesenchymal progenitors. (a) Bone marrow CFU-F assays for wild type (WT) versus PNN mutant littermates. (b) AP staining after 8 days in osteogenic medium following CFU-F assays. (c) AP quantitative assay for high-density BMSCs cultures in osteogenic medium. (d) Real-time PCR of osteoblast markers in BMSCs from 15-week-old mice cultured for 10 days in regular medium. (e) Phase-contrast pictures of high-density BMSCs cultures in adipogenic medium. Note lipid droplets only in wild type cells. (f) CFU-F assays for wild type BMSCs with or without 1 μM DAPT. Arrow denotes a typical “type I” CFU-F. (g) AP staining immediately following CFU-F assays of wild type BMSCs in f. (h) Representative “type II” CFU-Fs from g. *: p<0.05, n=3.
A decrease in mesenchymal progenitors would predict reduced differentiation capacity for the PNN BMSCs. To examine whether this was the case, we further cultured the BMSCs in osteogenic media following formation of CFU-Fs, and found that the PNN cells produced far fewer osteoblast-lineage cells (AP-positive) than normal (Fig. 4b). In addition, when BMSCs from 8- or 15-week-old mice were cultured at a higher density in either osteogenic or regular medium, the PNN samples were markedly impaired in osteoblast differentiation, as indicated by the low AP activity (Fig. 4c), as well as the low mRNA levels for several common osteoblast markers (Fig. 4d). Similarly, when BMSCs were cultured in an adipogenic medium, the PNN cells produced virtually no adipocytes identifiable by the characteristic lipid droplets (Fig. 4e), even though Notch signaling was previously shown to be dispensable for adipocyte differentiation 25. These results are therefore consistent with a severe deficit in bone marrow mesenchymal progenitors in PNN mice.
To corroborate further that Notch controls the fate of bone marrow mesenchymal progenitors, we examined whether DAPT (N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester), a γ-secretase inhibitor that blocks all Notch signaling, alters the frequency of CFU-Fs among wild type BMSCs. DAPT completely eliminated “type I” CFU-Fs (Fig. 4f), although it did not alter the number of “type II” CFU-Fs (∼20 per 106 marrow cells). Interestingly, when assayed directly for AP activity without being further cultured in osteogenic conditions, nearly all “type II” CFU-Fs from the DAPT-treated samples stained strongly positive, with many containing only AP-positive cells, whereas in the control samples the clusters contained only a small number of positive cells (Fig. 4g–h). Thus, Notch signaling (inhibited by DAPT) negatively regulates osteoblastogenesis, and this finding further supports the conclusion that the lack of osteoblast differentiation from the PNN cultures (see Fig. 4b) was most likely due to the loss of precursors instead of deficiency in the osteogenic program per se. Overall, these results support the notion that Notch signaling normally maintains mesenchymal progenitors while suppressing osteoblast differentiation.
Progressive bone loss in mature PNN mice
The severe reduction of mesenchymal progenitors in adolescent mice, along with the increased osteoclast numbers and total activity, predicts that PNN mice may exhibit a net loss of bone when they age. To test this possibility, we took advantage of the fact that the PNN mice did not die prematurely (unlike PPS mice in this regard), and examined the long bones by X-ray and μCT techniques at progressively older ages. Because the increase in trabecular bone mass at 8 weeks was most dramatic at the metaphyseal region (immediately below the growth plate) (see Fig. 2b), we focused our analyses on this region in the older animals. Remarkably, although the PNN mice still maintained a slightly higher trabecular bone mass than controls at 13 weeks of age (data not shown), the magnitude was significantly reduced when compared to that at 8 weeks. Moreover, at 14 and 15 weeks the mutant mice became progressively more osteopenic, and at 26 weeks they reached a normalized bone mass (bv/tv) only 10% of the control (Fig. 5a, and Supplementary Table 1), indicating that the PNN mice rapidly lost bone mass as they aged. The age-related bone phenotype was observed in both male and female mice. In agreement with the μCT data showing a rapid decrease in trabeculae number (Tb. N* in Supplementary Table I), histology of bone sections confirmed a marked reduction of bone trabeculae at the metaphyseal region in 26-week-old PNN mice compared to the control (Fig. 5b). Furthermore, the osteoblast density on trabecular bone surfaces (No. OB/mm) was significantly reduced in the mutant at this age (cuboidal OB down by 15%, flat OB down by 40%, p<0.05), indicating a marked decrease in the total number of trabecular osteoblasts in the aged PNN mice. In addition, the elongated region of hypertrophic cartilage, characteristic of the PNN animals in the embryo and at 8 weeks (see Fig. 3a and Fig. 2c, respectively), was no longer present at 26 weeks. Instead, the growth plate of the PNN mice was notably shorter, disorganized, and with significantly fewer chondrocytes than normal (Fig. 5b). The shortening of the hypertrophic region indicates that the increased total osteoclast activity in the mutant animals eventually overtook the accumulation of hypertrophic cartilage at the older age. Overall, the data indicate that sustaining a proper pool of mesenchymal progenitors through Notch signaling is critical for bone homeostasis in adult life, and that Notch signaling is also required for maintaining a normal growth plate.
Figure 5.
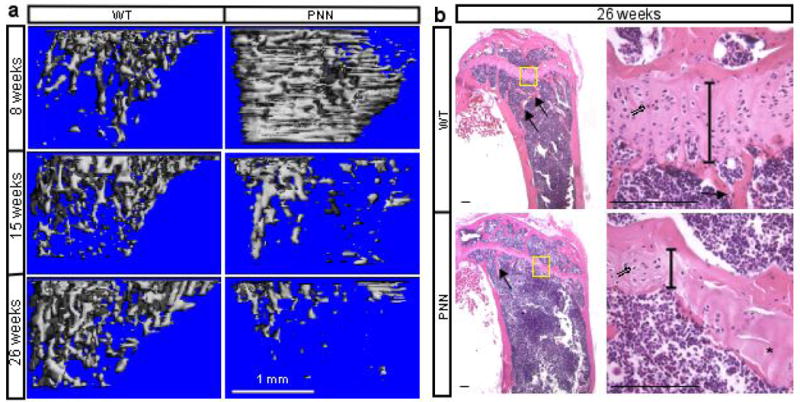
Progressive bone loss in mature PNN mice. (a) 3-D reconstruction of metaphyseal trabecular bone in wild type (WT) versus PNN mice at different ages. (b) H&E staining of medial longitudinal sections through the tibia of 26-week-old mice. Arrows: bone trabeculae. Right panels: higher magnification of boxed regions in corresponding left panels. Double-lined arrows: chondrocytes. Vertical lines denote height of growth plate. Asterisk: area devoid of chondrocytes.
Mechanisms for Notch function in bone
Because the PNN mice formed more osteoclasts even though Prx1Cre did not target this lineage, we hypothesized that Notch-deficient osteoblasts may affect osteoclastogenesis. Because osteoblasts are known to balance osteoclastogenesis through production of the positive regulator Rankl (Tnfsf11) and the antagonist Opg (Tnfrsf11b) 26, we examined mRNA levels in the tibia of 8-week-old mice by real-time PCR. Western analyses confirmed that Notch2 (both the full-length and the NICD form) was reduced by >90% in the long bones of PNN mice, although Notch1-NICD was reduced by only ∼60% (Fig. 6a). The Notch1 antibody failed to detect a distinct full-length protein in either wild type or PNN bones in this experiment (data not shown). Importantly, the PNN tibia RNA showed a 5.5-fold reduction in Rankl, but a 2-fold increase in Opg (Fig. 6b). Consequently, the Rankl/Opg ratio was 11 fold higher than normal, explaining the increased osteoclast number in the mutant animals. Thus, Notch signaling in long-bone osteoblasts indirectly regulates osteoclast differentiation by controlling the expression of Rankl and Opg.
Figure 6.
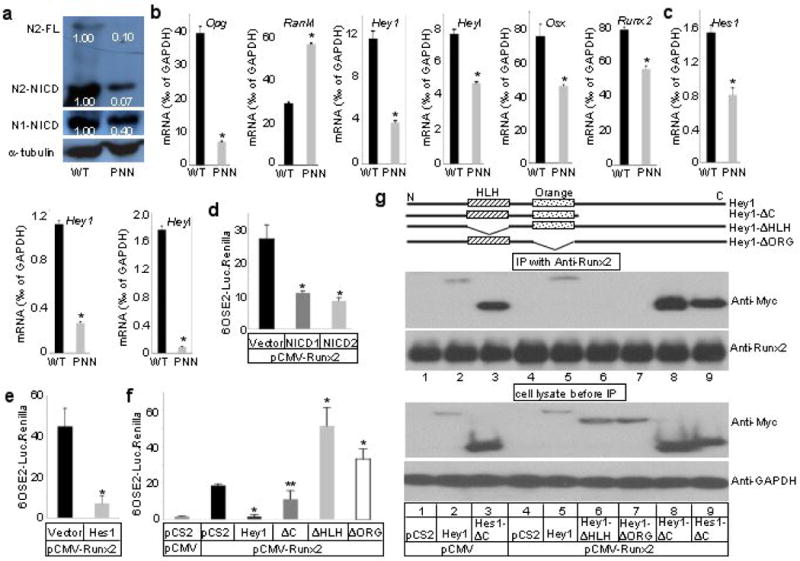
Mechanisms for Notch function in bone. (a) Western analyses for Notch1 and 2 in protein extracts from tibiae and femora of 8-week-old PNN versus wild type (WT) mice. Signal intensity normalized to α-tubulin. (b) Real-time PCR with RNA from tibiae of 8-week-old mice. (c) Real-time PCR with RNA from BMSCs of 15-week-old mice. (d-f) Luciferase reporter assays in CHO cells. *p < 0.01, ** p = 0.05, n=3, p values in f calculated against samples transfected with pCS2+MT empty vector and pCMV-Runx2 (black bar). (g) Co-immunoprecipitation of Hes1 or Hey1 with Runx2 in ST2 cells.
To probe the molecular mechanism through which Notch maintained mesenchymal progenitors, we examined the expression of the Hes/Hey family of molecules, known mediators of canonical Notch signaling in other tissues. Of the six potential targets of Notch, Hes1, 5, 7 and Hey1, 2, l27, Hey1 and Heyl were found to be significantly downregulated in the long bones of PNN mice at 8 weeks of age (Fig. 6b), whereas Hes1 was not changed and the others were not detected at any significant levels (<0.01‰ of GAPDH) (data not shown). Similarly, BMSCs from 15-week-old animals expressed only Hes1, Hey1 and Heyl at significant levels, and all three were markedly reduced in the PNN samples (Fig. 6c). These results therefore identify Hes1, Hey1 and Heyl as potential mediators of Notch signaling in osteoblast-lineage cells.
We next examined whether Notch controls osteoblast differentiation by affecting Runx2 and Osx (Sp7), two critical transcription activators of the lineage. We first determined whether Runx2 and/or Osx expression was increased in the long bones of 8-week-old PNN mice. Real-time PCR assays showed that both genes were, on the contrary, downregulated in the mutant bones (containing a higher bone mass) (Fig. 6b), indicating that changes in Runx2 and Osx levels are unlikely to be the primary mechanism through which Notch suppresses osteoblast differentiation. To test whether Notch signaling could inhibit the transactivating ability of Runx2, we performed transient transfection assays. In comparison to cells expressing Runx2 alone, co-expression of either NICD1 (from Notch1), NICD2 (from Notch2), Hes1 or Hey1 significantly reduced the expression of a Runx2-responsive reporter in both CHO cells (Fig. 6d–f), and the murine bone marrow stromal cell line ST2 cells (Supplementary Data Fig. S3, and data not shown). To understand further the regulation of Runx2 activity by Hes/Hey proteins, we performed deletion studies with Hey1 as an example. In transfection experiments, deletion products missing either the helix-loop-helix (Hey1-ΔHLH) or the Orange domain (Hey1-ΔORG) failed to repress Runx2 activity, but instead enhanced it (Fig. 6f), indicating that these constructs may have a dominant-negative effect. On the other hand, Hey1-ΔC, which lacked a C-terminal portion of Hey1, retained some but not all of the inhibitory function on Runx2. Thus, the HLH and Orange domains are critical for the inhibition whereas the C-terminal region may also play a role. Overall, Notch signaling may inhibit osteoblast differentiation through Hes/Hey proteins that impede the function of Runx2.
To determine whether Hes/Hey proteins may physically associate with Runx2, we performed co-immunoprecipitation experiments. Here, ST2 cells were transfected to overexpress Hey1 or its variant (all Myc-tagged), either alone or with Runx2; the cell lysates were immunoprecipitated with a Runx2 antibody and then probed with a Myc antibody following SDS-PAGE. The full-length Hey1 was detected in the Runx2-immunoprecipitate, at similar levels with or without Runx2 overexpression (Fig. 6g, lanes 5 and 2, respectively), indicating that Hey1 interacts with endogenous Runx2 and that Hey1 is likely to be the limiting factor in the interaction. Consistent with the transfection data that the HLH and Orange domains were critical for the inhibition of Hey1 on Runx2, neither Hey1-ΔHLH nor Hey1-ΔORG interacted with Runx2 (Fig. 6g, lanes 6 and 7). On the other hand, Hey1-ΔC retained the interaction with Runx2 (Fig. 6g, lane 8). The stronger signal with Hey1-ΔC than full-length Hey1 was likely due to the fact that the former was expressed at a much higher level in these experiments (Fig. 6g, lower panel). Similarly, Hes1-ΔC interacted with Runx2 with or without Runx2 overexpression (Fig. 6g, lanes 3 and 9). Thus, Hes/Hey proteins physically interact with Runx2 in the cell through the HLH and Orange domains.
In summary, the present study demonstrates that Notch signaling in osteoblast-lineage cells performs critical functions for bone homeostasis. First, it maintains bone marrow mesenchymal progenitors through inhibition of osteoblast differentiation. Second, it regulates osteoclastogenesis from bone marrow macrophage precursors by modulating the production of Rankl and Opg by osteoblasts. In our model, Notch deficiency leads to excessive osteoblastogenesis, and therefore more bone and fewer bone marrow mesenchymal progenitors in adolescent mice. On the other hand, in older animals, diminution of the progenitor pool causes a deficit in osteoblast production, which, when combined with heightened osteoclast differentiation, leads to precipitous bone loss associated with age. The model is consistent with our finding that the total number of trabecular osteoblasts in the PNN mice was greater than the control at a young age (increased bone surface but no change in osteoblast density on surface), but was reduced to below the control level when they grew older (decreased bone surface and decreased osteoblast density). However, it should be noted that our methodology does not allow direct assessment of mesenchymal progenitors and their differentiation in vivo. Thus the exact relationship between the progenitors and the age-related osteopenic phenotype remains to be further elucidated.
The finding that Notch inhibits osteoblastogenesis is consistent with a number of in vitro studies 28-30 but seems to be at odds with several others that suggested a stimulatory role 31,32. As most of these in vitro studies relied on overexpression of either NICD or the ligands, the discrepancy may be caused by differences in the level and/or the duration of Notch activation achieved in each study. In addition, because the stimulatory effect of Notch was observed only in cells co-treated with BMP 31,32, Notch might have enhanced proliferation of BMP-induced osteoblasts, without affecting differentiation per se, in those studies.
To investigate whether Notch signaling is required in committed osteoblasts, we attempted to ablate Notch1 and Notch2 using ColI-Cre, which expresses Cre under the control of a 2.3-kb promoter sequence of the murine Col1a1 gene and was previously shown to function effectively in these cells 33. Animals with the genotype of ColI-Cre; N1c/c; N2c/c (C1NN mutants) were viable but in contrast to PPS and PNN mutants, did not show any obvious skeletal phenotype at 8 weeks of age (Supplementary Data Fig. S4a–f). Western analyses revealed that Notch1 (>90%), but not Notch2 (∼20%), was efficiently deleted in the long bones of C1NN mutants at 8 weeks of age (Supplementary Data Fig. S4g). Thus, we cannot rule out the possibility that the lack of bone phenotype at this age may be due to incomplete deletion of Notch2. Interestingly, μCT analyses revealed that the C1NN mice had a lower trabecular bone mass than their sex-matched littermates at 5 months of age (data not shown), although the phenotype was less severe than that in the same-age PNN mice. We investigated this observation in a separate study and found that loss of Notch1 alone in committed osteoblasts increased osteoclastogenesis secondary to dysregulation of Rankl and Opg, similar to what is described for the PNN mice in this study (reported elsewhere). These findings confirm that Notch not only controls osteoblastogenesis early in the lineage, but also acts in more mature osteoblast-lineage cells to regulate osteoclastogenesis indirectly.
The importance of Notch signaling in bone homeostasis is consistent with Notch activation in the osteoblast lineage. By using a transgenic Notch reporter (TNR) mouse that drives expression of GFP under the control of repeated RBP-Jκ-response elements 34, we have consistently observed GFP-positive cells in live bone fragments free of marrow cells (data not shown). To specifically monitor Notch1 lineages, we took advantage of the N1∷Cre mouse that allows Cre recombination specifically in cells that have experienced Notch1 activation 35. In mice carrying both the N1∷Cre and the Rosa26-LacZ reporter (R26R) allele 36, LacZ-positive cells were found both on the trabecular bone surface and within the bone matrix (Supplementary Data Fig. S5a). Intriguingly, none of the growth plate chondrocytes were LacZ-positive, indicating either that Notch1 is not activated in the chondrocyte lineage, or that the level of activation was below the sensitivity threshold of this approach as previously discussed 35. If the former is true, the chondrocyte phenotype in PNN mice could be due to either loss of Notch2 signaling in chondrocytes, or to potential non-cell-autonomous effects caused by loss of Notch1 and/or Notch2 signaling in other cell types; future studies are necessary to discern these possibilities. Furthermore, a subset of BMSCs from the N1∷Cre;R26R mice expressed LacZ, indicative of active or historical Notch1 signaling in these cells (Supplementary Data Fig. S5b–c). Whereas the present work identifies Notch2 as the predominant regulator of osteoblastogenesis from bone marrow mesenchymal progenitors, our other study demonstrates a principle role for Notch1 in osteoclast precursors (reported elsewhere). It is of great interest in the future to examine the basis for these differences.
We concur with previous findings that Hes/Hey family molecules may mediate Notch inhibition on osteoblast differentiation through inhibition of Runx2 activity. Whereas a previous study identified Hey1 and Hey2 as potential mediators in aortic valves 37, we find that Hes1, Hey1 and Heyl are likely candidates in bone marrow mesenchymal progenitors. In keeping with previous reports that Hey1 inhibited the transcriptional activity of Runx2 in either COS7 cells 37, or cultured osteoblastic cells 38, we show that both Hes1 and Hey1 inhibit Runx2 in CHO and ST2 cells. Moreover, the inhibition by Hey1 requires the HLH and Orange domains, both of which are also necessary for the physical interaction between Hey1 and Runx2 in the cell. A definitive answer to the role of Hes/Hey proteins in osteoblastogenesis will require analyses of compound mutant mice.
The present study indicates that a short pulse of Notch inhibitor treatment may enhance bone formation by stimulating osteoblastogensis from bone marrow mesenchymal progenitors in vivo. On other hand, both this and our other study indicate that sustained inhibition of Notch signaling in osteoblasts leads to overproduction of osteoclasts and bone resorption. Finally, activating Notch signaling in bone marrow mesenchymal progenitors may help to maintain their phenotype and to expand their population in vitro.
Methods
Mouse strains
The N1n/+ 39, N1c/c 40, N2c/c 41, PS1c/c 42, PS2n/n 8, Col1-Cre 33 and Prx1Cre 22 mouse strains are as previously described. The Animal Studies Committee at Washington University approved all animal procedures.
Analyses of mice
We obtained radiographs of mouse hindlimbs using a Faxitron X-ray system (Faxitron X-ray Corp), and used Micro computed tomography (μCT 40, Scanco Medical AG) for three-dimensional reconstruction, and quantification of bone parameters. For Fig. 5a, we reconstructed each image from 100 of 16-μm slices immediately below the growth plate, with a threshold of 200. We performed histology on paraffin sections, following decalcification for postnatal samples. For dynamic histomorphometry of postnatal mice, we injected calcein (Sigma) intraparitoneally at 7.5 mg/kg on days 7 and 2 prior to sacrifice, and sectioned tibias in methyl-methacrylate. We performed in situ hybridization and BrdU labeling as previously described 43, and performed quantitative histomorphometry on paraffin sections using the Osteomeasure Analysis System (Osteometrics).
Cell cultures and in vitro assays
We harvested bone marrow cells and performed high density cultures as previously described 44. For CFU-F assays, we seeded single-cell suspension of nucleated bone marrow cells at 3×106 on T25 flasks and cultured the cells in DMEM (Sigma) containing 20% lot-selected FBS (Hyclone) and 1-Thioglycerol (Sigma) for 11-14 days without change of medium. For CFU-F assays, we scored colonies with more than 50 small, round or spindle-shaped cells in direct contact with each other as “type I” CFU-Fs, and counted clusters with more than 100 cells including large cells with multiple processes as “type II” CFU-Fs. We induced differentiation of BMSCs by either osteogenic (50 μg/ml ascorbic acid, 50 mM β-glycerol-2-phosphate) or adipogenic (0.5 μM IBMX, 60 μM indomethacine, 0.5 μM hydrocortisone) DMEM containing 10% FBS (Atlas).
We extracted bone proteins from tibiae and femora after bone marrow cells were flushed away. The Notch1 antibody was as previously described 45, and the Notch2 antibody (C651.6DbHN) was from Developmental Studies Hybridoma Bank at the University of Iowa.
We extracted total RNA with Trizol (Invitrogen) either from bone fragments after being flushed to remove marrow cells, or from cultured BMSCs. We performed Real-time PCR using SYBR green (Biorad) in an ABI-7500 machine.
We performed transient transfections using Lipofectamine (Invitrogen). For luciferase assays, we seeded ST2 and CHO cells in 24-well plates at 3×104 and 4.5×104 cells, respectively, and co-transfected the NICD- or Hes1/Hey1-expressing construct or the corresponding empty vector (0.13μg) along with pCMV-Runx2 (0.07 μg, Dr. Roberto Civitelli), p6OSE2-Luc (0.1μg, Dr. Roberto Civitelli), and pRL-Renilla (0.01μg, Promega) for 8 hrs using Lipofectamine (0.5μl/well), before further culturing the cells for 48 hrs and finally harvesting them for dual luciferase activity assays (Promega). The NICD1- or NICD2-expressing construct is as previously descibed 46. pSV2-CMV-Hes1 was originally from Dr. Ryoichiro Kageyama (Kyoto University). We generated pCS2-Hey1, pCS2-Hey1-ΔHLH, pCS2-Hey1-ΔORG, pCS2-Hey1-ΔC and pCS2-Hes1-ΔC by cloning the appropriate cDNAs into the pCS2+MT vector (containing a 6×Myc tag). We generated cDNAs encoding the truncated proteins by PCR from full-length mouse cDNA clones (ATCC): Hes1-ΔC: deletion of a.a. 161-282; Hey1-ΔC: deletion of a.a. 177-299; Hey1-ΔHLH: deletion of a.a. 66-107; Hey1-ΔORG: deletion of a.a. 121-166. For co-immunoprecipitation experiments, we transiently transfected ST2 cells with the Hes1 or Hey1 constructs, in combination with pCMV-Runx2 or the empty vector; we immunoprecipitated the cell lysate with a Runx2 antibody (Santa Cruz Biotechnology), and resolved the precipitate by SDS-PAGE before probing the membrane with a Myc antibody (Santa Cruz Biotechnology). The GAPDH antibody is from Chemicon.
Supplementary Material
Acknowledgments
This work was supported in part by US National Institutes of Health grants DK065789 (F.L.), HD044056 (R.K.), AR046852 (F.P.R.), AR046523 (S.L.T.) and 5T32AR07033 (M.J.H.). We thank Pamela Robey and Sergei Kuznetsov for advice on bone marrow CFU-F assays, Jie Shen (Harvard Medical School) and Thomas Gridley (The Jackson Laboratory) for mouse strains. We also thank Dwight Towler and Matthew Silva for their help with the response to reviewers' comments.
References
- 1.Bianco P, Riminucci M, Gronthos S, Robey PG. Bone marrow stromal stem cells: nature, biology, and potential applications. Stem Cells. 2001;19:180–92. doi: 10.1634/stemcells.19-3-180. [DOI] [PubMed] [Google Scholar]
- 2.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–6. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 3.Chiba S. Notch signaling in stem cell systems. Stem Cells. 2006;24:2437–47. doi: 10.1634/stemcells.2005-0661. [DOI] [PubMed] [Google Scholar]
- 4.Schroeter EH, Kisslinger JA, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393:382–6. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- 5.Honjo T. The shortest path from the surface to the nucleus: RBP-J kappa/Su(H) transcription factor. Genes Cells. 1996;1:1–9. doi: 10.1046/j.1365-2443.1996.10010.x. [DOI] [PubMed] [Google Scholar]
- 6.Kopan R, Goate A. A common enzyme connects notch signaling and Alzheimer's disease. Genes Dev. 2000;14:2799–806. doi: 10.1101/gad.836900. [DOI] [PubMed] [Google Scholar]
- 7.Donoviel DB, et al. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 1999;13:2801–10. doi: 10.1101/gad.13.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herreman A, et al. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc Natl Acad Sci U S A. 1999;96:11872–7. doi: 10.1073/pnas.96.21.11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krebs LT, et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000;14:1343–52. [PMC free article] [PubMed] [Google Scholar]
- 10.Hamada Y, et al. Mutation in ankyrin repeats of the mouse Notch2 gene induces early embryonic lethality. Development. 1999;126:3415–24. doi: 10.1242/dev.126.15.3415. [DOI] [PubMed] [Google Scholar]
- 11.Swiatek PJ, Lindsell CE, del Amo FF, Weinmaster G, Gridley T. Notch1 is essential for postimplantation development in mice. Genes Dev. 1994;8:707–19. doi: 10.1101/gad.8.6.707. [DOI] [PubMed] [Google Scholar]
- 12.Domenga V, et al. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev. 2004;18:2730–5. doi: 10.1101/gad.308904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hrabe de Angelis M, McIntyre J, 2nd, Gossler A. Maintenance of somite borders in mice requires the Delta homologue DII1. Nature. 1997;386:717–21. doi: 10.1038/386717a0. [DOI] [PubMed] [Google Scholar]
- 14.Duarte A, et al. Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev. 2004;18:2474–8. doi: 10.1101/gad.1239004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gale NW, et al. Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc Natl Acad Sci U S A. 2004;101:15949–54. doi: 10.1073/pnas.0407290101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue Y, et al. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet. 1999;8:723–30. doi: 10.1093/hmg/8.5.723. [DOI] [PubMed] [Google Scholar]
- 17.Jiang R, et al. Defects in limb, craniofacial, and thymic development in Jagged2 mutant mice. Genes Dev. 1998;12:1046–57. doi: 10.1101/gad.12.7.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dunwoodie SL, et al. Axial skeletal defects caused by mutation in the spondylocostal dysplasia/pudgy gene Dll3 are associated with disruption of the segmentation clock within the presomitic mesoderm. Development. 2002;129:1795–806. doi: 10.1242/dev.129.7.1795. [DOI] [PubMed] [Google Scholar]
- 19.Shen J, et al. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89:629–39. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- 20.Wong PC, et al. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature. 1997;387:288–92. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- 21.Pan Y, Liu Z, Shen J, Kopan R. Notch1 and 2 cooperate in limb ectoderm to receive an early Jagged2 signal regulating interdigital apoptosis. Dev Biol. 2005;286:472–82. doi: 10.1016/j.ydbio.2005.08.037. [DOI] [PubMed] [Google Scholar]
- 22.Logan M, et al. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33:77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- 23.Owen M, Friedenstein AJ. Stromal stem cells: marrow-derived osteogenic precursors. Ciba Found Symp. 1988;136:42–60. doi: 10.1002/9780470513637.ch4. [DOI] [PubMed] [Google Scholar]
- 24.Kuznetsov SA, et al. The interplay of osteogenesis and hematopoiesis: expression of a constitutively active PTH/PTHrP receptor in osteogenic cells perturbs the establishment of hematopoiesis in bone and of skeletal stem cells in the bone marrow. J Cell Biol. 2004;167:1113–22. doi: 10.1083/jcb.200408079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nichols AM, et al. Notch pathway is dispensable for adipocyte specification. Genesis. 2004;40:40–4. doi: 10.1002/gene.20061. [DOI] [PubMed] [Google Scholar]
- 26.Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell. 2002;2:389–406. doi: 10.1016/s1534-5807(02)00157-0. [DOI] [PubMed] [Google Scholar]
- 27.Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol. 2003;194:237–55. doi: 10.1002/jcp.10208. [DOI] [PubMed] [Google Scholar]
- 28.Deregowski V, Gazzerro E, Priest L, Rydziel S, Canalis E. Notch 1 overexpression inhibits osteoblastogenesis by suppressing Wnt/beta-catenin but not bone morphogenetic protein signaling. J Biol Chem. 2006;281:6203–10. doi: 10.1074/jbc.M508370200. [DOI] [PubMed] [Google Scholar]
- 29.Sciaudone M, Gazzerro E, Priest L, Delany AM, Canalis E. Notch 1 impairs osteoblastic cell differentiation. Endocrinology. 2003;144:5631–9. doi: 10.1210/en.2003-0463. [DOI] [PubMed] [Google Scholar]
- 30.Shindo K, et al. Osteogenic differentiation of the mesenchymal progenitor cells, Kusa is suppressed by Notch signaling. Exp Cell Res. 2003;290:370–80. doi: 10.1016/s0014-4827(03)00349-5. [DOI] [PubMed] [Google Scholar]
- 31.Nobta M, et al. Critical regulation of bone morphogenetic protein-induced osteoblastic differentiation by Delta1/Jagged1-activated Notch1 signaling. J Biol Chem. 2005;280:15842–8. doi: 10.1074/jbc.M412891200. [DOI] [PubMed] [Google Scholar]
- 32.Tezuka K, et al. Stimulation of osteoblastic cell differentiation by Notch. J Bone Miner Res. 2002;17:231–9. doi: 10.1359/jbmr.2002.17.2.231. [DOI] [PubMed] [Google Scholar]
- 33.Miao D, et al. Osteoblast-derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1-34. J Clin Invest. 2005;115:2402–11. doi: 10.1172/JCI24918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duncan AW, et al. Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nat Immunol. 2005;6:314–22. doi: 10.1038/ni1164. [DOI] [PubMed] [Google Scholar]
- 35.Vooijs M, et al. Mapping the consequence of Notch1 proteolysis in vivo with NIP-CRE. Development. 2007;134:535–44. doi: 10.1242/dev.02733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–1. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 37.Garg V, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–4. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 38.Zamurovic N, Cappellen D, Rohner D, Susa M. Coordinated activation of notch, Wnt, and transforming growth factor-beta signaling pathways in bone morphogenic protein 2-induced osteogenesis. Notch target gene Hey1 inhibits mineralization and Runx2 transcriptional activity. J Biol Chem. 2004;279:37704–15. doi: 10.1074/jbc.M403813200. [DOI] [PubMed] [Google Scholar]
- 39.Conlon RA, Reaume AG, Rossant J. Notch1 is required for the coordinate segmentation of somites. Development. 1995;121:1533–45. doi: 10.1242/dev.121.5.1533. [DOI] [PubMed] [Google Scholar]
- 40.Pan Y, et al. gamma-secretase functions through Notch signaling to maintain skin appendages but is not required for their patterning or initial morphogenesis. Dev Cell. 2004;7:731–43. doi: 10.1016/j.devcel.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 41.McCright B, Lozier J, Gridley T. Generation of new Notch2 mutant alleles. Genesis. 2006;44:29–33. doi: 10.1002/gene.20181. [DOI] [PubMed] [Google Scholar]
- 42.Yu H, et al. APP processing and synaptic plasticity in presenilin-1 conditional knockout mice. Neuron. 2001;31:713–26. doi: 10.1016/s0896-6273(01)00417-2. [DOI] [PubMed] [Google Scholar]
- 43.Hilton MJ, Tu X, Cook J, Hu H, Long F. Ihh controls cartilage development by antagonizing Gli3, but requires additional effectors to regulate osteoblast and vascular development. Development. 2005;132:4339–51. doi: 10.1242/dev.02025. [DOI] [PubMed] [Google Scholar]
- 44.Tu X, et al. Noncanonical Wnt Signaling through G Protein-Linked PKCdelta Activation Promotes Bone Formation. Dev Cell. 2007;12:113–27. doi: 10.1016/j.devcel.2006.11.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huppert SS, et al. Embryonic lethality in mice homozygous for a processing-deficient allele of Notch1. Nature. 2000;405:966–70. doi: 10.1038/35016111. [DOI] [PubMed] [Google Scholar]
- 46.Ong CT, et al. Target selectivity of vertebrate notch proteins. Collaboration between discrete domains and CSL-binding site architecture determines activation probability. J Biol Chem. 2006;281:5106–19. doi: 10.1074/jbc.M506108200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.