

Abstract
Molecular targeting of apoptotic signaling pathways has been extensively studied in recent years and directed towards the development of effective therapeutic modalities for treating advanced androgen-independent prostate tumors. The majority of therapeutic agents act through intrinsic or mitochondrial pathways to induce programmed cell death. The induction of apoptosis through endoplasmic reticulum (ER) stress pathways may provide an alternative to treat patients. The functional interaction between the BCL-2 family members and regulation of calcium homeostasis in the ER provides a critical link to the life or death outcome of the cell. Apoptosis induction mediated by ER stress-inducing agents is just beginning to be exploited for therapeutic targeting of prostate tumors. Insightful dissection of recently discovered apoptotic signaling pathways that function through the endoplasmic reticulum may identify novel molecules that could effectively target both androgen-dependent and androgen-independent prostate tumors. In this review, we focus on linking ER stress-induced apoptosis to therapeutic targeting of prostate tumors and dissect its cross-talk with the intrinsic and extrinsic apoptotic pathways.
Keywords: prostate cancer, apoptosis, BCL-2 family, ESR, UPR
INTRODUCTION
Prostate cancer is a major cause of mortality in American men and ranks as the second highest contributor of all male cancer mortality [1]. Charles Huggins first advocated androgen ablation monotherapy through medical or surgical castration for the treatment of prostate cancer sixty years ago. Today, androgen ablation remains a primary strategy to combat metastatic disease via apoptosis induction among androgen-dependent prostate cancer cells [2]. Androgens play a critically recognized role in the growth of androgen-dependent prostate tumors by blocking apoptosis [3]. Although reducing circulating androgen levels is initially effective in eliciting cell death and in reducing the cancer burden, this strategy affords a median patient survival of less than 36 months [4]. Prostate cancer cell survival may be maintained due to the fact that current hormone ablation treatment regimens, although effective in reducing circulating levels of hormone, only decrease tissue androgen levels by 75% [5]. By further understanding the apoptotic signaling pathways utilized by prostate tumors, new therapeutic targets may be identified to combat metastatic prostatic cancer that are independent of, or work in combination with, the current hormonal ablation regimens.
Signaling Apoptosis Execution: Key Players and Key Pathways
There are two well-understood pathways by which a cell can initiate its own death: intrinsic (mitochondrial) and extrinsic apoptosis. The extrinsic apoptotic pathway is a receptor-mediated mechanism that involves binding of a death ligand to its corresponding death receptor. Death ligands are members of the tumor necrosis factor (TNF) family of ligand, which include FasL, TNF-α-related apoptosis inducing ligand (TRAIL) and the weak apoptosis inducer, TNF-α. These ligands bind to members of the tumor necrosis factor (TNFR) family of death receptors, specifically FAS, DR4/DR5 and TNFR-1, respectively [6]. A functional death ligand – death receptor interaction results in receptor clustering drove most likely by plasma membrane localized ceramide [7,8]. Clustering of death receptors allows for the formation of the death-induced signaling complex (DISC). The DISC is comprised of Fas-associated death domain (FADD), procaspase-8 and the apoptotic inhibitor c-FLIP [9]. While associated with FADD through their death effector domains (DED) [10–15], procaspase-8 is autocatalytically processed to form an active p10/p18 homodimer [16]. Active caspase-8 leaves the DISC where it is free to cleave effector caspases-3 and -7, as well as other substrates, inducing the extrinsic apoptotic pathway.
A cell can activate the intrinsic apoptotic pathway in response to a number of cellular cues, including DNA damage, defective cell cycling, extracellular matrix detachment, hypoxia, survival factor loss or other types of severe cell distress [6]. Mitochondrial outer membrane permeabilization (MOMP) is the key event in the execution of intrinsic apoptosis and is regulated by members of the BCL-2 family of proteins. Once mitochondrial permeabilization occurs there is release of cytochrome-c, SMAC/DIABLO and other proapoptotic proteins into the cytoplasm. Cytochrome-c binding to Apaf-1 leads to the oligomerization and formation of a cytochrome-c/Apaf-1 complex which upon association with three mediators (PHAPI, CAS and HSP-70) forms the apoptosome. [17]. Procaspase-9 binds to the central ring of the apoptosome, autoactivates and subsequently cleaves effector caspases-3 and -7 resulting in apoptosis. Smac/DIABLO release is critical for promoting intrinsic apoptosis by binding to and inhibiting the inhibitor of apoptosis proteins (IAP) [18,19].
The BCL-2 oncoprotein was originally defined as a player in the survival of IL-3-deprived lymphoid cells [20–22]. BCL-2’s key role in sustaining cellular homeostasis was substantiated by the in vivo evidence that Bcl-2 null mice suffered from excessive apoptosis in lymphocytes, melanocytes and developmental renal cells [23–25]. The BCL-2 family members are divided into three groups: the anti-apoptotic group, including BCL-2; the proapoptotic “multi-BH domain” group, including BAX and BAK; and the proapoptotic “BH3-only” group, including Bid. The activity of BCL-2 family members is multi-faceted. BCL-2 helps maintain both mitochondrial membrane homeostasis and calcium homeostasis within the cell. BAX and BAK can respond to an intrinsic death stimulus by translocating (BAX), oligomerizing and inserting into the outer mitochondrial membrane to promote permeabilization. Proteolytic Bid cleavage mediated by caspase-8 leads to Bid myristoylation, mitochondrial localization and MOMP induction, an event that suggests a crosstalk event between the two major apoptotic pathways. It is the ratio of pro- and anti-apoptotic proteins that most likely controls the cellular vulnerability to apoptosis.
In addition to the major intrinsic and extrinsic apoptotic pathways, the endoplasmic reticulum (ER) can act as a functional forum for apoptosis induction and regulation. The BCL-2 members BAK and BAX can localize to the ER [26]. It is here where they maintain Ca2+ homeostasis by regulating its release. Analysis of Bax/Bak double knockout mice has shown reduced Ca2+ release from the ER, resulting in reduced mitochondrial Ca2+ uptake [27]. This evidence provides a potential mechanism via which BAX and BAK contribute to the induction and propagation of the intrinsic apoptotic response. In this model, proapoptotic BAX/BAK responds to an apoptotic stimulus by inducing Ca2+ efflux from the ER. Ca2+ from the ER is then taken up by the mitochondria, resulting in Ca2+ overload, which triggers mitochondrial localized BAX/BAK to induce MOMP.
Sensing and Transducing Stress Signals by the Endoplasmic Reticulum
Covering approximately half of the total membrane area and a third of newly translated proteins in a typical eukaryotic cell, the endoplasmic reticulum (ER) plays a major role in a number of cellular processes, including protein synthesis, targeting, folding and secretion [28]. It is also a critical site for vesicle trafficking, lipid and membrane biogenesis, steroid biosynthesis and intra-cellular calcium storage [29]. In addition, this organelle possesses a unique capacity to sense various cellular insults. In response to these perturbations an endoplasmic reticulum stress response (ESR) can be mounted. The ESR can be triggered by the accumulation of misfolded, unfolded or excessive protein, fluctuations in calcium levels, deprivation of glucose, and ER lipid or glycolipid imbalance [28,30–32]. Experimentally, the ESR is commonly initiated by thapsigargin and tunicamycin, drugs that affect intra-cellular calcium levels and protein trafficking, respectively [33,34]. The ESR serves to restore homeostasis through a set of three primary mechanisms, together termed the unfolded protein response (UPR). The UPR deals with the accumulation of unfolded proteins by folding them with the aid of additional chaperone proteins [32,35]. Another strategy is to eliminate the unfolded proteins by activating proteasome-mediated degradation through a unique reverse-translocation process referred to as ER-associated degradation (ERAD) [36–39]. Since the 26S proteasomes are only located in cytoplasm and nucleus, proteins targeted for ubiquitination must be pumped out of the ER prior to degradation through a process that is regulated, at least in part, by proteins upregulated by the UPR. Reduction of global protein translation is the third important activity that effectively reduces the excessive protein burden in the ER [40,41]. Together, these UPR activities normally succeed in restoring ER homeostasis and ensure cellular survival.
Much of what is known regarding the specific ESR pathways has been learned in the yeast model. In S. cerevisiae, the ER-resident transmembrane dual endoribonuclease/kinase Ire1p is solely responsible for sensing ER stress and coordinating the ESR [42,43]. Once ER stress is detected by Ire1p (through the amino-terminal luminal region), the protein dimerizes, undergoes a conformational shift and becomes auto-phosphorylated [44,45]. In this active state, a non-conventional endoribonuclease activity is turned on which acts on the transcription factor HAC1 mRNA, its only known substrate [46]. Ire1p splicing removes an intron in the 3′ region of HAC1, thereby conferring robust translation with the subsequent transcription of Hac1p-dependent genes responsible for carrying out the ESR [47,48]. In contrast to single-celled organisms, the ESR in metazoans is significantly more complex and has evolved considerably. Flies and worms express a single copy of three ER-resident transmembrane proteins that serve as ER stress sensors, which include IRE1, ATF and PERK [49]. Similar to yeast, the C. elegans and D. melanogaster IRE1 homolog alone remains sufficient to induce the complete UPR program in response to ER stress [50,51]. In mammals, an additional level of complexity arises, as two forms of each stress-sensing gene exist. IRE1α and β are both localized to the ER membrane, but only IRE1α is ubiquitously expressed and essential for normal development [52,53]. IRE1β is expressed exclusively in the gut and its loss can lead to intestinal colitis. Similar to the yeast homolog, only one target, X-box binding protein (Xbp-1) mRNA, exists for metazoan endoribonuclease activity [54]. When activated by ER stress, mammalian IRE1 removes a 26 nucleotide region from XBP-1, a process that creates an alternative reading frame ultimately allowing a transcriptional activation domain to be translated after the DNA binding domain. Therefore, two forms of XBP-1 exist, one that is unspliced and contains only a DNA-binding domain (uXBP-1) and a second that can bind to DNA and activate transcription (sXBP-1) [54]. In contrast to yeast, mammalian IRE1 is not the primary UPR regulator. Instead, recent work demonstrates that the ER stress sensor ATF6 performs these critical functions [55]. The mammalian transcription factor ATF6 is also present in two isoforms (α and β) that are ubiquitously expressed [49]. When ER stress is sensed, ATF6 is released from the chaperone GRP78. This exposes the Golgi localization signal sequence, permitting ATF6 to travel to the Golgi [56,57]. At the Golgi, site-1 (S1P) and site-2 (S2P) proteases cleave ATF6 [57]. The resulting N-terminal bZIP domain (ATF6 (N)) is capable of nuclear entry and transcription of the ER-stress inducible genes CHOP, GRP78, GRP94, HRD1 and EDEM [55,58]. The use of ATF6α null mouse embryonic fibroblasts (MEFs) led to the discovery that the ER stress response is carried out by the activity of ATF6α alone or by dimerizing with sXBP-1 [55].
PERK is a third ER stress-sensing protein that plays a major role in stifling protein synthesis initiation and global translation. In response to an ER insult, PERK oligomerizes and autophosphorylates to an active kinase. Activated PERK is capable of phosphorylating the α-subunit of eukaryotic translation initiation factor 2 (eIF2α), which subsequently down regulates global translation [59–61]. Phosphorylation of eIF2α also arrests cells in the G1 cell cycle phase via cyclin D1 loss, which prevents ER stressed cells from propagating [62].
One central regulator that ties together each UPR pathway is the chaperone protein GRP78. This protein interacts with each stress sensor in the inactivated state and functionally serves as a negative regulator [57,63]. When unfolded and/or misfolded proteins accumulate in the ER, they compete for GRP78 binding and, thereby, the chaperone interaction with the transmembrane protein sensors is abrogated.
Apoptotic Signaling in the Endoplasmic Reticulum: Stress Response Goes Deadly
As outlined above, three ER-resident transmembrane proteins (IRE1, ATF6 and PERK) can sense ER stress. The ESR carried out by the individual sensor molecules provides a protective feature to ensure survival until the stimulus is eliminated (Fig. 1). For example, PERK and ATF6 null MEFs die much more readily to ER stress induced by thapsigargin and tunicamycin [55,59]. When the ER stress perturbation is too great, however, apoptosis can result [29,64,65]. While the precise event driving this effect is unclear, one line of thought suggests that either a prolonged or a sufficiently great induction of one or more ER stress sensor proteins triggers an irreversible apoptotic cascade through induction of the ATF/XBP transcription factors (as shown in Fig. 1). CHOP/Gadd153 is an ER stress target that is transcriptionally upregulated by ATF6 [66] and encodes for a transcription factor with apoptotic activity. Its proapoptotic function can be blocked by GRP78 overexpression, thus placing CHOP apoptotic pathways as downstream from the ER [67]. Mechanistically, CHOP may control apoptosis via both intrinsic and extrinsic pathways by downregulating BCL-2, while upregulating DR5 expression [68,69].
Fig. 1.
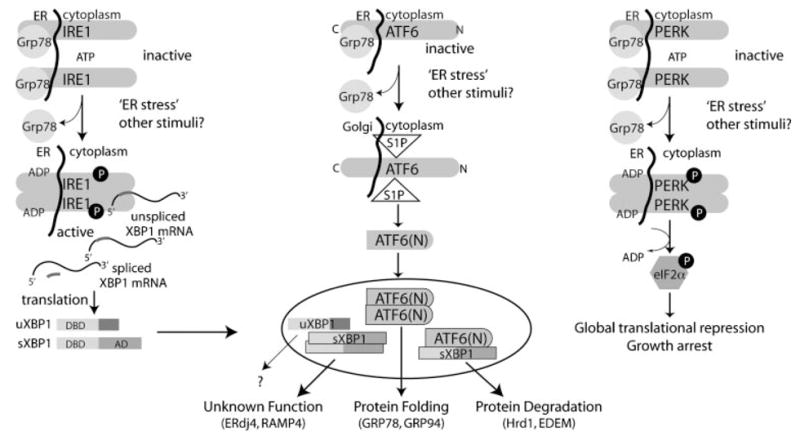
Sensing ER stress and subsequent signal transduction. Three main ER-resident transmembrane proteins (IRE1, ATF6, and PERK) in mammals can act as ER stress sensors. When ER stress is sensed, GRP78 becomes disassociated with the proteins allowing their activation. PERK responds to the ER stress by decreasing global translation. Signaling through IRE1 and ATF6 results in a transcription response to the ER stress mediatedbyXBP-1 and ATF6. These genes can act alone or in concert to increase protein folding machinery and initiate proteasomal degradation ofER-resident proteins. The full role for unspliced and spliced forms of XBP1 (uXBP1 and sXBP1) in confronting the ER stress is not yet understood.
In addition to translational repression, PERK has also been shown to have apoptotic activities mediated through eIF2α activity. The PERK-associated eIF2α has two phosphatase cofactors, Gadd34 and CReP, which can mediate apoptotic signaling downstream from the ER. Gadd34−/− cells in the presence of an ER stress exhibit constant eIF2α phosphorylation, resulting in fewer misfolded proteins in the ER lumen [70–72]. Furthermore, the Gadd34−/− mice are resistant to the toxicity induced upon tunicamycin treatment [71]. Likewise, CReP knockdown provides protection from ER stress [73]. In addition to these genetic studies, pharmacological eIF2α inhibition by the agent salubrinal provides protection from ER stress-mediated apoptosis [74]. Another potential mechanism by which ER stress may induce apoptosis is dependent on IRE1, but does not rely on its endoribonuclease activity. IRE1 can interact with the TRAF2 adaptor protein to recruit ASK1, a proapoptotic kinase. This interaction leads to the formation of an IRE1/TRAF2/ASK1 complex that activates JNK [75]. As ASK1−/− cells are partially resistant to apoptosis, this ternary complex may potentially regulate ESR-induced cell death [76]. Moreover, BAX and BAK can physically associate with IRE1 in the ER and positively regulate their proapoptotic activity [77].
Integration of the ER-Specific Death Signal to the Apoptotic Machinery
Due to their central role in dictating cellular life and death decisions, it is no surprise that the BCL-2 family can functionally control ESR-induced apoptosis. The regulation of calcium homeostasis by the ER provides a direct link to BCL-2 activity. ER Ca2+ dynamics is a principal avenue whereby BCL-2 phosphorylation alters susceptibility to apoptosis. When BCL-2 is phosphorylated, Ca2+ discharge from the ER is increased, with a secondary increase in mitochondrial Ca2+ uptake, thus enabling a mechanism through which posttranslational modification of BCL-2 inhibits its anti-apoptotic activity [78]. Moreover, BAX and BAK overexpression is sufficient to enhance Ca2+ release into the mitochondria, leading to permeabilization and increased cytochrome c release. Furthermore, MEFs lacking BAX and BAK exhibit notable resistance to ER stress-induced apoptosis, a finding which is substantiated by Bax−/−/Bak−/− cells exhibiting reduced Ca2+ release from the ER in response to arachidonic acid and oxidative stress [26,27,79]. Other studies involving ER stress stimuli, established that the presence of BAX and BAK at both the ER and mitochondria is indispensable for normal apoptosis execution [27] and proceeds via an Apaf-1-independent pathway [80].
As in the other apoptotic signaling pathways, NF-κB has recently found its connection to the ER stress-induced apoptotic cell death. In MCF-7 cells, NF-κB regulation suppresses ER stress-induced apoptosis [81]. Interestingly, IRE1-dependent NF-κB regulation also has been associated with TRAF2 down-regulation, upregulation of TNF-α expression and signaling through TNF receptor 1. Therefore, NF-κB may enable a cross-talk between ER-induced apoptosis and the death receptor mechanism [82].
The class of initiator caspases activated is a hallmark of the signaling pathway that the cell engages towards apoptosis induction. For instance, caspase-9 is an initiator for the intrinsic pathway, while caspase-8 and -10 are activated upon extrinsic stimuli. Two lines of evidence have suggested that caspase-12 is the initiator caspase for the ESR apoptotic response. First, caspase-12 is found in the ER membrane and can directly interact with IRE1 and the adaptor protein TRAF2 [83]. Second, caspase-12 is selectively processed through ER stress and not by other apoptotic stimuli [83]. The involvement of caspase-12 in ESR-induced apoptosis remains controversial due, in part, to the absence of a human homolog (caspase-12 is expressed in rodents and not in primates). The recent association of human caspase-4 as the caspase-12 human homolog, as well as its similar localization to ER membrane, points to a functional overlap between the two caspases in ER stress-induced apoptosis [84]. However, the role of both caspase-4 and -12 in the apoptotic ESR has not been supported in cells lacking these genes [85]; this study demonstrated that caspase-9 was the sole initiator caspase responsible for apoptosis induction in response to ER stress stimuli. A potential interactive scenario is illustrated in Figure 2, in which all three apoptosis signaling mechanisms, the mitochondrial, death receptor and ER stress apoptotic pathways are recruited by the cell towards its apoptotic execution when triggered by various ER stress stimuli.
Fig. 2.

ER stress-induced apoptotic signal transduction. This cartoon highlights how hyperactive ER stress stimuli sensed by IRE1, ATF6, and PERKmight result in apoptosis induction. A cross-talk between the mitochondrial, death receptor and ER stress apoptotic pathways may contribute to apoptosis mediated by chemical agents or by the accumulation of unfolded proteins.
Targeting ESR-Induced Apoptosisin Prostate Cancer: Current Value
Therapeutic exploitation of ESR apoptotic pathways has recently been attempted for the treatment of advanced prostate cancer. The theory that the SERCA pump could be a therapeutic target used against prostate cancer merits close examination. The sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) pump is present in all cell types and serves to transfer Ca2+ from the cytosol to the sarcoplasmic lumen. SERCA pump inhibition often leads to ER Ca2+depletion and elevation of cytoplasmic Ca2+. Thapsigargin, a known pharmacological inducer of ER stress and an effective apoptosis-inducing agent, inhibits the SERCA pump. In order to control targeting specificity and prevent bystander cytotoxicity, a PSA-cleavable, inactive thapsigargin derivative was synthesized [86]. In theory, proteolysis in PSA-producing sites will hydrolyze this pro-drug into an active bioavailable agent at prostate tumor sites.
Although the use of PSA to cleave pro-drugs may allow the induction of ER stress-induced apoptosis in prostate cells, there is yet to be identified a specific biochemical activity, biological process or prostate-specific gene to suggest that targeting prostate cancer cells through ESR pathways may have high therapeutic value compared to other apoptotic routes. Targeting the ESR/UPR pathways may be the ultimate mechanism to treat prostate cancer, but much needs to be learned first and fast. One problem is that the UPR has been best characterized in yeast and in the mouse model. Due to the evolution of the ER stress pathway, the exact biochemical pathways that each ER stress gene activates in human cells must be characterized. Second, while prostate tumors are highly heterogeneous, they are primarily composed of epithelial cells and one has to consider that much of the mammalian ESR research has exploited fibroblast models that may not accurately reflect pathways utilized in prostate cancer cells. For example, studies using mouse plasma cells demonstrated a function of XBP-1 that was unknown in fibroblasts [87]. Considering that mice lacking ATFα and β, IRE1α, and XBP-1 are not viable, one must assume that these players have multiple vital functions in other cell types that remain to be identified. Finally, the challenge will be to dissect the role of the individual stress sensors in the process of prostate tumor progression such that drugs can be designed to selectively target the proper ESR regulatory molecules, with the therapeutic promise recently shown by the targeting of the extrinsic death receptor pathway [88].
Acknowledgments
This work was supported by an Edwin Beer Award (S.R.S.) and an NIH R01 DK 53525-12 Grant (N.K.). Eric W. Lin was a recipient of a Markey Cancer Center Summer Research Fellowship and Dustin T. Gayheart is a GCRC Scholar from the University of Kentucky College of Medicine.
Grant sponsor: NIH; Grant number: R01 DK 53525–12.
Abbreviations
- PSA
prostate-specific antigen
- DISC
death-inducing signaling complex
- SERCA
sarcoplasmic/endoplasmic reticulum Ca2+ ATPase
- BCL-2
B-cell lymphoma 2
- p53
tumor protein 53
- Smac/DIABLO
second mitochondria-derived activator of caspases
- Apaf-1
apoptotic protease activating factor 1
- IAP
inhibitor of apoptosis proteins
- ESR
endoplasmic reticulum stress response
- OMI/HTRA1
high temperature requirement protein A2
- AIF
apoptosis inducing factor
- IL-3
interleukin 3
- BH
BCL-2 homology regions
- NGF
nerve growth factor
- TNFR
tumor necrosis factor receptor
- XIAP
X-linked inhibitor of apoptosis protein
- FASL
Fas ligand
- TRADD
TNF receptor-associated death domain
- FADD
Fas-associated death domain protein
- TRAIL
TNF-related apoptosis-inducing ligand
- NF-κB
nuclear factor of κB
- JNK
JUN N-terminal kinase
- eIF2α
eukaryotic translation initiation factor 2
- UPR
unfolded protein response
- ERAD
ER-associated degradation
- CHOP
CCAAT/-enhancer binding protein homologous protein
- GRP78
glucose regulated protein 78
- ATF6
activating transcription factor 6
- PERK
protein kinase-like endoplasmic reticulum kinase
- S1P
site 1 protease
- XBP-1
X-box binding protein 1
- TRAF2
TNF receptor associated factor 2
- IRE1
inositol-requiring kinase 1
- bZIP
basic leucine zipper
- ASK1
apoptosis signal-regulating kinase 1
- GADD
growth arrest and DNA damage inducible protein
- CReP
constitutive repressor of eIF2α
- MEFs
mouse embryonic fibroblasts
- MCF-7
Michigan Cancer Foundation 7 cells
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics. CA Cancer J Clin. 2008;58(2):71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Carter BS, Beaty TH, Steinberg GD, Childs B, Walsh PC. Mendelian inheritance of familial prostate cancer. Proc Natl Acad Sci USA. 1992;89(8):3367–3371. doi: 10.1073/pnas.89.8.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Colombel M, Gil Diez S, Radvanyi F, Buttyan R, Thiery JP, Chopin D. Apoptosis in prostate cancer. Molecular basis to study hormone refractory mechanisms. Ann N Y Acad Sci. 1996;784:63–69. doi: 10.1111/j.1749-6632.1996.tb16228.x. [DOI] [PubMed] [Google Scholar]
- 4.Glass TR, Tangen CM, Crawford ED, Thompson I. Metastatic carcinoma of the prostate: Identifying prognostic groups using recursive partitioning. J Urol. 2003;169(1):164–169. doi: 10.1016/S0022-5347(05)64059-1. [DOI] [PubMed] [Google Scholar]
- 5.Mostaghel EA, Page ST, Lin DW, Fazli L, Coleman IM, True LD, Knudsen B, Hess DL, Nelson CC, Matsumoto AM, Bremner WJ, Gleave ME, Nelson PS. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer Res. 2007;67(10):5033–5041. doi: 10.1158/0008-5472.CAN-06-3332. [DOI] [PubMed] [Google Scholar]
- 6.Ashkenazi A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat Rev Cancer. 2002;2(6):420–430. doi: 10.1038/nrc821. [DOI] [PubMed] [Google Scholar]
- 7.Scheel-Toellner D, Wang K, Assi LK, Webb PR, Craddock RM, Salmon M, Lord JM. Clustering of death receptors in lipid rafts initiates neutrophil spontaneous apoptosis. Biochem Soc Trans. 2004;32(Pt 5):679–681. doi: 10.1042/BST0320679. [DOI] [PubMed] [Google Scholar]
- 8.Grassme H, Cremesti A, Kolesnick R, Gulbins E. Ceramide-mediated clustering is required for CD95-DISC formation. Oncogene. 2003;22(35):5457–5470. doi: 10.1038/sj.onc.1206540. [DOI] [PubMed] [Google Scholar]
- 9.Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003;10(1):26–35. doi: 10.1038/sj.cdd.4401186. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Chun HJ, Wong W, Spencer DM, Lenardo MJ. Caspase-10 is an initiator caspase in death receptor signaling. Proc Natl Acad Sci USA. 2001;98(24):13884–13888. doi: 10.1073/pnas.241358198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sprick M, Weigand MA, Rieser E, Rauch CT, Juo P, Blenis J, Krammer PH, Walczak H. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity. 2000;12:599–609. doi: 10.1016/s1074-7613(00)80211-3. [DOI] [PubMed] [Google Scholar]
- 12.Kischkel F, Lawrence DA, Chuntharapai A, Schow P, Gazdar A, Blenis J, Arnott D, Ashkenazi A. Death receptor recruitment of endogenous caspase-10 and apoptosis initiation in the absence of caspase-8. J Biol Chem. 2001;276:46639–46646. doi: 10.1074/jbc.M105102200. [DOI] [PubMed] [Google Scholar]
- 13.Kischkel F, Lawrence DA, Chuntharapai A, Schow P, Kim KJ, Ashkenazi A. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity. 2000;12:611–620. doi: 10.1016/s1074-7613(00)80212-5. [DOI] [PubMed] [Google Scholar]
- 14.Kischkel F, Helbardt S, Behrmann I, Germer M, Pawlita M, Kramer PH, Peter ME. Cytotoxicity-dependent APO-1, (Fas/CD95)-associated proteins from a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bodmer J, Holler N, Reynard S, Vinciguerra P, Schneider P, Juo P, Blenis J, Tschopp J. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat Cell Biol. 2000;2:241–243. doi: 10.1038/35008667. [DOI] [PubMed] [Google Scholar]
- 16.Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, Peter ME. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC) EMBO J. 1997;16(10):2794–2804. doi: 10.1093/emboj/16.10.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim HE, Jiang X, Du F, Wang X. PHAPI CAS, and Hsp70 promote apoptosome formation by preventing Apaf-1 aggregation and enhancing nucleotide exchange on Apaf-1. Mol Cell. 2008;30(2):239–247. doi: 10.1016/j.molcel.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 18.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 19.Verhagen A, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to an antagonizing IAP protein. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 20.Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985;228:1440–1443. doi: 10.1126/science.3874430. [DOI] [PubMed] [Google Scholar]
- 21.Tsujimoto Y. Stress-resistance conferred by high level of bcl-2alpha protein in human B lypmphoblastoid cell. Oncogene. 1989;4:1331–1336. [PubMed] [Google Scholar]
- 22.Vaux D, Cory S, Adams JM. bcl-2 gene promotes haematopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1998;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 23.Veis D, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell. 1993;75:229–240. doi: 10.1016/0092-8674(93)80065-m. [DOI] [PubMed] [Google Scholar]
- 24.Nakayama K, Negishi I, Kuida K, Sawa H, Loh DY. Targeted disruption of Bcl-2alpha-beta in mice: Occurrence of gray hair, polycystic kidney disease, and lymphocytopenia. Proc Natl Acad Sci USA. 1994;91:3700–3704. doi: 10.1073/pnas.91.9.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamada S, Shimono A, Shinto Y, Tsujimura T, Takahashi T, Noda T, Kitamura Y, Kondoh H, Tsujimoto Y. Bcl-2 deficiency in mice leads to pleiotropic abnormalities: Accelerated lymphoid cell death in thymus and spleen, polycystic kidney, hair hypopigmentation, and distorted small intestine. Caner Res. 1995;55:354–359. [PubMed] [Google Scholar]
- 26.Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, Thompson CB. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol. 2003;162(1):59–69. doi: 10.1083/jcb.200302084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science. 2003;300(5616):135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 28.Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes Dev. 1999;13(10):1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 29.Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11(4):372–380. doi: 10.1038/sj.cdd.4401378. [DOI] [PubMed] [Google Scholar]
- 30.Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 2000;101(5):451–454. doi: 10.1016/s0092-8674(00)80855-7. [DOI] [PubMed] [Google Scholar]
- 31.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 32.Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569(1–2):29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 33.Treiman M, Caspersen C, Christensen SB. A tool coming of age: Thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca(2+)-ATPases. Trends Pharmacol Sci. 1998;19(4):131–135. doi: 10.1016/s0165-6147(98)01184-5. [DOI] [PubMed] [Google Scholar]
- 34.Duksin D, Mahoney WC. Relationship of the structure and biological activity of the natural homologues of tunicamycin. J Biol Chem. 1982;257(6):3105–3109. [PubMed] [Google Scholar]
- 35.Rutkowski DT, Kaufman RJ. A trip to the ER: Coping with stress. Trends Cell Biol. 2004;14(1):20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 36.Hampton RY. ER-associated degradation in protein quality control and cellular regulation. Curr Opin Cell Biol. 2002;14(4):476–482. doi: 10.1016/s0955-0674(02)00358-7. [DOI] [PubMed] [Google Scholar]
- 37.Jarosch E, Lenk U, Sommer T. Endoplasmic reticulum-associated protein degradation. Int Rev Cytol. 2003;223:39–81. doi: 10.1016/s0074-7696(05)23002-4. [DOI] [PubMed] [Google Scholar]
- 38.Plemper RK, Wolf DH. Retrograde protein translocation: ERADication of secretory proteins in health and disease. Trends Biochem Sci. 1999;24(7):266–270. doi: 10.1016/s0968-0004(99)01420-6. [DOI] [PubMed] [Google Scholar]
- 39.Tsai B, Ye Y, Rapoport TA. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat Rev Mol Cell Biol. 2002;3(4):246–255. doi: 10.1038/nrm780. [DOI] [PubMed] [Google Scholar]
- 40.Brostrom CO, Brostrom MA. Regulation of translational initiation during cellular responses to stress. Prog Nucleic Acid Res Mol Biol. 1998;58:79–125. doi: 10.1016/s0079-6603(08)60034-3. [DOI] [PubMed] [Google Scholar]
- 41.Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and, translational control in the Mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–599. doi: 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]
- 42.Cox JS, Shamu CE, Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 1993;73(6):1197–1206. doi: 10.1016/0092-8674(93)90648-a. [DOI] [PubMed] [Google Scholar]
- 43.Mori K, Ma W, Gething MJ, Sambrook J. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell. 1993;74(4):743–756. doi: 10.1016/0092-8674(93)90521-q. [DOI] [PubMed] [Google Scholar]
- 44.Shamu CE, Walter P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 1996;15(12):3028–3039. [PMC free article] [PubMed] [Google Scholar]
- 45.Welihinda AA, Kaufman RJ. The unfolded protein response pathway in Saccharomyces cerevisiae. Oligomerization and transphosphorylation of Ire1p (Ern1p) are required for kinase activation. J Biol Chem. 1996;271(30):18181–18187. doi: 10.1074/jbc.271.30.18181. [DOI] [PubMed] [Google Scholar]
- 46.Sidrauski C, Walter P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell. 1997;90(6):1031–1039. doi: 10.1016/s0092-8674(00)80369-4. [DOI] [PubMed] [Google Scholar]
- 47.Kawahara T, Yanagi H, Yura T, Mori K. Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response. Mol Biol Cell. 1997;8(10):1845–1862. doi: 10.1091/mbc.8.10.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mori K, Kawahara T, Yoshida H, Yanagi H, Yura T. Signalling from endoplasmic reticulum to nucleus: transcription factor with a basic-leucine zipper motif is required for the unfolded protein-response pathway. Genes Cells. 1996;1(9):803–817. doi: 10.1046/j.1365-2443.1996.d01-274.x. [DOI] [PubMed] [Google Scholar]
- 49.Bernales S, Papa FR, Walter P. Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol. 2006;22:487–508. doi: 10.1146/annurev.cellbio.21.122303.120200. [DOI] [PubMed] [Google Scholar]
- 50.Shen X, Ellis RE, Sakaki K, Kaufman RJ. Genetic interactions due to constitutive and inducible gene regulation mediated by the unfolded protein response in C. elegans. PLoS Genet. 2005;1(3):e37. doi: 10.1371/journal.pgen.0010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313(5783):104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 52.Bertolotti A, Wang X, Novoa I, Jungreis R, Schlessinger K, Cho JH, West AB, Ron D. Increased sensitivity to dextran sodium sulfate colitis in IRE1beta-deficient mice. J Clin Invest. 2001;107(5):585–593. doi: 10.1172/JCI11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002;16(4):452–466. doi: 10.1101/gad.964702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 55.Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. 2007;13(3):365–376. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 56.Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3(1):99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 57.Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6(6):1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 58.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23(21):7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5(5):897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 60.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397(6716):271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 61.Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC. Identification and, characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol. 1998;18(12):7499–7509. doi: 10.1128/mcb.18.12.7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brewer JW, Diehl JA. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc Natl Acad Sci U S A. 2000;97(23):12625–12630. doi: 10.1073/pnas.220247197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2(6):326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 64.Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene. 2003;22(53):8608–8618. doi: 10.1038/sj.onc.1207108. [DOI] [PubMed] [Google Scholar]
- 65.Ferri KF, Kroemer G. Organelle-specific initiation of cell death pathways. Nat Cell Biol. 2001;3(11):E255–263. doi: 10.1038/ncb1101-e255. [DOI] [PubMed] [Google Scholar]
- 66.Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7(6):1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- 67.Wang XZ, Lawson B, Brewer JW, Zinszner H, Sanjay A, Mi LJ, Boorstein R, Kreibich G, Hendershot LM, Ron D. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153) Mol Cell Biol. 1996;16(8):4273–4280. doi: 10.1128/mcb.16.8.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21(4):1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem. 2004;279(44):45495–45502. doi: 10.1074/jbc.M406933200. [DOI] [PubMed] [Google Scholar]
- 70.Kojima E, Takeuchi A, Haneda M, Yagi A, Hasegawa T, Yamaki K, Takeda K, Akira S, Shimokata K, Isobe K. The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress: Elucidation by GADD34-deficent mice. FASEB J. 2003;17(11):1573–1575. doi: 10.1096/fj.02-1184fje. [DOI] [PubMed] [Google Scholar]
- 71.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18(24):3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Novoa I, Zhang Y, Zeng H, Jungreis R, Harding HP, Ron D. Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 2003;22(5):1180–1187. doi: 10.1093/emboj/cdg112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jousse C, Oyadomari S, Novoa I, Lu P, Zhang Y, Harding HP, Ron D. Inhibition of a constitutive translation initiation factor 2alpha phosphatase, CReP, promotes survival of stressed cells. J Cell Biol. 2003;163(4):767–775. doi: 10.1083/jcb.200308075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307(5711):935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 75.Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16(11):1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell. 1998;2(3):389–395. doi: 10.1016/s1097-2765(00)80283-x. [DOI] [PubMed] [Google Scholar]
- 77.Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B, Brandt GS, Iwakoshi NN, Schinzel A, Glimcher LH, Korsmeyer SJ. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006;312(5773):572–576. doi: 10.1126/science.1123480. [DOI] [PubMed] [Google Scholar]
- 78.Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004;23(5):1207–1216. doi: 10.1038/sj.emboj.7600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wei M, Zong WZ, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rao RV, Castro-Obregon S, Frankowski H, Schuler M, Stoka V, del Rio G, Bredesen DE, Ellerby HM. Coupling endoplasmic reticulum stress to the cell death program. An Apaf-1-independent intrinsic pathway. J Biol Chem. 2002;277(24):21836–21842. doi: 10.1074/jbc.M202726200. [DOI] [PubMed] [Google Scholar]
- 81.Jiang H, Wek SA, McGrath BC, Scheuner D, Kaufman RJ, Cavener DR, Wek RC. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-KB in response to diverse cellular stresses. Mol Cell Biol. 2003;25:5651–5663. doi: 10.1128/MCB.23.16.5651-5663.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-KB activation and down-regulation of TRAF2 expression. Mol Cell Biol. 2006;26:3071–3084. doi: 10.1128/MCB.26.8.3071-3084.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, Tohyama M. Activation of caspase-12, an endoplasmic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem. 2001;276:13940–13935. doi: 10.1074/jbc.M010677200. [DOI] [PubMed] [Google Scholar]
- 84.Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, Tsujimoto Y, Tohyama M. Involvement of caspase-4 in endoplasmic reticulum st. J Cell Biol. 2004;165:347–356. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Obeng EA, Boise LH. Caspase-12 and caspase-4 are not required for caspase-dependent endoplasmic reticulum stress-induced apoptosis. J Biol Chem. 2005;280(33):29578–29587. doi: 10.1074/jbc.M502685200. [DOI] [PubMed] [Google Scholar]
- 86.Denmeade SR, Isaacs JT. The SERCA pump as a therapeutic target: making a “smart bomb” for prostate cancer. Cancer Biol Ther. 2005;4(1):14–22. doi: 10.4161/cbt.4.1.1505. [DOI] [PubMed] [Google Scholar]
- 87.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4(4):321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- 88.Garrison JB, Kyprianou N. Doxazosin induces apoptosis of benign and malignant prostate cells via a death receptor-mediated pathway. Cancer Res. 2006;66(1):464–472. doi: 10.1158/0008-5472.CAN-05-2039. [DOI] [PMC free article] [PubMed] [Google Scholar]