

Abstract
The role of cholesterol in Alzheimer's disease (AD) has been linked to the generation of toxic amyloid β peptides (Aβ). Using genetic mouse models of cholesterol loading, we examined whether mitochondrial cholesterol regulates Aβ neurotoxicity and AD pathology. Isolated mitochondria from brain or cortical neurons of transgenic mice overexpressing SREBP-2 (sterol regulatory element binding protein 2) or NPC1 (Niemann-Pick type C1) knock-out mice exhibited mitochondrial cholesterol accumulation, mitochondrial glutathione (mGSH) depletion and increased susceptibility to Aβ1–42-induced oxidative stress and release of apoptogenic proteins. Similar findings were observed in pharmacologically GSH-restricted rat brain mitochondria, while selective mGSH depletion sensitized human neuronal and glial cell lines to Aβ1–42-mediated cell death. Intracerebroventricular human Aβ delivery colocalized with mitochondria resulting in oxidative stress, neuroinflammation and neuronal damage that were enhanced in Tg-SREBP-2 mice and prevented upon mGSH recovery by GSH ethyl ester coinfusion, with a similar protection observed by intraperitoneal administration of GSH ethyl ester. Finally, APP/PS1 (amyloid precursor protein/presenilin 1) mice, a transgenic AD mouse model, exhibited mitochondrial cholesterol loading and mGSH depletion. Thus, mitochondrial cholesterol accumulation emerges as a novel pathogenic factor in AD by modulating Aβ toxicity via mGSH regulation; strategies boosting the particular pool of mGSH may be of relevance to slow down disease progression.
Introduction
Alzheimer's disease (AD) is a major neurodegenerative disorder characterized by progressive memory loss and cognitive impairment due to neuronal death mainly in cortex and hippocampus (Mattson, 2004; Zlokovic, 2005; Selkoe, 2007). Given the lack of effective therapy and its association with aging, AD is one of the most pressing health challenges worldwide. Although AD pathogenesis is still unknown, accumulation or faulty brain clearance of neurotoxic amyloid β peptides (Aβ) is thought to play a key role in AD (Tanzi et al., 2004; Walsh and Selkoe, 2004; Deane and Zlokovic, 2007; Haass and Selkoe, 2007). Indeed, recent data have showed that Aβ dimers isolated directly from AD brains impair synaptic plasticity and memory (Shankar et al., 2008).
In addition to aging, epidemiological observations identified hypercholesterolemia in mid-life as a major risk factor for AD (Notkola et al., 1998; Wolozin et al., 2000; Anstey et al., 2008). Despite that studies examining statin usage have produced mixed conclusions, the epidemiological evidence linking cholesterol to AD has been supported by in vivo (Schönknecht et al., 2002; Bandaru et al., 2007) and in vitro studies (Fassbender et al., 2001; Kalvodova et al., 2005; Osenkowski et al., 2008), indicating that membrane cholesterol promotes the amyloidogenic processing of the membrane bound amyloid precursor protein (APP) by the β-secretase BACE1 (β-site APP-cleaving enzyme 1) and γ-secretase to generate toxic Aβ.
Well before plaques are observed, intracellular aggregates of Aβ develop early in AD transgenic mice, which congregate in synaptic compartments and correlate with cognitive impairment (Oddo et al., 2003). Current evidence indicates the targeting of Aβ to intracellular sites, most notably mitochondria, causing oxidative stress, mitochondrial dysfunction and cell death (Casley et al., 2002; Lustbader et al., 2004; Caspersen et al., 2005; Lin and Beal, 2006; Manczak et al., 2006).
Mitochondria do not synthesize glutathione (GSH) de novo and mitochondrial GSH (mGSH) originates by the transport of cytosol GSH into mitochondria by a specific carrier whose sensitivity to changes in membrane fluidity has been previously characterized (for review, see Fernandez-Checa and Kaplowitz, 2005). Cholesterol-mediated changes in membrane dynamics impairs the mitochondrial transport of GSH, resulting in the depletion of mGSH, a critical antioxidant defense that determines the susceptibility to stimuli that induce mitochondrial oxidative stress, such as TNF-α, Fas, or hypoxia (Colell et al., 1997; Marí et al., 2006, 2008; Lluis et al., 2007). However, the specific role of mGSH in the regulation of Aβ toxicity has not been previously described. Thus, we addressed whether mitochondrial cholesterol accumulation modulates Aβ neurotoxicity and AD pathology, using genetic models of cholesterol loading, namely, transgenic mice overexpressing SREBP-2 and NPC1−/− mice (Yu et al., 2005). Overall, these findings expand the role of cholesterol in AD, indicating that in addition to fostering Aβ production, mitochondrial cholesterol loading determines Aβ neurotoxicity and AD-like pathology via mGSH depletion. Although total GSH depletion and altered GSH redox cycle have been described in AD (Boyd-Kimball et al., 2005; Resende et al., 2008), the fact that the pool of mGSH rather than cytosol GSH determines Aβ susceptibility implies that efforts should be directed to specifically replenish mGSH to slow down disease progression, using strategies that bypass the block of GSH transport into mitochondria imposed by cholesterol deposition.
Materials and Methods
Tg-SREBP-2, NPC1−/− and Tg-APP/PS1 mice.
Breeding pairs of B6;SJL-Tg(rPEPCKSREBF2)788Reh/J, BALB/cJ NPC1NIH and B6C3-Tg(APPswe,PSEN1dE9)85Dbo/J mice were obtained from The Jackson Laboratory. At the time of weaning (21 d), mice were genetically identified by PCR using DNA prepared from tail-tips and following the genotyping protocols provided by the supplier. All procedures involving animals and their care were approved by the ethics committee of the University of Barcelona and were conducted in accordance with institutional guidelines in compliance with national and international laws and policies.
Cell cultures.
SH-SY5Y human neuroblastoma (ECACC) and C6 rat glioma cell lines were grown in Ham's F12 (Sigma) containing 10% FBS and 2 mm glutamine. U118 human glioma and neuroglioma H4 cell lines were maintained in DMEM medium (Invitrogen) supplemented with 10% FBS and 2 mm glutamine. Mice neuron-rich cultures were obtained from the cerebral cortex of embryos (day 16) using standard techniques. Dissociated cells were suspended in Neurobasal medium (Invitrogen) with 2.5% (v/v) B27 supplement (Invitrogen), 0.5 mm glutamine, and 10% FBS and plated onto poly-d-lysine coated plates at a cell density of 2 × 105/cm2. The culture medium was changed 3 h after plating and every other day thereafter. After 24 h in vitro, the culture medium was replaced with serum-free medium. Experiments were performed at 6 d in vitro (DIV). The cellular composition of the cultures was evaluated at this time by immunohistochemical analysis using antibodies to neuron- and glial-specific markers and was found to consist of >95% neurons.
Mitochondrial isolation.
Cerebral cortices were isolated and homogenized in 210 mm Mannitol, 60 mm Sucrose, 10 mm KCl, 10 mm Sodium succinate, 1 mm ADP, 0.25 mm DTT, 0.1 mm EGTA, 10 mm HEPES, pH 7.4. The homogenates were centrifuged at 600 × g for 10 min, with the resulting supernatant being centrifuged at 10,300 × g for 15 min. The resulting pellet was suspended in 2 ml, loaded onto 8 ml of 30% (v/v) Percoll gradient and centrifuged at 95,000 × g for 30 min. The mitochondrial pellet was then rinsed twice by centrifuging 15 min at 7000 × g. Cortical neurons were fractionated into cytosol and mitochondria by digitonin permeabilization followed by oil gradient centrifugation as described previously in detail (Fernandez-Checa et al., 1987).
GSH and cholesterol measurements.
GSH levels in homogenates or cytosol and mitochondria were analyzed by the recycling method (Tietze, 1969). For total cholesterol determination, 10 mg of protein was saponified with alcoholic KOH in a 60°C heating block for 30 min. After the mixture had cooled, 10 ml of hexane and 3 ml of distilled water were added and shaken to ensure complete mixing. Appropriate aliquots of the hexane layer were evaporated under nitrogen and used for cholesterol measurement. HPLC analysis was made using a Waters μBondapak C18 10 μm reversed-phase column (30 cm × 4 mm inner diameter), with the mobile phase being 2-propanol/acetonitrile (50:50, v/v) and the flow rate of 1 ml/min. The amount of cholesterol was calculated from standard curves and the identity of the peaks was confirmed by spiking the sample with known standards.
Preparation of Aβ peptides.
Human recombinant Aβ1–42 and Aβ42–1 peptides (Bachem) were dissolved to 1 mm in hexafluoroisopropanol (HFIP) and aliquoted into microcentrifuge tubes, then the HFIP was evaporated, and the peptides were stored at 20°C until use. For oligomeric assembly, concentrated peptides were resuspended in DMSO (5 mm) and then diluted to 100 μm in phenol red-free media and incubated at 4°C for 24 h. Western blot analysis demonstrated the presence of soluble oligomeric forms of Aβ (supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
Aβ infusion.
The infusion model was adopted from previous work on the rat infusion model (Frautschy et al., 1996). Briefly, stainless steel cannula attached to micro-osmotic pumps (model 1004, Alzet) were stereotaxically implanted into the right lateral cerebral ventricle (at coordinates −1.0 mm mediolateral and −0.5 mm anterioposterior from bregma; −2 mm dorsal-ventral from skull). Pumps contained either oligomeric human Aβ1–42 (0.45 μg/μl) in vehicle [4 mm HEPES pH 8.0 + 0.21 μg/μl human high-density lipoprotein (HDL)] or vehicle alone. HDL was used in the pump to reduce Aβ aggregation and enhance neuropil delivery. The pump contents were released over a period of 28 d at a rate of ∼0.11 μl/h (50 ng/h Aβ; 26 ng/h HDL), which corresponds to 36 pmol/g brain/h. ELISA analyses indicated that the human recombinant Aβ content found in the brain after this infusion period was 4.3 ± 0.6 ng/g, which is in the range of the levels found in transgenic mice and humans (Hsiao et al., 1996; Pirttilä et al., 1997). At postoperative day 28, mice were anesthetized and perfused with PBS buffer containing a protease inhibitor cocktail. The brains were then removed and longitudinally bisected. The right half of the brain was fixed and embedded for histological examination, while the left hemisphere was snap frozen for biochemical evaluation. Levels of endogenous or human recombinant Aβ1–42 peptide in brain extracts were determined with duplicate measurements by the Aβ1–42 ELISA kits from Biosource International, according to the manufacturer's instructions. In some cases, GSH ethyl ester was either added to the infusion solution (0.5 mmol/ml) as described previously (Zeevalk et al., 2007), or injected intraperitoneally (1.25 mmol/kg/d) every 12 h over the last 2 weeks of the infusion period as described previously (Mårtensson et al., 1993) to test its efficacy in the protection against Aβ-mediated neuroinflammation and injury.
Immunohistochemistry.
Cryopreserved sections (10 μm) from –1.2 mm through –2.4 mm from bregma were processed according to the avidin–biotin–peroxidase (ABC, Vectastain; Vector Laboratories) staining method. After antigen retrieval treatment, the endogenous peroxidase was blocked by exposure to 2% H2O2 in methanol for 30 min. The sections were treated with the Avidin/Biotin blocking kit from Vector Laboratories according to the manufacturer's instructions. The primary antibodies: mouse anti-Aβ (1:300, Sigma), rabbit anti-GFAP (1:500, Dako), and rat anti-F4/80 (1:200, Santa Cruz Biotechnology) were incubated overnight at 4°C. After washing with PBS, sections were then incubated for 1 h with appropriated biotinylated secondary antibodies at a dilution of 1:400, followed by ABC staining for 1 h. The immunoreaction was visualized with diaminobenzidine (DAB enhanced liquid substrate system, Sigma). Sections were counterstained with hematoxylin (Dako).
Immunofluorescence and laser confocal imaging.
Cortical neurons were fixed for 15 min with 3.7% paraformaldehyde before permeabilization with 0.1% saponin in blocking buffer (1% BSA + 20 mm Glycine in PBS) for 15 min. Then, cells were incubated in the presence of anti-cytochrome c monoclonal antibody (1:200, clone 6H2.B4; PharMingen) diluted in blocking buffer with 0.025% saponin for 1 h at 37°C after 45 min incubation with the secondary antibody, FITC-conjugated goat anti-mouse IgG (1:400, Caltag Laboratories). Filipin (50 mg/ml, Sigma) was added during the secondary antibody incubation. Dewaxed hippocampal sections were first boiled in citrate buffer (10 mm sodium citrate pH 6.0) and incubated with 0.1 m glycine/PBS for 20 min to reduce autofluorescence. Tissues processed for Aβ staining were pretreated with 99% formic acid for 7 min. The sections were incubated overnight at 4°C with monoclonal anti-Aβ1–42 (1:300, Sigma) rabbit anti-cytochrome c (1:100, Santa Cruz Biotech), rat anti-F4/80 (1:200, Santa Cruz Biotechnology), and rabbit anti-GFAP (1:500, Dako) antibodies diluted in antibody diluent with background-reducing components from Dako. After washing in PBS, the sections were incubated for 45 min with the secondary antibodies diluted in the same vehicle solution. The secondary antibodies used were: FITC-conjugated goat anti-mouse IgG (1:400, Caltag Laboratories), Cy3-conjugated anti-rabbit IgG (1:400, Jackson ImmunoResearch), and Cy3-conjugated anti-rat IgG (1:400, Jackson ImmunoResearch). In some cases, nuclei were stained with Hoechst 33258 (2 μg/ml) before mounting the samples in Fluoromount-G (Southern Biotechnology). Confocal images were collected using a Leica SP2 laser scanning confocal microscope equipped with UV excitation, an argon laser, a 633/1.32 OIL PH3 CS objective and a confocal pinhole set at 1 Airy unit. All the confocal images shown were single optical sections.
Caspase-3 activity and mitochondrial membrane potential (ΔΨm).
Caspase activation in cell extracts was determined spectrofluorometrically using the specific fluorescent substrate Ac-DEVD-AMC (100 μm, Bachem). The release of 7-amino-4-trifluoromethyl coumarin (AMC) was continuously recorded with excitation at 355 nm and emission at 460 nm. The mitochondrial membrane potential was measured using tetramethylrhodamine ethyl ester (TMRE, 50 nm). The dye retention was analyzed with a Becton-Dickinson FACScan flow cytometer in FL2.
Electron microscopy.
Pellets containing the mitochondria purified fraction were fixed in 3% glutaraldehyde, 10 mm sodium phosphate buffer, pH 7.2, postfixed in 1% osmium tetroxide, dehydrated in graded steps of ethanol through propylene oxide, and embedded in Spurr (Sigma). Finally, ultrathin sections were stained with uranyl acetate and lead citrate and photographed in a JEOL JEM 1010 electron microscope.
Statistics.
Results are expressed as means ± SD. Statistical significance was performed using an ANOVA followed by Dunnett's post hoc tests for multiple comparisons or by the unpaired two-tailed Student's t test as indicated in the figure legends. A p < 0.05 value was considered statistically significant.
Results
Tg-SREBP-2 mice exhibit brain mitochondrial cholesterol accumulation, mGSH depletion, and enhanced susceptibility to Aβ neurotoxicity
To examine whether brain mitochondrial cholesterol modulates Aβ toxicity we used transgenic mice overexpressing the truncated active form of the transcription factor SREBP-2 (Tg-SREBP-2), which controls cholesterol synthesis (Horton et al., 2002). We first assessed whether Tg-SREBP-2 mice displays the cholesterol accumulation in brain which has been reported in liver and adipose tissue (Horton et al., 1998). The quality of brain mitochondria from Tg-SREBP-2 mice was assessed by electron microscopy, and lack of contamination from endoplasmic reticulum, plasma membrane and endosomes was estimated from Western blot analysis of PERK, Na+/K+ ATPase α1, and Rab5A, respectively (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Biochemical analyses indicated enhanced cholesterol levels in the homogenate and mitochondrial fraction of Tg-SREBP-2 mice with respect to wild-type (WT) mice (Fig. 1 A). mGSH is a critical mitochondrial antioxidant, which is negatively regulated by cholesterol accumulation due to impaired transport of cytosol GSH into mitochondria via modulation of membrane dynamics (Colell et al., 1997; Marí et al., 2006). Mitochondria from Tg-SREBP-2 mice exhibited lower mGSH levels compared with wild-type mice, whereas the content of GSH in the homogenate remained unaffected (Fig. 1 A). Next, we evaluated the mitochondrial response to Aβ-mediated oxidative stress. Soluble oligomeric Aβ1–42 (1 μm for 2 h) induced a ∼2-fold higher DCF (2′,7′-dichlorodihydrofluorescein) fluorescence, reflecting ROS generation in mitochondria from Tg-SREBP-2 mice compared with wild-type mice (Fig. 1 B), which was accompanied by enhanced release of cytochrome c and Smac/DIABLO (Fig. 1 C). To further assess whether the increased sensitivity of isolated mitochondria to Aβ-induced oxidative stress impacts on neuronal death, we isolated cortical neurons from wild-type and Tg-SREBP-2 embryos followed by Aβ exposure. Immunostaining analyses using the neuronal marker microtubule-associated protein 2 (MAP-2) confirmed the neuronal-enrichment of the cultures (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Laser confocal microscopy indicated higher cholesterol levels in cortical neurons from Tg-SREBP-2 mice compared with wild-type mice revealed by staining with filipin, a fluorescent polyene antibiotic that binds specifically to the 3β-hydroxyl group of sterols (Norman et al., 1972; Marí et al., 2006), which extensively colocalized with mitochondria stained with cytochrome c antibody (Fig. 1 D). These findings were accompanied by lower GSH levels in mitochondria isolated from cortical neurons of Tg-SREBP-2 mice compared with wild-type mice (2.1 ± 0.4 vs 3.3 ± 0.5 nmol/mg protein), with the sparing of cytosol GSH content (data not shown). Finally, cortical neurons from Tg-SREBP-2 mice exhibited enhanced susceptibility to Aβ1–42 (0.5 nm-1 μm for 48 h) mediated cell death compared with that of wild-type mice (Fig. 1 E). Importantly, this sensitivity in Tg-SREBP-2 mice is observed at a concentration of Aβ1–42 as low as 5 nm.
Figure 1.
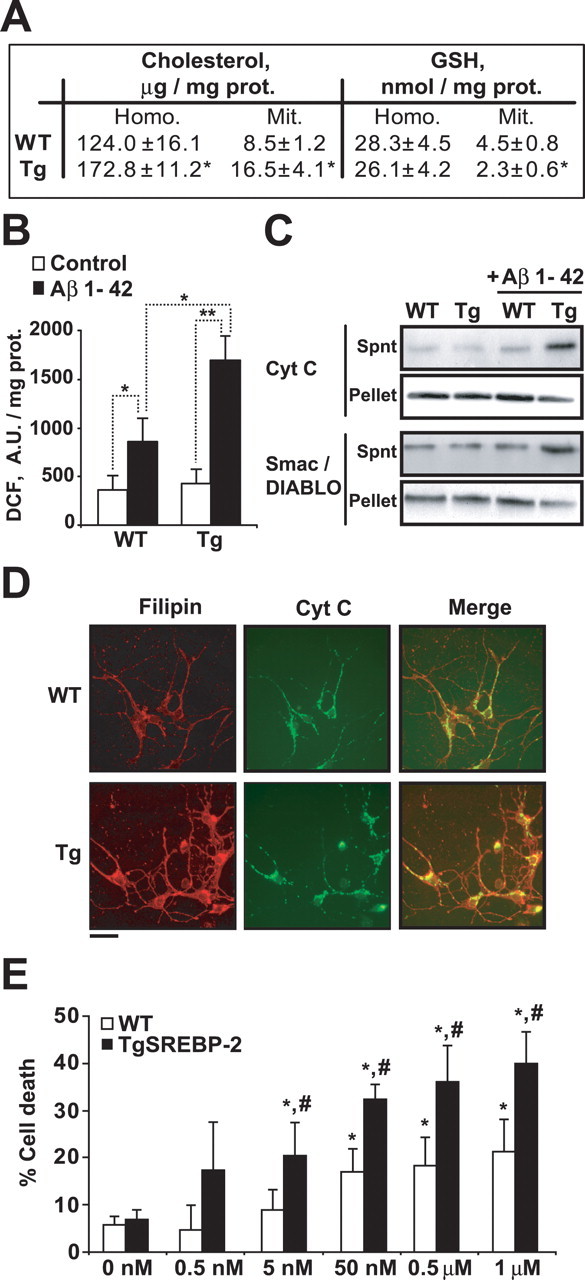
Increased susceptibility to Aβ1–42 exposure of brain mitochondria and cortical neurons from SREBP-2 transgenic mice. A, Total cholesterol levels and GSH levels of brain homogenate (Homo.) and isolated mitochondria (Mit.) from WT and SREBP-2 transgenic (Tg) mice; *p < 0.05 (n = 6–8 per genotype). B, C, WT and transgenic (TgSREBP-2) mitochondria (1 mg/ml) were exposed to Aβ1–42 (1 μm) for 2 h. B, Hydrogen peroxide generation determined by DCF fluorescence (A.U.: arbitrary units). *p < 0.05, **p < 0.01 (n = 6–8 per genotype). C, After Aβ treatment mitochondria were pelleted and the resulting supernatants (Spnt) and pellets were analyzed for the presence of apoptogenic proteins. Shown are representative immunoblottings of cytochrome c (Cyt C) and Smac/DIABLO (n = 4). D, E, Primary neurons isolated from WT and transgenic (TgSREBP-2) cerebral cortices at 6 DIV. D, Representative confocal images of cortical neurons stained with filipin (red) and mouse anti-cytochrome c antibody followed by the appropriated FITC-conjugated secondary antibody (Cyt C, green). Scale bar: 10 μm. E, Cell death of cortical neurons after exposure to increasing doses of Aβ1–42 as indicated for 48 h. *p < 0.05 versus untreated cells (0 nm), # p < 0.01 versus WT cells (n = 4–6). Values are mean ± SD; mean differences were compared by unpaired Student's t test.
Sensitivity of brain mitochondria from NPC1−/− mice to Aβ-induced oxidative stress and release of proapoptotic proteins
NPC1 is a late endosomal protein involved in the intracellular transport of cholesterol, whose ablation results in cholesterol and sphingolipids loading and progressive neurodegeneration (Paul et al., 2004). Recently, elevated mitochondrial cholesterol levels and mitochondrial dysfunction has been reported in the liver (Marí et al., 2006) and brain (Yu et al., 2005) from NPC1−/− mice. Cholesterol content was higher in brain mitochondria from NPC1−/− mice compared with wild-type mice (Fig. 2 A), which was accompanied by 50–60% depletion of mGSH stores (Fig. 2 A), without changes in cytosol GSH (data not shown). To examine the susceptibility of brain mitochondria from NPC1−/− mice to oligomeric Aβ, we first determined the generation of mitochondrial ROS after exposure to Aβ 1–42 (1 μm for 2 h). As seen, DCF fluorescence in response to Aβ was higher in brain mitochondria from NPC1−/− mice compared with mitochondria from NPC1+/+ brain (Fig. 2 B). Moreover, Aβ incubation induced an enhanced release of cytochrome c and Smac/DIABLO in mitochondria from NPC1−/− mice (Fig. 2 C). Collectively the preceding findings in Tg-SREBP-2 and NPC1−/− mice reveal the correlation between mitochondrial cholesterol loading and susceptibility to Aβ-induced oxidative stress and neurotoxicity, effects that are accompanied by selective mGSH depletion.
Figure 2.
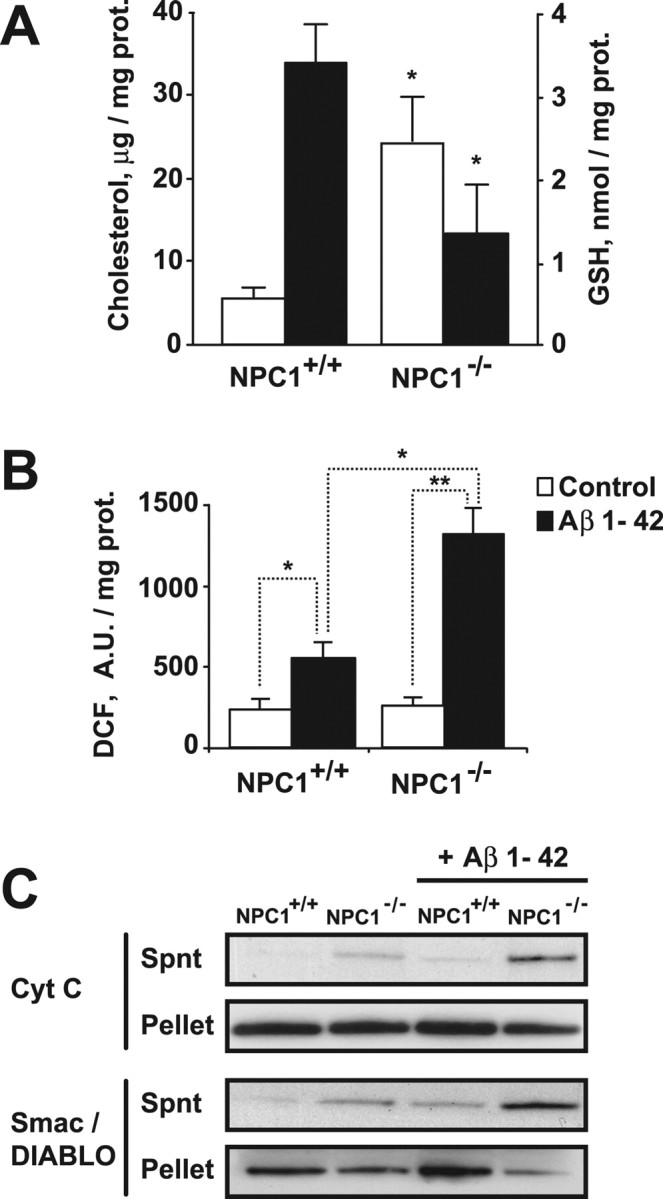
Isolated brain mitochondria from NPC1 knock-out mice show an increased sensitivity to Aβ1–42. A, Total cholesterol levels (white bars) and GSH levels (black bars) of isolated mitochondria from NPC1+/+ and NPC1−/− brains. *p < 0.05 (n = 3–4 per genotype). B, Hydrogen peroxide generation determined by DCF fluorescence of mitochondria (1 mg/ml) incubated with Aβ1–42 (1 μm) for 2 h. *p < 0.05, **p < 0.01 (n = 3–4 per genotype). C, After 2 h treatment with Aβ1–42 (1 μm) mitochondria were pelleted and the resulting supernatants (Spnt) and pellets were analyzed by Western blotting for the presence of cytochrome c (Cyt C) and Smac/DIABLO. Representative immunoblottings are shown. Values are mean ± SD; mean differences were compared by unpaired Student's t test. prot., Protein; A.U., arbitrary units.
mGSH-dependent susceptibility of brain mitochondria to Aβ toxicity
Early GSH depletion and altered GSH redox cycle have been recently shown in AD transgenic mice (Resende et al., 2008). Although GSH levels protect against Aβ neurotoxicity and Aβ has been shown to stimulate ROS generation from mitochondria (Cardoso et al., 2001; Boyd-Kimball et al., 2005; Lin and Beal, 2006), however, the particular role of mGSH depletion on Aβ-mediated toxicity has not been specifically addressed before. To investigate this question, mitochondria from rat brain were depleted of GSH by ethacrynic acid (EA), which covalently binds to GSH. As shown, EA (5 μm for 15 min) depleted mGSH levels (Fig. 3 A) and enhanced DCF fluorescence by Aβ1–42, indicative of higher ROS generation (Fig. 3 B), an effect not observed when mitochondria were exposed to the nontoxic peptide Aβ 42–1 (Fig. 3 B). A stepwise increase in mitochondrial DCF fluorescence by Aβ1–42 peptide was observed as a function of mGSH depletion by EA, that was accompanied by enhanced mitochondrial lipid peroxidation, estimated from the fluorescence loss of cis-parinaric-stained mitochondria (supplemental Fig. S4A–C, available at www.jneurosci.org as supplemental material). To rule out nonspecific effects of EA, we depleted mGSH in vivo using buthionine sulfoximine (BSO), a specific inhibitor of γ-GCS (γ-glutamylcysteine synthetase), the rate-limiting enzyme in GSH biosynthesis (Jain et al., 1991). Brain mitochondria from BSO-treated rats exhibited mGSH depletion (Fig. 3 A), recapitulating the effects observed with EA on Aβ-induced ROS generation (Fig. 3 B). In addition, mGSH depletion by both strategies enhanced the mitochondrial release of cytochrome c (Fig. 3 C) and Smac/DIABLO (data not shown) after exposure to Aβ. To further analyze the source of ROS induced by Aβ, mitochondrial electron flow was blocked at different respiratory complexes (Fig. 3 D). Inhibition of complex III by antimycin A potentiated ROS generation by Aβ in GSH-depleted mitochondria, while preincubation with rotenone and TTFA (thenoyltrifluoroacetone), which block complex I and II, respectively, prevented the Aβ-mediated increase of DCF fluorescence, pointing to complex III as the major site of ROS generation by Aβ. These findings indicate that mGSH controls the mitochondrial oxidative stress generation by Aβ.
Figure 3.
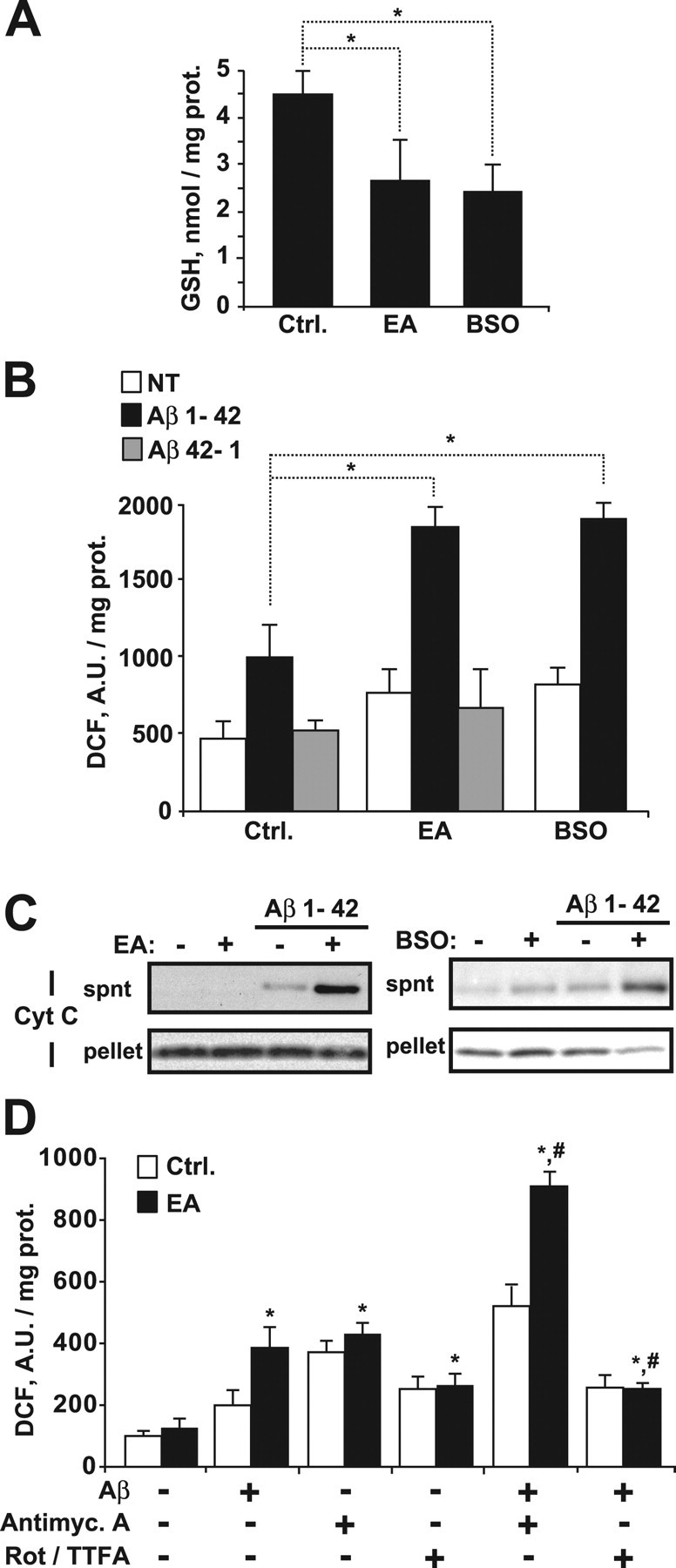
Mitochondrial GSH regulates Aβ-induced oxidative stress. Depletion of mitochondrial GSH levels was assessed in vitro by incubation of isolated mitochondria (1 mg/ml) with 5 μm ethacrynic acid (EA) for 15 min or in vivo by inhibiting the GSH synthesis with buthionine sulfoximine treatment (BSO, 3 mmol/kg). A, GSH levels after EA or BSO treatment. *p < 0.05 (n = 4–6). B, C, Mitochondria from rat brain pretreated with EA or BSO were exposed to Aβ 1–42 (5 μm) or the inactive form Aβ 42–1 (5 μm) for 2 h. B, Hydrogen peroxide generation determined by DCF fluorescence (A.U.: arbitrary units; NT: untreated mitochondria). *p < 0.05 (n = 4–6). C, After Aβ treatment mitochondria were pelleted and the resulting supernatants (spnt) and pellets were analyzed for the presence of cytochrome c (Cyt C) and Smac/DIABLO. Shown are representative immunoblottings of three independent experiments. D, Effect of blocking the electron transport chain at complex I, II, or III on the generation of hydrogen peroxide by Aβ. After EA treatment mitochondria were incubated with rotenone and TTFA (Rot/TTFA, 20 and 15 μm, respectively), antimycin A (Antimyc. A, 5 μm), and/or Aβ (5 μm) during 60 min. The blockers of the respiratory complexes were added 15 min before exposure to Aβ. Hydrogen peroxide generation was assessed by DCF fluorescence. *p < 0.05 versus untreated mitochondria; # p < 0.01 versus Aβ-treated mitochondria. (n = 3). Values are mean ± SD; mean differences were compared by Dunnett's test. prot., Protein; Ctrl., control.
Selective mGSH depletion with the spare of cytosol GSH sensitizes human neuronal and glial cells to Aβ neurotoxicity
We next asked whether selective mGSH depletion modulates Aβ neurotoxicity. To address this question, human neuroblastoma SH-SY5Y cells were exposed to 3-hydroxy-4-pentenoate (HP), a fatty acid derivative which is biotransformed in mitochondria into a Michael electrophile and then conjugated with GSH, resulting in its depletion (Marí et al., 2006, 2008, García-Ruiz et al., 2003). As seen, HP decreased mGSH levels with the sparing of GSH cytosol stores (Fig. 4 A), sensitizing SH-SY5Y cells to Aβ-induced cell death (Fig. 4 B). Similar results were observed in glial-derived H4, C6 and U118 cell lines after selective mGSH depletion by HP and Aβ challenge (supplemental Fig. S5A,B, available at www.jneurosci.org as supplemental material), indicating that the role of mGSH in regulating Aβ cytotoxicity is not restricted to neuronal cells. mGSH depletion in SH-SY6Y cells by HP potentiated Aβ-induced DCF fluorescence, indicative of enhanced ROS generation, and loss of mitochondrial membrane potential (supplemental Fig. S6A,B, available at www.jneurosci.org as supplemental material). Dying SH-SY5Y cells after HP plus Aβ exposure exhibited apoptotic features, including chromatin condensation/nuclear fragmentation (Fig. 4 C) and increased caspase-3 activity (Fig. 4 D). Interestingly, preincubation with the pancaspase inhibitor qVD-OPH (quinoline-Val-Asp-CH2-difluorophenoxy) prevented the morphological changes in chromatin condensation (Fig. 4 C) and caspase-3 activation (Fig. 4 D) induced by HP plus Aβ challenge, but failed to protect against cell death (Fig. 4 E). Pretreatment with antioxidants such as N-acetyl-cysteine (NAC) and butylated hydroxyanisole (BHA) significantly decreased ROS generation induced by HP plus Aβ exposure (Fig. 4 F) and the combined pretreatment with NAC and qVD-OPH protected mGSH-depleted SH-SY5Y cells against cell death triggered by Aβ (Fig. 4 E). Overall, these findings indicate that selective mGSH depletion sensitizes neuronal and glial cells to Aβ by stimulating oxidant-dependent cell death and caspase-independent apoptosis.
Figure 4.
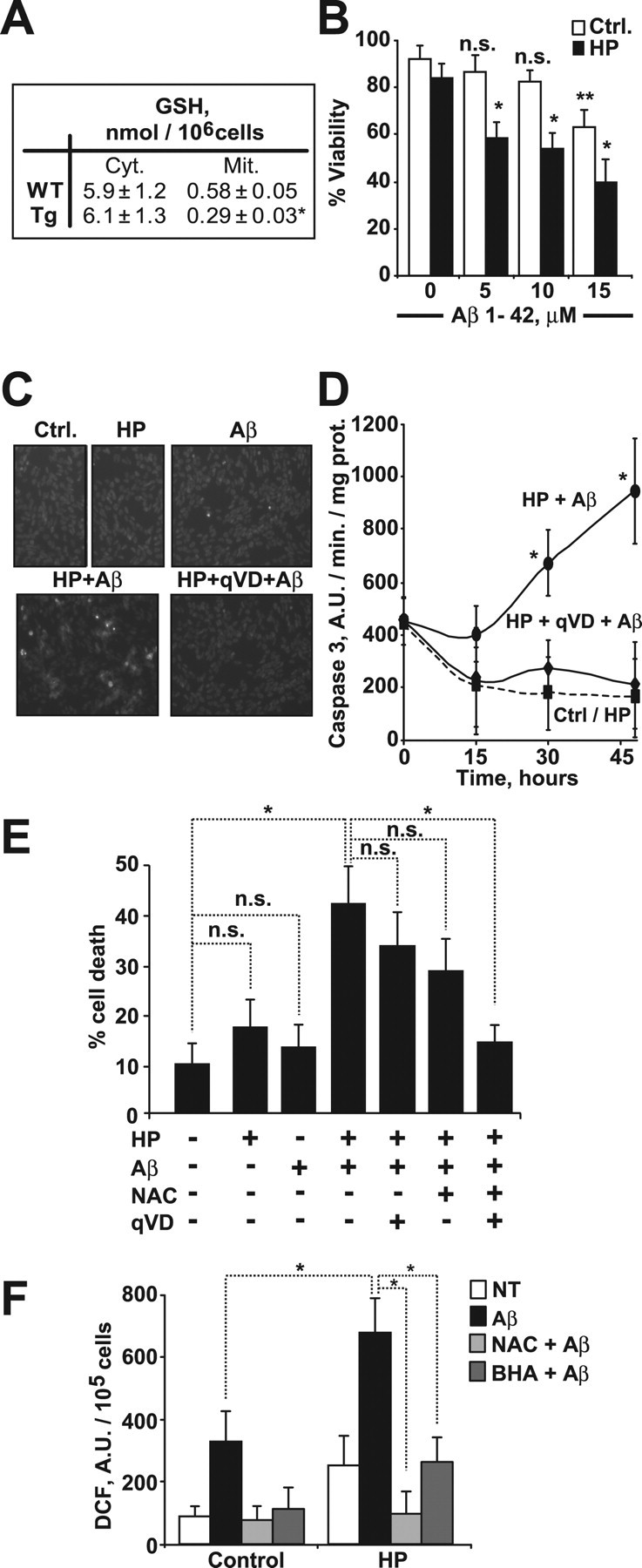
Selective depletion of mitochondrial GSH in human neuroblastoma SH-SY5Y cells enhances the apoptotic cell death induced by Aβ. Mitochondrial GSH depletion was assessed by incubating cells with (S)-3-hydroxy-4-pentenoate (HP, 5 mm) for 10 min. A, GSH levels. *p = 0.002 (n = 4). Cyt., Cytosol; Mit., mitochondria. B, Cell viability after 24 h incubation with increasing concentrations of Aβ1–42. *p < 0.01 versus HP-treated cells at 0 μm, **p = 0.02 versus control (Ctrl.) cells at 0 μm; n.s., not significant (n = 4). C, Chromatin morphology of cells exposed to Aβ1–42 (5 μm, 24 h) analyzed by Hoechst-33258 staining. Original magnification, 200× (n = 4). D, Caspase-3 activity of cell extracts from cells exposed to Aβ1–42 (5 μm, 24 h) with or without the pancaspase inhibitor qVD-OPH (20 μm). *p < 0.05 versus HP-treated cells. (n = 3). E, F, After preincubation with the antioxidant agents N-acetyl-cysteine (NAC, 3 mm) or butylated hydroxyanisole (BHA, 20 μm) and/or the caspase inhibitor qVD-OPH (20 μm) for 2 h, mitochondrial GSH was depleted by HP treatment and cells were exposed to Aβ 1–42 (5 μm). (n = 3–4). E, Cell death analyzed by PI staining at 24 h exposure to Aβ1–42. *p < 0.01. F, Hydrogen peroxide generation after 2 h incubation with Aβ1–42. *p < 0.05 (A.U.: arbitrary units). Values are mean ± SD; mean differences were compared by Dunnett's test (B, E, and F) or unpaired Student's t test.
Neuroinflammation in Tg-SREBP-2 mice after in vivo infusion of human Aβ
Having shown that brain mitochondrial cholesterol modulates Aβ toxicity by regulating mGSH, we next evaluated the onset of pathological features of AD such as neuroinflammation after intracerebroventricular infusion of human Aβ1–42 in Tg-SREBP-2 mice. Staining with an antibody that recognizes human Aβ (but not the endogenous mouse Aβ) demonstrated diffuse amyloid deposition in the hippocampus of Aβ-infused mice (for 28 d) (Fig. 5 A). Quantitative determination by ELISA revealed equivalent Aβ loading in wild-type mice and Tg-SREBP-2 mice (54.2 ± 14.6 and 66 ± 11.2 nmol/mg protein, respectively). Moreover, confocal analysis showed a significant colocalization of Aβ with cytochrome c staining (Fig. 5 B), indicating that a fraction of the infused Aβ targeted mitochondria. Of note, endogenous brain Aβ1–42 levels were similar in WT and Tg-SREBP-2 mice (supplemental Fig. S7, available at www.jneurosci.org as supplemental material).
Figure 5.
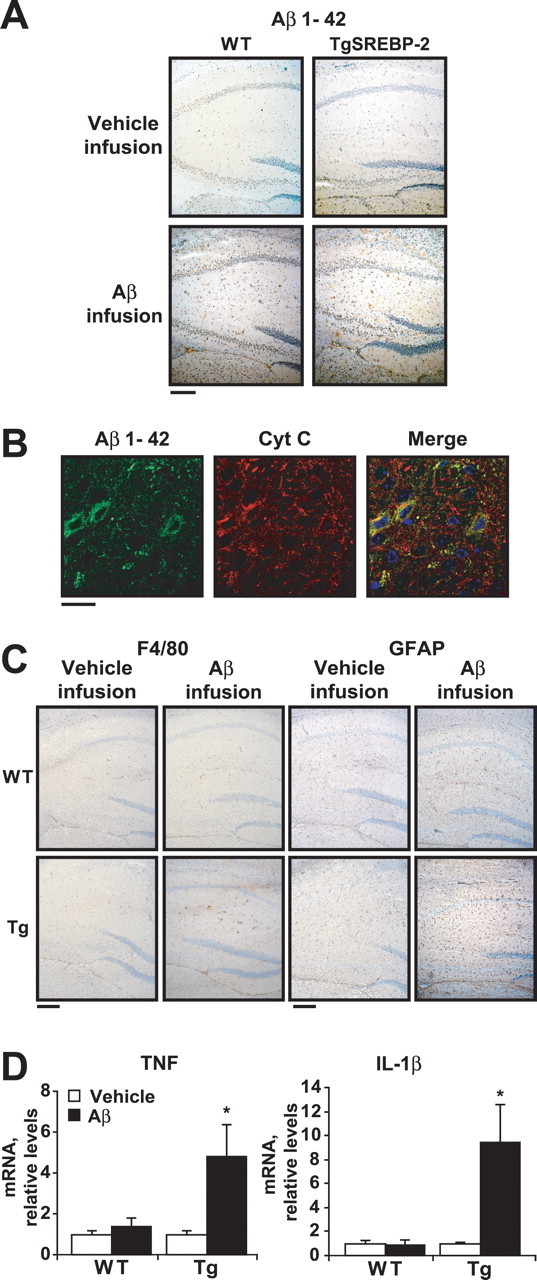
Increased neuroinflammation in SREBP-2 transgenic mice after Aβ1–42 infusion. WT and SREBP-2 transgenic (TgSREBP-2) mice were subjected to continuous intracerebroventricular infusion of vehicle or human Aβ1–42 solution (1.2 μg/d) for 28 d; then, half of the brain was processed for immunohistochemistry and the other hemisphere used for biochemical analysis. n = 6–8 mice per group. A, Aβ1–42 immunohistochemical staining. Representative photomicrographs of hippocampus from WT and transgenic mice after infusion are shown. Scale bar: 100 μm. B, Confocal analysis of Aβ internalization. Shown are representative images of Aβ1–24 (green) and cytochrome c (red) immunofluorescence from hippocampal sections of TgSREBP-2 brains after the infusion period. Merged image indicates partial colocalization of Aβ with the mitochondrial marker cytochrome c (Cyt C, yellow). Nuclei were stained with Hoechst-33258 (shown only in merged image; blue). Scale bar: 10 μm. C, Activation of microglia and astrocytes analyzed by F4/80 and GFAP immunostaining, respectively. Shown are representative photomicrographs of F4/80 and GFAP immunoreactivity as indicated, in sections of hippocampus from infused WT and SREBP-2 transgenic (Tg) mice. Scale bar: 100 μm. D, TNF-α and IL-1β mRNA expression analyzed by real-time PCR from WT and SREBP-2 transgenic (Tg) brain after vehicle or Aβ infusion. Shown are relative values after normalization against 18 s expression and are representative of three experiments ± SD, *p < 0.01 versus vehicle-infused TgSREBP-2 mice. Mean differences were compared by unpaired Student's t test.
Neuroinflammation, manifested as activated microglia and astrocytes close to Aβ depositions, is an invariant characteristic of AD brain, which plays a key role in the progression of the disease by further amplifying Aβ generation and neuronal damage (Heneka and O'Banion, 2007). Greater microglia activation, determined by F4/80 immunostaining, was observed in the hippocampus of Tg-SREBP-2 mice infused with Aβ compared with vehicle-infused or Aβ-infused wild-type mice (Fig. 5 C). Astrocyte activation, manifested as GFAP immunoreactivity, was also elevated in hippocampal regions of Aβ-infused Tg-SREBP-2 mice (Fig. 5 C). Moreover, confocal analysis of Aβ-infused Tg-SREBP-2 brains revealed the presence of microglia and astrocytes colocalizing with Aβ in amyloid deposits (supplemental Fig. S8, available at www.jneurosci.org as supplemental material). These histological analyses were complemented by quantitative measurements of mRNA levels of proinflammatory cytokines such as TNF-α and IL-1β. Ιncreased expression of TNF-α and IL-1β was only observed in Tg-SREBP-2 brains infused with Aβ (Fig. 5 D). Thus, Tg-SREBP-2 mice are more susceptible to Aβ-induced neuroinflammation.
Increased oxidative stress, synaptodendritic degeneration and neuronal damage in Tg-SREBP-2 mice after Aβ infusion
We next assessed the susceptibility of Tg-SREBP-2 mice to Aβ-induced oxidative stress and neurodegeneration. Malondialdehyde (MDA), an end product of lipid peroxidation, and accumulation of oxidized proteins assessed by carbonyl protein immunoblotting were significantly higher in the brain of Aβ-infused Tg-SREBP-2 mice compared with Aβ-infused wild-type mice, with no signs of oxidative stress detected in brains of vehicle-infused wild-type or Tg-SREBP-2 mice (Fig. 6 A,B). Previous studies have shown that synaptic injury is an early event in the progression of AD, which correlates with the severity of cognitive deficits in AD patients (Masliah et al., 1994). As shown, the levels of the presynaptic protein synaptophysin were significantly lower in Tg-SREBP-2 mice infused with Aβ compared with wild-type mice or vehicle-infused brains (Fig. 6 C), indicating extensive synaptic loss. In addition, neuronal death and apoptosis estimated by Fluoro-Jade B-positive degenerating neurons and TUNEL staining, respectively, were higher in Tg-SREBP-2 mice after intracerebroventricular Aβ infusion compared with wild-type mice (Fig. 6 D,E). These data underscore the susceptibility of Tg-SREBP-2 mice to Aβ-induced neurodegeneration.
Figure 6.
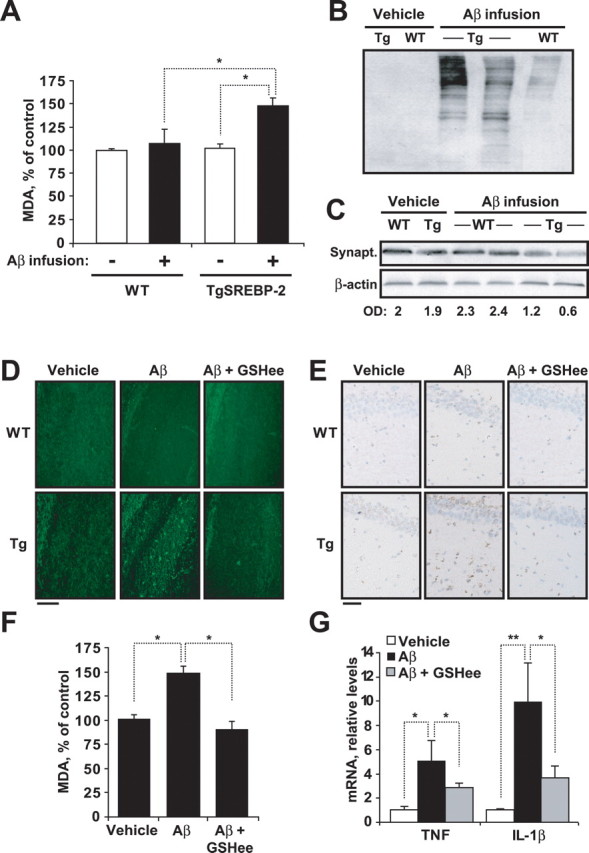
Aβ-infused TgSREBP-2 mice display enhanced oxidative stress and neuronal damage which is prevented by GSH ethyl ester (GSHee) coinfusion. WT and TgSREBP-2 mice were subjected to continuous infusion of vehicle or human Aβ1–42 solution (1.2 μg/d) for 28 d. n = 6–8 mice per group. A, Lipid peroxidation estimated as production of malondialdehyde (MDA). *p < 0.03. B, Representative immunoblotting showing presence of carbonyl proteins in WT and TgSREBP-2 (Tg) brains after infusion. C, Representative immunoblotting showing synaptophysin (Synapt.) protein levels after infusion. Densitometric values of the bands representing synaptophysin immunoreactivity were normalized with the values of the corresponding β-actin bands (O.D.: normalized optical density). D, Representative images of degenerated neurons in hippocampal regions by Fluoro-Jade B staining. Scale bar: 50 μm. E, Representative images of apoptotic cells in hippocampus by terminal deoxynucleotidyl transferase-mediated nick-end labeling. Scale bar: 25 μm. F, Lipid peroxidation in infused TgSREBP-2 brains estimated as malondialdehyde levels (MDA). *p < 0.01 (n = 6). G, TNF-α and IL-1β mRNA expression analyzed by real-time PCR from infused TgSREBP-2 brains. Relative values were normalized against 18 s expression. *p < 0.05, **p < 0.01 (n = 6). Values are mean ± SD; mean differences were compared by unpaired Student's t test.
GSH ethyl ester coinfusion restores mGSH and protects against Aβ-induced neurodegeneration in Tg-SREBP-2 mice
To address whether the exacerbated inflammation and neuronal death of Tg-SREBP-2 mice after Aβ infusion is promoted by cholesterol-mediated mGSH depletion, GSH ethyl ester (GSHee), a membrane-permeable form of GSH, was coinfused with Aβ. GSHee significantly increased the mitochondrial pool of GSH in Tg-SREBP-2 mice (3.2 ± 0.2 vs 1.4 ± 0.5 nmol/mg protein), preventing Aβ-induced increase in MDA levels (Fig. 6 F) and protein oxidation estimated by carbonyl protein immunoreactivity (supplemental Fig. S9A, available at www.jneurosci.org as supplemental material). Moreover, reactive astrogliosis and microgliosis, determined by GFAP and F4/80 immunoreactivity, respectively, were also reduced in Tg-SREBP-2 mice when GSHee was coinfused with Aβ (supplemental Fig. S9B, available at www.jneurosci.org as supplemental material). This outcome correlated with a significant decrease in the mRNA expression of the proinflammatory cytokines TNF-α and IL-1β (Fig. 6 G). In addition, neuronal damage, assessed by Fluoro-Jade B staining and TUNEL assay, in the hippocampal regions of Aβ-infused Tg-SREBP-2 brains was prevented by GSHee coinfusion (Fig. 6 D,E). We further analyzed whether intraperitoneal administration of GSHee was therapeutic against Aβ-induced neuroinflammation and neuronal loss. As seen, intraperitoneal GSHee administration during the Aβ infusion period significantly increased mGSH content (Fig. 7 A), attenuating MDA levels (Fig. 7 B) and carbonyl protein immunoblotting (Fig. 7 C), indicative of oxidative stress induced by Aβ in Tg-SREBP-2 mice. In addition, the enhanced presence of reactive astrocytes (Fig. 7 D) and the upregulated expression of TNF-α (Fig. 7 E) observed in Aβ-infused transgenic mice were significantly prevented by GSHee therapy, which correlates with a significant decrease in neuronal damage (Fig. 7 F). Thus, these data indicate that strategies that boost mGSH such as GSHee protect against Aβ-induced neurodegeneration.
Figure 7.
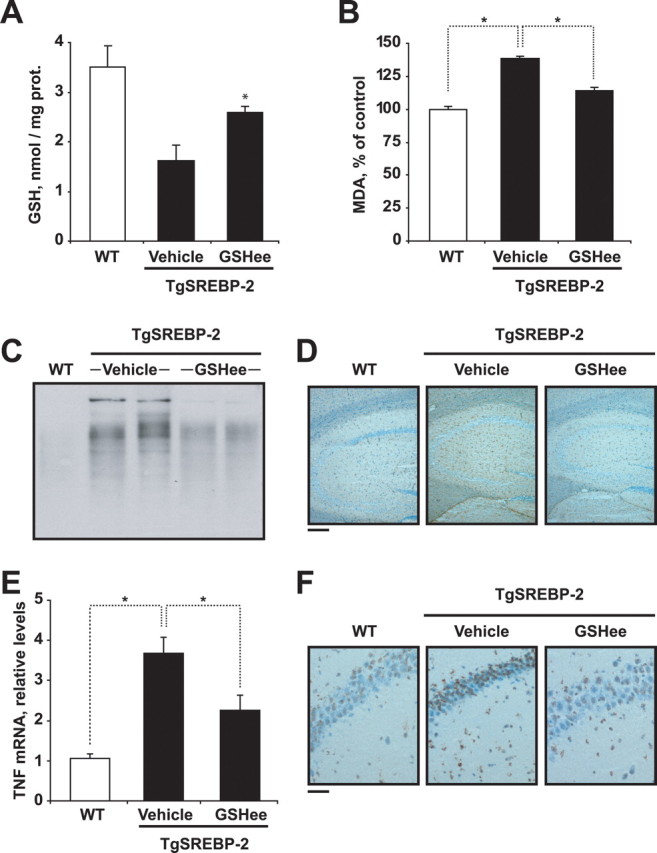
GSH ethyl ester (GSHee) intraperitoneal therapy protects against Aβ-induced neurodegeneration in Tg-SREBP-2 mice. WT and TgSREBP-2 mice were subjected to continuous infusion of human Aβ1–42 solution (1.2 μg/d) for 28 d. GSH ethyl ester (1.25 mmol/kg/d) or vehicle alone were administered intraperitoneally over the last 2 week of the infusion period. n = 4–6 mice per group. A, Mitochondrial GSH levels. *p = 0.01. prot., Protein. B, Lipid peroxidation estimated as production of malondialdehyde (MDA). *p < 0.01. C, Representative immunoblotting showing presence of carbonyl proteins. D, Activation of astrocytes analyzed by GFAP immunostaining. Shown are representative photomicrographs GFAP immunoreactivity in hippocampal sections. Scale bar: 100 μm. E, TNF-α expression analyzed by real-time PCR. Relative values were normalized against 18 s expression. *p < 0.05. F, Representative images of apoptotic cells in hippocampus by terminal deoxynucleotidyl transferase-mediated nick-end labeling. Scale bar: 25 μm. Values are mean ± SD; mean differences were compared by unpaired Student's t test.
Enhanced mitochondrial cholesterol loading and mGSH depletion in APP/PS1 transgenic mice
To address the relevance of the preceding findings in AD, we examined the mitochondrial cholesterol and GSH content in a genetic mouse model of AD, namely, APP/PS1 transgenic mice. Brain mitochondria were isolated from 4-, 7-, and 10-month-old APP/PS1 transgenic mice. As seen (Fig. 8 A), mitochondrial cholesterol levels were unchanged in 4- and 7-month-old transgenic mice, increasing twice as much in 10-month-old APP/PS1 transgenic mice compared with wild-type mice of the same age. As expected from our findings in Tg-SREBP-2 and NPC1−/− mice, the mGSH levels decreased only in 10-month-old APP/PS1 transgenic mice (Fig. 8 B). Preceding the mitochondrial cholesterol loading and the subsequent mGSH depletion, we observed a significant generation of Aβ detected by ELISA in 4-month-old APP/PS1 mice (data not shown). Furthermore, either SREBP-2 mRNA and protein levels were significantly elevated in Tg-APP/PS1 mice (Fig. 8 C,D), indicating that activation of SREBP-2 may contribute to the increase in cholesterol content. Thus, these findings lend further support for the relevance of enhanced mitochondrial cholesterol levels in AD.
Figure 8.
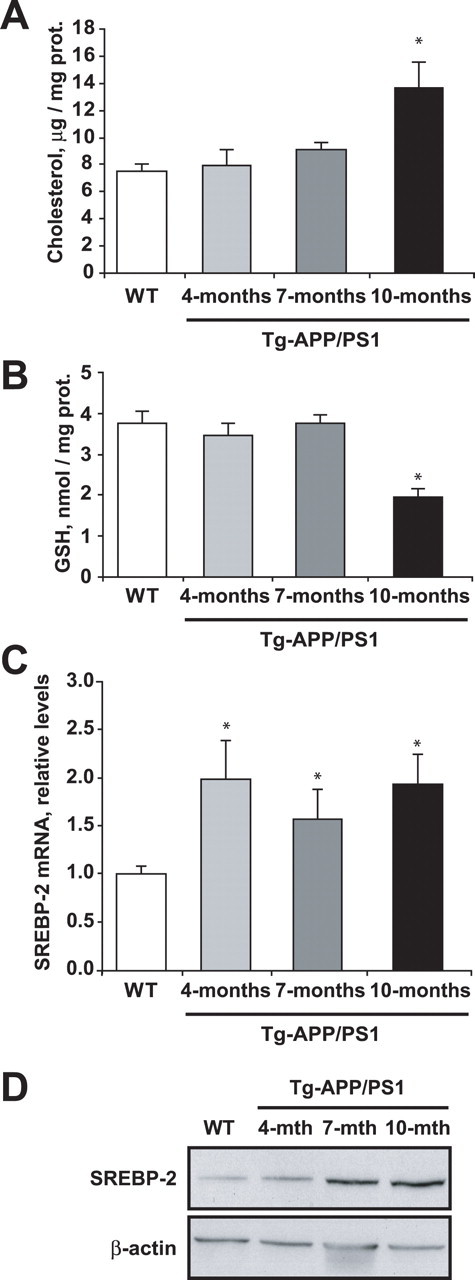
Brain mitochondria from 10-month-old AD transgenic mice exhibit increased cholesterol and depleted GSH levels. A, Total cholesterol levels. B, GSH levels of isolated brain mitochondria from WT mice and AD transgenic mice (Tg-APP/PS1) at the indicated ages. *p < 0.05 (n = 3–4). C, D, SREBP-2 expression in brain extracts from WT mice and Tg-APP/PS1 transgenic mice at the indicated ages. C, SREBP-2 mRNA expression analyzed by real-time PCR. Relative values were normalized against 18 s expression. *p < 0.05 (n = 3–4). D, Representative immunoblotting showing SREBP-2 protein levels. β-Actin levels were analyzed as a loading control. Values are mean ± SD; mean differences were compared by unpaired Student's t test. prot., Protein.
Discussion
Epidemiological, genetic and biochemical studies have identified cholesterol, Aβ and mitochondria as key factors in AD pathogenesis, but the specific relationship of this triad has not been addressed before. In particular, the role of cholesterol in AD examined thus far has been limited to the amyloidogenic processing of APP at the plasma membrane to generate Aβ. In this regard, varying proportions of the presenilins and other components of the γ-secretase complex (nicastrin, Aph-1 and Pen-2), along with γ-secretase activity and the β-secretase BACE1, are present in detergent-resistant cholesterol-enriched lipid rafts (Taylor and Hooper, 2007). In addition, the activities of BACE1 and γ-secretase are stimulated by rafts lipid components, such as glycosphingolipids and cholesterol (Kalvodova et al., 2005; Ariga et al., 2008; Osenkowski et al., 2008). Intriguingly, although the presence of APP in detergent-resistant rafts is relatively minor (Lee et al., 1998), once localized to the raft, the APP may be rapidly cleaved by β- and γ-secretases, thus contributing to Aβ generation. Moreover, previous studies have shown that cholesteryl-ester levels directly correlate with Aβ production (Puglielli et al., 2001) and that statins inhibit the dimerization of β-secretase via both isoprenoid- and cholesterol-mediated mechanisms (Parsons et al., 2006).
In the current study, we examined the role of cholesterol in AD from an unusual perspective and provide evidence linking mitochondrial cholesterol to Aβ neurotoxicity, consistent with and expanding the recognized role of mitochondrial dysfunction in AD (Casley et al., 2002; Lustbader et al., 2004; Caspersen et al., 2005; Lin and Beal, 2006; Manczak et al., 2006). In addition to being targets of Aβ toxicity, it is conceivable that mitochondria may constitute an additional source of Aβ generation, in agreement with the presence of APP in mitochondria from AD-vulnerable brain regions (Devi et al., 2006), and the finding of functional complexes of APP with γ-secretase and IDE in mitochondria (Hansson et al., 2004; Leissring et al., 2004), arguing that these organelles may contribute to the amyloidogenic APP processing and hence Aβ generation. Regardless of the source of Aβ generation either via the more established pathway of APP processing at the plasma membrane, particularly in cholesterol-enriched domains, or its in situ generation in mitochondria, we provide evidence that mitochondrial cholesterol accumulation stands as a critical determinant of Aβ toxicity both in vitro and in vivo using Tg-SREBP-2 and NPC1−/− mice.
It has been described that SREBP-2 downregulates the expression of genes which control brain Aβ clearance, such as LDLR-related protein 1 (also known as LRP) (Deane et al., 2004). Moreover, recent studies in AD patients and two mouse models of AD show that overexpression of serum response factor and myocardin in cerebral vascular smooth muscle cells (VSMC) generates an Aβ nonclearing VSMC phenotype through transactivation of SREBP-2, which is associated with Aβ accumulation (Bell et al., 2009); thus, it is conceivable that Tg-SREBP-2 may exhibit increased brain Aβ content secondary to lower LRP1 expression. Although we did not examine the expression level of LRP1, we did observe that the content of endogenous Aβ was similar in WT and Tg-SREBP-2 mice. These unexpected findings are consistent with evidence indicating that LRP1 promotes APP endocytosis and its processing to Aβ production (Ulery et al., 2000; Zerbinatti et al., 2004).
In agreement with the downregulation of mGSH by increased cholesterol-mediated impairment of mGSH transport from cytosol (Marí et al., 2006, 2008; Colell et al., 1997), we observed that isolated mitochondria from both Tg-SREBP-2 mice and NPC1 knock-out mice exhibit mGSH depletion and enhanced susceptibility to Aβ-mediated toxicity and mitochondrial membrane permeabilization. Although GSH exists in both cytosol and mitochondria, this latter pool has been shown to control the susceptibility to stimuli that trigger mitochondrial oxidative stress including TNF, Fas or hypoxia (Marí et al., 2006, 2008; Colell et al., 1997; Lluis et al., 2007). However, although total GSH depletion and altered GSH redox cycle has been described in AD, the particular pool of mGSH in modulating Aβ toxicity has not been specifically addressed. Two lines of evidence indicate that the mGSH depletion specifically accounted for the increased susceptibility to Aβ observed in these genetic models. First, the sensitivity of rat brain mitochondria to Aβ-mediated oxidative stress and release of intermembrane apoptotic proteins was dependent on the levels of mGSH. Second, selective pharmacological depletion of mGSH with the spare of cytosol GSH in human neuroblastoma and glial cells sensitized to Aβ-mediated cell death. Hence these findings expand the role of cholesterol in Aβ neurotoxicity, indicating that in addition to fostering the amyloidogenic processing of APP, mitochondrial cholesterol modulates Aβ neurotoxicity via mGSH regulation. Interestingly, recent findings have shown that hydrogen peroxide promotes Aβ production through JNK-dependent activation of γ-secretase (Shen et al., 2008), implying that in addition to the modulation of Aβ toxicity, enhanced mitochondrial cholesterol-mediated mGSH depletion may activate an autoamplification loop by stimulating oxidative stress to further generate Aβ, thus establishing a toxic vicious cycle.
Although increased cholesterol levels have been described in membranes from brain tissue samples of AD patients (Cutler et al., 2004; Bandaru et al., 2007), the specific pool of mitochondrial cholesterol has not been previously reported to the best of our knowledge. However, enhanced immunocytochemical localization of steroidegenic acute regulatory protein (StAR) has been described in the pyramidal hippocampal neurons of AD-affected patients (Webber et al., 2006). Given the role of StAR in the mitochondrial transport of cholesterol (Soccio and Breslow, 2004) and hence in the modulation of mitochondrial cholesterol levels (Montero et al., 2008), the increased expression of StAR in AD patients, would strongly suggest that mitochondrial cholesterol accumulation may actually occur in patients with AD. Indeed, in the current study, we observed mitochondrial cholesterol loading and subsequent mGSH depletion in APP/PS1 transgenic mice, a genetic mouse model of AD, and interestingly, these changes in mitochondria were preceded by Aβ accumulation in the brain of APP/PS1 mice. Based on this temporal relationship, it is tempting to speculate that Aβ may regulate cholesterol homeostasis and/or trafficking. Although this needs to be specifically addressed, consistent with this possibility, previous studies underscored that Aβ 1–40/1–42 ratio altered lipid homeostasis, and that familial presenilin mutations decreased the Aβ1–40/1–42 ratio resulting in enhanced cholesterol levels (Grimm et al., 2005), suggesting that Aβ generation contributed to increased cholesterol content. A complication in this possible scenario of mutual regulation between cholesterol and Aβ is the observation that cholesteryl-esters unlike free cholesterol modulated Aβ generation (Puglielli et al., 2001).
Previous studies described the requirement for functional mitochondria in Aβ-mediated neurotoxicity and that Aβ interacts with the regulatory heme of complex IV leading to mitochondrial dysfunction and ROS production (Cardoso et al., 2001; Atamna and Boyle, 2006). Through inhibition of electron flow at different mitochondrial respiratory complexes, we show that complex III is a major site for mitochondrial ROS by Aβ, whose levels increased if mGSH levels are low, resulting in mitochondrial depolarization, release of cytochrome c, and caspase-3 activation. Interestingly caspase inhibition failed to protect mGSH-dependent sensitization of SH-SY5Y cells to Aβ-mediated cell death despite efficiently preventing the acquisition of apoptotic features, suggesting that Aβ stimulated both an oxidant-dependent cell death pathway and caspase-independent apoptosis pathways, which are modulated by mGSH levels.
Our in vivo studies underscore a higher sensitivity of Tg-SREBP-2 mice to infusion of human Aβ, resulting in enhanced lipid peroxidation, oxidative modification of proteins, glia activation, indicative of neuroinflammation, and neuronal damage. Although cholesterol upregulation in Tg-SREBP-2 did not modulate endogenous Aβ generation, we observed that a significant fraction of infused human Aβ colocalized with mitochondria, pointing to the mitochondria as a target of the Aβ toxicity in vivo. To substantiate that the increased susceptibility of Tg-SREBP-2 mice to in vivo Aβ infusion was mediated by mitochondrial cholesterol-induced mGSH depletion, we tested the role of coinfusing Aβ with GSH ethyl ester. GSH ethyl ester is a membrane permeable form of GSH, which unlike GSH precursors such as N-acetylcysteine increases mGSH independently of the transport of GSH into mitochondria (Marí et al., 2006, 2008). This implies that GSH ethyl ester bypasses the block of cytosol GSH transport into mitochondria imposed by cholesterol-mediated membrane dynamics, thus leading to mGSH replenishment (Marí et al., 2006, 2008; Colell et al., 1997). The downregulation of oxidative stress, neuroinflammation and protection against in vivo infusion of Aβ in Tg-SREBP-2 via mGSH recovery highlights the relevance of mitochondrial cholesterol-mediated regulation of mGSH in Aβ-induced neurodegeneration. From a therapeutical perspective, the fact that the particular pool of mGSH rather than cytosol GSH determines Aβ susceptibility implies that efforts should focus to specifically replenish mGSH to slow down disease progression. The therapeutic usefulness of this strategy is illustrated here by the ability of intraperitoneal administration of GSH ethyl ester in protecting against Aβ-induced neuroinflammation and neuronal loss. In addition to directly modulating mGSH by GSH ethyl ester, targeting the increased pool of mitochondrial cholesterol may be of relevance in AD, as it would be expected to restore mGSH levels. In line with this possibility, it was shown in obese ob/ob mice that atorvastatin therapy was useful in preventing susceptibility to TNF-mediated steatohepatitis by preventing mitochondrial cholesterol loading which subsequently resulted in increased mGSH levels (Marí et al., 2006). Taking this precedent as well as the previous suggestions for the use of statins in AD (Wolozin et al., 2000; Fassbender et al., 2001; Parsons et al., 2006), the therapeutic combination of statins plus GSH ethyl ester may be of potential therapeutic significance in AD.
Footnotes
This work was supported in part by the Plan Nacional de Investigación Científica, Desarrollo e Innovación Tecnológica Grants SAF2005-03923 and SAF2006-06780, by Research Center for Liver and Pancreatic Diseases Grant P50 AA 11999 funded by the National Institute on Alcohol Abuse and Alcoholism, and by the Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas supported by the Instituto de Salud Carlos III. We are grateful to Dr. M. Guzman from Complutense University, Madrid, for kindly providing the glia-derived cell lines. The technical assistance of Alberto García-Martínez is highly appreciated. We thank the Servicio Científico-técnico of Institut d'Investigacions Biomèdiques August Pi i Sunyer for EM, confocal imaging, and flow cytometry analysis assistance, Dr. C. Sanmartin from Navarra University for the HP synthesis, and Dr. S. Pons for kindly advising on neuron cultures. We thank Drs. C. Garcia-Ruiz, M. Mari, and A. Morales for critical reading of this manuscript. A.C. is supported by the Ramon y Cajal Research Program (Ministerio de Educación y Ciencias). This work is dedicated to patients suffering from Alzheimer's disease, in particular Faustina Torres Muñoz, who recently passed away as a victim of this devastating disease.
References
- Anstey KJ, Lipnicki DM, Low LF. Cholesterol as a risk factor for dementia and cognitive decline: a systematic review of prospective studies with meta-analysis. Am J Geriatr Psychiatry. 2008;16:343–354. doi: 10.1097/JGP.0b013e31816b72d4. [DOI] [PubMed] [Google Scholar]
- Ariga T, McDonald MP, Yu RK. Role of ganglioside metabolism in the pathogenesis of Alzheimer's disease–a review. J Lipid Res. 2008;49:1157–1175. doi: 10.1194/jlr.R800007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atamna H, Boyle K. Amyloid-beta peptide binds with heme to form a peroxidase: relationship to the cytopathologies of Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:3381–3386. doi: 10.1073/pnas.0600134103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandaru VV, Troncoso J, Wheeler D, Pletnikova O, Wang J, Conant K, Haughey NJ. ApoE4 disrupts sterol and sphingolipid metabolism in Alzheimer's but not normal brain. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Deane R, Chow N, Long X, Sagare A, Singh I, Streb JW, Guo H, Rubio A, Van Nostrand W, Miano JM, Zlokovic BV. SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat Cell Biol. 2009;11:143–153. doi: 10.1038/ncb1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd-Kimball D, Sultana R, Abdul HM, Butterfield DA. Gamma-glutamylcysteine ethyl ester-induced up-regulation of glutathione protects neurons against Abeta(1–42)-mediated oxidative stress and neurotoxicity: implications for Alzheimer's disease. J Neurosci Res. 2005;79:700–706. doi: 10.1002/jnr.20394. [DOI] [PubMed] [Google Scholar]
- Cardoso SM, Santos S, Swerdlow RH, Oliveira CR. Functional mitochondria are required for amyloid beta-mediated neurotoxicity. FASEB J. 2001;15:1439–1441. doi: 10.1096/fj.00-0561fje. [DOI] [PubMed] [Google Scholar]
- Casley CS, Canevari L, Land JM, Clark JB, Sharpe MA. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem. 2002;80:91–100. doi: 10.1046/j.0022-3042.2001.00681.x. [DOI] [PubMed] [Google Scholar]
- Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- Colell A, García-Ruiz C, Morales A, Ballesta A, Ookhtens M, Rodés J, Kaplowitz N, Fernández-Checa JC. Transport of reduced glutathione in hepatic mitochondria and mitoplasts from ethanol-treated rats: effect of membrane physical properties and S-adenosyl-L-methionine. Hepatology. 1997;26:699–708. doi: 10.1002/hep.510260323. [DOI] [PubMed] [Google Scholar]
- Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Zlokovic BV. Role of blood brain barrier in the pathogenesis of Alzheimer's disease. Curr Alzheimer Res. 2007;4:191–197. doi: 10.2174/156720507780362245. [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006;26:9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassbender K, Simons M, Bergmann C, Stroick M, Lutjohann D, Keller P, Runz H, Kuhl S, Bertsch T, von Bergmann K, Hennerici M, Beyreuther K, Hartmann T. Simvastatin strongly reduces levels of Alzheimer's disease β-amyloid peptides Aβ 42 and Aβ 40 in vitro and in vivo. Proc Natl Acad Sci U S A. 2001;98:5856–5861. doi: 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Checa JC, Kaplowitz N. Hepatic mitochondrial glutathione: transport and role in disease and toxicity. Toxicol Appl Pharmacol. 2005;204:263–273. doi: 10.1016/j.taap.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Fernandez-Checa JC, Ookhtens M, Kaplowitz N. Effect of chronic ethanol feeding on rat hepatocytic glutathione: compartmentation, efflux and response to incubation with ethanol. J Clin Invest. 1987;80:57–62. doi: 10.1172/JCI113063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frautschy SA, Yang F, Calderón L, Cole GM. Rodent models of Alzheimer's disease: rat Aβ infusion approaches to amyloid deposits. Neurobiol Aging. 1996;17:311–321. doi: 10.1016/0197-4580(95)02073-x. [DOI] [PubMed] [Google Scholar]
- García-Ruiz C, Colell A, Marí M, Morales A, Calvo M, Enrich C, Fernández-Checa JC. Defective TNF-alpha-mediated hepatocellular apoptosis and liver damage in acidic sphingomyelinase knockout mice. J Clin Invest. 2003;111:197–208. doi: 10.1172/JCI16010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm MO, Grimm HS, Pätzold AJ, Zinser EG, Halonen R, Duering M, Tschäpe JA, De Strooper B, Müller U, Shen J, Hartmann T. Regulation of cholesterol and sphingomyelin metabolism by amyloid beta and presenilin. Nat Cell Biol. 2005;7:1118–1123. doi: 10.1038/ncb1313. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hansson CA, Frykman S, Farmery MR, Tjernberg LO, Nilsberth C, Pursglove SE, Ito A, Winblad B, Cowburn RF, Thyberg J, Ankarcrona M. Nicastrin, presenilin, APH-1, and PEN-2 form active gamma-secretase complexes in mitochondria. J Biol Chem. 2004;279:51654–51660. doi: 10.1074/jbc.M404500200. [DOI] [PubMed] [Google Scholar]
- Heneka MT, O'Banion MK. Inflammatory processes in Alzheimer's disease. J Neuroimmunol. 2007;184:69–91. doi: 10.1016/j.jneuroim.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Horton JD, Shimomura I, Brown MS, Hammer RE, Goldstein JL, Shimano H. Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J Clin Invest. 1998;101:2331–2339. doi: 10.1172/JCI2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Jain A, Mårtensson J, Stole E, Auld PA, Meister A. Glutathione deficiency leads to mitochondrial damage in brain. Proc Natl Acad Sci U S A. 1991;88:1913–1917. doi: 10.1073/pnas.88.5.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvodova L, Kahya N, Schwille P, Ehehalt R, Verkade P, Drechsel D, Simons K. Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J Biol Chem. 2005;280:36815–36823. doi: 10.1074/jbc.M504484200. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Liyanage U, Bickel PE, Xia W, Lansbury PT, Jr, Kosik KS. A detergent-insoluble membrane compartment contains Aβ in vivo. Nat Med. 1998;4:730–734. doi: 10.1038/nm0698-730. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Farris W, Wu X, Christodoulou DC, Haigis MC, Guarente L, Selkoe DJ. Alternative translation initiation generates a novel isoform of insulin-degrading enzyme targeted to mitochondria. Biochem J. 2004;383:439–446. doi: 10.1042/BJ20041081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Lluis JM, Buricchi F, Chiarugi P, Morales A, Fernandez-Checa JC. Dual role of mitochondrial reactive oxygen species in hypoxia signaling: activation of nuclear factor-{kappa} B via c-SRC and oxidant-dependent cell death. Cancer Res. 2007;67:7368–7377. doi: 10.1158/0008-5472.CAN-07-0515. [DOI] [PubMed] [Google Scholar]
- Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A{beta} accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- Marí M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, Enrich C, Fernandez-Checa JC, García-Ruiz C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4:185–198. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Marí M, Colell A, Morales A, Caballero F, Moles A, Fernández A, Terrones O, Basañez G, Antonsson B, García-Ruiz C, Fernández-Checa JC. Mechanism of mitochondrial glutathione-dependent hepatocellular susceptibility to TNF despite NF-kB activation. Gastroenterology. 2008;134:1507–1520. doi: 10.1053/j.gastro.2008.01.073. [DOI] [PubMed] [Google Scholar]
- Mårtensson J, Han J, Griffith OW, Meister A. Glutathione ester delays the onset of scurvy in ascorbate-deficient guinea pigs. Proc Natl Acad Sci U S A. 1993;90:317–321. doi: 10.1073/pnas.90.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, DeTeresa R, Alford M, Terry R. Synaptic and neuritic alterations during the progression of Alzheimer's disease. Neurosci Lett. 1994;174:67–72. doi: 10.1016/0304-3940(94)90121-x. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero J, Morales A, Llacuna L, Lluis JM, Terrones O, Basañez G, Antonsson B, Prieto J, García-Ruiz C, Colell A, Fernández-Checa JC. Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma. Cancer Res. 2008;68:5246–5256. doi: 10.1158/0008-5472.CAN-07-6161. [DOI] [PubMed] [Google Scholar]
- Norman AW, Demel RA, de Kruyff B, van Deenen LL. Studies on the biological properties of polyene antibiotics. Evidence for the direct interaction of filipin with cholesterol. J Biol Chem. 1972;247:1918–1929. [PubMed] [Google Scholar]
- Notkola IL, Sulkava R, Pekkanen J, Erkinjuntti T, Ehnholm C, Kivinen P, Tuomilehto J, Nissinen A. Serum total cholesterol, apolipoprotein E 4 allele, and Alzheimer's disease. Neuroepidemiology. 1998;17:14–20. doi: 10.1159/000026149. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles:intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Osenkowski P, Ye W, Wang R, Wolfe MS, Selkoe DJ. Direct and potent regulation of gamma-secretase by its lipid microenvironment. J Biol Chem. 2008;283:22529–22540. doi: 10.1074/jbc.M801925200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons RB, Price GC, Farrant JK, Subramaniam D, Adeagbo-Sheikh J, Austen BM. Statins inhibit the dimerization of b-secretase via both isoprenoid and cholesterol-mediated mechanisms. Biochem J. 2006;399:205–214. doi: 10.1042/BJ20060655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul CA, Boegle AK, Maue RA. Before the loss: neuronal dysfunction in Niemann-Pick Type C disease. Biochim Biophys Acta. 2004;1685:63–76. doi: 10.1016/j.bbalip.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Pirttilä T, Soininen H, Mehta PD, Heinonen O, Lehtimäki T, Bogdanovic N, Paljärvi L, Kim KS, Kosunen O, Winblad B, Riekkinen P, Sr, Wisniewski HM. Apolipoprotein E genotype and amyloid load in Alzheimer disease and control brains. Neurobiol Aging. 1997;18:121–127. doi: 10.1016/s0197-4580(96)00204-7. [DOI] [PubMed] [Google Scholar]
- Puglielli L, Konopka G, Pack-Chung E, MacKenzie Ingano LA, Berezovska O, Hyman BT, Chang TY, Tanzi RE, Kovacs DM. Acyl-coenzyme A:cholesterol acyltransferase modulates the generation of the amyloid b-peptide. Nat Cell Biol. 2001;3:905–912. doi: 10.1038/ncb1001-905. [DOI] [PubMed] [Google Scholar]
- Resende R, Moreira PI, Proença T, Deshpande A, Busciglio J, Pereira C, Oliveira CR. Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radic Biol Med. 2008;44:2051–2057. doi: 10.1016/j.freeradbiomed.2008.03.012. [DOI] [PubMed] [Google Scholar]
- Schönknecht P, Lütjohann D, Pantel J, Bardenheuer H, Hartmann T, von Bergmann K, Beyreuther K, Schröder J. Cerebrospinal fluid 24S-hydroxycholesterol is increased in patients with Alzheimer's disease compared to healthy controls. Neurosci Lett. 2002;324:83–85. doi: 10.1016/s0304-3940(02)00164-7. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Developing preventive therapies for chronic diseases: lessons learned from Alzheimer's disease. Nutr Rev. 2007;65:S239–243. doi: 10.1111/j.1753-4887.2007.tb00370.x. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C, Chen Y, Liu H, Zhang K, Zhang T, Lin A, Jing N. Hydrogen peroxide promotes Aβ production through JNK-dependent activation of g-secretase. J Biol Chem. 2008;283:17721–17730. doi: 10.1074/jbc.M800013200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soccio RE, Breslow JL. Intracellular cholesterol transport. Arterioscler Thromb Vasc Biol. 2004;24:1150–1160. doi: 10.1161/01.ATV.0000131264.66417.d5. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Moir RD, Wagner SL. Clearance of Alzheimer's Ab peptide: The many roads to perdition. Neuron. 2004;43:605–608. doi: 10.1016/j.neuron.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Taylor DR, Hooper NM. Role of lipid rafts in the processing of the pathogenic prion and Alzheimer's amyloid-beta proteins. Semin Cell Dev Biol. 2007;18:638–648. doi: 10.1016/j.semcdb.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Tietze F. Enzymatic method for quantitative determination of nano-gram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- Ulery PG, Beers J, Mikhailenko I, Tanzi RE, Rebeck GW, Hyman BT, Strickland DK. Modulation of beta-amyloid precursor protein processing by the low density lipoprotein receptor-related protein (LRP). Evidence that LRP contributes to the pathogenesis of Alzheimer's disease. J Biol Chem. 2000;275:7410–7415. doi: 10.1074/jbc.275.10.7410. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron. 2004;44:181–193. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Webber KM, Stocco DM, Casadesus G, Bowen RL, Atwood CS, Previll LA, Harris PL, Zhu X, Perry G, Smith MA. Steroidogenic acute regulatory protein (StAR): evidence of gonadotropin-induced steroidogenesis in Alzheimer disease. Mol Neurodegener. 2006;1:14. doi: 10.1186/1750-1326-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439–1443. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- Yu W, Gong JS, Ko M, Garver WS, Yanagisawa K, Michikawa M. Altered cholesterol metabolism in Niemann-Pick type C1 mouse brains affects mitochondrial function. J Biol Chem. 2005;280:11731–11739. doi: 10.1074/jbc.M412898200. [DOI] [PubMed] [Google Scholar]
- Zeevalk GD, Manzino L, Sonsalla PK, Bernard LP. Characterization of intracellular elevation of glutathione (GSH) with glutathione monoethyl ester and GSH in brain and neuronal cultures: relevance to Parkinson's disease. Exp Neurol. 2007;203:512–520. doi: 10.1016/j.expneurol.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbinatti CV, Wozniak DF, Cirrito J, Cam JA, Osaka H, Bales KR, Zhuo M, Paul SM, Holtzman DM, Bu G. Increased soluble amyloid-beta peptide and memory deficits in amyloid model mice overexpressing the low-density lipoprotein receptor-related protein. Proc Natl Acad Sci U S A. 2004;101:1075–1080. doi: 10.1073/pnas.0305803101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci. 2005;28:202–208. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]