

Abstract
Background
Elevated C-reactive protein (CRP) may contribute to elevated arterial pressure in Ang II-dependent hypertension. However, the in vivo effects of Ang II and of mineralocorticoid receptor (MR) antagonism on CRP during Ang II-dependent hypertension have not been examined. In addition, urinary CRP excretion as a method to monitor the progression of Ang II-induced inflammation has not been evaluated.
Methods
Urine samples were collected from three groups (n = 10/group) of rats: 1) normotensive control, 2) angiotensin II infused (Ang II; 60 ng/min), and 3) Ang II + eplerenone (epl; 25 mg/d). A diet containing epl (0.1 %) was provided after 1 week of Ang II infusion.
Results
After 28 d, Ang II increased SBP from 136 ± 5 to 207 ± 8 mmHg; this response in SBP was not altered following MR antagonism (215 ± 6 mmHg). Ang II-infusion increased plasma CRP from 14 ± 2 to 26 ± 3 μg/mL and increased urinary CRP excretion nearly 8-fold (143 ± 26 vs 1102 ± 115 ng/d). Treatment with eplerenone reduced plasma CRP by 25 % and urinary immunoreactive CRP (irCRP) by 34 % in Ang II-infused rats suggesting that aldosterone contributes to the CRP-associated inflammatory response in Ang II-dependent hypertension.
Conclusions
The increase in SBP preceded the increase in irCRP excretion by at least 4 days suggesting that CRP does not significantly contribute to increased arterial blood pressure in Ang II-dependent hypertension. The blockade of MR reduced plasma CRP and urinary irCRP excretion demonstrating the contribution of aldosterone to the Ang II-induced generation of CRP. Furthermore, urinary CRP may serve as a non-invasive index for monitoring cardiovascular inflammation during hypertension.
Keywords: aldosterone, eplerenone, inflammation, mineralocorticoids, spironolactone
Introduction
C-reactive protein (CRP) is an acute phase protein that has emerged as an important risk factor for assessing cardiovascular inflammation.1–5 Studies have also suggested that CRP contributes to the development of hypertension,6 while others have shown reductions in CRP with specific treatments independent of changes in systolic blood pressure.7, 8 In studies using double transgenic rats expressing human renin and angiotensinogen genes (dTGR), induction of the transgenes was associated with a 52% increase in SBP and a 10-fold increase in serum CRP 9 suggesting that increased SBP is associated with a parallel increase in plasma CRP when Ang II is inappropriately elevated. However, a low, subpressor dosage (30 ng/min) of Ang II increased mean plasma CRP by 26.5% after 7d independent of an increase in SBP suggesting that Ang II increases CRP independent of increased blood pressure.10 Thus, the in vivo effects of Ang II-infusion on CRP are not well characterized. Because aldosterone and Ang II have been shown to play significant roles in the progression of pro-inflammatory pathways 11–14 and hypertension,11, 15 elevated aldosterone levels induced by Ang II infusion may contribute to elevated levels of CRP. However, the effects of chronic mineralocorticoid receptor (MR) antagonism during Ang II-dependent hypertension on CRP have not been examined.
While most of the data regarding CRP levels in CVD and hypertension have utilized plasma concentrations, a single urinary CRP value was reported from a sample taken from a fetal urinary bladder at 15 weeks of gestation providing evidence that CRP or its metabolized fragments are excreted in the urine and are detectable.16 Thus, we used the protocol of Volanakis et al. 17, 18 to extract urinary CRP fragments and to quantify them as a means of monitoring the progression of the CRP-associated inflammatory response during Ang II-dependent hypertension and to further evaluate the effects of chronic MR antagonism on CRP. Using urinary CRP excretion, we were able to address the hypothesis that Ang II infusion increases CRP and that chronic aldosterone receptor antagonism reduces CRP during Ang II-dependent hypertension.
Methods
All protocols used were approved by the Institutional Animal Care and Use Committee of Tulane University Health Sciences Center.
Animals and study procedures
Male, Sprague-Dawley rats (200–225 g; Charles River, Wilmington, MA) were randomly distributed among three groups (n = 10/group): 1) normotensive controls, 2) Ang II-infused hypertensives, and 3) Ang II hypertensives treated with eplerenone (0.1% in the diet). At this percentage, each animal received approximately 25 mg/d (100 mg/kg/d). This dosage has been shown to result in optimal pharmacokinetic characteristics for effective in vivo inhibition of MR in the rat11, in addition to being the most commonly used dosage of eplerenone in studies with rats.11, 13, 19–23 More importantly, this dosage has been shown to produce significant physiological benefits independent of completely reducing the hypertension in most rodent studies.11, 13, 20–23 At the onset, animals were either sham operated (controls) or implanted with osmotic mini-pumps (Durect Corp., Cupertino, CA; model 2004) containing Ang II (Phoenix Pharmaceuticals, Belmont, CA; 60 ng/min for 28 d). Following these procedures animals were placed in metabolic cages designed to facilitate the collection of daily urine voids. Animals were allowed to acclimate to the cages for 3 days prior to the initiation of data collection. Arterial blood pressure measurements were taken weekly on days 6, 13, 20, and 27 by tail-cuff plethysmography, and published previously24, but reported here to confirm induction of hypertension.
The diet (Purina rodent chow #5002) of the epl-treated animals was switched to a feed containing 0.1 % eplerenone (Pfizer, St. Louis, MO) on the eighth day. The other animals were maintained on the same rodent chow (#5002) but without the added epl throughout the study. Urine output was recorded daily for the first 16 d and then intermittently on days 19, 22, 25, and 28. After urine volume was recorded, an aliquot was collected for analysis of urinary immunoreactive CRP (irCRP), 8-isoprostane (8-iso), total protein and creatinine. Urinary total protein and creatinine were measured from samples collected on each day, and urinary irCRP and 8-iso were measured from samples collected on days 4, 7, 8, 10, 15, 19, 22, 25 and 28.
Dissections
Following final measurements on day 28, animals were decapitated and trunk blood was collected into chilled vials containing 5 mM EDTA plus protein inhibitor cocktail (pH = 7.4, 12.5 mM 1–10-phenanthroline, 22 μM pepstatin, 10 μM PMSF, 20 μM enalaprilat) for plasma CRP measurements.
CRP Separation and Measurement
Separation of immunoreactive CRP (irCRP) in urine was accomplished by using phosphorylcholine affinity resin in the presence of high Ca2+ as described originally by Volanakis et al.17 and later by Oliveira et al.25 for use in mammals. Disposable affinity columns (Pierce, Rockford, IL; part no. 29) were loaded with 0.5 mL of immobilized ρ-aminophenyl phosphoryl choline gel (Pierce; part #20307) manufactured specifically for the isolation of CRP. Once the gel settled in the column, the column was equilibrated with 1.0 mL of high Ca2+, binding buffer (0.1 M Tris, 0.15 M NaCl, 10 mM CaCl2, pH 8.0) before 0.5 mL of urine was loaded and allowed to incubate for 1 hr. The gel was washed with 2.5 mL of binding buffer and eluted with 1.5 mL of high EDTA, elution buffer (0.1 M Tris, 0.15 M NaCl, 10 mM EDTA, pH 8.0). The eluted fraction was collected in 2.0 mL conical vials and dried in a concentrating centrifuge. Dried samples were reconstituted in 0.5 mL of storage buffer (0.01 M sodium phosphate, 0.15 M NaCl) and run in duplicate in a commercially available ELISA kit (Helica BioSystems, Fullerton, CA). Plasma was diluted 1:1000 in assay buffer (supplied with the kit) prior to extraction on the column, and run with the same assay kit. Intact CRP has a molecular weight of approximately 120 kDa,17, 25–27 and thus, is not readily filterable by the kidney, but it consists of two primary subunits of approximately 20 to 24 kDa,17, 25–27 which should be freely filterable. Therefore, recognizing we are measuring CRP fragments and not entire protein, urinary CRP fragments are referred to as urinary immunoreactive CRP (irCRP) for simplicity.
8-Isoprostane, Total Protein & Creatinine Analyses
For determination of urinary 8-isoprostane, urine samples were diluted 1:10 in assay buffer (supplied with the kit) and samples were measured in duplicate (Cayman Chemicals, MI). Urine samples were diluted 1:100 prior to measurement of total protein by photospectrometry (BioRad, Hercules, CA). Creatinine in urine samples were measured directly by photospectrometry using an automated autoanalyzer (Hitachi 912; Roche Diagnostics, Indianapolis, IN). Urinary excretion values were the product of concentration and urine volume.
Statistics
Means (± SE) of daily measurements were compared by analysis of variance adjusted for repeated measures over time. If significant (P < 0.05) group × time interactions were detected, one-way analysis of variance was performed to determine differences on specific days. Means for plasma CRP values were compared by one-way analysis of variance. For all cases, if significance (P < 0.05) was detected, a Fisher’s Protected Least Significant Difference test was applied post-hoc. Statistics were performed using Statview software (SAS, Cary, NC).
Results
Blood Pressure
Ang II increased (p < 0.01) mean SBP from 133 ± 4 mmHg in Controls to 170 ± 5 mmHg and 149 ± 6 mmHg in Ang II-infused and Ang II-infused + epl groups, respectively, by day 6. A further increase (p < 0.05) of 16 % in Ang II-infused and 24 % in Ang II-infused rats treated with eplerenone was observed by day 13. Mean SBP remained elevated (p < 0.001) above control (136 ± 5 vs. 207 ± 8 mmHg) levels for the remainder of the study. No sustained significant effect of eplerenone on SBP was observed (215 ± 5 mmHg) on 27 d.
Plasma and Urinary CRP
Ang II infusion increased (p < 0.01) mean plasma CRP 79 % (14.3 ± 0.8 vs 25.6 ± 3.4 μg/mL), and treatment with eplerenone reduced (p < 0.05) mean plasma CRP 25 % (19.2 ± 0.9 μg/mL) (Figure 1A, insert). Although eplerenone treatment reduced mean plasma CRP by 25%, this level was still 34% higher than control (p < 0.05) (Figure 1A, insert). After 28 days of Ang II infusion, urinary irCRP excretion was increased (p < 0.001) nearly 8-fold (143 ± 26 vs 1102 ± 115 ng/d) (Figure 1B). Treatment with eplerenone reduced (p < 0.05) urinary irCRP excretion by 34 % (723 ± 58 ng/d) (Figure 1B). On day 28 when plasma and urine samples were collected, plasma CRP and urinary irCRP excretion were positively and significantly correlated (urinary irCRP excretion = 62*plasma CRP – 367; R = 0.559; p < 0.01).
Figure 1.
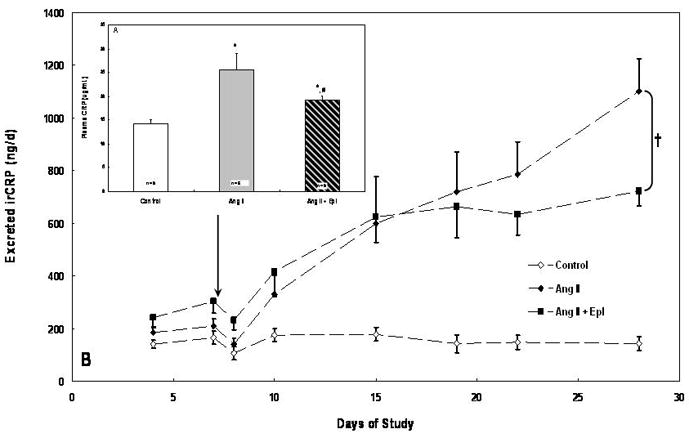
A) Insert: Mean (± SE) plasma C-reactive protein (CRP) in Control, Angiotensin II (Ang II) and Ang II + eplerenone treated animals (n =10/group) following 28 d. B) Mean (± SE) urinary immunoreactive C-reactive protein (irCRP) excretion in Control, Angiotensin II (Ang II) and Ang II + eplerenone treated animals (n = 10/group) over 28 d. Chronic infusion of Ang II (60 ng/min) began on day 1 and eplerenone treatment (25 mg/d) began on day 8 (arrow). * denotes significantly (p < 0.01) difference from Control; # denotes significantly (p < 0.05) different from Ang II. † denotes significant group × time interaction between Ang II and Ang II + Epl groups (p < 0.05). Both Ang II groups were significantly (p < 0.01) different from Control.
Urinary 8-isoprostane, Total Protein & Creatinine Excretion
Ang II infusion increased (p < 0.05) urinary 8-isoprostane excretion by 62 % by day 8 and excretion levels remained elevated for the remainder of the study with the exception of day 19 (Figure 2). Treatment with eplerenone did not significantly reduce urinary 8-isoprostane excretion (Figures 2). Ang II infusion increased urinary total protein excretion by 3.3-fold by day 28 (Figure 3). A significant group effect was not observed in urinary total protein excretion prior to epl treatment in Ang II-infused animals (p > 0.05); however a significant (p < 0.01) group effect was detected following epl treatment corresponding with a 54 % decrease in mean total protein excretion when compared to Ang II-infused animals (Figure 3). Neither a significant group nor group X time effect was observed in urinary creatinine excretion. At the end of the study (day 28), urinary creatinine excretion was not different among the 3 groups (control: 100 ± 9, Ang II: 97 ± 16, Ang II + epl: 85 ± 16 μmol/d).
Figure 2.

Mean (± SE) urinary 8-isoprostane excretion in Control, Angiotensin II (Ang II) and Ang II + eplerenone treated animals (n = 10/group) over 28 d. Chronic infusion of Ang II (60 ng/min) began on day 1 and eplerenone treatment (25 mg/d) began on day 8 (arrow). * denotes significantly (p < 0.05) different from Control. Both Ang II groups were significantly different from Control; however, no treatment effect was detected.
Figure 3.

Mean (± SE) urinary total protein excretion in Control, Angiotensin II (Ang II) and Ang II + eplerenone treated animals (n = 10/group) over 28 d. Chronic infusion of Ang II (60 ng/min) began on day 1 and eplerenone treatment (25 mg/d) began on day 8 (arrow). * denotes significant group × time interaction between Ang II and Ang II + Epl groups (p < 0.01). # denotes significant group × time interaction between Control and Ang II + Epl groups (p < 0.05).
Discussion
C-reactive protein is an important biomarker for evaluating the risk of cardiovascular disease and hypertension,1–4, 28–30 but recently has emerged as a contributing factor in the promotion of CVD. 6, 31 Recent studies have suggested that CRP has a direct effect in promoting atherosclerosis and endothelial inflammation,32–34 suggesting that CRP is not just a link to cardiovascular disease, but may be a direct mediator manifesting the deleterious condition.28, 31, 35, 36 Elevated Ang II promotes atherosclerosis37–40 and has been shown in vitro to generate CRP from VSMCs, underscoring the importance of Ang II in facilitating VSMC inflammation via CRP during the manifestation of atherosclerosis.10 However, the effects of elevated Ang II and aldosterone on CRP during Ang II-dependent hypertension are not well described. An important and novel finding of the current study is that aldosterone contributes to the Ang II-induced increase in CRP observed during Ang II-dependent hypertension. The present study also demonstrates that Ang II induces an increase in circulating CRP in an in vivo model of hypertension. In addition, we have developed a method for measuring urinary CRP excretion as a means to routinely monitor the progression of the Ang II-induced increase in CRP.
The contribution of aldosterone to the increase in CRP during Ang II-dependent hypertension had not been previously evaluated. The ability of MR antagonism to partially reduce the Ang II-induced increase in both plasma CRP (by 25%) and urinary irCRP excretion (by 34%) indicates that aldosterone contributes to the manifestation and maintenance of the CRP response to Ang II infusion. The only other study known to us that examined the effects of chronic MR antagonism on CRP levels did not report a change in circulating CRP levels following 1 to 3 mo of treatment with spironolactone in four separate groups of heart failure or angina patients.41 The chronic MR blockade in the present study did not completely alleviate the increase in plasma CRP induced by Ang II infusion suggesting that Ang II is a more potent stimulant of CRP generation than aldosterone. The reduction in plasma CRP observed in dTGR rats treated with losartan for 3 weeks was much greater than that observed in the present study in which rats were infused with Ang II and treated with eplerenone for 3 weeks further suggesting that Ang II contributes to increased CRP to a greater extent than aldosterone.
The infusion of Ang II increased irCRP that was detected in as early as 7 days in the Ang II + eplerenone group prior to the initiation of their eplerenone treatment. By day 10, urinary irCRP excretion was increased in both Ang II-infused groups. The present study reports the seminal finding of urinary irCRP excretion used to monitor the progression of CRP changes during a specific exogenous manipulation. The application of this developed method for assessing urinary irCRP excretion has important clinical relevance for non-invasively monitoring the progression of the inflammation associated with CVD, CHD and/or hypertension in patients over time. More importantly, it provides a non-invasive method to monitor the effectiveness of therapies on alleviating CRP-associated pathogenesis.
The associations among CRP, SBP and Ang II are not well characterized. In patients with coronary heart disease (CHD)42 or essential hypertension43, treatment with an angiotensin receptor blocker (ARB; losartan [2 mo] or valsartan [3 mo], respectively) reduced SBP, but not CRP suggesting that Ang II signaling does not influence CRP production under these conditions. In addition, elevated Ang II was not associated with elevated CRP in patients with Bartter’s/Gitelman’s syndromes.44 However, treatment with valsartan for 12 months reduced CRP levels in heart failure patients5 suggesting that Ang II signaling must be disrupted for sufficient time to affect CRP levels. In contrast, treatment with losartan for 3 weeks in dTGR rats (an Ang II-dependent model of hypertension) abolished the hypertension and the increase in serum CRP suggesting that increased CRP is associated with both hypertension and Ang II. 9 In constrast, Peng et al.10 demonstrated an increase in plasma CRP with a subpressor dosage of Ang II (7d) suggesting that the Ang II-induced increase in CRP is independent of an increase in SBP and may not necessarily contribute to increased SBP. The present study also demonstrates in vivo that Ang II infusion (28d) increases plasma CRP and urinary irCRP excretion; however the Ang II dosage was sufficient to increase SBP. Collectively, these data suggest that CRP responds to cardiovascular events and/or Ang II differentially. The in vitro10 and in vivo9, 10 studies on the effects of Ang II on CRP generation provide evidence that Ang II is a potent stimulant of CRP regardless of the changes in arterial blood pressure.
The ability of losartan to greatly reduce the arterial blood pressure in dTGR rats may have also contributed to the fact that a greater reduction in circulating CRP was also observed since MR antagonism in the present study did not reduce SBP. This is important because recently it was shown that transgenic mice over expressing rabbit CRP exhibited elevated SBP and decreased vascular angiotensin receptor 2 (AT2) expression indicating that CRP directly induces hypertension via downregulation of AT2.6 The present study demonstrates that the increase in SBP preceded the increase in urinary irCRP excretion by about 4 days suggesting that the increase in CRP did not contribute to the manifestation of the hypertension in this model. Furthermore, the lack of a decrease in SBP despite decreases in plasma and urinary CRP suggests that CRP may not contribute significantly to the maintenance of the hypertension in the Ang II-infused model.
While the present study did not examine the intracellular mechanisms by which aldosterone contributes to the generation of CRP, Peng et al.10 demonstrated that Ang II induces an increase in CRP via a MAP kinase signaling pathway, which may be the mechanism by which aldosterone stimulates CRP production. The cellular mechanisms by which aldosterone stimulate CRP production warrant further investigation and may prove to be clinically relevant from a therapeutic perspective.
Consistent with previous studies, Ang II induced an increase in proteinuria that was nearly ameliorated by chronic MR blockade. In a mild form of hypertension (2-kidney, 1-clip) in which peak mean SBP was only 130 mmHg, MR antagonism reduced SBP and proteinuria in parallel19 suggesting that increased aldosterone in concert with increased arterial pressure contribute to renal damage. However, the present study demonstrates that treatment with eplerenone initiated after the induction of the Ang II-dependent hypertension (which was more intense than that in the 2-K, 1-C model19) was sufficient to ameliorate the proteinuria, independent of the sustained elevation in arterial blood pressure suggesting that aldosterone promotes renal damage independent of the hypertension, similar to that observed in other models of hypertension.12, 21
That Ang II stimulates oxidative stress 45–47 and inflammation 48, 49 is well established; however the contributions of aldosterone to Ang II-induced oxidative stress are not as well defined. Treatment with spironolactone (150 mg/kg/d) in Ang II-infused rats reduced plasma 8-isoprostane by approximately 30% after 4 weeks of treatment; however SBP was not reduced50 suggesting that aldosterone may contribute to oxidative stress independent of increased arterial pressure. In contrast, spironolactone did not reduce urinary 8-isoprostane levels in dopamine D2 receptor deficient mice despite normalizing the SBP51 suggesting that aldosterone does not contribute significantly to the oxidative stress in a non-Ang II model of hypertension. Treatment with eplerenone in the present study also did not reduce urinary 8-isoprostane levels and SBP; however, the dosage and treatment duration were greater in the previous Ang II study50, which may have contributed to the disparate results. The increase in urinary 8-isoprostane over the course of the Ang II infusion in both of the infused groups is consistent with the effects of Ang II on oxidative stress. Treatment with the superoxide dismutase mimetic, tempol, reduced mean arterial pressure (MAP) in Ang II-infused rats suggesting that oxidative stress contributes to the increase in MAP in Ang II-dependent hypertension52. Thus, the lack of parallel decreases in both SBP and urinary 8-iso excretion following treatment with an MR antagonist in the present study suggests that the effects of aldosterone on increased arterial pressure may involve mechanisms other than those associated with oxidative stress in Ang II-dependent hypertension.
Furthermore the present study demonstrates that while the CRP-mediated inflammatory response is partially aldosterone-dependent, the oxidative stress response is not aldosterone mediated suggesting that the inflammatory and oxidative stress pathways in Ang II-mediated hypertension may be independent. Nonetheless, the present study clearly demonstrates that Ang II is a potent stimulant of both inflammatory and oxidative stress pathways, while aldosterone only contributes partially to the Ang II-mediated inflammatory response.
In conclusion, the present study demonstrates that Ang II induces an increase in plasma CRP and urinary irCRP excretion in vivo independent of an increase in SBP. The decrease in plasma CRP and urinary irCRP following treatment with an MR blocker indicates that aldosterone contributes to the increase in Ang II-induced generation of CRP in vivo. However, because MR antagonism did not completely alleviate the increase in CRP associated with infusion of Ang II, it is possible that the CRP contributed to the observed sustained elevation in arterial blood pressure. The correlation between plasma and urinary CRP levels suggest that urinary irCRP excretion reflects circulating levels and thus may serve as a relevant marker for monitoring the progression of CRP-associated inflammatory response as well as a non-invasive means for assessing the utility of anti-inflammatory drugs without obtaining frequent blood samples. The present study revealed that aldosterone contributes to the CRP-associated inflammatory response and proteinuria in Ang II-dependent hypertension.
Acknowledgments
We thank Drs. M. Graciano and M. Prieto-Carrasquero for their assistance throughout the study, and Mrs. D. Conte for her excellent technical assistance.
Grants & other support: RMO was partially supported by an NIH NRSA (F32 HL076985) from NHLBI. Research was supported by NHLBI grant (HL 26371 to LGN), NIH COBRE grant (P20 RR017659) from the Institutional Award (IdeA) program of the National Center for Research Resources, the Millennium Health Excellence Fund from the Louisiana Board of Regents, the University of California, and the University of California, Merced Graduate Research Committee Award.
Disclosures
The authors would like to thank Drs. M. Heron and E. McMahon of Pfizer (St. Louis, MO) for their generous contribution of eplerenone (SC-66110). None of the authors need to disclose any potential conflicts of interest regarding any aspect of this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Koenig W, Sund M, Frohlich M, et al. C-Reactive Protein, a Sensitive Marker of Inflammation, Predicts Future Risk of Coronary Heart Disease in Initially Healthy Middle-Aged Men: Results From the MONICA (Monitoring Trends and Determinants in Cardiovascular Disease) Augsburg Cohort Study, 1984 to 1992. Circulation. 1999;99:237–242. doi: 10.1161/01.cir.99.2.237. [DOI] [PubMed] [Google Scholar]
- 2.Morrow D, Ridker P. C-reactive protein, inflammation, and coronary risk. Med Clin North Am. 2000;84:149–161. doi: 10.1016/s0025-7125(05)70211-x. [DOI] [PubMed] [Google Scholar]
- 3.Ridker PM, Hennekens CH, Buring JE, Rifai N. C-Reactive Protein and Other Markers of Inflammation in the Prediction of Cardiovascular Disease in Women. N Engl J Med. 2000;342:836–843. doi: 10.1056/NEJM200003233421202. [DOI] [PubMed] [Google Scholar]
- 4.Sesso H, Buring J, Rifai N, Blake G, Gaziano J, Ridker P. C-reactive protein and the risk of developing hypertension. J Am Med Assoc. 2003;290:2945–2951. doi: 10.1001/jama.290.22.2945. [DOI] [PubMed] [Google Scholar]
- 5.Anand IS, Latini R, Florea VG, et al. C-Reactive Protein in Heart Failure: Prognostic Value and the Effect of Valsartan. Circulation. 2005;112:1428–1434. doi: 10.1161/CIRCULATIONAHA.104.508465. [DOI] [PubMed] [Google Scholar]
- 6.Vongpatanasin W, Thomas G, Schwartz R, et al. C-reactive protein causes downregulation of vascular angiotensin subtype 2 receptors and systolic hypertension in mice. Circulation. 2007;115:1020–1028. doi: 10.1161/CIRCULATIONAHA.106.664854. [DOI] [PubMed] [Google Scholar]
- 7.Kim J-S, Kang TS, Kim J-B, et al. Significant association of C-reactive protein with arterial stiffness in treated non-diabetic hypertensive patients. Atherosclerosis. 2007;192:401–406. doi: 10.1016/j.atherosclerosis.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 8.King D, Egan B, Mainous A, Geesey M. The effects of extended-release metoprolol succinate on C-reactive protein levels in persons with hypertension. Journal of Clinical Hypertension. 2006;8:257–260. doi: 10.1111/j.1524-6175.2005.05248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shagdarsuren E, Wellner M, Braesen J-H, et al. Complement Activation in Angiotensin II-Induced Organ Damage. Circ Res. 2005;97:716–724. doi: 10.1161/01.RES.0000182677.89816.38. [DOI] [PubMed] [Google Scholar]
- 10.Peng N, Liu J-t, Gao D-f, Lin R, Li R. Angiotensin II-induced C-reactive protein generation: Inflammatory role of vascular smooth muscle cells in atherosclerosis. Atherosclerosis. 2007;193:292–298. doi: 10.1016/j.atherosclerosis.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 11.Blasi ER, Rocha R, Rudolph AE, Blomme EAG, Polly ML, McMahon EG. Aldosterone/salt induces renal inflammation and fibrosis in hypertensive rats. Kidney Int. 2003;63:1791–1800. doi: 10.1046/j.1523-1755.2003.00929.x. [DOI] [PubMed] [Google Scholar]
- 12.Rocha R, Chander PN, Khanna K, Zuckerman A, Stier CT., Jr Mineralocorticoid blockade reduces vascular injury in stroke-prone hypertensive rats. Hypertension. 1998;31:451–458. doi: 10.1161/01.hyp.31.1.451. [DOI] [PubMed] [Google Scholar]
- 13.Rocha R, Martin-Berger CL, Yang P, Scherrer R, Delyani J, McMahon E. Selective aldosterone blockade prevents angiotensinII/salt-induced vascular inflammation in the rat heart. Endocrinology. 2002;143:4828–4836. doi: 10.1210/en.2002-220120. [DOI] [PubMed] [Google Scholar]
- 14.Struthers AD, MacDonald TM. Review of aldosterone- and angiotensin II-induced target organ damage and prevention. Cardiovasc Res. 2004;61:663–670. doi: 10.1016/j.cardiores.2003.11.037. [DOI] [PubMed] [Google Scholar]
- 15.Virdis A, Neves MF, Amiri F, Viel E, Touyz RM, Schiffrin EL. Spironolactone Improves Angiotensin-Induced Vascular Changes and Oxidative Stress. Hypertension. 2002;40:504–510. doi: 10.1161/01.hyp.0000034738.79310.06. [DOI] [PubMed] [Google Scholar]
- 16.Raio L, Ghezzi F, Mueller M, McDougall J, Malek A. Evidence of fetal C-reactive protein urinary excretion in early gestation. Obstet Gynecol. 2003;101:1062–1063. doi: 10.1016/s0029-7844(02)02251-2. [DOI] [PubMed] [Google Scholar]
- 17.Volanakis JE, Clements WL, Schrohenloher RE. C-reactive protein: purification by affinity chromatography and physicochemical characterization. J Immunol Met. 1978;23:285–295. [Google Scholar]
- 18.Volanakis J, Kaplan M. Specificity of C-reactive protein for choline phosphate residues of pneumococcal C-polysaccharide. Proc Soc Exp Biol Med. 1971;136:612–614. doi: 10.3181/00379727-136-35323. [DOI] [PubMed] [Google Scholar]
- 19.Hao L, Kanno Y, Fukushima R, Watanabe Y, Ishida Y, H S. Effects of Eplerenone on Heart and Kidney in Two-Kidney, One-Clip Rats. Am J Nephrol. 2004;24:54–60. doi: 10.1159/000075945. [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi N, Hara K, Tojo A, et al. Eplerenone Shows Renoprotective Effect by Reducing LOX-1-Mediated Adhesion Molecule, PKC{epsilon}-MAPK-p90RSK, and Rho-Kinase Pathway. Hypertension. 2005;45:538–544. doi: 10.1161/01.HYP.0000157408.43807.5a. [DOI] [PubMed] [Google Scholar]
- 21.Rocha R, Stier CT, Jr, Kifor I, et al. Aldosterone: A mediator of myocardial necrosis and renal arteriopathy. Endocrinology. 2000;141:3871–3878. doi: 10.1210/endo.141.10.7711. [DOI] [PubMed] [Google Scholar]
- 22.Sanz-Rosa D, Oubina MP, Cediel E, et al. Eplerenone Reduces Oxidative Stress and Enhances eNOS in SHR: Vascular Functional and Structural Consequences. Antioxid Redox Signaling. 2005;7:1294–1301. doi: 10.1089/ars.2005.7.1294. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki J, Iwai M, Mogi M, et al. Eplerenone With Valsartan Effectively Reduces Atherosclerotic Lesion by Attenuation of Oxidative Stress and Inflammation. Arterioscler Thromb Vasc Biol. 2006;26:917–921. doi: 10.1161/01.ATV.0000204635.75748.0f. [DOI] [PubMed] [Google Scholar]
- 24.Ortiz RM, Graciano ML, Seth D, Awayda MS, Navar LG. Aldosterone Receptor Antagonism Exacerbates Intrarenal Angiotensin II Augmentation in Ang II-dependent Hypertension. Am J Physiol: Renal Physiol. 2007;293:F139–F147. doi: 10.1152/ajprenal.00504.2006. [DOI] [PubMed] [Google Scholar]
- 25.Oliveira EB, Gotschlich EC, Liu TY. Comparative studies on the binding properties of human and rabbit C- reactive proteins. J Immunol. 1980;124:1396–1402. [PubMed] [Google Scholar]
- 26.Oliveira E, Gotschlich E, Liu T-Y. Primary structure of human C-reactive protein. Proc Natl Acad Sci. 1977;74:3148–3151. doi: 10.1073/pnas.74.8.3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Volanakis JE. Human C-reactive protein: expression, structure, and function. Mol Immunol. 2001;38:189–197. doi: 10.1016/s0161-5890(01)00042-6. [DOI] [PubMed] [Google Scholar]
- 28.de Ferranti SD, Rifai N. C-reactive protein: a nontraditional serum marker of cardiovascular risk. Cardiovasc Pathol. 2007;16:14–21. doi: 10.1016/j.carpath.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 29.Reinehr T, Stoffel-Wagner B, Roth CL, Andler W. High-sensitive C-reactive protein, tumor necrosis factor [alpha], and cardiovascular risk factors before and after weight loss in obese children. Metabolism. 2005;54:1155–1161. doi: 10.1016/j.metabol.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 30.Sesso H, Wang L, Buring J, Ridker P, Gaziano J. Comparison of interleukin-6 and C-reactive protein for the risk of developing hypertension in women. Hypertension. 2007;49:304–310. doi: 10.1161/01.HYP.0000252664.24294.ff. [DOI] [PubMed] [Google Scholar]
- 31.Wilson AM, Ryan MC, Boyle AJ. The novel role of C-reactive protein in cardiovascular disease: Risk marker or pathogen. Int J Cardiol. 2006;106:291–297. doi: 10.1016/j.ijcard.2005.01.068. [DOI] [PubMed] [Google Scholar]
- 32.Pasceri V, Chang J, Willerson JT, Yeh ETH. Modulation of C-Reactive Protein-Mediated Monocyte Chemoattractant Protein-1 Induction in Human Endothelial Cells by Anti-Atherosclerosis Drugs. Circulation. 2001;103:2531–2534. doi: 10.1161/01.cir.103.21.2531. [DOI] [PubMed] [Google Scholar]
- 33.Pasceri V, Willerson JT, Yeh ETH. Direct Proinflammatory Effect of C-Reactive Protein on Human Endothelial Cells. Circulation. 2000;102:2165–2168. doi: 10.1161/01.cir.102.18.2165. [DOI] [PubMed] [Google Scholar]
- 34.Zwaka TP, Hombach V, Torzewski J. C-Reactive Protein-Mediated Low Density Lipoprotein Uptake by Macrophages: Implications for Atherosclerosis. Circulation. 2001;103:1194–1197. doi: 10.1161/01.cir.103.9.1194. [DOI] [PubMed] [Google Scholar]
- 35.Li J-J, Fang C-H. C-reactive protein is not only an inflammatory marker but also a direct cause of cardiovascular diseases. Med Hypoth. 2004;62:499–506. doi: 10.1016/j.mehy.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 36.Yeh ETH, Anderson HV, Pasceri V, Willerson JT. C-Reactive Protein: Linking Inflammation to Cardiovascular Complications. Circulation. 2001;104:974–975. doi: 10.1161/01.cir.104.9.974. [DOI] [PubMed] [Google Scholar]
- 37.Daugherty A, Cassis L. Chronic Angiotensin II Infusion Promotes Atherogenesis in Low Density Lipoprotein Receptor -/- Mice. Ann NY Acad Sci. 1999;892:108–118. doi: 10.1111/j.1749-6632.1999.tb07789.x. [DOI] [PubMed] [Google Scholar]
- 38.Keidar S, Heinrich R, Kaplan M, Hayek T, Aviram M. Angiotensin II Administration to Atherosclerotic Mice Increases Macrophage Uptake of Oxidized LDL: A Possible Role for Interleukin-6. Arterioscler Thromb Vasc Biol. 2001;21:1464–1469. doi: 10.1161/hq0901.095547. [DOI] [PubMed] [Google Scholar]
- 39.Nobuhiko A, Suganuma E, Babaev VR, et al. Angiotensin II Amplifies Macrophage-Driven Atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:2143–2148. doi: 10.1161/01.ATV.0000145607.03879.e0. [DOI] [PubMed] [Google Scholar]
- 40.Takaya T, Kawashima S, Shinohara M, et al. Angiotensin II type 1 receptor blocker telmisartan suppresses superoxide production and reduces atherosclerotic lesion formation in apolipoprotein E-deficient mice. Atherosclerosis. 2006;186:402–410. doi: 10.1016/j.atherosclerosis.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 41.Godfrey V, Farquharson CAJ, Macdonald JE, Yee K-M, Struthers AD. Effect of spironolactone on C-reactive protein levels in patients with heart disease. Int J Cardiol. 2007;117:282–284. doi: 10.1016/j.ijcard.2006.05.069. [DOI] [PubMed] [Google Scholar]
- 42.Prasad A, Koh KK, Schenke WH, et al. Role of angiotensin II type 1 receptor in the regulation of cellular adhesion molecules in atherosclerosis. Am Heart J. 2001;142:248–253. doi: 10.1067/mhj.2001.116699. [DOI] [PubMed] [Google Scholar]
- 43.Manabe S, Okura T, Watanabe S, Fukuoka T, Higaki J. Effects of Angiotensin II Receptor Blockade with Valsartan on Pro-Inflammatory Cytokines in Patients with Essential Hypertension. J Cardiovasc Pharmacol. 2005;46:735–739. doi: 10.1097/01.fjc.0000185783.00391.60. [DOI] [PubMed] [Google Scholar]
- 44.Davis PA, Mussap M, Pagnin E, et al. Early markers of inflammation in a high angiotensin II state--results of studies in Bartter’s/Gitelman’s syndromes. Nephrol Dial Transplant. 2006;21:1697–1701. doi: 10.1093/ndt/gfl112. [DOI] [PubMed] [Google Scholar]
- 45.Agarwal R, Campbell R, Warnock D. Oxidative stress in hypertension and chronic kidney disease: role of angiotensin II. Semin Nephrol. 2004;24:101–14. doi: 10.1016/j.semnephrol.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 46.Kopkan L, Castillo A, Navar LG, Majid DSA. Enhanced superoxide generation modulates renal function in ANG II-induced hypertensive rats. Am J Physiol Renal Physiol. 2006;290:F80–86. doi: 10.1152/ajprenal.00090.2005. [DOI] [PubMed] [Google Scholar]
- 47.Reckelhoff JF, Romero JC. Role of oxidative stress in angiotensin-induced hypertension. Am J Physiol Regul Integr Comp Physiol. 2003;284:R893–912. doi: 10.1152/ajpregu.00491.2002. [DOI] [PubMed] [Google Scholar]
- 48.Ferrario C, Strawn W. Role of the renin-angiotensin-aldosterone system and proinflammatory mediators in cardiovascular disease. Am J Cardiol. 2006;98:121–128. doi: 10.1016/j.amjcard.2006.01.059. [DOI] [PubMed] [Google Scholar]
- 49.Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin II. Int J Biochem Cell Biol. 2003;35:881–900. doi: 10.1016/s1357-2725(02)00271-6. [DOI] [PubMed] [Google Scholar]
- 50.Zhao W, Ahokas RA, Weber KT, Sun Y. ANG II-induced cardiac molecular and cellular events: role of aldosterone. Am J Physiol Heart Circ Physiol. 2006;291:H336–343. doi: 10.1152/ajpheart.01307.2005. [DOI] [PubMed] [Google Scholar]
- 51.Armando I, Wang X, Villar VAM, et al. Reactive Oxygen Species-Dependent Hypertension in Dopamine D2 Receptor-Deficient Mice. Hypertension. 2007;49:672–678. doi: 10.1161/01.HYP.0000254486.00883.3d. [DOI] [PubMed] [Google Scholar]
- 52.Nishiyama A, Fukui T, Fujisawa Y, et al. Systemic and Regional Hemodynamic Responses to Tempol in Angiotensin II-Infused Hypertensive Rats. Hypertension. 2001;37:77–83. doi: 10.1161/01.hyp.37.1.77. [DOI] [PubMed] [Google Scholar]