

Abstract
On one-electron oxidation all molecules including DNA bases become more acidic in nature. For the GC base pair experiments suggest that a facile proton transfer takes place in the G•+-C base pair from N1 of G•+ to N3 of cytosine. This intra-base pair proton transfer reaction has been extensively considered using theoretical methods for the gas phase and it is predicted that the proton transfer is slightly unfavorable in disagreement with experiment. In the present study, we consider the effect of the first hydration layer on the proton transfer reaction in G•+-C by the use of density functional theory (DFT), B3LYP/6-31+G** calculations of the G•+-C base pair in the presence of 6 and 11 water molecules. Under the influence of hydration of 11 waters, a facile proton transfer from N1 of G•+ to N3 of C is predicted. The zero point energy (ZPE) corrected forward and backward energy barriers, for the proton transfer from N1 of G•+ to N3 of C, was found to be 1.4 and 2.6 kcal/mol, respectively. The proton transferred G•-(H+)C + 11H2O was found to be 1.2 kcal/mol more stable than G•+-C + 11H2O in agreement with experiment. The present calculation demonstrates that the inclusion of the first hydration shell around G•+-C base pair has an important effect on the internal proton transfer energetics.
The direct interaction of ionizing radiation with DNA creates “holes” and electrons which are able to transfer through the stacked bases in DNA.1– 3 Holes are primarily captured by guanine which has the lowest oxidation potential4 while the electron is captured by thymine and cytosine as a result of their higher electron affinities. The one-electron oxidized DNA bases become considerably more acidic in nature in comparison to their neutral state. For guanine its radical cation (G•+) deprotonates from its N1-H site in the aqueous environment. While for the base pair there is a consensus that within the G•+-C base pair, G•+ only partially transfers it’s N1 proton (H+) to the N3 site on cytosine (C) forming the proton transferred species, G•-(H+)C, (see scheme 1).3a, 5 – 7 The degree of proton transfer is significant since in DNA, G•+-C and G•-(H+)C are suggested to have different hole transfer rates and reactivity. For example, only the guanine cation (G•+) is thought to react with water and then further undergo irreversible reaction with oxygen to produce 8-oxoguanine and other oxidized compounds.3a, 8 The prototropic equilibrium within G•+-C is also a critical factor in moderating long range hole transfer in DNA. The inter-base proton transfer equilibrium within the G•+-C base pair is critical to the description of the hole transfer dynamics in DNA because in the proton transferred state the hole tends to localize. Using solution phase data Steenken5 estimated the pKa of guanosine radical cation (dG•+) as 3.9. Based on the pKa value of G•+, Steenken5 proposed a facile proton transfer reaction from N1 of guanine to hydrogen bonded cytosine as shown in scheme 1. Using density functional PW91 and triple-ζ STO basis set with polarization function Thorp et al.9 also calculated the pKa of G•+ as 4.01. In addition to experiments3, 5 – 7 the inter-base proton transfer reaction in neutral and oxidized G-C base pair was also studied using theory.10 – 15 In Table 1, we present the zero point energy (ZPE) corrected forward and backward activation energy (ΔE) barriers (in kcal/mol) for proton transfer in G•+-C base pair in gas phase as well as in the presence of 6 and 11 waters along with the changes in enthalpy (ΔH) and free energy (ΔG) calculated at 298 K as shown in scheme 1. Recently, Li et al.14 calculated ZPE-corrected forward and backward energy barriers for proton transfer as 2.96 and 1.58 kcal/mol using the B3LYP/6-31+G* method and concluded this reaction to be unfavorable for the proton transfer since the G•-(H+)C was found to be 1.4 kcal/mol less stable than G•+-C (see Table 1). Thus, a disagreement between experiment3a, 5 – 7 and theory10 – 15 exists for this proton transfer reaction. The effect of surrounding environment was neglected in these earlier calculations.10 – 15 All previous theoretical calculations11–14 which did not include waters of hydration predict a positive ΔH and ΔG (see Table 1) in contradiction to experimental observations.
Scheme 1.
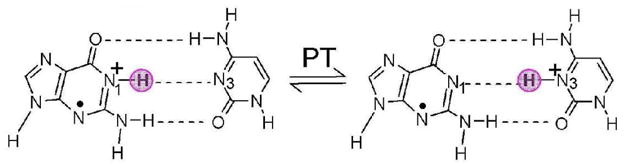
Schematic diagram showing proton transfer (PT) reaction in one electron oxidized G•+–C base pair. The highlighted pink circle shows the locations of proton (H+) in the base pair.
Table 1.
Proton transfer activation energya (ΔE), enthalpy (ΔH) and free energy (ΔG) (in kcal/mol) calculated using different methods with zero point energy (ZPE) correction.
| System | Activation barrier (ΔE) |
ΔHb | ΔGb | |
|---|---|---|---|---|
| forward | backward | |||
| G•+-C+11H2Oc | 1.42 | 2.59 | −1.32 | −0.65 |
| G•+-C+6H2Oc | 2.60 | 1.67 | 1.04 | 0.90 |
| G•+-Cc | 2.21 | 0.76 | 1.45 | 1.38 |
| G•+-Cd | 2.96 | 1.58 | 1.38 | 1.38 |
| G•+-Ce | 4.26e | 3.04e | 1.30 | 1.30 |
| G•+-Cf | - | - | 1.6f | |
| G•+-Cg | - | - | - | 1.18g |
Energies were calculated considering G•+-C as reference, see Figure 1.
Enthalpy (ΔH) and free energy (ΔG) changes calculated at 298 K with ZPE correction in kcal/mol.
Present calculation using B3LYP/6-31+G**.
B3LYP/6-31+G* (ref. 14).
B3LYP/6-31G** and without ZPE correction (ref. 11).
ZPE corrected ΔE calculated using UB3LYP/D95*//UHF/6-31G* (ref. 10).
HF/3-21G* (ref. 13).
From recent studies,16, 17 it is evident that the surrounding environment (hydration) is essential to properly predict the experimental results. For example, using B3LYP/6-31G* level of calculation and considering the hydration surrounding the guanosine radical cation (dG•+), we were able to predict N1-H site of dG•+ as the preferred site for deprotonation in aqueous solution as observed by electron spin resonance (ESR).16a In this letter, we report theoretical results that suggest hydration indeed has a pronounced effect that favors the proton transfer reaction within the GC base pair as shown in scheme 1.
In this present study, the geometries of G•+-C, G•-(H+)C (PT) and the transition state (TS) [G•---H+---C] for proton transfer reaction in gas phase (without hydration) and in the presence of 6 and 11 water molecules were fully optimized using density functional theory (DFT) B3LYP and 6–31+G** basis set. Frequency analyses was also performed, at the same level of theory and basis set, to ensure the existence of G•+-C and G•-(H+)C (PT) as local minimum structures and for TS [G•---H+---C] a negative frequency was found. The calculations were carried out using Gaussian 03 suite of programs18a and spin densities were plotted using GaussView18b and Molekel18c molecular modeling softwares. Based on solvation densities (defining the locations of waters around G-C base pair) from molecular dynamics simulation studies19 the initial geometries of hydrated, i.e., G-C + 6H2O and G-C + 11H2O in neutral state were generated as follows: (i) In the G-C + 6H2O, six water molecules were placed near N7(G), O6(G), N3(G), N2(G), O2(C), and N4(C) atoms surrounding the G-C base pair in the hydrogen bonding conformation and (ii) in addition to the initial G-C + 6H2O structure, we placed five more waters near the remaining exterior atoms of the G-C base pair (see Figure S4 for atom numbers). The neutral GC + 6H2O and GC + 11H2O thus generated were fully optimized at B3LYP/6-31+G** level of theory. Further, these optimized neutral geometries were considered as the starting geometries for optimization in their radical cation state. The optimized geometries are presented in the supporting information, see Figures S4 – S9.
For G•+-C and G•+-C + 6H2O, we found that proton transfer process is not favored. G•+-C was found to be 1.5 kcal/mol more stable than the G•-(H+)C in gas phase, while, with six waters (G•+-C+ 6H2O) the corresponding stability was slightly lower at 1.0 kcal/mol (see Table 1). Six waters only partially solvates the G•+-C pair which is clearly evident from the optimized geometries, see Figures S6 and S7 in supporting information.
The optimized structures of G•+-C in the presence of 11 waters are shown in Figures S4 and S5, respectively, in the supporting information. From the optimized geometries, we found that on both sides of the G-C pair water molecules form a contiguous hydrogen bonding network with themselves and with the base pair extending from N4(C) to C8(G) on one side and from N1(C) to N9(G) on the other side of the base pair, see Figures S4 and S5.
In Figure 1 we see that guanine is nearly completely surrounded by the water molecules, whereas, the hydrophobic C5C6 bond area of cytosine remains free of water (see also Figures S4 and S5). The B3LYP/6-31+G** calculated forward and backward energy (ΔE) barriers for the proton transfer without ZPE correction were found to be 3.85 and 5.51 kcal/mol, respectively (see Figure S2 in the supporting information). This shows that G•-(H+)C is 1.7 kcal/mol more stable than G•+-C. The inclusion of ZPE correction substantially reduces the forward barrier height to 1.42 kcal/mol while the backward barrier was found to be 2.59 kcal/mol (see Figures 1, S3 and Table 1). After ZPE correction G•-(H+)C was found to be 1.2 kcal/mol more stable than G•+-C. The B3LYP/6-31+G** calculated positive vibrational frequencies for G•+-C + 11H2O and G•-(H+)C + 11 H2O also confirm the existence of these local minimum structures (see Figures S10 and S11 in the supporting information). A negative frequency (−1241 cm−1) also confirms the transition state [G•---H+---C] for the proton transfer (see Figures 1 and S12). It is also evident from Figure 1 that before, during and after PT spin densities are localized only on guanine. The bulk effect of solvation was considered using the polarized continuum model (PCM) for the bare and the 11 water hydrated G•+-C. The PCM model had a negligible effect on the fully hydrated equilibrium (0.1 kcal/mol) but for the non hydrated system the effect was more substantial and proton transfer became energetically favorable (see supporting information). In the present study, it is important to note for the validity of this study that the total number of hydrogen bonds in reactant, TS and product are same, i.e., 11 in G•+-C + 6H2O and 20 in G•+-C + 11H2O complexes. Also the orientation of water molecules are almost unchanged, see Figures 1 and S4 – S7 in the supporting information.
Figure 1.
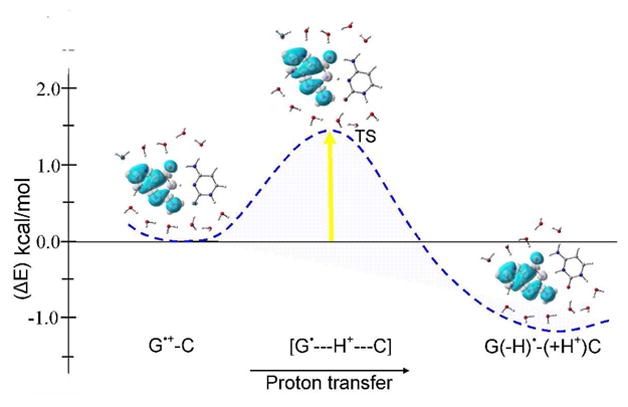
The B3LYP/6-31+G** calculated potential energy surface (PES) of proton transfer (PT) in G•+-C in the presence of 11 waters with zero point energy (ZPE) correction. Energy is given in kcal/mol. Spin density distributions during proton transfer are also shown.
From B3LYP/6-31+G** optimized TS structure of G•+-C, G•+-C + 6H2O and G•+-C + 11H2O we found that during proton transfer G and C bases are drawn more closely together, see Figures S4 – S9. The hydrogen bonding distances between G and C in G•+-C + 11H2O at TS are 1.779, 2.643 and 1.677 Å, respectively, however, the corresponding distances in G•+-C + 11H2O are 2.005, 2.855 and 1.630 Å, respectively. From the vibrational frequency analyses of G•+-C + 11H2O (see Figures S10 – S12 in the supporting information), we found that in-plane ring vibration of G along N1-H direction occurs at 537 cm−1 while the in-plane ring vibration of C occurs at 613 cm−1. These in-plane vibrational modes with the G•+-C base pair would clearly couple to the proton transfer from G to C (see Figure 1). Also, a vibration at 2751 cm−1 is assigned to the G(N1-H) stretching mode in G•+-C + 11H2O. After PT the corresponding C(N3-H) vibration occurs at 2849 cm−1, a blue shift of 98 cm−1.
Our calculations demonstrate that under the influence of hydration, which mimic the hydrated environment surrounding the G•+-C base pair in DNA, a facile proton transfer from G•+ to C is predicted. The negligible effect of the bulk solvent (0.1 kcal/mol) on fully hydrated equilibrium, shown in Figure 1, clearly shows that the stability of the deprotonated G•-(H+)C + 11H2O largely comes from the interactions with the surrounding 11 waters which mimic the first hydration shell (see details in the supporting information). –While at low temperatures the equilibrium would be shifted completely towards the proton transferred side, G•-(H+)C, at room temperature the G•+-C would be present in equilibrium. We also note that at elevated temperatures the dynamics of the surrounding waters, not considered in this study, could at times favor the back proton transfer process. Indeed the presence of the cation radical form of G in DNA is critical to explain the chemistry and the migratory nature of the DNA hole.
Supplementary Material
Acknowledgments
This work was supported by the NIH NCI under grant no. R01CA045424. Authors are thankful to the Arctic Region Supercomputing Center (ARSC) and NSF grant CHE-072689 for providing computational facilities.
Footnotes
Supporting Information Available: Full reference 18, figures showing PT scheme, optimized geometries, spin densities and vibrational spectra. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Sevilla MD, Becker D. Electron Paramagn Reson. 2004;19:243. [Google Scholar]
- 2.Swiderek P. Angew Chem Int Ed. 2006;45:4056. doi: 10.1002/anie.200600614. [DOI] [PubMed] [Google Scholar]
- 3.(a) Ghosh AK, Schuster GB. J Am Chem Soc. 2006;128:4172. doi: 10.1021/ja0573763. [DOI] [PubMed] [Google Scholar]; (b) Lewis FD, Daublain P, Cohen B, Vura-Weis J, Wasielewski MR. Angew Chem Int Ed. 2008;47:3798. doi: 10.1002/anie.200705903. [DOI] [PubMed] [Google Scholar]; (c) Shao F, Augustyn K, Barton JK. J Am Chem Soc. 2005;127:17445. doi: 10.1021/ja0563399. [DOI] [PubMed] [Google Scholar]; (d) Takada T, Kawai K, Fujitsuka M, Majima T. Proc Natl Acad Sci USA. 2004;101:14002. doi: 10.1073/pnas.0402756101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Conwell EM, Basko DM. J Am Chem Soc. 2001;123:11441. doi: 10.1021/ja015947v. [DOI] [PubMed] [Google Scholar]; (f) Giese B, Amaudrut J, Köhler AK, Spormann M, Wessely S. Nature. 2001;412:318. doi: 10.1038/35085542. [DOI] [PubMed] [Google Scholar]; (g) Ratner M. Nature. 1999;397:480. doi: 10.1038/17232. [DOI] [PubMed] [Google Scholar]
- 4.Sevilla MD, Besler B, Colson AO. J Phys Chem. 1995;99:1060. [Google Scholar]
- 5.(a) Steenken S. Chem Rev. 1989;89:503. [Google Scholar]; (b) Candeias LP, Steenken S. J Am Chem Soc. 1989;111:1094. [Google Scholar]
- 6.(a) Yamagami R, Kobayashi K, Tagawa S. J Am Chem Soc. 2008;130:14772. doi: 10.1021/ja805127e. [DOI] [PubMed] [Google Scholar]; (b) Kobayashi K, Tagawa S. J Am Chem Soc. 2003;125:10213. doi: 10.1021/ja036211w. [DOI] [PubMed] [Google Scholar]; (c) Kobayashi K, Yamagami R, Tagawa S. J Phys Chem B. 2008;112:10752. doi: 10.1021/jp804005t. [DOI] [PubMed] [Google Scholar]
- 7.Nir E, Kleinermanns K, de Vries MS. Nature. 2000;408:949. doi: 10.1038/35050053. [DOI] [PubMed] [Google Scholar]
- 8.Burrows CJ, Muller JG. Chem Rev. 1998;98:1109. doi: 10.1021/cr960421s. [DOI] [PubMed] [Google Scholar]
- 9.Baik MH, Silverman JS, Yang IV, Ropp PA, Szalai VA, Yang W, Thorp HH. J Phys Chem B. 2001;105:6437. [Google Scholar]
- 10.Hutter M, Clark T. J Am Chem Soc. 1996;118:7574. [Google Scholar]
- 11.Bertran J, Oliva A, Rodríguez-Santiago L, Sodupe M. J Am Chem Soc. 1998;120:8159. [Google Scholar]
- 12.Rak J, Makowska J, Voityuk AA. Chem Phys. 2006;325:567. [Google Scholar]
- 13.Colson AO, Besler B, Sevilla MD. J Phys Chem. 1992;96:9787. [Google Scholar]
- 14.Li X, Cai Z, Sevilla MD. J Phys Chem B. 2001;105:10115. [Google Scholar]
- 15.Florián J, Leszczyński J. J Am Chem Soc. 1996;118:3010. [Google Scholar]
- 16.(a) Adhikary A, Kumar A, Becker D, Sevilla MD. J Phys Chem B. 2006;110:24171. doi: 10.1021/jp064361y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Adhikary A, Kumar A, Khanduri D, Sevilla MD. J Am Chem Soc. 2008;130:10282. doi: 10.1021/ja802122s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kumar A, Sevilla MD. Chem Phys Chem. 2009 doi: 10.1002/cphc.200900025. [DOI] [Google Scholar]
- 17.(a) Guerra CF, Bickelhaupt FM, Snijders JG, Baerends EJ. J Am Chem Soc. 2000;122:4117. [Google Scholar]; (b) Witwicki M, Jezierska J, Ozarowski A. Chem Phys Lett. 2009;473:160. [Google Scholar]
- 18.(a) Frisch MJ, et al. Gaussian03, revision B.04. Gaussian, Inc; Pittsburgh, PA: 2003. [Google Scholar]; (b) GaussView. Gaussian, Inc; Pittsburgh, PA: 2003. [Google Scholar]; (c) Molekel 4.3. www.cscs.ch/molekel/
- 19.(a) Makarov V, Pettitt BM, Feig M. Acc Chem Res. 2002;35:376. doi: 10.1021/ar0100273. [DOI] [PubMed] [Google Scholar]; (b) Auffinger P, Westhof E. J Mol Biol. 2000;300:1113. doi: 10.1006/jmbi.2000.3894. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.