

Abstract
Current therapies for Parkinson’s disease significantly improve the quality of life for patients suffering from this neurodegenerative disease, yet none of the current therapies has been convincingly shown to slow or prevent the progression of disease. Much has been learned about the pathophysiology of Parkinson’s disease in recent years, and these discoveries offer a variety of potential targets for protective therapy. Mechanisms implicated in the disease process include oxidative stress, mitochondrial dysfunction, protein aggregation and misfolding, inflammation, excitotoxicity, and apoptosis. At the same time, the involvement of these diverse processes makes modeling the disease and evaluation of potential treatments difficult. In addition, available clinical tools are limited in their ability to monitor the progression of the disease. In this review, we summarize the different pathogenic mechanisms implicated in Parkinson’s disease and neuroprotective strategies targeting these mechanisms currently under clinical study or under preclinical development, with a view towards strategies that seem most promising.
Keywords: oxidative stress, neuroinflammation, excitotoxicity, apoptosis, trophic factors, alpha-synuclein, clinical trials
Parkinson’s disease (PD) is a progressive neurodegenerative disease causing tremor, rigidity, bradykinesia, and gait impairment. These motor deficits have been mainly attributed to the progressive loss of dopaminergic neurons in the substantia nigra (SN), and consequently the focus of current PD therapy has been on the replacement of dopamine. This approach has, in some respects, been remarkably effective and has led to treatments which can substantially improve both the duration and quality of life of patients affected by PD. Ultimately, however, most patients develop motor complications which are difficult to control with currently available treatments. In addition, they develop a variety of non-motor symptoms, including anosmia, sleep disorders, autonomic impairment, and cognitive impairment. These non-motor symptoms are refractory to most current treatments, and frequently become the most significant source of disability. The complexity of these late-stage complications points to the importance of developing neuroprotective strategies which could be applied early in the disease in order to prevent or delay the later complications of the disorder.
Pathological studies have shown that dopaminergic degeneration is only a small part of a much broader spectrum of PD-related neurodegeneration that may begin with pathology in the brainstem and then progress beyond the SN to cortical and subcortical regions [1]. In this current view, the overall duration of PD spans decades from the first signs to the late stages, with involvement of the motor system only in the middle stages. The long, slow course of neurologic impairment in PD offers an important opportunity: if treatments that delay or prevent the neurodegenerative process are developed, they could be employed at an early stage of the disease to prevent the severely debilitating complications of advanced PD. Indeed, it is likely that technologies that can detect the disease process before the onset of visible symptoms will become practical in the near future; the main barrier to progress against PD is the lack of a meaningful neuroprotective treatment that can be applied after the disease is identified.
In this review, we define “neuroprotective” therapies as those that slow or prevent further neurodegeneration of neuronal populations involved in PD, both dopaminergic and non-dopaminergic. Our definition for “neuroprotection” does not include “neurorestorative” strategies that aim to replace neuronal elements after they are lost (for example, fetal tissue or stem-cell-based transplants). Currently, there are no treatments that are clearly established as neuroprotective in PD, and the discovery of such treatments is an important goal of much work in the field.
Mechanisms for PD pathogenesis and targets for neuroprotection
An understanding of the mechanisms underlying the development and progression of PD pathology is critical for the development of neuroprotective therapies. A complex interplay of multiple environmental and genetic factors has been implicated in PD, suggesting that PD likely represents a syndrome instead of a single disorder with the same primary cause in all cases. Several mechanisms have been implicated as crucial to PD pathogenesis: oxidative stress, mitochondrial dysfunction, protein aggregation and misfolding, inflammation, excitotoxicity, apoptosis and other cell death pathways, and loss of trophic support (Table 1). No one mechanism appears to be primary in all cases of PD, and these pathogenic mechanisms likely act synergistically through complex interactions to promote neurodegeneration. Discussion of these mechanisms is briefly reviewed here in reference to their implications for the development of neuroprotective therapies. The reader is referred to other reviews in this issue that address PD pathogenesis in more detail.
Table 1.
Mechanisms of PD pathogenesis and targets for therapy
| PD pathogenic mechanism | Targets for neuroprotection |
|---|---|
| Oxidative stress and mitochondrial dysfunction | Inhibitors of dopamine metabolism (e.g. MAO inhibitors, dopamine receptor agonists) Electron transport enhancers (e.g. CoQ10) Other antioxidants (e.g. vitamin E, uric acid) Glutathione promoters (e.g. selenium) |
| Protein aggregation and misfolding | Inhibitors of α-syn aggregation Agents that reduce α-syn protein levels Enhancers of parkin function Enhancers of UCH-L1 function Enhancers of proteosomal or lysosomal pathways |
| Neuroinflammation | Anti-inflammatory agents (e.g. NSAIDs, statins, minocycline) |
| Excitotoxicity | NMDA receptor antagonists Calcium channel antagonists |
| Apoptosis and cell death pathways | Anti-apoptotic agents |
| Loss of trophic factors | Neurotrophic factors (e.g. GDNF, neurturin) |
Oxidative stress and mitochondrial dysfunction
Oxidative stress results from an overabundance of reactive free radicals secondary to either an overproduction of reactive species or a failure of cell buffering mechanisms that normally limit their accumulation. Excess reactive species can react with cellular macromolecules and thereby disrupt their normal functions. Oxidative damage to proteins, lipids, and nucleic acids has been found in the SN of patients with PD [2–4]. Both overproduction of reactive species and failure of cellular protective mechanisms appear to be operative in PD.
Dopamine metabolism promotes oxidative stress through the production of quinones, peroxides, and other reactive oxygen species (ROS) [5, 6]. Mitochondrial dysfunction is another source for the production of ROS, which can then further damage mitochondria. Complex I activity is diminished in the SN of PD patients [7], while inhibitors of complex I, such as MPP+ and rotenone, cause a Parkinsonian syndrome in animal models [8–10]. The mechanisms responsible for mitochondrial dysfunction in PD are not well understood, but inherited or acquired mutations in mitochondrial DNA may contribute [11–13]. Increased iron levels seen in the SN of PD patients [14, 15] also promote free radical damage, particularly in the presence of neuromelanin.
There is also evidence of impairment of endogenous protective mechanisms in PD. The antioxidant protein glutathione is reduced in postmortem PD nigra [16–18]. Several of the genes linked to familial forms of PD appear to be involved in protection against oxidative stress, including PTEN-induced putative kinase (PINK1) and DJ-1 [19–22].
Several different strategies have been proposed to limit oxidative stress in PD. These strategies include inhibitors of monoamine oxidase, a key enzyme involved in dopamine catabolism; enhancers of mitochondrial electron transport, such as Coenzyme Q10; compounds that can directly quench free radicals, such as vitamin E; and molecules that can promote endogenous mechanisms to buffer free radicals, such as selenium. The advantage of many of these agents is that they are well tolerated with few adverse effects, although convincing clinical evidence for the effectiveness of this approach is still lacking.
Protein aggregation and misfolding
Protein aggregation and misfolding have emerged as important mechanisms in many neurodegenerative disorders, including PD, Alzheimer’s disease, and Huntington’s disease. While the proteins involved in these disorders are different, each is associated with characteristic aggregates of misfolded protein, and these abnormal aggregates appear to acquire toxic properties.
In PD, the primary aggregating protein is alpha-synuclein (α-syn), whose link to PD was first identified through rare families with autosomal dominant PD caused by mutations in this protein [23–26]. While mutations in α-syn are found in a very small number of inherited PD cases, α-syn is the major component of Lewy bodies and Lewy neurites found in sporadic PD [27, 28]. Gene duplication of the α-syn locus also causes PD, further supporting the central role of this protein in PD pathogenesis [29]. Point mutations [30–33], overexpression [34], and oxidative damage of α-syn [35] have all been postulated to promote self-aggregation.
While abundant evidence links α-syn to PD, the mechanism by which overabundance or aggregation of α-syn causes neuronal injury is not understood. Likewise, it is unclear which molecular form of α-syn is toxic. Hypotheses include toxic effects of oligomers on cell membranes or proteosomal function, effects of α-syn on gene transcription or regulation, interactions of α-syn with cell signaling and cell death cascades, alterations in dopamine storage and release, and α-syn-mediated activation of inflammatory mechanisms [36–46].
Recent studies implicating parkin and ubiquitin carboxyl-terminal hydrolase L1 (UCH-L1) in genetic forms of PD reinforce the connection between protein aggregation and PD pathogenesis [47, 48]. Parkin is an E3 ubiquitin ligase involved in targeting misfolded proteins for degradation, and mutations of parkin found in genetic forms of PD disrupt its E3 ubiquitin ligase activity [49–51]. Interestingly, native α-syn does not appear to be a substrate for parkin, although modified forms may be [52, 53], and brains from patients with parkin-associated PD do not usually contain Lewy bodies [54–57]. Parkin does appear to have a variety of other substrates that may play a role in protein turnover and degradation, including HSP70, which is known to modulate α-syn toxicity [58, 59]. UCH-L1 serves as an ubiquitin recycling enzyme in neurons, and its dysfunction promotes aggregation of damaged proteins, including α-syn [60–62].
Together, these observations suggest that overproduction or impaired clearance of α-syn, resulting in aggregation, may be a central mechanism for PD. Therefore, therapeutic strategies to prevent protein aggregation or to enhance the clearance of misfolded proteins are the subject of intensive study at present. Inhibitors of α-syn aggregation could serve as potential neuroprotective therapies, although a clearer understanding of the toxic form of α-syn is important. Molecules that promote protein clearance could also have therapeutic potential. Promoters of protein clearance could include enhancers of parkin or UCH-L1 function, or molecules that promote proteosomal or lysosomal degradation pathways.
Neuroinflammation
Neuroinflammation has been increasingly recognized as a primary mechanism involved in PD pathogenesis [63–65]. Activation of microglia has been demonstrated in SN and striatum from postmortem PD brains and in PD animal models [66–70]. Pro-inflammatory cytokines, such as IL-1β, IL-6, and TNF-α, are elevated in the CSF and basal ganglia in PD patients [71, 72]. The complement system is also implicated in PD pathogenesis, as elevated serum levels of complement proteins and the presence of complement proteins in Lewy bodies have been detected in PD [73, 74]. How microglia become activated in PD is not fully understood, but both cytokines and α-syn aggregation can promote microglia activation [65]. In vitro, both aggregated and nitrated forms of α-syn can directly trigger a microglial response and release of cytotoxic factors [75–77]. In mouse models, α-syn or modified forms of the protein can trigger both microglial and humoral responses [45, 46], and inhibition of NF-KB signaling is neuroprotective [78].
Given the evidence for neuroinflammation in PD, agents with anti-inflammatory effects have been investigated for their neuroprotective potential. Many of these anti-inflammatory drugs are already in common use for other indications. Non-steroidal anti-inflammatory agents (NSAIDs) reduce dopaminergic cell death in animal and culture PD models (reviewed in [64]), and epidemiological studies have suggested that certain NSAIDs and statin drugs may reduce PD risk [79–85]. Minocycline is another agent with anti-inflammatory capabilities that is currently being investigated in human trials [86].
Excitotoxicity
Excitotoxicity has been implicated as a pathogenic mechanism in several neurodegenerative disorders, including PD. Glutamate is the primary excitatory transmitter in the mammalian central nervous system and a primary driver of the excitotoxic process. Dopaminergic neurons in the SN have high levels of glutamate receptors and receive glutamatergic innervation from the subthalamic nucleus and cortex. Excessive NMDA receptor activation by glutamate could increase intracellular calcium levels that then activate cell death pathways [87]. Calcium influx produced by excessive glutamate receptor activation can also promote peroxynitrite production through the activation of nitric oxide synthase [88]. Levels of 3-nitrotyrosine, a marker of peroxynitrite formation, are increased in postmortem SN from PD patients [89]. NMDA receptor antagonists protect against dopaminergic cell loss in MPTP models [90, 91], but a major limitation to clinical application is the low potency and poor tolerability of currently available agents. Riluzole has glutamate antagonist properties, but a small clinical trial failed to show any neuroprotective effect [92]. Neuroprotective effects have also been attributed to amantadine, which has modest NMDA antagonist properties as well as other actions [93]. Human dopaminergic neurons have NMDA receptors with an unusual subunit composition characterized by abundant expression of the NR2D subunits, which have physiological and pharmacological properties distinct from the receptor types found in many other regions of the brain [94]. The most promising opportunity for the use of glutamate antagonists as neuroprotective agents for PD may lie in the development of NMDA antagonists with selectivity for specific channel subunits [95].
Another approach to modifying excitotoxicity is to address the downstream processes, which include intracellular calcium and related signaling systems. An important recent development is the discovery of the calcium-dependent pacemaking properties of nigral dopaminergic neurons, which expose the cells to high levels of intracellular calcium in the course of normal physiological activity (reviewed in [96]). This reliance on calcium channels results in a large energetic burden for these neurons, as intracellular calcium has to be sequestered regularly into the endoplasmic reticulum and mitochondria to prevent the activation of cell death pathways. The consequence is nigral cell aging through the production of free radicals and ROS. Blockade of L-type Cav1.3 calcium channels leads to a reversion of the cells to a “juvenile” Na+-dependent form of pacemaking and resistance to MPTP and 6-OHDA toxicity in mice [97]. Epidemiological studies have suggested that patients treated with dihydropyridines for hypertension have a lower incidence of PD [98]. The dihydropyridine isradipine is currently being investigated as a potential neuroprotective agent.
Apoptosis
Apoptosis, or programmed cell death, is a mechanism that has been demonstrated to participate in neural development and to play a role in some forms of neural injury. There has been controversy as to whether apoptosis is directly involved in PD. Several pathological studies have revealed signs of both apoptotic and autophagic cell death in the SN of PD brains [99–103], although the extent is limited, perhaps because of the slow process of cell death which underlies PD. Apoptotic cell death has also been observed in animal PD models (reviewed in [104–106]). Alterations in cell death pathways are unlikely to be the primary cause for PD, but both apoptotic and autophagic cell death pathways are hypothesized to become activated in PD through oxidative stress, protein aggregation, excitotoxicity, or inflammatory processes. Activation of these cell death pathways most likely represents end-stage processes in PD neurodegeneration. Therefore, inhibitors of these cell death pathways have been proposed as potential neuroprotective agents regardless of the initial cause for neurodegeneration in PD. Two different compounds that inhibit apoptotic signaling have been recently tested in human PD trials (see below; [107, 108]).
Loss of trophic factors
The loss of neurotrophic factors has been implicated as a potential contributor to cell death observed in PD. The neurotrophic factors brain-derived neurotrophic factor (BDNF), glial-derived neurotrophic factor (GDNF), and nerve growth factor (NGF) have all been demonstrated to be reduced in the nigra in PD [109–111]. As a result, treatment with growth factors has been proposed as a potential neuroprotective therapy in PD. Indeed, the potent ability of these agents to stimulate growth and arborization of dopaminergic neurons suggests that they may be useful neuroprotective treatments, even if deficiency of the factors is not the primary cause of the disease process. GDNF and a related growth factor, neurturin, are both protective against neurodegeneration in animal models [112–116]. GDNF has been evaluated in human trials [117–120], and neurturin is currently being investigated in a phase II trial (see below).
Current state of neuroprotection in clinical studies
While many drugs have shown promise in animal PD models, this promise has not yet translated into therapies that are clearly neuroprotective in human PD. A recent practice parameter from the American Academy of Neurology concluded that “no treatment has been shown to be neuroprotective” [121]. The barriers to establishing a neuroprotective therapy stem from the complexity of the disease process as well as the limits of the clinical tools available to monitor the progression of the disease and to observe the effects of an intervention. Recent studies seeking a neuroprotective effect illustrate the importance of both of these factors. Readers are referred to the recent review on neuroprotection clinical trials [122].
Levo-dopa as a neuroprotective agent: the ELLDOPA Trial
Levodopa (L-dopa) is one of the oldest and most effective therapies for the symptoms of PD. Although widely used, until recently there was little data on the impact of L-dopa therapy on the long-term progression of PD. Because dopamine catabolism produces free radicals, there has been concern that treatment of PD patients with the precursor L-dopa could potentially promote neurodegeneration in PD [123, 124]. On the other hand, a variety of preclinical data has suggested a neuroprotective effect [125, 126].
The Earlier versus Later L-Dopa (ELLDOPA) trial was a placebo-controlled, double-blind study designed to evaluate whether treatment of PD patients with L-dopa modified disease progression as compared to those treated with placebo [127]. 361 patients were treated with placebo or L-dopa at 150 mg/day, 300 mg/day, or 600 mg/day for 40 weeks, followed by a two week washout period. The primary outcome measure was change in UPDRS score from baseline. Patients on L-dopa, particularly those on the highest dose, showed a smaller change in UPDRS score from baseline than those on placebo. This result suggests that L-dopa does not have a deleterious effect, but instead is neuroprotective. This interpretation has been controversial, as the finding could be explained by the possibility that the effects of L-dopa outlasted the two-week washout period. In addition, a secondary outcome measure of the trial, β-CIT neuroimaging, suggested the opposite conclusion, with patients on the highest dose of L-dopa showing the greatest decline in the uptake of β-CIT, a dopamine transporter ligand.
The ELLDOPA trial illustrates some of the most confounding issues in studies of PD neuroprotection. The potent effect of many dopaminergic agents on the symptoms of the disease can make assessment of the rate of progression difficult. Neuroimaging techniques are a more direct measure of the physiology of brain dopamine systems, but the reliability of these techniques as measures for PD progression remains uncertain [128, 129]. At this time, it is not clear whether L-dopa is neuroprotective, but fears of its potential toxicity have been somewhat allayed by the ELLDOPA trial.
Dopamine receptor agonists
Dopamine receptor agonists have been hypothesized as potentially neuroprotective by acting at D2 autoreceptors found on dopaminergic SN terminals to suppress dopamine release and thus reduce oxidative stress. Indeed, in vitro and animal studies have shown that dopamine receptor agonists can reduce dopaminergic cell death [130–137]. Certain agonists, such as pramipexole, may also act as direct antioxidants because of their hydroxylated benzyl ring structure [130, 138].
Two large-scale clinical trials have attempted to assess the neuroprotective properties of dopamine agonist drugs using neuroimaging strategies. The CALM-PD trial compared pramipexole to L-dopa treatment in patients with early PD [139]. In this study all the patients received an active treatment: 301 patients were randomized to pramipexole or L-dopa. In a subset of 82 patients, the uptake of radiolabeled β-CIT was assessed. Patients treated with pramipexole did show less of a decline in β-CIT uptake compared to those treated with L-dopa alone. A similar result was obtained in the REAL-PET trial, which compared patients with early PD treated with ropinirole to those treated with L-dopa [140]. 162 patients had fluorodopa (18F-dopa) PET imaging to assess 18F-dopa uptake in the putamen at four weeks and at 24 months after initiating drug treatment. Patients on ropinirole showed less of a decrease in putaminal 18F-dopa uptake. While both of these studies suggest a neuroprotective effect of dopamine agonists, there are several important limitations, including lack of a placebo control and lack of an extended washout of the medications. In addition, clinical outcome measures revealed either no difference or even less of an improvement in UPDRS in patients treated with the dopamine agonist as compared to L-dopa. The most substantial limitation of these studies is the difficulty of establishing that the neuroimaging measures reflect long-term neuroprotection of dopamine systems, and not short-term pharmacological modification of the uptake of the radiotracers [128, 129].
Antioxidant therapies
Given the role of oxidative stress in PD pathogenesis, several agents with antioxidant properties have been studied in clinical trials, including selegiline, vitamin E, and rasagiline. Selegiline reduces dopamine oxidation by inhibiting monoamine oxidase B (MAO-B) and is the antioxidant drug most studied in clinical trials. The DATATOP trial was the largest clinical trial to investigate the neuroprotective potential of selegiline, along with vitamin E, in patients with early PD [141, 142]. 800 patients were randomized to placebo, vitamin E, selegiline, or the combination of vitamin E and selegiline, and evaluated every three months up to a maximum of 24 months. The primary outcome was the time to clinical decision to treat with L-dopa. Selegiline did significantly delay the time of onset of L-dopa treatment. Those treated with vitamin E showed no difference compared to placebo-treated patients, and vitamin E did not appear to add any benefit to selegiline’s effects. While this study suggests that selegiline may delay disease progression, the unanticipated confounder was that selegiline itself likely has mild symptomatic effects that improve motor symptoms in PD.
Rasagiline is a newer MAO-B inhibitor that is more potent than selegiline and has metabolites with potential antioxidant properties. It has been studied using a delayed-start clinical trial design intended to reduce the confounding effect of symptomatic efficacy (Fig. 1). The TEMPO study enrolled 404 patients with early PD who were treated with placebo or rasagiline for six months and then all were placed on rasagiline for another six months [143]. The primary outcome was change in UPDRS score from baseline at 12 months. Those treated initially with rasagiline had a smaller increase in UPDRS scores compared to those first started on placebo. Because all patients were on rasagiline at the end of the study, it is assumed that symptomatic effects of rasagiline were similar between the two groups. This study does suggest that early treatment confers a long-lasting improvement, but the overall duration of this study was relatively short, and the group sizes were modest. A larger study (ADAGIO) to verify these initial results in a larger sample and over a longer time course is currently ongoing.
Figure 1.
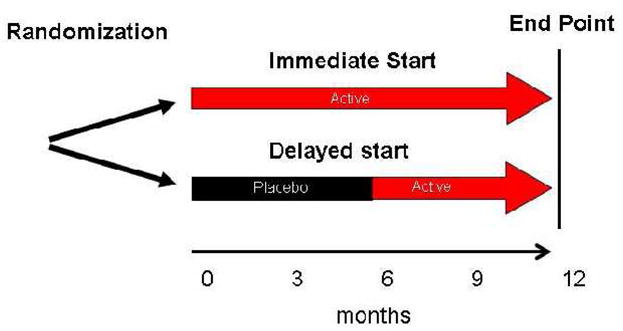
Schematic description of the delayed-start design. Patients are randomized to either immediate or delayed initiation of the therapeutic agent. Patients in the immediate group start active therapy at the onset of the study and stay on active therapy for the duration of the trial. Those randomized to delayed initiation take placebo initially for a predetermined time period and then are switched to active therapy.
Coenzyme Q10 (CoQ10) is a cofactor in the electron transport chain in mitochondria and has been shown to reduce dopaminergic neurodegeneration in mouse PD models [144]. A small pilot study of 80 patients compared patients treated with CoQ10 at 300mg, 600mg, or 1200mg to patients treated with placebo [145]. Patients were followed for 16 months or until they required L-dopa treatment. The primary outcome was change in total UPDRS compared to baseline. There was a trend towards reduction in UPDRS score change in patients treated with CoQ10 compared to placebo (p=0.09), and secondary analysis suggested that most of the benefit was in the group treated with 1200mg of CoQ10. More recently, CoQ10 has been examined using a “futility study” [146]. This design employs small group sizes and is intended to identify treatments which clearly do not have a significant effect on disease progression and, therefore, should be excluded from further study [147–149]. In this paradigm, CoQ10 did not meet the criteria for futility, and a larger, long-term study comparing high doses of CoQ10 to placebo has recently been initiated.
Creatine promotes mitochondrial ATP production and has been shown to be neuroprotective in animal models [150]. A pilot study compared 60 patients with early PD treated with either creatine or placebo for two years [151]. No difference was seen in UPDRS score or in β-CIT uptake between the control and creatine groups. Subanalysis of the individual UPDRS subscales showed a difference in the “mentation, behavior, and mood” section. Creatine has also been examined in the futility study paradigm and could not be rejected as futile [86]. A large phase III trial examining creatine’s effect on disease progression in a diverse population of patients with different stages of PD is in progress.
While none of these clinical studies have convincingly shown that these antioxidant therapies slow disease progression, several of these trials indicate that selegiline, rasagiline, and CoQ10 may be promising. In addition, all of the antioxidant approaches have a good safety record, a desirable property in a neuroprotective therapy that may need to be administered to a large number of patients for many years.
Anti-apoptotic agents
Several anti-apoptotic agents have been examined in controlled clinical trials. The propargylamine TCH346 is an anti-apoptotic factor that inhibits the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which can initiate apoptosis. Although TCH346 was shown in both 6-OHDA and MPTP animal models to reduce dopaminergic cell loss [152, 153], a double-blind, randomized trial involving 301 patients over 12 to 18 months failed to show a significant difference in clinical outcome [107]. CEP-1347, an inhibitor of mixed lineage kinases that can activate the c-Jun N-terminal kinase (JNK) pathway involved in cell death, is another anti-apoptotic agent that showed promise in preclinical studies [154–156]. The PRECEPT trial involved 806 patients with early PD randomized to placebo or CEP-1347, and the primary outcome was time to need for dopaminergic therapy [108]. This study was terminated early when an interim analysis revealed futility of the experimental treatment.
The failure of these two anti-apoptotic drugs has raised a number of important questions. Both were very effective in commonly used neurotoxin-based animal models of PD, raising the question of the predictive validity of these models. These failures have also raised the questions of whether anti-apoptotic strategies are an effective approach when applied at the relatively late stage of the disease when symptoms are clinically evident and whether anti-apoptotic therapies would need to be combined with trophic support [157].
Trophic factors
Several neurotrophic factors have been examined in human clinical trials. Glial-derived neurotrophic factor (GDNF) is a potent neurotrophic factor that supports the survival of dopaminergic nigral neurons and has been shown to be neuroprotective in animal models for PD [112, 113, 158]. So far, it has been delivered primarily by direct infusion of the protein into the brain. A small, open-label trial suggested effectiveness [117], but a larger controlled study involving 34 PD patients was halted because of lack of efficacy [118]. In addition, serious, device-related adverse events occurred in three patients, and several patients developed serum antibodies to GDNF. After the onset of the human trial, a toxicology study revealed that some monkeys infused with high doses of GDNF showed cerebellar cell loss, raising further concerns regarding the safety of GDNF infusions [118].
An alternative to direct infusion of trophic factors is delivery of these agents by gene transfer approaches [159]. Recent gene therapy efforts have used neurturin, a neurotrophic factor related to GDNF that also promotes dopaminergic neuronal survival in culture [160, 161]. Delivery of an adeno-associated viral (AAV-2) vector containing neurturin (CERE-120) protected against dopaminergic cell loss and improved motor function in MPTP-treated monkeys [116] and in 6-OHDA-treated rats [114, 115]. CERE-120 has been tested for its safety in an open-label clinical study involving 12 patients with advanced PD [162]. The treatment was well tolerated, and there was evidence for a substantial clinical effect: off-medication UPDRS motor subscore was significantly reduced and mean on time was increased at one year following injection. While these findings are promising, data from open-label trials must be interpreted cautiously. A larger double-blind study is in progress.
Neuroimmunophilins, intracellular receptor proteins that bind immunosuppressive drugs like cyclosporine and FK506, are highly expressed in the brain and can promote neuronal growth independently of their immunosuppressive effects [163–165]. Neuroimmunophilin ligands have shown some evidence of neuroprotection in 6-OHDA and MPTP animal models [166–168], although neurodegeneration has not been reduced in all PD animal models [169, 170]. The mechanism of action is unknown but may involve the induction of neurotrophic factors [171] or the antioxidant glutathione [172, 173]. A recent phase II trial of the neuroimmunophilin ligand GPI-1485 showed no significant difference of motor symptoms in patients with mild to moderate PD treated with GPI-1485 for six months compared to those treated with placebo [174]. However, there was a non-significant trend of increased β-CIT uptake in those patients taking the highest dose of GPI-1485 [174].
Lessons from Clinical Trials of Neuroprotective Therapy
Despite the promise of several different neuroprotective strategies, none of the human clinical trials completed so far have shown any therapy to have clear disease-modifying effects. These human studies have not focused only on a single pathogenic mechanism, but a wide range of potential pathogenic mechanisms have been explored. Some of the agents studied do appear to have potential neuroprotective effects, and our current trial designs may be preventing us from adequately detecting neuroprotection in humans. What are some of the limitations of testing for neuroprotection that may have lead to these “failures”?
Limitation of clinical measures of disease
One major issue in determining whether a therapy is neuroprotective in human PD is how to test neuroprotection in a living patient. Traditionally, clinical measures based on neurological exam have been used to assess progression (or lack of progression) of PD. The UPDRS scale involves clinical examination of motor function combined with scales rating patient’s subjective view of function in daily activities. There are several limitations to this scale [175, 176]. First, available symptomatic treatments have a large effect on the UPDRS scale, which may obscure evidence of neuroprotection. The scale is also heavily weighted towards motor dysfunction, particularly tremor-related symptoms. Much of the disability associated with PD is not considered in the scale, such as that related to autonomic dysfunction. A new version of the UPDRS is currently under development, which may address some of these limitations [177].
To overcome the potentially confounding symptomatic effects of a treatment, many trials have incorporated a washout period before assessing the primary outcome measure. However, these symptomatic drug effects may be longstanding and outlast the washout period [178]. An alternative approach has been to use a delayed-start design where one group of patients is started on the therapy several months before the comparative group (Fig. 1) [178]. This approach is based on the assumption that the symptomatic effects would be similar across the groups at the end of the study. However, there are still some potential problems with this approach [143]. For example, longer treatment may result in increased sensitivity to drug. The delayed-start paradigm may also result in different rates of drop out between groups, as patients randomized to placebo start are more likely to drop out. Many investigators regard the delayed-start design as the best available approach for evaluating neuroprotection.
Limitations of neuroimaging strategies
In laboratory studies the “gold standard” for evaluating neuroprotection is direct counts of surviving dopamine neurons, an approach not currently feasible in living patients. SPECT and PET imaging have been used as default measures of dopamine cell numbers, yet these imaging techniques, which are based on dopamine neurochemistry, have their limitations [128, 129]. β-CIT SPECT imaging measures the binding of the radioactive tracer to the dopamine transporter on dopaminergic terminals in the striatum, while [18F]fluorodopa PET imaging measures the conversion of the radioactive tracer into fluorodopamine within the terminals. Although they rely on different aspects of dopamine neurochemistry, a concern with both approaches is that the underlying chemistry may be altered by the pharmacological effects of the treatments under investigation such that imaging changes may not necessarily reflect changes in dopaminergic neuron counts [128]. None of these imaging techniques have yet been validated as appropriate surrogate measures of neuroprotection through correlation with neuropathology and/or clinical symptoms [129, 179, 180], and considerably more long-term data with these methods is required.
Limitations of Animal Models
The failure of so many potential neuroprotective therapies could also be explained by the limitations of animal models. Currently, there is no one animal model for PD that mimics the full pathology and clinical symptomology of the illness. Traditionally, preclinical studies have focused on toxin-based models using 6-OHDA or MPTP. Both of these models show degeneration of SN neurons, but the time course and pathological features of these models are different from human disease [122, 181]. Perhaps more importantly, there is no substantial evidence for a role of either of these toxins in human PD. With the discovery of α-syn, parkin, and other proteins identified through genetic PD studies, genetic-based models have been developed as alternatives to toxin-based models. These models incorporate some additional features of the disease, but still fall short of an authentic re-creation. For example, transgenic mice that either express mutant α-syn (A30P or A53T) or overexpress wildtype α-syn show variable motor deficits, α-syn inclusions, and dopamine terminal loss, but none of these transgenic animals show actual loss of dopaminergic neurons [182, 183].
The predictive power of animal models of PD will be confirmed only if there is some success in demonstrating neuroprotection in humans. In the meantime, most in the field rely on examining effectiveness of potential treatments in several different animal models, with the hope that treatments exhibiting a broad effect in these diverse models are the ones mostly likely to exhibit effectiveness against human disease.
Heterogeneity and time course of the disease
The discovery of genes that cause PD has emphasized the diversity of the disorder: not only does it involve anatomical sites outside the dopaminergic system, but it likely also has a variety of interrelated causes. Most of the single-gene mutations discovered so far are rare and unlikely to play a significant role in broad-based clinical trials; however, parkin mutations are relatively frequent in young-onset patients [48, 184, 185], and LRRK2 mutations may account for 2% of a general clinical population of PD [186–189]. A more problematic issue is that there are likely a number of genetic risk factors for PD, which could alter the response to putative neuroprotective strategies. Therefore, defining subgroups of PD may be essential in order to establish neuroprotective efficacy. In addition, as PD involves many different pathogenic mechanisms, several agents may need to be combined to block multiple pathways in order to achieve neuroprotection.
For a potential neuroprotective agent to be most effective, patients need to be treated early, before much cell loss has occurred. By the time most patients develop the typical clinical symptoms of PD, it is estimated that at least 60% of nigral dopamine neurons have degenerated [190–192]. They may already have a variety of non-motor symptoms, including sleep disturbance and autonomic dysfunction [193]. Such earlier symptoms are guiding the ongoing search for methods to detect “pre-symptomatic” PD, ranging from simple tests of olfaction to sophisticated neuroimaging studies.
The future of neuroprotection
In 2003, the NIH-appointed Committee to Identify Neuroprotective Agents in Parkinson’s (CINAPS) published an assessment of potential neuroprotective compounds and prioritized 12 compounds to be studied further in clinical trials [194]. Since then, the list of potential therapies has grown longer, but a convincing success in human trials is still awaited. Of those strategies still under active study, several of the most promising are listed below. A common theme among several of these strategies is that interest is driven largely by the results of human genetic, pathological, and epidemiological studies – perhaps reflecting a concern in the field that an animal model with useful predictive properties in terms of neuroprotection has not yet been identified. The wealth of information regarding PD pathogenesis from genetic studies is leading to novel approaches to neuroprotection based on the biology of α-syn, LRRK2, and others. While much more research is needed before translating such theoretical therapies into clinical trials, these approaches may lead to therapies that protect not only against dopaminergic cell loss but also against the loss of other neuronal populations at risk in PD and related disorders.
Adenosine receptor antagonists
Epidemiological studies have indicated that caffeine may reduce the incidence of PD, at least in men [195, 196]. As caffeine mediates its action by antagonizing adenosine receptors, this finding has led to interest in evaluating adenosine receptor antagonists as potential neuroprotective agents (reviewed in [197]). In the striatum, the A2A receptor can heterodimerize with the D2 receptor to inhibit dopamine signaling [198, 199], while inhibition of the A2A receptor can promote dopamine function. Two small clinical trials of the A2a antagonist istradefylline (KW-6002) has demonstrated potential symptomatic effects in advanced PD [200, 201]. More recent research has suggested that A2A antagonists not only improve symptomatic function in PD but may also be neuroprotective. Caffeine and istradefylline are both neuroprotective in the MPTP model [202, 203], and caffeine (and related A2a antagonists) has been identified by CINAPS as a priority agent to be evaluated for neuroprotection in clinical trials [194]. A potential advantage of A2a antagonists is that they have also been shown to be neuroprotective in non-dopaminergic brain areas in animal models [204–206] so that they may protect against PD neurodegeneration found in regions besides the SN.
Anti-inflammatory agents
The role of inflammation in PD has become more recognized recently. Activation of microglia, increased cytokine production, and increased complement protein levels have been demonstrated in PD (reviewed in [63–65]). As a means to slow disease progression, anti-inflammatory agents, including NSAIDs and minocycline, have been pursued as potential disease-modifying treatments for PD. Several studies in culture and in animal models have shown that certain NSAIDs, such as aspirin, have neuroprotective qualities, although there is conflicting data regarding which NSAIDs, what dosing, and what timing provides the best neuroprotection (reviewed in [64]). Epidemiological studies examining the association of regular NSAID use with the risk of PD have provided conflicting results. An initial study by Chen et al. showed that NSAID use lowers the risk of PD by 45% [81], and a followup study by the same group showed that only ibuprofen had this neuroprotective effect [80]. Other epidemiological studies examining this association have shown nonsignificant trends or have shown this association only in men [79, 82, 83]. At this time, it is unclear that any of the currently available NSAIDs have genuine neuroprotective properties in PD, but the more general strategy of targeting the mechanisms of neuroinflammation seems very promising.
An example of an alternative approach to targeting neuroinflammation may be the use of statins (3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors). In addition to lowering cholesterol, these drugs have anti-inflammatory effects, including reduction of TNFα, nitric oxide, and superoxide production by microglia [207]. Statins may also act to scavenge free radicals [208]. Simvastatin has been shown to reduce dopamine loss in MPTP animal models [207]. Recent epidemiological studies showed that statin use, particularly simvastatin, is associated with reduced PD incidence [84, 85]. A limitation of these studies is that the use of statins was not randomized. Other studies have suggested low LDL cholesterol levels increased PD risk [209, 210], so that the increased use of statins among controls may just reflect high LDL levels that would be protective against PD. These issues need to be explored in a prospective, randomized study. Minocycline, a second generation tetracycline long used as an antimicrobial agent, has anti-inflammatory effects independent of its antimicrobial activity.
Minocycline blocks microglial activation and may also have anti-apoptotic activity in culture [211, 212]. It protects against dopaminergic cell loss in both the MPTP and 6-OHDA animal models [213–215]. A recent futility study showed that minocycline was well tolerated and could not be rejected as futile, setting the stage for larger phase III trials [86].
Other antioxidants
Epidemiological studies have pointed to uric acid as a potential neuroprotective agent in PD. Uric acid acts as an antioxidant by scavenging reactive oxygen and nitrogen species [216]. Studies have shown a decreased incidence of PD among subjects with high serum urate levels [217–219] and among subjects with gout [220]. In patients with early PD, higher plasma urate levels correlate with slower disease progression [221]. Uric acid can reduce dopaminergic cell death in response to rotenone and homocysteine in culture [222]. A recent study showed that subjects on diets that promote high urate levels have a reduced risk of developing PD [223]. Such a urate-rich diet could serve as a neuroprotective therapy in PD. However, the potential benefits of a urate-rich diet have to be weighed against the risk of developing gout and cardiovascular disease. A large-scale clinical trial of the effectiveness of elevating urate in patients with PD is in the planning stages.
Alpha-synuclein-directed therapies
Although its mechanism for inducing neurotoxicity is not well understood, α-syn appears to be an important mediator of toxicity in PD. Disruption of α-syn aggregation has been the focus of research to develop novel therapies against PD. Alpha-synuclein aggregation can be reduced at several levels: 1) by reducing α-syn protein production; 2) by increasing α-syn clearance; 3) by preventing or reducing chemical modifications that can promote aggregated species; or 4) by directly interfering with aggregation. Potential methods to reduce α-syn protein production include small-molecule modifiers of transcription and RNAi-based methods to knock down translation. Increased α-syn clearance could become enhanced by activation of proteosomal or lysosomal pathways. Augmentation of parkin or UCH-L1 activity could promote the clearance of α-syn and other aggregated proteins. Indeed, parkin overexpression can rescue cells from α-syn pathology in animal models [224–226]. Promotion of chaperone function could also promote α-syn clearance. The chaperone protein Hsp70 can reduce insoluble α-syn aggregates in vitro and in vivo [58], and the compound geldanamycin can reduce α-syn aggregation in vitro by increasing Hsp70 levels [227]. Activation of lysosomal degradation could also induce α-syn clearance; the lysosomal enzyme cathepsin D reduces α-syn aggregation and toxicity in culture and in animal models [228]. In addition, vaccine-based therapies have been pursued as potential strategies for increasing α-syn clearance. Alpha-synuclein transgenic mice vaccinated against α-syn showed decreased α-syn accumulation secondary to increased degradation via lysosomal pathways [229]. As oxidative modification and phosphorylation of α-syn both promote aggregation of α-syn [35, 230, 231], antioxidant therapies and kinase inhibitors could also help reduce α-syn aggregation and toxicity.
Finally, direct blockers of α-syn aggregation could be developed into therapeutic strategies for PD. Peptide-based inhibitors have been developed that can block α-syn self-association and aggregation [232–234]. The chaperone-like proteins, β-syn and 14-3-3 proteins, have also been demonstrated to reduce α-syn aggregation [235–237], and increasing their expression and/or function could serve as a basis for therapeutic intervention. All of these strategies are at a relatively early stage of development and await further study before proceeding to human intervention trials.
Kinase inhibitors
The most common genetic cause of PD to date is mutation in the gene LRRK2, which causes about 2% of all cases of PD [186–189] and up to 40% in historically isolated populations [238–241]. The LRRK2 gene codes for a large protein, also known as dardarin, which contains a serine/threonine kinase domain and a GTPase domain. The native function of this protein is currently poorly understood. The most common pathogenic mutation of LRRK2 is associated with increased kinase activity [242–244]. Evidence that pathogenic mutations increase this activity makes the kinase activity of LRRK2 an important target for neuroprotective therapy. Kinases are generally good targets for small molecule therapies, and indeed certain therapies in other diseases are based on inhibition of kinase activity. At this time, however, the endogenous substrates for LRRK2 are unknown, making it difficult to devise the development of LRRK2 kinase inhibitors. As the function of LRRK2 is further characterized, a clearer therapeutic strategy based on LRRK2 biology should become more apparent.
Trophic Factors
Trophic factor approaches are one of the older strategies for neuroprotective therapy in PD, but also an approach with substantial promise for the future (see above). A strength of this approach is that the biology of the factors themselves is well known, and it does not rely on a detailed understanding of the mechanisms of cell death in PD. Trophic factors may enhance dopaminergic survival, regardless of the mechanism of cell death. On the other hand, at least as presently conceived, they directly target the dopaminergic deficit and not the non-dopaminergic aspects of the disease. The studies of gene therapy delivery of neurturin are in an advanced stage, and if successful will not only provide an immediate treatment for many patients with PD, but may also open to the door to a wider range of gene therapy approaches.
Conclusions
Neuroprotection in PD remains an important but elusive goal. A successful neuroprotective treatment could transform PD from a relentlessly progressive and disabling disease to a problem that can be managed with only a modest effect on quality of life. Current barriers include a lack of knowledge of the basic mechanisms of PD, and deficiencies in the methodology used to study disease progression. Overall, however, the activity aimed at understanding and treating PD has grown exponentially and should ultimately result in better therapies for PD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 2.Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, Jenner P, Halliwell B. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem. 1997;69:1196–1203. doi: 10.1046/j.1471-4159.1997.69031196.x. [DOI] [PubMed] [Google Scholar]
- 3.Dexter DT, Holley AE, Flitter WD, Slater TF, Wells FR, Daniel SE, Lees AJ, Jenner P, Marsden CD. Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: an HPLC and ESR study. Mov Disord. 1994;9:92–97. doi: 10.1002/mds.870090115. [DOI] [PubMed] [Google Scholar]
- 4.Dexter DT, Sian J, Rose S, Hindmarsh JG, Mann VM, Cooper JM, Wells FR, Daniel SE, Lees AJ, Schapira AH, et al. Indices of oxidative stress and mitochondrial function in individuals with incidental Lewy body disease. Ann Neurol. 1994;35:38–44. doi: 10.1002/ana.410350107. [DOI] [PubMed] [Google Scholar]
- 5.Hastings TG, Lewis DA, Zigmond MJ. Reactive dopamine metabolites and neurotoxicity: implications for Parkinson’s disease. Adv Exp Med Biol. 1996;387:97–106. doi: 10.1007/978-1-4757-9480-9_13. [DOI] [PubMed] [Google Scholar]
- 6.Sulzer D, Zecca L. Intraneuronal dopamine-quinone synthesis: a review. Neurotox Res. 2000;1:181–195. doi: 10.1007/BF03033289. [DOI] [PubMed] [Google Scholar]
- 7.Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet. 1989;1:1269. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- 8.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 9.Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 10.Langston JW, Forno LS, Rebert CS, Irwin I. Selective nigral toxicity after systemic administration of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyrine (MPTP) in the squirrel monkey. Brain Res. 1984;292:390–394. doi: 10.1016/0006-8993(84)90777-7. [DOI] [PubMed] [Google Scholar]
- 11.Wooten GF, Currie LJ, Bennett JP, Harrison MB, Trugman JM, Parker WD., Jr Maternal inheritance in Parkinson’s disease. Ann Neurol. 1997;41:265–268. doi: 10.1002/ana.410410218. [DOI] [PubMed] [Google Scholar]
- 12.Schapira AH. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008;7:97–109. doi: 10.1016/S1474-4422(07)70327-7. [DOI] [PubMed] [Google Scholar]
- 13.Cantuti-Castelvetri I, Lin MT, Zheng K, Keller-McGandy CE, Betensky RA, Johns DR, Beal MF, Standaert DG, Simon DK. Somatic mitochondrial DNA mutations in single neurons and glia. Neurobiol Aging. 2005;26:1343–1355. doi: 10.1016/j.neurobiolaging.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 14.Dexter DT, Wells FR, Lees AJ, Agid F, Agid Y, Jenner P, Marsden CD. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J Neurochem. 1989;52:1830–1836. doi: 10.1111/j.1471-4159.1989.tb07264.x. [DOI] [PubMed] [Google Scholar]
- 15.Riederer P, Sofic E, Rausch WD, Schmidt B, Reynolds GP, Jellinger K, Youdim MB. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J Neurochem. 1989;52:515–520. doi: 10.1111/j.1471-4159.1989.tb09150.x. [DOI] [PubMed] [Google Scholar]
- 16.Perry TL, Yong VW. Idiopathic Parkinson’s disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neurosci Lett. 1986;67:269–274. doi: 10.1016/0304-3940(86)90320-4. [DOI] [PubMed] [Google Scholar]
- 17.Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, Jenner P, Marsden CD. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36:348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- 18.Sofic E, Lange KW, Jellinger K, Riederer P. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson’s disease. Neurosci Lett. 1992;142:128–130. doi: 10.1016/0304-3940(92)90355-b. [DOI] [PubMed] [Google Scholar]
- 19.Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 20.Kim RH, Smith PD, Aleyasin H, Hayley S, Mount MP, Pownall S, Wakeham A, You-Ten AJ, Kalia SK, Horne P, Westaway D, Lozano AM, Anisman H, Park DS, Mak TW. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc Natl Acad Sci U S A. 2005;102:5215–5220. doi: 10.1073/pnas.0501282102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 22.Yokota T, Sugawara K, Ito K, Takahashi R, Ariga H, Mizusawa H. Down regulation of DJ-1 enhances cell death by oxidative stress, ER stress, and proteasome inhibition. Biochem Biophys Res Commun. 2003;312:1342–1348. doi: 10.1016/j.bbrc.2003.11.056. [DOI] [PubMed] [Google Scholar]
- 23.Athanassiadou A, Voutsinas G, Psiouri L, Leroy E, Polymeropoulos MH, Ilias A, Maniatis GM, Papapetropoulos T. Genetic analysis of families with Parkinson disease that carry the Ala53Thr mutation in the gene encoding alpha-synuclein. Am J Hum Genet. 1999;65:555–558. doi: 10.1086/302486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 25.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 26.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 27.Irizarry MC, Growdon W, Gomez-Isla T, Newell K, George JM, Clayton DF, Hyman BT. Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson’s disease and cortical Lewy body disease contain alpha-synuclein immunoreactivity. J Neuropathol Exp Neurol. 1998;57:334–337. doi: 10.1097/00005072-199804000-00005. [DOI] [PubMed] [Google Scholar]
- 28.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 29.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 30.El-Agnaf OM, Jakes R, Curran MD, Wallace A. Effects of the mutations Ala30 to Pro and Ala53 to Thr on the physical and morphological properties of alpha-synuclein protein implicated in Parkinson’s disease. FEBS Lett. 1998;440:67–70. doi: 10.1016/s0014-5793(98)01419-7. [DOI] [PubMed] [Google Scholar]
- 31.Narhi L, Wood SJ, Steavenson S, Jiang Y, Wu GM, Anafi D, Kaufman SA, Martin F, Sitney K, Denis P, Louis JC, Wypych J, Biere AL, Citron M. Both familial Parkinson’s disease mutations accelerate alpha-synuclein aggregation. J Biol Chem. 1999;274:9843–9846. doi: 10.1074/jbc.274.14.9843. [DOI] [PubMed] [Google Scholar]
- 32.Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 33.Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 35.Souza JM, Giasson BI, Chen Q, Lee VM, Ischiropoulos H. Dityrosine cross-linking promotes formation of stable alpha -synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J Biol Chem. 2000;275:18344–18349. doi: 10.1074/jbc.M000206200. [DOI] [PubMed] [Google Scholar]
- 36.Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- 37.Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flower TR, Chesnokova LS, Froelich CA, Dixon C, Witt SN. Heat shock prevents alpha-synuclein-induced apoptosis in a yeast model of Parkinson’s disease. J Mol Biol. 2005;351:1081–1100. doi: 10.1016/j.jmb.2005.06.060. [DOI] [PubMed] [Google Scholar]
- 39.Saha AR, Ninkina NN, Hanger DP, Anderton BH, Davies AM, Buchman VL. Induction of neuronal death by alpha-synuclein. Eur J Neurosci. 2000;12:3073–3077. doi: 10.1046/j.1460-9568.2000.00210.x. [DOI] [PubMed] [Google Scholar]
- 40.Smith WW, Jiang H, Pei Z, Tanaka Y, Morita H, Sawa A, Dawson VL, Dawson TM, Ross CA. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Genet. 2005;14:3801–3811. doi: 10.1093/hmg/ddi396. [DOI] [PubMed] [Google Scholar]
- 41.Volles MJ, Lansbury PT., Jr Zeroing in on the pathogenic form of alpha-synuclein and its mechanism of neurotoxicity in Parkinson’s disease. Biochemistry. 2003;42:7871–7878. doi: 10.1021/bi030086j. [DOI] [PubMed] [Google Scholar]
- 42.Cookson MR, van der Brug M. Cell systems and the toxic mechanism(s) of alpha-synuclein. Exp Neurol. 2008;209:5–11. doi: 10.1016/j.expneurol.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet. 2006;15:3012–3023. doi: 10.1093/hmg/ddl243. [DOI] [PubMed] [Google Scholar]
- 44.Yacoubian TA, Cantuti-Castelvetri I, Bouzou B, Asteris G, McLean PJ, Hyman BT, Standaert DG. Transcriptional dysregulation in a transgenic model of Parkinson disease. Neurobiol Dis. 2008;29:515–528. doi: 10.1016/j.nbd.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benner EJ, Banerjee R, Reynolds AD, Sherman S, Pisarev VM, Tsiperson V, Nemachek C, Ciborowski P, Przedborski S, Mosley RL, Gendelman HE. Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS ONE. 2008;3:e1376. doi: 10.1371/journal.pone.0001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Theodore S, McLean PJ, Standaert DG. Microglial activation following targeted over-expression of human Alpha-Synuclein in the mouse Substantia Nigra. Soc Neurosci Abstract Viewer/Itinerary Planner Program #50.3/M16. 2007 [Google Scholar]
- 47.Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH. The ubiquitin pathway in Parkinson’s disease. Nature. 1998;395:451–452. doi: 10.1038/26652. [DOI] [PubMed] [Google Scholar]
- 48.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 49.Imai Y, Soda M, Takahashi R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem. 2000;275:35661–35664. doi: 10.1074/jbc.C000447200. [DOI] [PubMed] [Google Scholar]
- 50.Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM. Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci U S A. 2000;97:13354–13359. doi: 10.1073/pnas.240347797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med. 2001;7:1144–1150. doi: 10.1038/nm1001-1144. [DOI] [PubMed] [Google Scholar]
- 53.Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson’s disease. Science. 2001;293:263–269. doi: 10.1126/science.1060627. [DOI] [PubMed] [Google Scholar]
- 54.Mori H, Kondo T, Yokochi M, Matsumine H, Nakagawa-Hattori Y, Miyake T, Suda K, Mizuno Y. Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology. 1998;51:890–892. doi: 10.1212/wnl.51.3.890. [DOI] [PubMed] [Google Scholar]
- 55.Takahashi H, Ohama E, Suzuki S, Horikawa Y, Ishikawa A, Morita T, Tsuji S, Ikuta F. Familial juvenile parkinsonism: clinical and pathologic study in a family. Neurology. 1994;44:437–441. doi: 10.1212/wnl.44.3_part_1.437. [DOI] [PubMed] [Google Scholar]
- 56.Hayashi S, Wakabayashi K, Ishikawa A, Nagai H, Saito M, Maruyama M, Takahashi T, Ozawa T, Tsuji S, Takahashi H. An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov Disord. 2000;15:884–888. doi: 10.1002/1531-8257(200009)15:5<884::aid-mds1019>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 57.Farrer M, Chan P, Chen R, Tan L, Lincoln S, Hernandez D, Forno L, Gwinn-Hardy K, Petrucelli L, Hussey J, Singleton A, Tanner C, Hardy J, Langston JW. Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol. 2001;50:293–300. doi: 10.1002/ana.1132. [DOI] [PubMed] [Google Scholar]
- 58.Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ. Hsp70 Reduces alpha-Synuclein Aggregation and Toxicity. J Biol Chem. 2004;279:25497–25502. doi: 10.1074/jbc.M400255200. [DOI] [PubMed] [Google Scholar]
- 59.Moore DJ, West AB, Dikeman DA, Dawson VL, Dawson TM. Parkin mediates the degradation-independent ubiquitination of Hsp70. J Neurochem. 2008;105:1806–1819. doi: 10.1111/j.1471-4159.2008.05261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McNaught KS, Mytilineou C, Jnobaptiste R, Yabut J, Shashidharan P, Jennert P, Olanow CW. Impairment of the ubiquitin-proteasome system causes dopaminergic cell death and inclusion body formation in ventral mesencephalic cultures. J Neurochem. 2002;81:301–306. doi: 10.1046/j.1471-4159.2002.00821.x. [DOI] [PubMed] [Google Scholar]
- 61.Chung KK, Dawson VL, Dawson TM. The role of the ubiquitin-proteasomal pathway in Parkinson’s disease and other neurodegenerative disorders. Trends Neurosci. 2001;24:S7–14. doi: 10.1016/s0166-2236(00)01998-6. [DOI] [PubMed] [Google Scholar]
- 62.Nishikawa K, Li H, Kawamura R, Osaka H, Wang YL, Hara Y, Hirokawa T, Manago Y, Amano T, Noda M, Aoki S, Wada K. Alterations of structure and hydrolase activity of parkinsonism-associated human ubiquitin carboxyl-terminal hydrolase L1 variants. Biochem Biophys Res Commun. 2003;304:176–183. doi: 10.1016/s0006-291x(03)00555-2. [DOI] [PubMed] [Google Scholar]
- 63.Tansey MG, McCoy MK, Frank-Cannon TC. Neuroinflammatory mechanisms in Parkinson’s disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp Neurol. 2007;208:1–25. doi: 10.1016/j.expneurol.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Esposito E, Di Matteo V, Benigno A, Pierucci M, Crescimanno G, Di Giovanni G. Non-steroidal anti-inflammatory drugs in Parkinson’s disease. Exp Neurol. 2007;205:295–312. doi: 10.1016/j.expneurol.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 65.McGeer EG, McGeer PL. The role of anti-inflammatory agents in Parkinson’s disease. CNS Drugs. 2007;21:789–797. doi: 10.2165/00023210-200721100-00001. [DOI] [PubMed] [Google Scholar]
- 66.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 67.McGeer PL, Schwab C, Parent A, Doudet D. Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration. Ann Neurol. 2003;54:599–604. doi: 10.1002/ana.10728. [DOI] [PubMed] [Google Scholar]
- 68.Orr CF, Rowe DB, Mizuno Y, Mori H, Halliday GM. A possible role for humoral immunity in the pathogenesis of Parkinson’s disease. Brain. 2005;128:2665–2674. doi: 10.1093/brain/awh625. [DOI] [PubMed] [Google Scholar]
- 69.Cicchetti F, Brownell AL, Williams K, Chen YI, Livni E, Isacson O. Neuroinflammation of the nigrostriatal pathway during progressive 6-OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. Eur J Neurosci. 2002;15:991–998. doi: 10.1046/j.1460-9568.2002.01938.x. [DOI] [PubMed] [Google Scholar]
- 70.Sherer TB, Betarbet R, Kim JH, Greenamyre JT. Selective microglial activation in the rat rotenone model of Parkinson’s disease. Neurosci Lett. 2003;341:87–90. doi: 10.1016/s0304-3940(03)00172-1. [DOI] [PubMed] [Google Scholar]
- 71.Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, Nagatsu T. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett. 1994;180:147–150. doi: 10.1016/0304-3940(94)90508-8. [DOI] [PubMed] [Google Scholar]
- 72.Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett. 1994;165:208–210. doi: 10.1016/0304-3940(94)90746-3. [DOI] [PubMed] [Google Scholar]
- 73.Yamada T, McGeer PL, McGeer EG. Lewy bodies in Parkinson’s disease are recognized by antibodies to complement proteins. Acta Neuropathol. 1992;84:100–104. doi: 10.1007/BF00427222. [DOI] [PubMed] [Google Scholar]
- 74.Goldknopf IL, Sheta EA, Bryson J, Folsom B, Wilson C, Duty J, Yen AA, Appel SH. Complement C3c and related protein biomarkers in amyotrophic lateral sclerosis and Parkinson’s disease. Biochem Biophys Res Commun. 2006;342:1034–1039. doi: 10.1016/j.bbrc.2006.02.051. [DOI] [PubMed] [Google Scholar]
- 75.Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhou Y, Hong JS, Zhang J. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 2005;19:533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]
- 76.Reynolds AD, Glanzer JG, Kadiu I, Ricardo-Dukelow M, Chaudhuri A, Ciborowski P, Cerny R, Gelman B, Thomas MP, Mosley RL, Gendelman HE. Nitrated alpha-synuclein-activated microglial profiling for Parkinson’s disease. J Neurochem. 2008;104:1504–1525. doi: 10.1111/j.1471-4159.2007.05087.x. [DOI] [PubMed] [Google Scholar]
- 77.Reynolds AD, Kadiu I, Garg SK, Glanzer JG, Nordgren T, Ciborowski P, Banerjee R, Gendelman HE. Nitrated alpha-synuclein and microglial neuroregulatory activities. J Neuroimmune Pharmacol. 2008;3:59–74. doi: 10.1007/s11481-008-9100-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ghosh A, Roy A, Liu X, Kordower JH, Mufson EJ, Hartley DM, Ghosh S, Mosley RL, Gendelman HE, Pahan K. Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2007;104:18754–18759. doi: 10.1073/pnas.0704908104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bower JH, Maraganore DM, Peterson BJ, Ahlskog JE, Rocca WA. Immunologic diseases, anti-inflammatory drugs, and Parkinson disease: a case-control study. Neurology. 2006;67:494–496. doi: 10.1212/01.wnl.0000227906.99570.cc. [DOI] [PubMed] [Google Scholar]
- 80.Chen H, Jacobs E, Schwarzschild MA, McCullough ML, Calle EE, Thun MJ, Ascherio A. Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Ann Neurol. 2005;58:963–967. doi: 10.1002/ana.20682. [DOI] [PubMed] [Google Scholar]
- 81.Chen H, Zhang SM, Hernan MA, Schwarzschild MA, Willett WC, Colditz GA, Speizer FE, Ascherio A. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch Neurol. 2003;60:1059–1064. doi: 10.1001/archneur.60.8.1059. [DOI] [PubMed] [Google Scholar]
- 82.Hernan MA, Logroscino G, Garcia Rodriguez LA. Nonsteroidal anti-inflammatory drugs and the incidence of Parkinson disease. Neurology. 2006;66:1097–1099. doi: 10.1212/01.wnl.0000204446.82823.28. [DOI] [PubMed] [Google Scholar]
- 83.Ton TG, Heckbert SR, Longstreth WT, Jr, Rossing MA, Kukull WA, Franklin GM, Swanson PD, Smith-Weller T, Checkoway H. Nonsteroidal anti-inflammatory drugs and risk of Parkinson’s disease. Mov Disord. 2006;21:964–969. doi: 10.1002/mds.20856. [DOI] [PubMed] [Google Scholar]
- 84.Wolozin B, Wang SW, Li NC, Lee A, Lee TA, Kazis LE. Simvastatin is associated with a reduced incidence of dementia and Parkinson’s disease. BMC Med. 2007;5:20. doi: 10.1186/1741-7015-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wahner AD, Bronstein JM, Bordelon YM, Ritz B. Statin use and the risk of Parkinson disease. Neurology. 2008;70:1418–1422. doi: 10.1212/01.wnl.0000286942.14552.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.A randomized, double-blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology. 2006;66:664–671. doi: 10.1212/01.wnl.0000201252.57661.e1. [DOI] [PubMed] [Google Scholar]
- 87.Mody I, MacDonald JF. NMDA receptor-dependent excitotoxicity: the role of intracellular Ca2+ release. Trends Pharmacol Sci. 1995;16:356–359. doi: 10.1016/s0165-6147(00)89070-7. [DOI] [PubMed] [Google Scholar]
- 88.Dawson VL, Dawson TM. Nitric oxide neurotoxicity. J Chem Neuroanat. 1996;10:179–190. doi: 10.1016/0891-0618(96)00148-2. [DOI] [PubMed] [Google Scholar]
- 89.Good PF, Hsu A, Werner P, Perl DP, Olanow CW. Protein nitration in Parkinson’s disease. J Neuropathol Exp Neurol. 1998;57:338–342. doi: 10.1097/00005072-199804000-00006. [DOI] [PubMed] [Google Scholar]
- 90.Turski L, Bressler K, Rettig KJ, Loschmann PA, Wachtel H. Protection of substantia nigra from MPP+ neurotoxicity by N-methyl-D-aspartate antagonists. Nature. 1991;349:414–418. doi: 10.1038/349414a0. [DOI] [PubMed] [Google Scholar]
- 91.Brouillet E, Beal MF. NMDA antagonists partially protect against MPTP induced neurotoxicity in mice. Neuroreport. 1993;4:387–390. doi: 10.1097/00001756-199304000-00011. [DOI] [PubMed] [Google Scholar]
- 92.Jankovic J, Hunter C. A double-blind, placebo-controlled and longitudinal study of riluzole in early Parkinson’s disease. Parkinsonism Relat Disord. 2002;8:271–276. doi: 10.1016/s1353-8020(01)00040-2. [DOI] [PubMed] [Google Scholar]
- 93.Kornhuber J, Weller M, Schoppmeyer K, Riederer P. Amantadine and memantine are NMDA receptor antagonists with neuroprotective properties. J Neural Transm Suppl. 1994;43:91–104. [PubMed] [Google Scholar]
- 94.Counihan TJ, Landwehrmeyer GB, Standaert DG, Kosinski CM, Scherzer CR, Daggett LP, Velicelebi G, Young AB, Penney JB., Jr Expression of N-methyl-D-aspartate receptor subunit mRNA in the human brain: mesencephalic dopaminergic neurons. J Comp Neurol. 1998;390:91–101. [PubMed] [Google Scholar]
- 95.Hallett PJ, Standaert DG. Rationale for and use of NMDA receptor antagonists in Parkinson’s disease. Pharmacol Ther. 2004;102:155–174. doi: 10.1016/j.pharmthera.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 96.Surmeier DJ. Calcium, ageing, and neuronal vulnerability in Parkinson’s disease. Lancet Neurol. 2007;6:933–938. doi: 10.1016/S1474-4422(07)70246-6. [DOI] [PubMed] [Google Scholar]
- 97.Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature. 2007;447:1081–1086. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- 98.Rodnitzky RL. Can calcium antagonists provide a neuroprotective effect in Parkinson’s disease? Drugs. 1999;57:845–849. doi: 10.2165/00003495-199957060-00001. [DOI] [PubMed] [Google Scholar]
- 99.Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 100.Tompkins MM, Basgall EJ, Zamrini E, Hill WD. Apoptotic-like changes in Lewy-body-associated disorders and normal aging in substantia nigral neurons. Am J Pathol. 1997;150:119–131. [PMC free article] [PubMed] [Google Scholar]
- 101.Hirsch EC, Hunot S, Faucheux B, Agid Y, Mizuno Y, Mochizuki H, Tatton WG, Tatton N, Olanow WC. Dopaminergic neurons degenerate by apoptosis in Parkinson’s disease. Mov Disord. 1999;14:383–385. doi: 10.1002/1531-8257(199903)14:2<383::aid-mds1037>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 102.Tatton NA. Increased caspase 3 and Bax immunoreactivity accompany nuclear GAPDH translocation and neuronal apoptosis in Parkinson’s disease. Exp Neurol. 2000;166:29–43. doi: 10.1006/exnr.2000.7489. [DOI] [PubMed] [Google Scholar]
- 103.Tatton WG, Chalmers-Redman R, Brown D, Tatton N. Apoptosis in Parkinson’s disease: signals for neuronal degradation. Ann Neurol. 2003;53(Suppl 3):S61–70. doi: 10.1002/ana.10489. discussion S70-62. [DOI] [PubMed] [Google Scholar]
- 104.Blum D, Torch S, Lambeng N, Nissou M, Benabid AL, Sadoul R, Verna JM. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson’s disease. Prog Neurobiol. 2001;65:135–172. doi: 10.1016/s0301-0082(01)00003-x. [DOI] [PubMed] [Google Scholar]
- 105.Jellinger KA. Cell death mechanisms in neurodegeneration. J Cell Mol Med. 2001;5:1–17. doi: 10.1111/j.1582-4934.2001.tb00134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mattson MP. Neuronal life-and-death signaling, apoptosis, and neurodegenerative disorders. Antioxid Redox Signal. 2006;8:1997–2006. doi: 10.1089/ars.2006.8.1997. [DOI] [PubMed] [Google Scholar]
- 107.Olanow CW, Schapira AH, LeWitt PA, Kieburtz K, Sauer D, Olivieri G, Pohlmann H, Hubble J. TCH346 as a neuroprotective drug in Parkinson’s disease: a double-blind, randomised, controlled trial. Lancet Neurol. 2006;5:1013–1020. doi: 10.1016/S1474-4422(06)70602-0. [DOI] [PubMed] [Google Scholar]
- 108.Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology. 2007;69:1480–1490. doi: 10.1212/01.wnl.0000277648.63931.c0. [DOI] [PubMed] [Google Scholar]
- 109.Howells DW, Porritt MJ, Wong JY, Batchelor PE, Kalnins R, Hughes AJ, Donnan GA. Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Exp Neurol. 2000;166:127–135. doi: 10.1006/exnr.2000.7483. [DOI] [PubMed] [Google Scholar]
- 110.Chauhan NB, Siegel GJ, Lee JM. Depletion of glial cell line-derived neurotrophic factor in substantia nigra neurons of Parkinson’s disease brain. J Chem Neuroanat. 2001;21:277–288. doi: 10.1016/s0891-0618(01)00115-6. [DOI] [PubMed] [Google Scholar]
- 111.Mogi M, Togari A, Kondo T, Mizuno Y, Komure O, Kuno S, Ichinose H, Nagatsu T. Brain-derived growth factor and nerve growth factor concentrations are decreased in the substantia nigra in Parkinson’s disease. Neurosci Lett. 1999;270:45–48. doi: 10.1016/s0304-3940(99)00463-2. [DOI] [PubMed] [Google Scholar]
- 112.Kordower JH, Emborg ME, Bloch J, Ma SY, Chu Y, Leventhal L, McBride J, Chen EY, Palfi S, Roitberg BZ, Brown WD, Holden JE, Pyzalski R, Taylor MD, Carvey P, Ling Z, Trono D, Hantraye P, Deglon N, Aebischer P. Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson’s disease. Science. 2000;290:767–773. doi: 10.1126/science.290.5492.767. [DOI] [PubMed] [Google Scholar]
- 113.Eslamboli A, Georgievska B, Ridley RM, Baker HF, Muzyczka N, Burger C, Mandel RJ, Annett L, Kirik D. Continuous low-level glial cell line-derived neurotrophic factor delivery using recombinant adeno-associated viral vectors provides neuroprotection and induces behavioral recovery in a primate model of Parkinson’s disease. J Neurosci. 2005;25:769–777. doi: 10.1523/JNEUROSCI.4421-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gasmi M, Brandon EP, Herzog CD, Wilson A, Bishop KM, Hofer EK, Cunningham JJ, Printz MA, Kordower JH, Bartus RT. AAV2-mediated delivery of human neurturin to the rat nigrostriatal system: long-term efficacy and tolerability of CERE-120 for Parkinson’s disease. Neurobiol Dis. 2007;27:67–76. doi: 10.1016/j.nbd.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 115.Gasmi M, Herzog CD, Brandon EP, Cunningham JJ, Ramirez GA, Ketchum ET, Bartus RT. Striatal delivery of neurturin by CERE-120, an AAV2 vector for the treatment of dopaminergic neuron degeneration in Parkinson’s disease. Mol Ther. 2007;15:62–68. doi: 10.1038/sj.mt.6300010. [DOI] [PubMed] [Google Scholar]
- 116.Kordower JH, Herzog CD, Dass B, Bakay RA, Stansell J, 3rd, Gasmi M, Bartus RT. Delivery of neurturin by AAV2 (CERE-120)-mediated gene transfer provides structural and functional neuroprotection and neurorestoration in MPTP-treated monkeys. Ann Neurol. 2006;60:706–715. doi: 10.1002/ana.21032. [DOI] [PubMed] [Google Scholar]
- 117.Gill SS, Patel NK, Hotton GR, O’Sullivan K, McCarter R, Bunnage M, Brooks DJ, Svendsen CN, Heywood P. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med. 2003;9:589–595. doi: 10.1038/nm850. [DOI] [PubMed] [Google Scholar]
- 118.Lang AE, Gill S, Patel NK, Lozano A, Nutt JG, Penn R, Brooks DJ, Hotton G, Moro E, Heywood P, Brodsky MA, Burchiel K, Kelly P, Dalvi A, Scott B, Stacy M, Turner D, Wooten VG, Elias WJ, Laws ER, Dhawan V, Stoessl AJ, Matcham J, Coffey RJ, Traub M. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol. 2006;59:459–466. doi: 10.1002/ana.20737. [DOI] [PubMed] [Google Scholar]
- 119.Nutt JG, Burchiel KJ, Comella CL, Jankovic J, Lang AE, Laws ER, Jr, Lozano AM, Penn RD, Simpson RK, Jr, Stacy M, Wooten GF. Randomized, double-blind trial of glial cell line-derived neurotrophic factor (GDNF) in PD. Neurology. 2003;60:69–73. doi: 10.1212/wnl.60.1.69. [DOI] [PubMed] [Google Scholar]
- 120.Slevin JT, Gerhardt GA, Smith CD, Gash DM, Kryscio R, Young B. Improvement of bilateral motor functions in patients with Parkinson disease through the unilateral intraputaminal infusion of glial cell line-derived neurotrophic factor. J Neurosurg. 2005;102:216–222. doi: 10.3171/jns.2005.102.2.0216. [DOI] [PubMed] [Google Scholar]
- 121.Suchowersky O, Gronseth G, Perlmutter J, Reich S, Zesiewicz T, Weiner WJ. Practice Parameter: neuroprotective strategies and alternative therapies for Parkinson disease (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2006;66:976–982. doi: 10.1212/01.wnl.0000206363.57955.1b. [DOI] [PubMed] [Google Scholar]
- 122.Hung AY, Schwarzschild MA. Clinical trials for neuroprotection in Parkinson’s disease: overcoming angst and futility? Curr Opin Neurol. 2007;20:477–483. doi: 10.1097/WCO.0b013e32826388d6. [DOI] [PubMed] [Google Scholar]
- 123.Fahn S. Is levodopa toxic? Neurology. 1996;47:S184–195. doi: 10.1212/wnl.47.6_suppl_3.184s. [DOI] [PubMed] [Google Scholar]
- 124.Barzilai A, Melamed E, Shirvan A. Is there a rationale for neuroprotection against dopamine toxicity in Parkinson’s disease? Cell Mol Neurobiol. 2001;21:215–235. doi: 10.1023/A:1010991020245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Murer MG, Dziewczapolski G, Menalled LB, Garcia MC, Agid Y, Gershanik O, Raisman-Vozari R. Chronic levodopa is not toxic for remaining dopamine neurons, but instead promotes their recovery, in rats with moderate nigrostriatal lesions. Ann Neurol. 1998;43:561–575. doi: 10.1002/ana.410430504. [DOI] [PubMed] [Google Scholar]
- 126.Datla KP, Blunt SB, Dexter DT. Chronic L-DOPA administration is not toxic to the remaining dopaminergic nigrostriatal neurons, but instead may promote their functional recovery, in rats with partial 6-OHDA or FeCl(3) nigrostriatal lesions. Mov Disord. 2001;16:424–434. doi: 10.1002/mds.1091. [DOI] [PubMed] [Google Scholar]
- 127.Fahn S, Oakes D, Shoulson I, Kieburtz K, Rudolph A, Lang A, Olanow CW, Tanner C, Marek K. Levodopa and the progression of Parkinson’s disease. N Engl J Med. 2004;351:2498–2508. doi: 10.1056/NEJMoa033447. [DOI] [PubMed] [Google Scholar]
- 128.Ravina B, Eidelberg D, Ahlskog JE, Albin RL, Brooks DJ, Carbon M, Dhawan V, Feigin A, Fahn S, Guttman M, Gwinn-Hardy K, McFarland H, Innis R, Katz RG, Kieburtz K, Kish SJ, Lange N, Langston JW, Marek K, Morin L, Moy C, Murphy D, Oertel WH, Oliver G, Palesch Y, Powers W, Seibyl J, Sethi KD, Shults CW, Sheehy P, Stoessl AJ, Holloway R. The role of radiotracer imaging in Parkinson disease. Neurology. 2005;64:208–215. doi: 10.1212/01.WNL.0000149403.14458.7F. [DOI] [PubMed] [Google Scholar]
- 129.Allain H, Bentue-Ferrer D, Akwa Y. Disease-modifying drugs and Parkinson’s disease. Prog Neurobiol. 2008;84:25–39. doi: 10.1016/j.pneurobio.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 130.Ogawa N, Tanaka K, Asanuma M, Kawai M, Masumizu T, Kohno M, Mori A. Bromocriptine protects mice against 6-hydroxydopamine and scavenges hydroxyl free radicals in vitro. Brain Res. 1994;657:207–213. doi: 10.1016/0006-8993(94)90969-5. [DOI] [PubMed] [Google Scholar]
- 131.Carvey PM, Pieri S, Ling ZD. Attenuation of levodopa-induced toxicity in mesencephalic cultures by pramipexole. J Neural Transm. 1997;104:209–228. doi: 10.1007/BF01273182. [DOI] [PubMed] [Google Scholar]
- 132.Kitamura Y, Kosaka T, Kakimura JI, Matsuoka Y, Kohno Y, Nomura Y, Taniguchi T. Protective effects of the antiparkinsonian drugs talipexole and pramipexole against 1-methyl-4-phenylpyridinium-induced apoptotic death in human neuroblastoma SH-SY5Y cells. Mol Pharmacol. 1998;54:1046–1054. [PubMed] [Google Scholar]
- 133.Iida M, Miyazaki I, Tanaka K, Kabuto H, Iwata-Ichikawa E, Ogawa N. Dopamine D2 receptor-mediated antioxidant and neuroprotective effects of ropinirole, a dopamine agonist. Brain Res. 1999;838:51–59. doi: 10.1016/s0006-8993(99)01688-1. [DOI] [PubMed] [Google Scholar]
- 134.Vu TQ, Ling ZD, Ma SY, Robie HC, Tong CW, Chen EY, Lipton JW, Carvey PM. Pramipexole attenuates the dopaminergic cell loss induced by intraventricular 6-hydroxydopamine. J Neural Transm. 2000;107:159–176. doi: 10.1007/s007020050014. [DOI] [PubMed] [Google Scholar]
- 135.Zou L, Xu J, Jankovic J, He Y, Appel SH, Le W. Pramipexole inhibits lipid peroxidation and reduces injury in the substantia nigra induced by the dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in C57BL/6 mice. Neurosci Lett. 2000;281:167–170. doi: 10.1016/s0304-3940(00)00853-3. [DOI] [PubMed] [Google Scholar]
- 136.Iravani MM, Haddon CO, Cooper JM, Jenner P, Schapira AH. Pramipexole protects against MPTP toxicity in non-human primates. J Neurochem. 2006;96:1315–1321. doi: 10.1111/j.1471-4159.2005.03625.x. [DOI] [PubMed] [Google Scholar]
- 137.Olanow CW, Jenner P, Brooks D. Dopamine agonists and neuroprotection in Parkinson’s disease. Ann Neurol. 1998;44:S167–174. doi: 10.1002/ana.410440725. [DOI] [PubMed] [Google Scholar]
- 138.Cassarino DS, Fall CP, Smith TS, Bennett JP., Jr Pramipexole reduces reactive oxygen species production in vivo and in vitro and inhibits the mitochondrial permeability transition produced by the parkinsonian neurotoxin methylpyridinium ion. J Neurochem. 1998;71:295–301. doi: 10.1046/j.1471-4159.1998.71010295.x. [DOI] [PubMed] [Google Scholar]
- 139.Dopamine transporter brain imaging to assess the effects of pramipexole vs levodopa on Parkinson disease progression. JAMA. 2002;287:1653–1661. doi: 10.1001/jama.287.13.1653. [DOI] [PubMed] [Google Scholar]
- 140.Whone AL, Watts RL, Stoessl AJ, Davis M, Reske S, Nahmias C, Lang AE, Rascol O, Ribeiro MJ, Remy P, Poewe WH, Hauser RA, Brooks DJ. Slower progression of Parkinson’s disease with ropinirole versus levodopa: The REAL-PET study. Ann Neurol. 2003;54:93–101. doi: 10.1002/ana.10609. [DOI] [PubMed] [Google Scholar]
- 141.Effect of deprenyl on the progression of disability in early Parkinson’s disease. The Parkinson Study Group. N Engl J Med. 1989;321:1364–1371. doi: 10.1056/NEJM198911163212004. [DOI] [PubMed] [Google Scholar]
- 142.Effects of tocopherol and deprenyl on the progression of disability in early Parkinson’s disease. The Parkinson Study Group. N Engl J Med. 1993;328:176–183. doi: 10.1056/NEJM199301213280305. [DOI] [PubMed] [Google Scholar]
- 143.A controlled, randomized, delayed-start study of rasagiline in early Parkinson disease. Arch Neurol. 2004;61:561–566. doi: 10.1001/archneur.61.4.561. [DOI] [PubMed] [Google Scholar]
- 144.Beal MF, Matthews RT, Tieleman A, Shults CW. Coenzyme Q10 attenuates the 1-methyl-4-phenyl-1,2,3,tetrahydropyridine (MPTP) induced loss of striatal dopamine and dopaminergic axons in aged mice. Brain Res. 1998;783:109–114. doi: 10.1016/s0006-8993(97)01192-x. [DOI] [PubMed] [Google Scholar]
- 145.Shults CW, Oakes D, Kieburtz K, Beal MF, Haas R, Plumb S, Juncos JL, Nutt J, Shoulson I, Carter J, Kompoliti K, Perlmutter JS, Reich S, Stern M, Watts RL, Kurlan R, Molho E, Harrison M, Lew M. Effects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol. 2002;59:1541–1550. doi: 10.1001/archneur.59.10.1541. [DOI] [PubMed] [Google Scholar]
- 146.A randomized clinical trial of coenzyme Q10 and GPI-1485 in early Parkinson disease. Neurology. 2007;68:20–28. doi: 10.1212/01.wnl.0000250355.28474.8e. [DOI] [PubMed] [Google Scholar]
- 147.Elm JJ, Goetz CG, Ravina B, Shannon K, Wooten GF, Tanner CM, Palesch YY, Huang P, Guimaraes P, Kamp C, Tilley BC, Kieburtz K. A responsive outcome for Parkinson’s disease neuroprotection futility studies. Ann Neurol. 2005;57:197–203. doi: 10.1002/ana.20361. [DOI] [PubMed] [Google Scholar]
- 148.Schwid SR, Cutter GR. Futility studies: spending a little to save a lot. Neurology. 2006;66:626–627. doi: 10.1212/01.wnl.0000204644.81956.65. [DOI] [PubMed] [Google Scholar]
- 149.Tilley BC, Palesch YY, Kieburtz K, Ravina B, Huang P, Elm JJ, Shannon K, Wooten GF, Tanner CM, Goetz GC. Optimizing the ongoing search for new treatments for Parkinson disease: using futility designs. Neurology. 2006;66:628–633. doi: 10.1212/01.wnl.0000201251.33253.fb. [DOI] [PubMed] [Google Scholar]
- 150.Matthews RT, Ferrante RJ, Klivenyi P, Yang L, Klein AM, Mueller G, Kaddurah-Daouk R, Beal MF. Creatine and cyclocreatine attenuate MPTP neurotoxicity. Exp Neurol. 1999;157:142–149. doi: 10.1006/exnr.1999.7049. [DOI] [PubMed] [Google Scholar]
- 151.Bender A, Koch W, Elstner M, Schombacher Y, Bender J, Moeschl M, Gekeler F, Muller-Myhsok B, Gasser T, Tatsch K, Klopstock T. Creatine supplementation in Parkinson disease: a placebo-controlled randomized pilot trial. Neurology. 2006;67:1262–1264. doi: 10.1212/01.wnl.0000238518.34389.12. [DOI] [PubMed] [Google Scholar]
- 152.Andringa G, Eshuis S, Perentes E, Maguire RP, Roth D, Ibrahim M, Leenders KL, Cools AR. TCH346 prevents motor symptoms and loss of striatal FDOPA uptake in bilaterally MPTP-treated primates. Neurobiol Dis. 2003;14:205–217. doi: 10.1016/s0969-9961(03)00125-6. [DOI] [PubMed] [Google Scholar]
- 153.Andringa G, van Oosten RV, Unger W, Hafmans TG, Veening J, Stoof JC, Cools AR. Systemic administration of the propargylamine CGP 3466B prevents behavioural and morphological deficits in rats with 6-hydroxydopamine-induced lesions in the substantia nigra. Eur J Neurosci. 2000;12:3033–3043. doi: 10.1046/j.1460-9568.2000.00181.x. [DOI] [PubMed] [Google Scholar]
- 154.Saporito MS, Brown EM, Miller MS, Carswell S. CEP-1347/KT-7515, an inhibitor of c-jun N-terminal kinase activation, attenuates the 1-methyl-4-phenyl tetrahydropyridine-mediated loss of nigrostriatal dopaminergic neurons In vivo. J Pharmacol Exp Ther. 1999;288:421–427. [PubMed] [Google Scholar]
- 155.Mathiasen JR, McKenna BA, Saporito MS, Ghadge GD, Roos RP, Holskin BP, Wu ZL, Trusko SP, Connors TC, Maroney AC, Thomas BA, Thomas JC, Bozyczko-Coyne D. Inhibition of mixed lineage kinase 3 attenuates MPP+-induced neurotoxicity in SH-SY5Y cells. Brain Res. 2004;1003:86–97. doi: 10.1016/j.brainres.2003.11.073. [DOI] [PubMed] [Google Scholar]
- 156.Lotharius J, Falsig J, van Beek J, Payne S, Dringen R, Brundin P, Leist M. Progressive degeneration of human mesencephalic neuron-derived cells triggered by dopamine-dependent oxidative stress is dependent on the mixed-lineage kinase pathway. J Neurosci. 2005;25:6329–6342. doi: 10.1523/JNEUROSCI.1746-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wang LH, Johnson EM., Jr Mixed lineage kinase inhibitor cep- 1347 fails to delay disability in early Parkinson disease. Neurology. 2008;71:462. doi: 10.1212/01.wnl.0000324506.93877.5e. author reply 462–463. [DOI] [PubMed] [Google Scholar]
- 158.Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- 159.Lewis TB, Standaert DG. Design of clinical trials of gene therapy in Parkinson disease. Exp Neurol. 2008;209:41–47. doi: 10.1016/j.expneurol.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 160.Akerud P, Alberch J, Eketjall S, Wagner J, Arenas E. Differential effects of glial cell line-derived neurotrophic factor and neurturin on developing and adult substantia nigra dopaminergic neurons. J Neurochem. 1999;73:70–78. doi: 10.1046/j.1471-4159.1999.0730070.x. [DOI] [PubMed] [Google Scholar]
- 161.Horger BA, Nishimura MC, Armanini MP, Wang LC, Poulsen KT, Rosenblad C, Kirik D, Moffat B, Simmons L, Johnson E, Jr, Milbrandt J, Rosenthal A, Bjorklund A, Vandlen RA, Hynes MA, Phillips HS. Neurturin exerts potent actions on survival and function of midbrain dopaminergic neurons. J Neurosci. 1998;18:4929–4937. doi: 10.1523/JNEUROSCI.18-13-04929.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Marks WJ, Jr, Ostrem JL, Verhagen L, Starr PA, Larson PS, Bakay RA, Taylor R, Cahn-Weiner DA, Stoessl AJ, Olanow CW, Bartus RT. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson’s disease: an open-label, phase I trial. Lancet Neurol. 2008;7:400–408. doi: 10.1016/S1474-4422(08)70065-6. [DOI] [PubMed] [Google Scholar]
- 163.Steiner JP, Connolly MA, Valentine HL, Hamilton GS, Dawson TM, Hester L, Snyder SH. Neurotrophic actions of nonimmunosuppressive analogues of immunosuppressive drugs FK506, rapamycin and cyclosporin A. Nat Med. 1997;3:421–428. doi: 10.1038/nm0497-421. [DOI] [PubMed] [Google Scholar]
- 164.Guo X, Dawson VL, Dawson TM. Neuroimmunophilin ligands exert neuroregeneration and neuroprotection in midbrain dopaminergic neurons. Eur J Neurosci. 2001;13:1683–1693. doi: 10.1046/j.0953-816x.2001.01542.x. [DOI] [PubMed] [Google Scholar]
- 165.Khan Z, Ferrari G, Kasper M, Tonge DA, Steiner JP, Hamilton GS, Gordon-Weeks PR. The non-immunosuppressive immunophilin ligand GPI-1046 potently stimulates regenerating axon growth from adult mouse dorsal root ganglia cultured in Matrigel. Neuroscience. 2002;114:601–609. doi: 10.1016/s0306-4522(02)00314-7. [DOI] [PubMed] [Google Scholar]
- 166.Steiner JP, Hamilton GS, Ross DT, Valentine HL, Guo H, Connolly MA, Liang S, Ramsey C, Li JH, Huang W, Howorth P, Soni R, Fuller M, Sauer H, Nowotnik AC, Suzdak PD. Neurotrophic immunophilin ligands stimulate structural and functional recovery in neurodegenerative animal models. Proc Natl Acad Sci U S A. 1997;94:2019–2024. doi: 10.1073/pnas.94.5.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Costantini LC, Cole D, Chaturvedi P, Isacson O. Immunophilin ligands can prevent progressive dopaminergic degeneration in animal models of Parkinson’s disease. Eur J Neurosci. 2001;13:1085–1092. doi: 10.1046/j.0953-816x.2001.01473.x. [DOI] [PubMed] [Google Scholar]
- 168.Zhang C, Steiner JP, Hamilton GS, Hicks TP, Poulter MO. Regeneration of dopaminergic function in 6-hydroxydopamine-lesioned rats by neuroimmunophilin ligand treatment. J Neurosci. 2001;21:RC156. doi: 10.1523/JNEUROSCI.21-15-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Emborg ME, Shin P, Roitberg B, Sramek JG, Chu Y, Stebbins GT, Hamilton JS, Suzdak PD, Steiner JP, Kordower JH. Systemic administration of the immunophilin ligand GPI 1046 in MPTP-treated monkeys. Exp Neurol. 2001;168:171–182. doi: 10.1006/exnr.2000.7592. [DOI] [PubMed] [Google Scholar]
- 170.Eberling JL, Pivirotto P, Bringas J, Steiner JP, Kordower JH, Chu Y, Emborg ME, Bankiewicz KS. The immunophilin ligand GPI-1046 does not have neuroregenerative effects in MPTP-treated monkeys. Exp Neurol. 2002;178:236–242. doi: 10.1006/exnr.2002.8023. [DOI] [PubMed] [Google Scholar]
- 171.Tanaka K, Fujita N, Ogawa N. Immunosuppressive (FK506) and non-immunosuppressive (GPI1046) immunophilin ligands activate neurotrophic factors in the mouse brain. Brain Res. 2003;970:250–253. doi: 10.1016/s0006-8993(03)02434-x. [DOI] [PubMed] [Google Scholar]
- 172.Tanaka K, Fujita N, Yoshioka M, Ogawa N. Immunosuppressive and non-immunosuppressive immunophilin ligands improve H(2)O(2)-induced cell damage by increasing glutathione levels in NG108-15 cells. Brain Res. 2001;889:225–228. doi: 10.1016/s0006-8993(00)02851-1. [DOI] [PubMed] [Google Scholar]
- 173.Tanaka K, Yoshioka M, Miyazaki I, Fujita N, Ogawa N. GPI1046 prevents dopaminergic dysfunction by activating glutathione system in the mouse striatum. Neurosci Lett. 2002;321:45–48. doi: 10.1016/s0304-3940(01)02547-2. [DOI] [PubMed] [Google Scholar]
- 174.Poulter MO, Payne KB, Steiner JP. Neuroimmunophilins: a novel drug therapy for the reversal of neurodegenerative disease? Neuroscience. 2004;128:1–6. doi: 10.1016/j.neuroscience.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 175.Ramaker C, Marinus J, Stiggelbout AM, Van Hilten BJ. Systematic evaluation of rating scales for impairment and disability in Parkinson’s disease. Mov Disord. 2002;17:867–876. doi: 10.1002/mds.10248. [DOI] [PubMed] [Google Scholar]
- 176.The Unified Parkinson’s Disease Rating Scale (UPDRS): status and recommendations. Mov Disord. 2003;18:738–750. doi: 10.1002/mds.10473. [DOI] [PubMed] [Google Scholar]
- 177.Goetz CG, Fahn S, Martinez-Martin P, Poewe W, Sampaio C, Stebbins GT, Stern MB, Tilley BC, Dodel R, Dubois B, Holloway R, Jankovic J, Kulisevsky J, Lang AE, Lees A, Leurgans S, LeWitt PA, Nyenhuis D, Olanow CW, Rascol O, Schrag A, Teresi JA, Van Hilten JJ, LaPelle N. Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): Process, format, and clinimetric testing plan. Mov Disord. 2007;22:41–47. doi: 10.1002/mds.21198. [DOI] [PubMed] [Google Scholar]
- 178.Kieburtz K. Issues in neuroprotection clinical trials in Parkinson’s disease. Neurology. 2006;66:S50–57. doi: 10.1212/wnl.66.10_suppl_4.s50. [DOI] [PubMed] [Google Scholar]
- 179.Morrish PK, Rakshi JS, Bailey DL, Sawle GV, Brooks DJ. Measuring the rate of progression and estimating the preclinical period of Parkinson’s disease with [18F]dopa PET. J Neurol Neurosurg Psychiatry. 1998;64:314–319. doi: 10.1136/jnnp.64.3.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Marek K, Innis R, van Dyck C, Fussell B, Early M, Eberly S, Oakes D, Seibyl J. [123I]beta-CIT SPECT imaging assessment of the rate of Parkinson’s disease progression. Neurology. 2001;57:2089–2094. doi: 10.1212/wnl.57.11.2089. [DOI] [PubMed] [Google Scholar]
- 181.Betarbet R, Sherer TB, Greenamyre JT. Animal models of Parkinson’s disease. Bioessays. 2002;24:308–318. doi: 10.1002/bies.10067. [DOI] [PubMed] [Google Scholar]
- 182.Hashimoto M, Rockenstein E, Masliah E. Transgenic models of alpha-synuclein pathology: past, present, and future. Ann N Y Acad Sci. 2003;991:171–188. [PubMed] [Google Scholar]
- 183.Maries E, Dass B, Collier TJ, Kordower JH, Steece-Collier K. The role of alpha-synuclein in Parkinson’s disease: insights from animal models. Nat Rev Neurosci. 2003;4:727–738. doi: 10.1038/nrn1199. [DOI] [PubMed] [Google Scholar]
- 184.Lohmann E, Periquet M, Bonifati V, Wood NW, De Michele G, Bonnet AM, Fraix V, Broussolle E, Horstink MW, Vidailhet M, Verpillat P, Gasser T, Nicholl D, Teive H, Raskin S, Rascol O, Destee A, Ruberg M, Gasparini F, Meco G, Agid Y, Durr A, Brice A. How much phenotypic variation can be attributed to parkin genotype? Ann Neurol. 2003;54:176–185. doi: 10.1002/ana.10613. [DOI] [PubMed] [Google Scholar]
- 185.Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med. 2000;342:1560–1567. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- 186.Deng H, Le W, Guo Y, Hunter CB, Xie W, Jankovic J. Genetic and clinical identification of Parkinson’s disease patients with LRRK2 G2019S mutation. Ann Neurol. 2005;57:933–934. doi: 10.1002/ana.20510. [DOI] [PubMed] [Google Scholar]
- 187.Gilks WP, Abou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ, Shaw K, Bhatia KP, Bonifati V, Quinn NP, Lynch J, Healy DG, Holton JL, Revesz T, Wood NW. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet. 2005;365:415–416. doi: 10.1016/S0140-6736(05)17830-1. [DOI] [PubMed] [Google Scholar]
- 188.Kachergus J, Mata IF, Hulihan M, Taylor JP, Lincoln S, Aasly J, Gibson JM, Ross OA, Lynch T, Wiley J, Payami H, Nutt J, Maraganore DM, Czyzewski K, Styczynska M, Wszolek ZK, Farrer MJ, Toft M. Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet. 2005;76:672–680. doi: 10.1086/429256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S, Ferreira JJ, Tolosa E, Kay DM, Klein C, Williams DR, Marras C, Lang AE, Wszolek ZK, Berciano J, Schapira AH, Lynch T, Bhatia KP, Gasser T, Lees AJ, Wood NW. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;7:583–590. doi: 10.1016/S1474-4422(08)70117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Riederer P, Wuketich S. Time course of nigrostriatal degeneration in parkinson’s disease. A detailed study of influential factors in human brain amine analysis. J Neural Transm. 1976;38:277–301. doi: 10.1007/BF01249445. [DOI] [PubMed] [Google Scholar]
- 191.Tissingh G, Bergmans P, Booij J, Winogrodzka A, van Royen EA, Stoof JC, Wolters EC. Drug-naive patients with Parkinson’s disease in Hoehn and Yahr stages I and II show a bilateral decrease in striatal dopamine transporters as revealed by [123I]beta-CIT SPECT. J Neurol. 1998;245:14–20. doi: 10.1007/s004150050168. [DOI] [PubMed] [Google Scholar]
- 192.Tissingh G, Booij J, Bergmans P, Winogrodzka A, Janssen AG, van Royen EA, Stoof JC, Wolters EC. Iodine-123-N-omega-fluoropropyl-2beta-carbomethoxy-3beta-(4-iod ophenyl)tropane SPECT in healthy controls and early-stage, drug-naive Parkinson’s disease. J Nucl Med. 1998;39:1143–1148. [PubMed] [Google Scholar]
- 193.Tolosa E, Compta Y, Gaig C. The premotor phase of Parkinson’s disease. Parkinsonism Relat Disord. 2007;13(Suppl):S2–7. doi: 10.1016/j.parkreldis.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 194.Ravina BM, Fagan SC, Hart RG, Hovinga CA, Murphy DD, Dawson TM, Marler JR. Neuroprotective agents for clinical trials in Parkinson’s disease: a systematic assessment. Neurology. 2003;60:1234–1240. doi: 10.1212/01.wnl.0000058760.13152.1a. [DOI] [PubMed] [Google Scholar]
- 195.Ross GW, Abbott RD, Petrovitch H, Morens DM, Grandinetti A, Tung KH, Tanner CM, Masaki KH, Blanchette PL, Curb JD, Popper JS, White LR. Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA. 2000;283:2674–2679. doi: 10.1001/jama.283.20.2674. [DOI] [PubMed] [Google Scholar]
- 196.Ascherio A, Zhang SM, Hernan MA, Kawachi I, Colditz GA, Speizer FE, Willett WC. Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women. Ann Neurol. 2001;50:56–63. doi: 10.1002/ana.1052. [DOI] [PubMed] [Google Scholar]
- 197.Schwarzschild MA, Agnati L, Fuxe K, Chen JF, Morelli M. Targeting adenosine A2A receptors in Parkinson’s disease. Trends Neurosci. 2006;29:647–654. doi: 10.1016/j.tins.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 198.Ferre S, Fuxe K. Dopamine denervation leads to an increase in the intramembrane interaction between adenosine A2 and dopamine D2 receptors in the neostriatum. Brain Res. 1992;594:124–130. doi: 10.1016/0006-8993(92)91036-e. [DOI] [PubMed] [Google Scholar]
- 199.Ferre S, O’Connor WT, Fuxe K, Ungerstedt U. The striopallidal neuron: a main locus for adenosine-dopamine interactions in the brain. J Neurosci. 1993;13:5402–5406. doi: 10.1523/JNEUROSCI.13-12-05402.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bara-Jimenez W, Sherzai A, Dimitrova T, Favit A, Bibbiani F, Gillespie M, Morris MJ, Mouradian MM, Chase TN. Adenosine A(2A) receptor antagonist treatment of Parkinson’s disease. Neurology. 2003;61:293–296. doi: 10.1212/01.wnl.0000073136.00548.d4. [DOI] [PubMed] [Google Scholar]
- 201.Hauser RA, Hubble JP, Truong DD. Randomized trial of the adenosine A(2A) receptor antagonist istradefylline in advanced PD. Neurology. 2003;61:297–303. doi: 10.1212/01.wnl.0000081227.84197.0b. [DOI] [PubMed] [Google Scholar]
- 202.Chen JF, Xu K, Petzer JP, Staal R, Xu YH, Beilstein M, Sonsalla PK, Castagnoli K, Castagnoli N, Jr, Schwarzschild MA. Neuroprotection by caffeine and A(2A) adenosine receptor inactivation in a model of Parkinson’s disease. J Neurosci. 2001;21:RC143. doi: 10.1523/JNEUROSCI.21-10-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Ikeda K, Kurokawa M, Aoyama S, Kuwana Y. Neuroprotection by adenosine A2A receptor blockade in experimental models of Parkinson’s disease. J Neurochem. 2002;80:262–270. doi: 10.1046/j.0022-3042.2001.00694.x. [DOI] [PubMed] [Google Scholar]
- 204.Phillis JW. The effects of selective A1 and A2a adenosine receptor antagonists on cerebral ischemic injury in the gerbil. Brain Res. 1995;705:79–84. doi: 10.1016/0006-8993(95)01153-6. [DOI] [PubMed] [Google Scholar]
- 205.Jones PA, Smith RA, Stone TW. Protection against hippocampal kainate excitotoxicity by intracerebral administration of an adenosine A2A receptor antagonist. Brain Res. 1998;800:328–335. doi: 10.1016/s0006-8993(98)00540-x. [DOI] [PubMed] [Google Scholar]
- 206.Monopoli A, Lozza G, Forlani A, Mattavelli A, Ongini E. Blockade of adenosine A2A receptors by SCH 58261 results in neuroprotective effects in cerebral ischaemia in rats. Neuroreport. 1998;9:3955–3959. doi: 10.1097/00001756-199812010-00034. [DOI] [PubMed] [Google Scholar]
- 207.Selley ML. Simvastatin prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced striatal dopamine depletion and protein tyrosine nitration in mice. Brain Res. 2005;1037:1–6. doi: 10.1016/j.brainres.2004.02.083. [DOI] [PubMed] [Google Scholar]
- 208.Di Napoli P, Taccardi AA, Oliver M, De Caterina R. Statins and stroke: evidence for cholesterol-independent effects. Eur Heart J. 2002;23:1908–1921. doi: 10.1053/euhj.2002.3236. [DOI] [PubMed] [Google Scholar]
- 209.de Lau LM, Koudstaal PJ, Hofman A, Breteler MM. Serum cholesterol levels and the risk of Parkinson’s disease. Am J Epidemiol. 2006;164:998–1002. doi: 10.1093/aje/kwj283. [DOI] [PubMed] [Google Scholar]
- 210.Huang X, Chen H, Miller WC, Mailman RB, Woodard JL, Chen PC, Xiang D, Murrow RW, Wang YZ, Poole C. Lower low-density lipoprotein cholesterol levels are associated with Parkinson’s disease. Mov Disord. 2007;22:377–381. doi: 10.1002/mds.21290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci. 2001;21:2580–2588. doi: 10.1523/JNEUROSCI.21-08-02580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol. 2001;166:7527–7533. doi: 10.4049/jimmunol.166.12.7527. [DOI] [PubMed] [Google Scholar]
- 213.Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, Triarhou LC, Chernet E, Perry KW, Nelson DL, Luecke S, Phebus LA, Bymaster FP, Paul SM. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2001;98:14669–14674. doi: 10.1073/pnas.251341998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22:1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.He Y, Appel S, Le W. Minocycline inhibits microglial activation and protects nigral cells after 6-hydroxydopamine injection into mouse striatum. Brain Res. 2001;909:187–193. doi: 10.1016/s0006-8993(01)02681-6. [DOI] [PubMed] [Google Scholar]
- 216.Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci U S A. 1981;78:6858–6862. doi: 10.1073/pnas.78.11.6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Davis JW, Grandinetti A, Waslien CI, Ross GW, White LR, Morens DM. Observations on serum uric acid levels and the risk of idiopathic Parkinson’s disease. Am J Epidemiol. 1996;144:480–484. doi: 10.1093/oxfordjournals.aje.a008954. [DOI] [PubMed] [Google Scholar]
- 218.de Lau LM, Koudstaal PJ, Hofman A, Breteler MM. Serum uric acid levels and the risk of Parkinson disease. Ann Neurol. 2005;58:797–800. doi: 10.1002/ana.20663. [DOI] [PubMed] [Google Scholar]
- 219.Weisskopf MG, O’Reilly E, Chen H, Schwarzschild MA, Ascherio A. Plasma urate and risk of Parkinson’s disease. Am J Epidemiol. 2007;166:561–567. doi: 10.1093/aje/kwm127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Alonso A, Rodriguez LA, Logroscino G, Hernan MA. Gout and risk of Parkinson disease: a prospective study. Neurology. 2007;69:1696–1700. doi: 10.1212/01.wnl.0000279518.10072.df. [DOI] [PubMed] [Google Scholar]
- 221.Schwarzschild MA, Schwid SR, Marek K, Watts A, Lang AE, Oakes D, Shoulson I, Ascherio A, Hyson C, Gorbold E, Rudolph A, Kieburtz K, Fahn S, Gauger L, Goetz C, Seibyl J, Forrest M, Ondrasik J. Serum urate as a predictor of clinical and radiographic progression in Parkinson disease. Arch Neurol. 2008;65:716–723. doi: 10.1001/archneur.2008.65.6.nct70003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Duan W, Ladenheim B, Cutler RG, Kruman, Cadet JL, Mattson MP. Dietary folate deficiency and elevated homocysteine levels endanger dopaminergic neurons in models of Parkinson’s disease. J Neurochem. 2002;80:101–110. doi: 10.1046/j.0022-3042.2001.00676.x. [DOI] [PubMed] [Google Scholar]
- 223.Gao X, Chen H, Choi HK, Curhan G, Schwarzschild MA, Ascherio A. Diet, urate, and Parkinson’s disease risk in men. Am J Epidemiol. 2008;167:831–838. doi: 10.1093/aje/kwm385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Haywood AF, Staveley BE. Parkin counteracts symptoms in a Drosophila model of Parkinson’s disease. BMC Neurosci. 2004;5:14. doi: 10.1186/1471-2202-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Lo Bianco C, Schneider BL, Bauer M, Sajadi A, Brice A, Iwatsubo T, Aebischer P. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2004;101:17510–17515. doi: 10.1073/pnas.0405313101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Yamada M, Mizuno Y, Mochizuki H. Parkin gene therapy for alpha-synucleinopathy: a rat model of Parkinson’s disease. Hum Gene Ther. 2005;16:262–270. doi: 10.1089/hum.2005.16.262. [DOI] [PubMed] [Google Scholar]
- 227.McLean PJ, Klucken J, Shin Y, Hyman BT. Geldanamycin induces Hsp70 and prevents alpha-synuclein aggregation and toxicity in vitro. Biochem Biophys Res Commun. 2004;321:665–669. doi: 10.1016/j.bbrc.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 228.Qiao L, Hamamichi S, Caldwell KA, Caldwell GA, Wilson S, Yacoubian T, Xie ZL, Speake L, Parks R, Crabtree D, Crimmins S, Uchiyama Y, Zhou Y, Peng L, Lu Y, Standaert DG, Walls KC, Shacka JJ, Roth K, Zhang J. A neuroprotective role of lysosomal enzyme cathepsin D against a-synuclein pathogenesis. Movement Disorders. 2008;23:S8. [Google Scholar]
- 229.Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, Games D, Schenk D. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron. 2005;46:857–868. doi: 10.1016/j.neuron.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 230.Chen L, Feany MB. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci. 2005;8:657–663. doi: 10.1038/nn1443. [DOI] [PubMed] [Google Scholar]
- 231.Smith WW, Margolis RL, Li X, Troncoso JC, Lee MK, Dawson VL, Dawson TM, Iwatsubo T, Ross CA. Alpha-synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J Neurosci. 2005;25:5544–5552. doi: 10.1523/JNEUROSCI.0482-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Bodles AM, El-Agnaf OM, Greer B, Guthrie DJ, Irvine GB. Inhibition of fibril formation and toxicity of a fragment of alpha-synuclein by an N-methylated peptide analogue. Neurosci Lett. 2004;359:89–93. doi: 10.1016/j.neulet.2003.12.077. [DOI] [PubMed] [Google Scholar]
- 233.El-Agnaf OM, Paleologou KE, Greer B, Abogrein AM, King JE, Salem SA, Fullwood NJ, Benson FE, Hewitt R, Ford KJ, Martin FL, Harriott P, Cookson MR, Allsop D. A strategy for designing inhibitors of alpha-synuclein aggregation and toxicity as a novel treatment for Parkinson’s disease and related disorders. FASEB J. 2004;18:1315–1317. doi: 10.1096/fj.03-1346fje. [DOI] [PubMed] [Google Scholar]
- 234.Amer DA, Irvine GB, El-Agnaf OM. Inhibitors of alpha-synuclein oligomerization and toxicity: a future therapeutic strategy for Parkinson’s disease and related disorders. Exp Brain Res. 2006;173:223–233. doi: 10.1007/s00221-006-0539-y. [DOI] [PubMed] [Google Scholar]
- 235.Hashimoto M, Rockenstein E, Mante M, Mallory M, Masliah E. beta-Synuclein inhibits alpha-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron. 2001;32:213–223. doi: 10.1016/s0896-6273(01)00462-7. [DOI] [PubMed] [Google Scholar]
- 236.Windisch M, Hutter-Paier B, Rockenstein E, Hashimoto M, Mallory M, Masliah E. Development of a new treatment for Alzheimer’s disease and Parkinson’s disease using anti-aggregatory beta-synuclein-derived peptides. J Mol Neurosci. 2002;19:63–69. doi: 10.1007/s12031-002-0012-8. [DOI] [PubMed] [Google Scholar]
- 237.Yacoubian TA, Standaert DG. Role of 14–3-3 proteins in alpha-synuclein-induced neurotoxicity. Soc Neurosci Abstract Viewer/Itinerary Planner Program #882.8/J1. 2007 [Google Scholar]
- 238.Lesage S, Durr A, Tazir M, Lohmann E, Leutenegger AL, Janin S, Pollak P, Brice A. LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N Engl J Med. 2006;354:422–423. doi: 10.1056/NEJMc055540. [DOI] [PubMed] [Google Scholar]
- 239.Lesage S, Ibanez P, Lohmann E, Pollak P, Tison F, Tazir M, Leutenegger AL, Guimaraes J, Bonnet AM, Agid Y, Durr A, Brice A. G2019S LRRK2 mutation in French and North African families with Parkinson’s disease. Ann Neurol. 2005;58:784–787. doi: 10.1002/ana.20636. [DOI] [PubMed] [Google Scholar]
- 240.Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M, Hunt AL, Klein C, Henick B, Hailpern SM, Lipton RB, Soto-Valencia J, Risch N, Bressman SB. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med. 2006;354:424–425. doi: 10.1056/NEJMc055509. [DOI] [PubMed] [Google Scholar]
- 241.Hulihan MM, Ishihara-Paul L, Kachergus J, Warren L, Amouri R, Elango R, Prinjha RK, Upmanyu R, Kefi M, Zouari M, Sassi SB, Yahmed SB, El Euch-Fayeche G, Matthews PM, Middleton LT, Gibson RA, Hentati F, Farrer MJ. LRRK2 Gly2019Ser penetrance in Arab-Berber patients from Tunisia: a case-control genetic study. Lancet Neurol. 2008;7:591–594. doi: 10.1016/S1474-4422(08)70116-9. [DOI] [PubMed] [Google Scholar]
- 242.West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 244.West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, Dawson TM. Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]