

Abstract
Haemophilus influenzae β-carbonic anhydrase (HICA) is hypothesized to be an allosteric protein that is regulated by the binding of bicarbonate ion to a non-catalytic (inhibitory) site that controls the ligation of Asp44 to the catalytically essential zinc ion. We report here the X-ray crystallographic structures of two variants (W39F and Y181F) involved in the binding of bicarbonate ion in the non-catalytic site and an active site variant (D44N) that is incapable of forming a strong zinc ligand. The alteration of Trp39 to Phe increases the apparent Ki for bicarbonate inhibition by 4.8-fold. While the structures of W39F and Y181F are very similar to the wild-type enzyme, the X-ray crystal structure of the D44N variant reveals that it has adopted an active site conformation nearly identical to that of non-allosteric β-carbonic anhydrases. We propose that the structure of the D44N variant is likely to be representative of the active conformation of the enzyme. These results lend additional support to the hypothesis that HICA is an allosteric enzyme that can adopt active and inactive conformations, the latter of which is stabilized by bicarbonate ion binding to a non-catalytic site.
Keywords: beta carbonic anhydrase, Haemophilus influenzae, allosteric site, bicarbonate, zinc metalloenzyme
Carbonic anhydrases (carbonate hydrolyase, EC 4.2.1.1) are metalloenzymes that catalyze the interconversion of CO2 and bicarbonate:
| (1) |
To date, there are five known convergently evolved forms of the enzyme: the α-form is found in animals and a few eubacteria (1); the β-form is found in bacteria, yeast, and plant chloroplasts (2); a γ-form is represented in a species from the Archaea domain (3); the δ- (4) and ζ-forms (5) have been identified in a marine diatom. Most are active as zinc-metalloenzymes, although the γ- and ζ-forms can utilize Fe2+ (6, 7) and Cd2+ (8) in vivo, respectively. X-ray crystallography (8-14) and/or EXAFS1 (15) reveals that most CA forms share a similar four-coordinate metal ion coordination sphere of the type His3(H2O) or Cys2His(H2O), where the ionizable water ligand presumably serves an essential role in the catalytic mechanism. However, X-ray crystallographic analysis of β-CAs reveal two distinct structural subclasses of this enzyme form. The first subclass, exemplified by the enzymes from Pisium sativum (PDB 1EKJ), Methanobacterium thermoautotrophicum (PDB 1G5C), and Mycobacterium tuberculosis Rv1284 (PDB 1YLK), has the canonical Cys2His(H2O) zinc coordination site. The second subclass of β-CAs, exemplified by the enzymes from Haemophilus influenzae (PDB 2A8C), Escherichia coli (PDB 1I6P), Porphyridium purpureum (PDB 1DDZ) and M. tuberculosis Rv3588c (PDB 1YM3), have a unique zinc coordination geometry, where the catalytically essential water molecule has been replaced by an Asp residue, resulting in a “closed” Cys2HisAsp coordination sphere. Despite the differences in X-ray crystal structures, kinetics studies of plant and bacterial β-CA (12, 16-22) show that both subclasses of enzyme are highly efficient catalysts for eq 1, with kcat values on the order of 105 s−1 and kcat/Km values on the order of 108 M −1 s−1. H. influenzae β-CA (HICA) has been shown to have highly cooperative pH-rate profiles for kcat and kcat/Km, and exhibits cooperative inhibition by HCO3− ion (12). In addition, X-ray crystallographic studies of both HICA (PDB 2A8D) and E. coli β-CA (ECCA, PDB 2ESF) reveal a noncatalytic binding site for bicarbonate ion. In contrast to HICA—a member of the second subclass of β-CAs—members of the first subclass of β-CA that have been thoroughly kinetically characterized (P. sativum (21), M. thrermoautotrophicum (22), and the highly sequence-homologous Arabidopsis thaliana (17) enzyme) are not inhibited by HCO3− and do not exhibit cooperative pH-rate profiles. Therefore it has been hypothesized that HICA and other members of the second structural subclass of β-CA represent a unique, allosterically regulated form of CA, in which the observed X-ray crystallographic structures probably represent the inactive, T-state (23) conformation of the enzyme (12). Presumably, the active, R-state (23) of these allosterically regulated β-CAs resembles the crystal structures of the first subclass of β-CAs. The working hypothesis is that enzyme activity of HICA and related proteins are regulated by a simple ligand exchange mechanism where Asp442 displaces the catalytically essential water molecule. The T-state conformation, where Asp44 is bound to the metal ion, is stabilized by the binding of HCO3− in the non-catalytic pocket, where it interacts with Trp39, Arg64, Tyr181, and the main chain carbonyl oxygen of Val47. The side chain of Val47 plays a critical role in displacing the noncatalytic HCO3− ion in the R state. (See Scheme 1). To date, no member of the HICA-like subclass of β-CA has been crystallized in a conformational state other than the hypothesized T-state, with a closed Cys2HisAsp metal coordination sphere. The existence of an R-state conformation for HICA is inferred from the observed catalytic kinetics, which is similar to other β-CAs, and the likely structure of the R-state can only indirectly inferred from the structures of the other, non-allosteric structural subclass of β-CA.
Scheme 1.
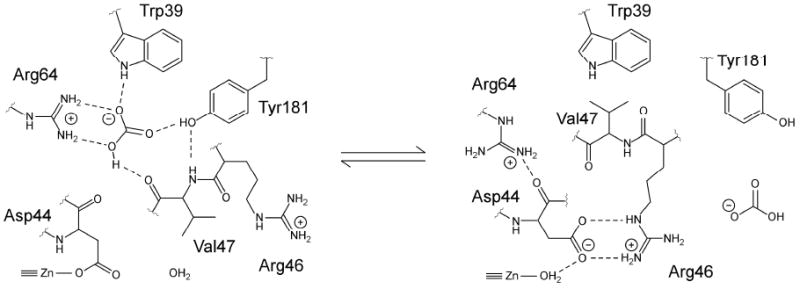
Structural schematic of key active site and noncatalytic bicarbonate binding site interactions in the hypothesized active (R-state) and inactive (T-state) conformations of HICA (12).
In order to further investigate the hypothesized allosteric regulation mechanism described by Scheme 1, we kinetically and structurally characterized some allosteric site variants of HICA, namely Trp39→Phe, Tyr181→Phe, and Asp44→Asn. We find that Trp39→Phe and Tyr181→Phe variants are catalytically competent and isostructural with wild-type HICA, and at least one of these binds HCO3− more weakly than the native enzyme. The Asp44→Asn variant, although inactive, appears to adopt an R-state-like conformation in the crystalline state. This is the first allosteric β-CA for which two distinct conformations have been observed in the same protein, and the results support the proposed allosteric regulation mechanism described by Scheme 1.
Experimental Procedures
Expression and Purification of Recombinant Enzymes
Site-directed mutations of the gene coding for HICA were constructed using megaprimer PCR (24) with Vent (New England Biolabs) or Pfu turbo (Stratagene) polymerase and commercial oligonucleotides (Integrated DNA Techonolgies). For variant W39F, a mutated oligonucleotide3 5′-AGCAACCAATGAAAAGGTAATGTG-3′ was paired with the 5′ oligonucleotide primer PHI1X (5′-TGCCCATGGATAAAATTAAACAACTCTTT-3) in the first PCR reaction to give a 129 bp product. This PCR product was used as a megaprimer in a second PCR reaction with the 3′ oligonucleotide primer PHI2X (5′-TGCCTGCAGTTATTATGTATTTTCAAGATG-3′) to create the final mutated HICA gene. The final PCR product was digested with NcoI and PstI (Promega) and ligated (Quick Ligase, New England Biolabs) into pTrc99a vector digested with the same enzymes. Variant D44N was constructed similarly using the mutated oligonucleotide 5′-GCACACGGCTATTAGAGCAACCA-3′. For variant Y181F, the mutated oligonucleotide 5′-ACGGCTGGGTATTTGATGTAAATG was combined with oligonucleotide primer PHI2X in the first round of PCR and the megaprimer product combined with PHI1X for the second round of PCR. Otherwise, construction of the Y181F expression plasmid was as described above. All expression plasmids were subjected to DNA sequencing (PE Biosystems ABI 310, BigDye 3.0 chemistry) of the entire HICA gene to verify introduction of the correct mutation.
Expression, purification, storage, and quantification of variant HICA proteins was exactly as described for the wild-type protein (12). Briefly, crude homogenates of overexpressed protein were purified to homogeneity by ion exchange (Q-Sepharose FF), hydrophobic interaction (butylsepharose FF), and gel exclusion chromatography (Superdex 200) using an AKTA FPLC (GE Healthcare), and quantified by ICP-OES (Perkin Elmer Optima SC 3000) for zinc at 213.856 nm.
Steady-State Kinetics Methods
Saturated solutions of CO2 were prepared by bubbling CO2 gas into water in a vessel maintained at 25.0 ± 0.1 °C, and dilutions prepared in the absence of air by coupling two gas-tight syringes as described by Khalifah (25). CO2 concentrations were calculated based on a 33.8 mM saturated solution at 25 °C (26).
All steady state kinetic measurements were made at 25 °C using a Hi-Tech SF-61DX2 stopped-flow spectrophotometer. Initial rates of CO2 hydration were measured using the changing pH-indicator method described previously (25, 27, 28). All stopped-flow kinetic studies were carried out in the presence of 250 mM Na2SO4, which was required for maximum enzyme stability and activity in dilute solution (12). Values of kcat and Km were determined by non-linear least squares fits to ν/[E] vs. [CO2] data using Origin 7.0 (Microcal). The kinetic constants kcat and kcat/Km are reported here on a per subunit basis. The pH-rate profiles of kcat and kcat/Km were fit to equations 2 or 3, as described previously. Equation 2 describes a pH dependence that requires the sequential loss of two protons to generate the active enzyme, where kobs is the observed value of kcat or kcat/Km, kmax is the maximal value of kcat or kcat/Km, and Ka1 and Ka2 correspond to the dissociation constants for the stepwise ionization of the enzyme, EH2 ⇆ EH− + H+ ⇆ E2− + 2 H+, where the doubly deprotonated form of the enzyme is the active species.
| (2) |
Equation 3 describes a pH dependence that requires the cooperative loss of four protons with identical pKa values to generate the active enzyme species, i.e., EH4 ⇆ E4− + 4 H+, where E 4− is the active species of enzyme, and kmax is the maximal value of kcat or kcat/Km.
| (3) |
Experimental data for kcat and kcat/Km were fit to the logarithmic forms of equations 2-3 using nonlinear least squares in Origin 7.0 (Microcal). Values of kcat and kcat/Km are reported here on a per subunit basis.
O-18 exchange kinetics methods
The rate of exchange of 18O between species of CO2 and water (Equations 4 and 5) is catalyzed by carbonic anhydrase (29):
| (4) |
| (5) |
Using an Extrel EXM-200 mass spectrometer with a membrane permeable to gases, we measured the isotopic exchange of 18O shown in Equations 2 and 3 at chemical equilibrium and 25 °C (29). Solutions contained 250 mM Na2SO4 for maximum enzyme stability and activity (12). No added buffer species were present.
This method determines two rates in the catalytic pathway. The first, defined as R1, is the rate of interconversion of CO2 and HCO3− at chemical equilibrium. The pH dependence of R1 in wild-type HICA is described by Equation 4,
| (6) |
where kmax is the maximal value of R1, KZn is the apparent acid dissociation constant of enzyme-bound Zn-OH2, and Ka is the apparent acid dissociation constant 4 groups with identical or nearly identical pKa values which must be deprotonated to generate the active species of enzyme (12).
For HICA-W39F, a modified version of equation 6 was required to model the data. Specifically, the apparent 4-proton dependence of the low pH limb of the data is apparently decoupled into two sequential two-proton events (12). Thus, in equation 7, the apparent Ka of equation 6 has been split into two separate Ka values, Ka1 and Ka2:
| (7) |
The 18O exchange method also determines a second rate, RH2O, which is the rate of release from the enzyme of water labeled with 18O. A proton from a donor group (BH+ in Equation 3) converts zinc-bound 18O-labeled hydroxide to zinc-bound 18O-labeled water, which readily exchanges with and is irreversibly diluted by unlabeled solvent H216O. For wild-type HICA the magnitude of RH2O can be interpreted in terms of the rate constant of proton transfer from a donor group to the zinc-bound hydroxide according to Equation 8, where kB is the maximal rate of proton transfer, KD is the apparent acid dissociation constant for groups that donate protons to the Zn-OH2 to enable the loss of 18O label to water, and KA has the same meaning as in equation 6 (12):
| (8) |
Experimental data for R1 and RH2O were fit to the logarithmic forms of equations 6-8 using nonlinear least squares in Origin 7.0 (Microcal). Values of R1/[E] and RH2O/[E] are reported here on a per subunit basis.
Substrate inhibition data for 18O exhange kinetics was fit to equation 9 as described previously (12) using non-linear least squares in Origin 7.0 (Microcal). In equation 9, kobs is the observed value of RH2O/[E], kmax is the maximal value of RH2O/[E], [S] is the combined concentration of CO2 + HCO3−, Keff is the CO2 + HCO3− concentration that yields half-maximal RH2O/[E], and Ki is the effective substrate/product inhibitor dissociation constant based on the concentration of CO2 + HCO3− present.
| (9) |
Crystallographic methods
Single, orthorhombic rods of HICA-W39F were grown overnight in 0.04 M ammonium phosphate, pH 4.3, 12.5 mg/mL protein at 22 °C using hanging drop vapor diffusion. Crystals were soaked in artificial mother liquor plus 30% glycerol for 30-60 seconds prior to flash cooling in liquid nitrogen. Data collection was performed with a rotating copper anode source (Rigaku RU-200) and an R-Axis IV image plate detector using 0.5° oscillations at a temperature of 95K. Data was processed using DENZO and SCALEPACK (30) (HKL Research). An initial molecular replacement solution was obtained by using wild-type HICA (PDB 2A8D) search model using EPMR (31). This initial solution was subjected to rigid-body refinement, simulated annealing, and multiple rounds of refinement using CNS 1.1 (32) and model-building in Coot (33). Final refinement was carried out using Refmac5 (34-36) using TLS (37, 38) with one TLS group per protein chain.
Single, monoclinic crystals of HICA-Y181F were obtained in 48 hr by the hanging drop method using 1.7 M (NH4)2SO4, 4% (v/v) PEG-400, 0.10 M HEPES, pH 7.50 and 12 mg/mL of protein at 4 °C. Individual crystals were transferred to artificial mother liquor supplemented with 25% glycerol for 30 seconds, followed by flash-cooling in liquid nitrogen. To prepare HICA-Y181F complexed with bicarbonate ion, individual crystals were soaked for 15 min in artificial mother liquor containing 25% glycerol and 100 mM NaHCO3. Data collection (PDB 3E28 and 3E2A) was performed with a rotating copper anode source (Rigaku RU-200) and an R-Axis IV image plate detector using 0.5° oscillations at a temperature of 95K. Data was processed using DENZO and SCALEPACK (30) (HKL Research). An initial molecular replacement solution was obtained by using wild-type HICA (PDB 2A8D) search model using EPMR (31). This initial solution was subjected to rigid-body refinement, simulated annealing, and multiple rounds of refinement using CNS 1.1 (32) and model-building in the program O (39) and Coot (33). Final refinement was carried out using Refmac5 (33) using TLS (37, 38) with one TLS group per protein chain.
Large, tetragonal crystals of HICA-D44N were obtained in 3 weeks by the hanging drop method using 0.8 M sodium acetate, 0.1 M MES, pH 6.5, 8 mg/mL protein at 22 °C. Individual crystals were transferred to artificial mother liquor supplemented with 30% glycerol (PDB 3E1V) or 30% glycerol and 100 mM NaHCO3 (PDB 3E1W) for 1-2 min, followed by flash-cooling in liquid nitrogen. Data collection for PDB 3E1V was performed with an Oxford Diffraction Xcalibur system using a PX Ultra Enhance Cu-Kα source and an Onyx CCD detector. Data was processed using MOSFLM (40) and SCALA (41). An initial molecular replacement solution was obtained by using wild-type HICA (PDB 2A8D) search model using EPMR. Data collection for PDB 3E1W was carried out at beamline F2 of CHESS at a wavelength of 0.95 Å. Data was processed using MOSFLM (40) and SCALA (41), and an initial molecular replacement solution as obtained by using a search model derived from a single dimer of wild-type HICA (PDB 2A8D) in Phaser (42). For both PDB 3E1V and 3E1W the initial solution was subjected to multiple rounds of refinement using Refmac5 (33) and model-building in Coot (33). Final refinement used TLS (33) with one TLS group per protein chain.
Data collection and refinement statistics are reported in Table 1.
Table 1.
Data collection and refinement statistics for HICA variantsa
| W39F (PDB 3E24) |
D44N (PDB 3E1V) |
Y181F (PDB 3E28) |
Y181F + 100 mM HCO3− (PDB 3E2A) |
|
|---|---|---|---|---|
| Data collection statistics | ||||
| Source | Rigaku RU-200 | Oxford Diffraction PX Enhance Ultra | Rigaku RU-200 | Rigaku RU-200 |
| Wavelength (Å) | 1.54 | 1.54 | 1.54 | 1.54 |
| Space Group | I212121 | P43212 | C2 | C2 |
| Cell parameters (Å) | 48.2, 129.6, 144.1 | 81.6, 81.6, 187.8 | 230.2, 144.9, 52.7 | 230.6, 145.5, 53.2 |
| (°) | 90, 90, 90 | 90, 90, 90 | 90, 93.8, 90 | 90, 93.8, 90 |
| Resolution (Å) | 29.5–2.30 (2.36-2.30) | 29.2–2.80 (2.87-2.80) | 29.8–2.50 (2.56-2.50) | 24.7–2.30 (2.36-2.30) |
| Unique reflections | 20243 | 16060 | 59536 | 77406 |
| Redundancy | 4.6 (4.3) | 10.2 | 4.0 (4.0) | 3.5 (3.4) |
| Completeness | 98.7 (98.4) | 98.6 (97.4) | 99.9 (100) | 100 (99.9) |
| Rsym (%) | 0.059 (0.314) | 0.093 (0.463) | 0.090 (0.520) | 0.065 (0.343) |
| 〈I〉/〈σI〉b | 22.2 (4.4) | 19.4 (4.1) | 19.6 (2.6) | 18.7 (3.6) |
| Refinement statistics | ||||
| Reflections in test set | 1990 | 812 | 2984 | 7808 |
| Rwork (%) | 0.191 | 0.194 | 0.206 | 0.205 |
| Rfree (%) | 0.236 | 0.233 | 0.256 | 0.244 |
| No. of atoms | ||||
| Protein | 2942 | 2890 | 9940 | 9906 |
| Ligand | 20 | 0 | 90 | 64 |
| Ion | 2 | 2 | 6 | 6 |
| Solvent | 138 | 21 | 172 | 158 |
| rmsd from idealc | ||||
| Bond distance (Å) | 0.010 | 0.012 | 0.012 | 0.011 |
| Bond angle (°) | 1.2 | 1.3 | 1.3 | 1.2 |
| Ramachandran plot outliers (%)d | 5.0 | 6.6 | 3.6 | 2.4 |
Results
Overexpression and purification of enzymes
Variant HICAs were expressed in E. coli at levels of 15-25% of total cellular protein as measured by scanning densitometry of crude lysates run out on SDS-PAGE gels. Elution of all variant proteins on a calibrated gel exclusion column (Superdex 200, GE Healthcare) was identical to that of wild-type HICA, indicating a tetrameric holoenzyme in solution with a molecular weight of approximately 100 kDa.
Structure of W39F, Y181F variants
Both HICA-W39F and Y181F are nearly identical in overall structure to wild-type HICA. Indeed these variants appear to adopt the T-state conformation of the wild-type enzyme. The biological unit appears to be a dimer of dimers. A dimerization interface extends the β-sheet core across a pair of monomers that define a fundamental dimer, and is in close proximity to the active site zinc ion. A tetramerization interface is orthogonal to the dimerization interface, and provides for the interaction and association of the fundamental dimers. This interface is characterized by a cluster of basic amino acids side chains: Arg160, Lys165, and Arg198 from one dimer, and Arg 124 from the neighboring dimer.
The asymmetric unit of HICA-W39F consists of one-half of each fundamental dimer in the biological unit (Figure 1A). There is no discernable electron density for the N-terminal α-helix (residues 1-33); presumably these residues are disordered in the crystal. As for wild-type HICA, the tetramerization interface shows two strong, approximately spherical electron density peaks concentrated in the region surrounded by the Arg160, Lys165, Arg198, and Arg 124′. These electron density peaks have been interpreted as phosphate ions, which are present in the crystallization solution (Figure 1B). Two additional, strong electron density peaks—also presumably phosphate ions—are also observed in special positions astride the dimerization interface and symmetry axis, near Arg64, Glu50, and a water molecule from each monomer (Figure 1C). The guanido group of Arg64 appears to occupy two alternate conformations around the phosphate ion, and is within hydrogen bonding distance of the phosphate oxygen atoms. A water molecule and the amide nitrogen of Ala49 also appear to stabilize phosphate ion binding in this pocket. Wild-type and W39F HICA have overall structures that superimpose to within 0.326 Å rms (43). The most significant structural deviations involve Ser45 and Arg46. The side chain of Ser45 has been reoriented by approximately 90° such that it protrudes somewhat into the bicarbonate binding site; as a consequence, the side chain of Arg46 has been reoriented 90° in the opposite direction so that it now protrudes more toward bulk solvent, away from the active site. The side chain of the variant residue Phe39 is oriented in the same plane and position as the wild-type Trp39. Asp44 remains ligated to the active site zinc ion, indicating that HICA-W39F has crystallized in the inactive, or T-state conformation. Because of the low pH of the crystallization conditions, it was not possible to soak HICA-W39F crystals with bicarbonate ion.
Figure 1.
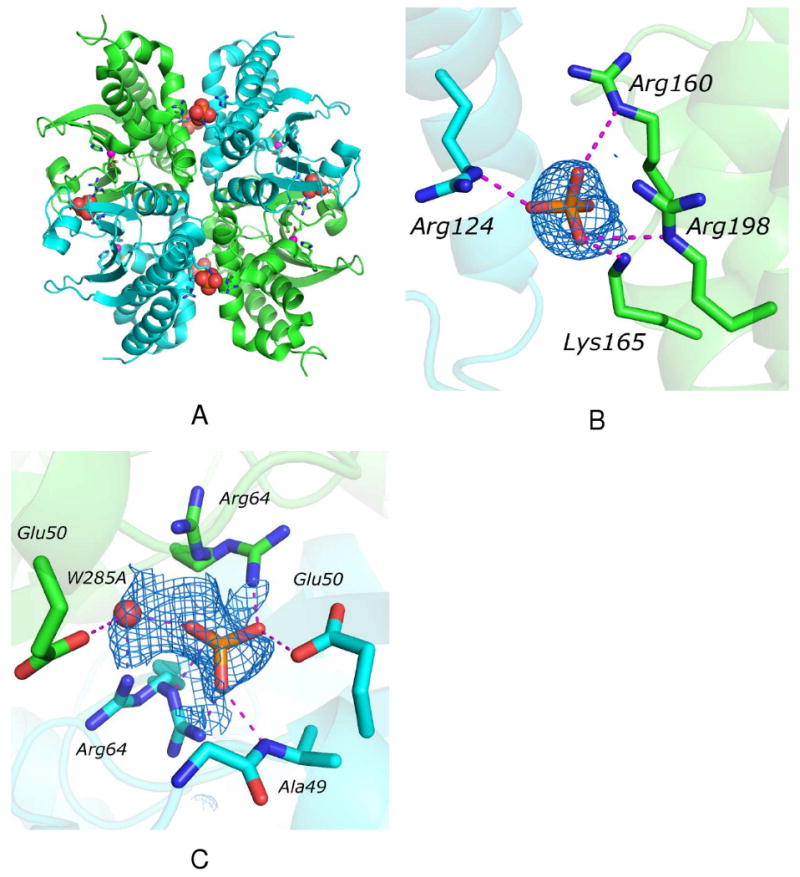
Crystal structure of HICA-W39F. Chain A is green, chain B is cyan. (A) Structure of the tetrameric biological unit. The tetramerization interface is oriented vertically; the dimerization interface is oriented horizontally. Phosphate ions are shown as space-filling models, zinc ions are magenta spheres, and residues interacting with phosphate and zinc ions are depicted as sticks. (B) Phosphate ion binding site on the tetramerization interface. Dashed magenta lines indicate heavy-atom distances <3.5 Å. The Fo-Fc omit map is contoured at 1.6 σ. (C) Phosphate ion binding site on the dimerization interface. Dashed magenta lines indicate heavy-atom distances <3.5 Å. The Fo-Fc omit map is contoured at 1.2 σ.
In the absence of HCO3−, HICA-Y181F (PDB 3E28) crystallizes in the same unit cell as wild-type HICA, with 1½ biological (tetrameric) units in the asymmetric unit (Figure 2A). Like HICA-W39F, HICA-Y181F shows six strong, approximately spherical electron density regions near Arg64 and Glu50 on the dimerization interface (2 on special positions between chains A and B), and an additional six, spherical electron density regions near the cluster of basic amino acid residues on the tetramerization interface). These have been interpreted as sulfate ions, which are present in the crystallization solution. In addition, there are six slightly weaker regions of spherical electron density in a hydrophobic pocket near the active site zinc ligand His98 formed by Val47, Val183, and Phe181. These regions appear to be modeled well by sulfate ions at half-occupancy, where the sulfate ion can accept hydrogen bonds from His98-Nδ1 and the main chain amide NH of Arg46. Wild-type and Y181F HICA have overall structures that superimpose to 0.374 Å rms (43). As for HICA-W39F, the most significant structure deviations involve the reorientation of the Ser45-Arg46 loop by about 90°, with the side chain of Ser45 protruding into the bicarbonate binding site, and Arg46 oriented more toward bulk solvent. The variant side chain, Phe181, is rotated out of the bicarbonate binding site toward the protein exterior compared to the wild type Tyr181. Asp44 remains ligated to the active site zinc ion, indicating that HICA-Y181F has crystallized in the inactive, or T-state conformation.
Figure 2.
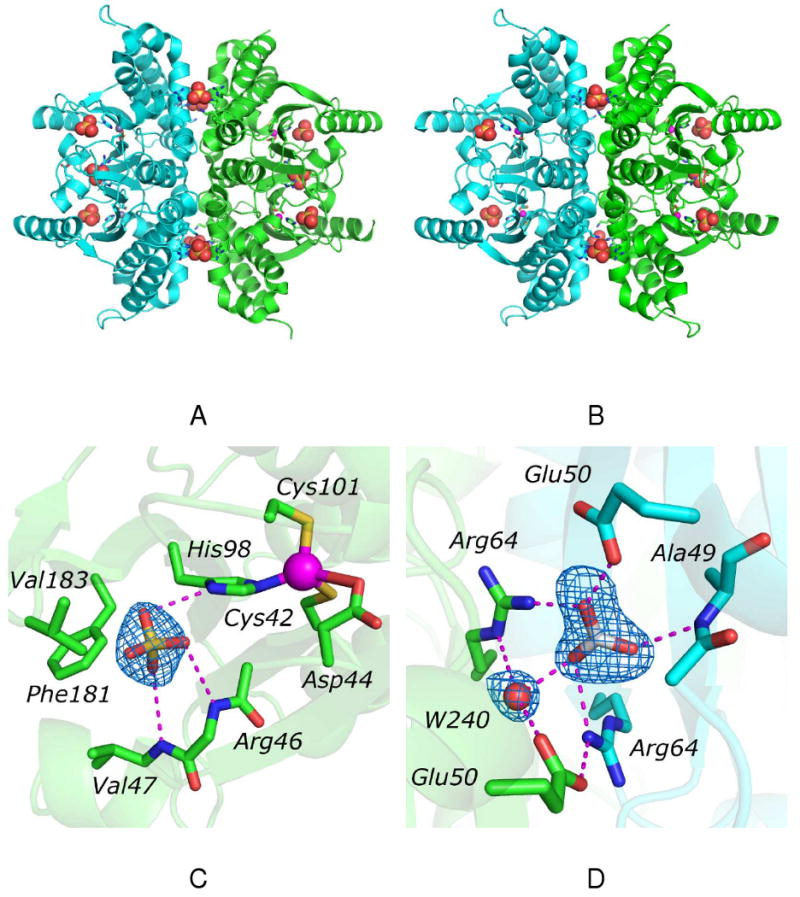
Crystal structure of HICA-Y181F. (A) Structure of the tetrameric biological unit in the absence of bicarbonate ion. The two dimers are colored green and cyan. The dimerization interface is oriented horizontally; the tetramerization interface is oriented vertically. Sulfate ions are shown as space-filling models, zinc ions are magenta spheres, and residues interacting with sulfate and zinc ions are depicted as sticks. (B) Structure of the tetrameric biological unit in the presence of bicarbonate ion. The two dimers are colored green and cyan. The dimerization interface is oriented horizontally; the tetramerization interface is oriented vertically. Sulfate and bicarbonate ions are shown as space-filling models, zinc ions are magenta spheres, and residues interacting with sulfate and zinc ions are depicted as sticks. (C) Sulfate ion binding site near the active site. Shown as sticks selected residues that interact with the sulfate or zinc ions. The zinc ion is depicted as a magenta sphere. The side chain of Arg46 is omitted for clarity. Dashed magenta lines indicate heavy-atom distances <3.5 Å. The Fo-Fc omit map is contoured at 2.0 σ (D) Bicarbonate ion binding site on the dimerization interface. Chain D is colored green; chain F is colored cyan. Bicarbonate ion and selected residues interacting with it are shown as sticks. Water-240 is depicted as a red sphere. Dashed magenta lines indicate heavy-atom distances <3.5 Å. The Fo-Fc omit map is contoured at 3.0 σ.
In the presence of 100 mM NaHCO3, the protein chain of Y181F HICA (PDB 3E2A) is isostructural with the same enzyme in the absence of HCO3− (Figure 2B). While the N-terminal helix can easily be visualized in the electron density map, residues 19-32, which are a bridging loop and short helical segment in wild-type HICA are poorly resolved, and presumably disordered in this crystal. Strong, spherical regions of electron density are identified and interpreted as sulfate ions. As for PDB 3E28, six sulfate ions are found on the tetramerization interface; another six sulfate ions are appear to be bound in the hydrophobic pocket near His98 (Figure 2C). All of these sulfate ions model well at full occupancy. A single, triangular, planar region of electron density, interpreted as a bicarbonate ion, is observed on the dimerization interface between chains D and F only (Figure 2D). No clear electron density interpretable as a bicarbonate ion is seen in any of the six allosteric sites in the asymmetric unit, nor in any of the other dimerization interfaces. The lone bicarbonate ion appears to interact with Arg64 from chains D and F. Glu50F interacts directly with the bicarbonate ion O3 atom, presumably acting as a hydrogen bond acceptor from the bicarbonate OH group, while Glu50E interacts with a water molecule (HOH 240) that is likely a hydrogen bond donor to the O1 atom of bicarbonate. The amide NH of Ala49 is positioned to donate a hydrogen bond to O2 of the bicarbonate ion.
Structure of D44N variant
The overall protein fold of D44N HICA (PDB 3E1V) is nearly identical to that of wild-type HICA, with an rms deviation of 0.407 Å. There do not appear to be any anions bound to the protein under these crystallization conditions. However, there are two regions of HICA-D44N where the polypeptide backbone differs markedly from the wild-type enzyme. The first of these is Asp185-Gly192, which in the wild type enzyme is the N-terminal half of the terminal, solvent-exposed β-strand in the extended β-sheet core of the protein dimer. In HICA-D44N, this stretch of residues has lost its β-sheet character and appears to be a less structured loop instead. This loop is remote located relatively far (17 Å) from the active site zinc ion or the allosteric binding site (10-18 Å), and is not likely to have any functional significance. The other region of HICA-D44N that differs markedly from the wild type enzyme is the segment Asp44-Pro48. In the wild type enzyme, this loop is organized to create space for a bicarbonate binding pocket site and an extensive hydrogen bonding network: Arg64, Tyr181, and Trp39 act as hydrogen bond donors to bicarbonate, and the carbonyl oxygen of Val47 acts as a hydrogen bond acceptor for the lone OH hydrogen of the bicarbonate ion (12). Compared to wild-type HICA, the side chain of Ser45 has swung away from the allosteric site toward bulk solvent, and Val47 has swung from the exterior of the protein directly into the bicarbonate binding pocket (Figure 3). The protrusion of Val47 into the bicarbonate binding pocket surely precludes bicarbonate binding because of its steric bulk. Indeed in HICA-D44N, the side chains of both Tyr181 and Arg64 have been evicted from the bicarbonate binding pocket, and the Nη2 of Arg64 is within hydrogen bonding distance (3.4 Å in 3E1V and 3.1 Å in 3E1W) of the carbonyl oxygen of Asn44. The position of Trp39 is relatively unchanged. Most significantly, Asn44 has detached from the zinc ion in HICA-D44N, and has apparently been replaced by a water molecule. This water molecule, HOH-231, is pinned between the amide NH of Gly-102 and the carbonyl (or possibly amide NH) of Asn44, and is a distant zinc ligand, with a Zn-O distance of 2.6 Å. (Figure 3). Crystals of D44N HICA soaked in 100 mM NaHCO3 (PDB 3E1W) show no additional electron density that can be interpreted as bicarbonate ion anywhere in the asymmetric unit. The structure obtained from this crystal is identical to that of D44N in the absence of HCO3− (data not shown).
Figure 3.
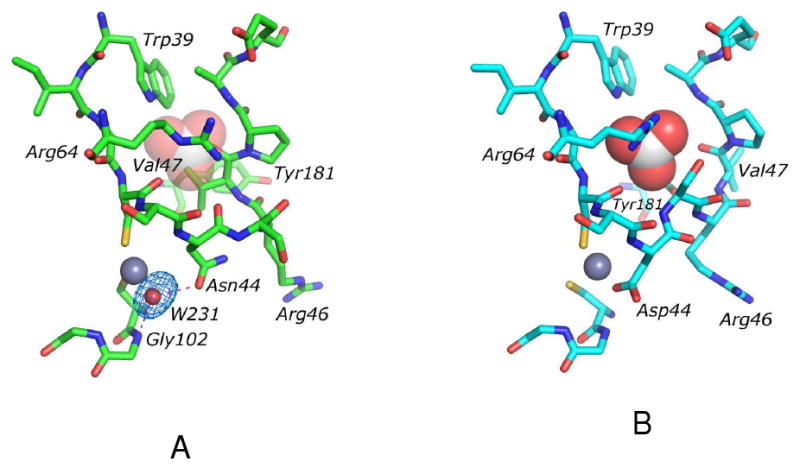
Identical views of the aligned crystal structures of (A) HICA-D44N (PDB 3E1V) and (B) wild-type HICA (PDB 2A8D). Residues in the allosteric and active sites are depicted as sticks. Zinc ions are depicted as grey spheres, water molecule (in HICA-D44N) is depicted as a red sphere, and bicarbonate ion (in wild type HICA) is depicted as space-filling model. For reference, the binding site of bicarbonate ion is depicted as partially transparent spheres in (A). Dashed magenta lines in (A) indicate heavy-atom distances <3.5 Å. The Fo-Fc omit map for water-231 is contoured at 3.0 σ. Key residues in the allosteric and active site are labeled. Structures were aligned using Pymol (43)
The Zn-O bond distance for HOH-231 in HICA-D44N is significantly longer (2.6 Å) than the mean value of 2.01 Å observed for zinc metalloenzymes with four-coordinate ligand spheres in protein structures determined to high resolution (44). However, it should be noted that there are 251 structures (about 10% of all tabulated structures) in the MESPEUS database (45) with zinc-water bond lengths of 2.6 Å or longer. While many of these examples include octahedral, pentacoordinate, or dinuclear zinc ligand spheres, there are at least three four-coordinate structures that are relevant to HICA-D44N. Human β-1 alcohol dehydrogenase (PDB 1HSZ) has a Cys2His(OH2) coordination sphere similar to HICA, with a reported zinc-water bond length of 2.8 Å. The human α-carbonic anhydrase II variants T200S (PDB 5CA2) (46) and V143G (PDB 7CA2) (47) have His3(OH2) zinc coordination spheres with reported zinc-water distances of 2.8 Å. For these latter two enzymes, it is postulated that the long Zn-O bond distance can be accounted for by partial occupancy of the zinc-water site by anions (possibly chloride or azide) present in the crystallization solution at small concentrations.
Thus, it seems there might be three potential explanations for the apparent 2.6 Å zinc-water bond length in HICA-D44N. The first, and simplest accounting of the observed electron density is that HICA-D44N has an unusually long zinc-water bond, perhaps stabilized by hydrogen bonding interactions with the amide NH of Gly102 (3.5 Å) and the carbonyl oxygen of Asn44 (2.8 Å). The second possibility is that acetate ion, present in the crystallization solution, is present at low occupancy at the open coordination site of zinc, and this serves to expand the electron density away from the zinc ion in a way that a water molecule refines to a longer than normal Zn-O distance. The third possibility is that Asn44 could be a weak, fourth ligand to zinc at low occupancy. Rotation of the Cα-Cβ bond of Asn44 by approximately 30° would bring the amide nitrogen within bonding distance of the zinc ion. The electron density of the amide group at low occupancy would likely cause refinement of the zinc-bound water to a longer than normal value. In any case, the electron density observed is adequately modeled by a solvent molecule at full occupancy, and it seems likely that water is the most probable ligand, and is present at high occupancy.
Steady-state kinetics
The pH-rate profiles of kcat and kcat/Km were measured for Y181F and W39F HICA (Figure 4). It was not possible to accurately measure the catalytic rate of HICA-Y181F below pH 8.25 because of low activity (as little as 1% or less of wild-type at pH values less than 8.25) of this variant. There was not a sufficient range of data available for quantification of the pH-rate data for HICA-Y181F. However, it should be noted that the enzyme does retain significant catalytic activity, especially at high pH. Therefore, it does not appear that the replacement of Tyr181 with Phe has directly affected the catalytic mechanism, although it may have had an indirect effect on the relative equilibrium distribution of the T-state of the enzyme, or the ability of the enzyme to be activated by sulfate ion. Sulfate ion at 200-250 mM is required for maximal activity of wild-type HICA in kinetic assays (12).
Figure 4.
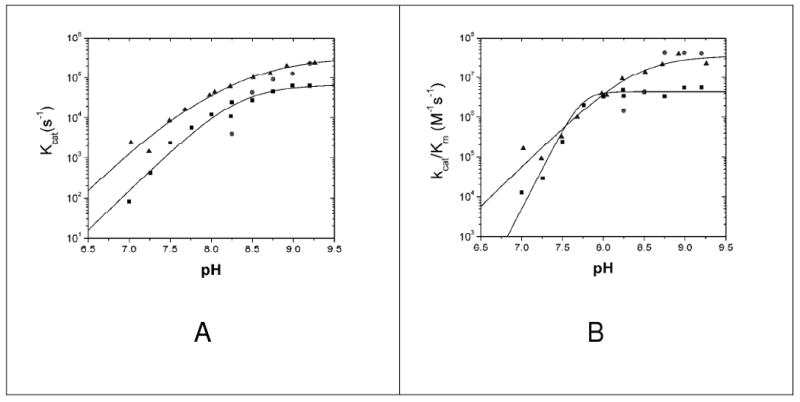
pH-rate profiles of kcat (A)and kcat/Km (B) for wild-type HICA (■), HICA-W39F (▲), and HICA-Y181F (●). Wild-type HICA data is from (12) where the kcat data was fit to equation 2, with kmax = 69 ± 29 ms−1, pKa1 = 8.3 ± 0.6, and pKa2 = 8.4 ± 0.6; kcat/Km data was fit to equation 3, where kmax = 4.3 ± 0.8 μM−1 s−1 and pKa = 7.74 ± 0.04. HICA-W39F kcat data was fit to equation 2, where kmax = 320 ± 140 ms−1, pKa1 = 7.5 ± 0.3, and pKa2 = 8.8 ± 0.3; kcat/Km data was also fit to equation 2, with kmax = 36 ± 20 μM−1 s−1, pKa1 = 8.4 ± 0.7, and pKa2 = 8.4 ± 0.8. Reaction conditions were 25 °C, 40 mM buffer (1-methylimidazole, pH 7.0-8.25; 1,2-dimethylimidazole, pH 8.25-9.25), 30-200 μM indicator dye (p-nitrophenol with 1-methylimidazole; m-cresol purple with 1,2-dimethylimidazole), 10 μM EDTA, 250 mM Na2SO4, and 0.4-15 μM enzyme)
The variant D44N had enzyme activity less than 0.2% of the wild-type enzyme (kcat < 200 s−1) at pH 7.0-9.25. The pH-rate profile of kcat for HICA-W39F is similar to wild-type enzyme, with a two-proton dependence that suggests that two H+ ions must be lost to form the active form of the enzyme (12). The pH-rate profile of kcat/Km for HICA-W39F does not show the highly cooperative pH dependence of wild-type HICA, but rather looks similar to the pH dependence of kcat. Both HICA-W39F and HICA-Y181F have maximal activities that are comparable to the wild-type enzyme.
Oxygen-18 Exchange kinetics
The W39F variant of HICA has a maximal values of R1/[E] and RH2O/[E], that are comparable to the wild type enzyme (Figure 5). As for wild-type enzyme, the pH profile of R1/[E] for W39F HICA can be interpreted in terms of two sets of two-proton ionizations at low pH, and a two-proton dependence at high pH (12). The pH profile of RH2O/[E] is also consonant with the wild-type enzyme (12). Under the optimal experimental conditions for wild-type HICA 18O kinetics HICA-Y181F has maximal R1/[E] and RH2O/[E] values < 1% of that of wild-type or W39F HICA. 18O exchange activity of HICA-D44N was not detectable under the experimental conditions used, and is estimated at < 0.2% of the wild type enzyme. Therefore, it was not possible to quantify meaningful 18O kinetics pH rate profiles for the Y181F or D44N variants.
Figure 5.
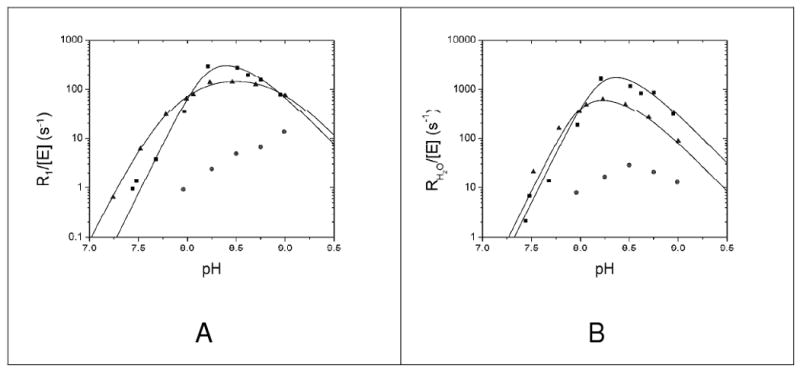
pH dependence of R1 (A) and RH2O (B) for wild-type HICA (■), HICA-W39F (▲), and HICA-Y181F (●). Wild-type HICA data is from (12); R1 data were fit to equation 6, where kmax = 490 ± 390 s−1, pKa = 8.2 ± 0.1, and pKZn = 8.6 ± 0.2; RH2O data were fit to equation 8, where kmax = 3200 ± 3700 s−1, pKa = 8.2 ± 0.1, and pKD = 8.5 ± 0.3. For HICA-W39F, R1 data were fit to equation 7, where kmax = 190 ± 20 s−1, pKa1 = 7.59 ± 0.04, pKa2 = 8.07 ± 0.04, and pKD = 8.91 ± 0.05; RH2O data were fit to equation 8 where kmax = 870 ± 530 s−1, pKa = 8.0 ± 0.1, and pKD = 8.5 ± 0.2. Reaction conditions were 25 °C, 2 mM CO2 + HCO3−, 10 μM EDTA, 250 mM Na2SO4, and 0.06-0.25 μM enzyme.
Substrate/product inhibition of RH2O (Figure 6) and R1 (not shown) are biphasic for both wild type HICA and HICA-W39F. At low CO2 + HCO3− concentrations, RH2O increases with substrate concentration, as expected, but at higher concentrations, a marked inhibitory effect is observed. This inhibitory component is cooperative, and is satisfactorily modeled by the cooperative binding of two molecules of inhibitor (presumably bicarbonate ion) to the enzyme (12). However, the apparent Ki for HCO3− for W39F for RH2O/[E] is 4.8 ± 1.1 times larger (76 ± 14 mM) than that for the wild-type enzyme (16 ± 2 mM) (12). This corresponds to a ΔΔG° at 25°C of 0.8-1.0 kcal/mol of apparent binding energy for bicarbonate ion between the wild type enzyme and HICA-W39F. This apparent change in ΔG° is consistent with the loss of a single stabilizing hydrogen bond contributed by Trp-39 to bicarbonate ion in the allosteric binding site. Values of RH2O (Figure 6) and R1 (not shown) for HICA-Y181F are very low under the assay conditions, less than 1% of wild-type at any substrate concentration.
Figure 6.
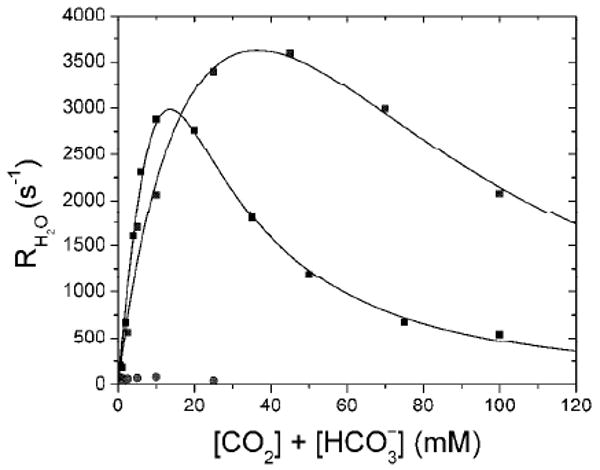
Substrate inhibition of R1 and RH2O for wild type HICA(■), HICA-W39F (▲), and HICA-Y181F (●). Data were fit to equation 9. Wild-type HICA data is from (12), where kmax = 34 ± 24 ms−1, Keff = 75 ± 55 mM, and Ki = 16 ± 2 mM. For HICA-W39F, where kmax = 7.1 ± 1.8 ms−1, Keff = 22 ± 9 mM, and Ki = 76 ± 14 mM. Reaction conditions were 25 °C, pH 8.75, 10 μM EDTA, 250 mM Na2SO4, and 0.06-0.25 μM enzyme.
Discussion
Anion binding to HICA variants
Both HICA-W39F and HICA-Y181F bind polyatomic anions (phosphate and sulfate, respectively) at a site on the tetramerization interface observed previously for wild-type HICA (12). However, these variants also bind phosphate or sulfate at a site in a crevice on the dimerization interface near the allosteric binding site, involving least one residue, Arg64, that is located near and interacts with bicarbonate in the proposed allosteric site in wild type HICA. In addition, HICA-Y181F can also bind sulfate ions a hydrophobic pocket near the active site. This latter sulfate ion binding site appears to be unique to HICA-Y181F. Figure 2C does not suggest any obvious role for this anion binding site in catalysis or inhibition of the enzyme.
HICA-Y181F does not appear to bind bicarbonate ion in the allosteric site at 100 mM, a concentration of bicarbonate ion that is observed to fully populate the allosteric binding site in wild-type HICA. This suggests that the alteration of Tyr181 to Phe at least qualitatively destabilizes the binding of bicarbonate to the allosteric site compared to the wild type enzyme. However, at 100 mM, bicarbonate ion apparently can bind to at least one of the anion binding sites on the dimerization interface, near residues Arg64 and Glu50 from each protein chain on the dimer interface. Although it is impossible to deduce this with certainty from a single structure, this binding feature is suggestive of a possible ingress route for bicarbonate ion from bulk solution to the allosteric site.
Activity of HICA-W39F
The overall catalytic efficiency of HICA-W39F at steady state is similar to that of wild type HICA, and the replacement of Trp39 with Phe does substantially change the pH-rate cooperativity of the enzyme. Specifically, the pH dependence of kcat appears to be controlled by the sequential ionization of two groups to generate the active form of the enzyme. In the wild type, the Ka values for these two groups are experimentally indistinguishable, but in In HICA-W39F these two pKa values have apparently diverged into two pKa values of approximately 7.5 and 8.8. A similar divergence of nearly coincident pKa values in wild-type enzyme is seen in the R1 data for HICA-W39F. The low pH limb of this data—which correlates to the pH dependence of the conformational changes that lead to active enzyme, not to the ionization state of catalytic residues (12)—is still highly cooperative for HICA-W39F. In wild-type HICA, the low pH limb of R1 (and RH2O) can be modeled by the cooperative loss of 4 protons with identical pKa values. It was previously suggested (12), based on the structure of the wild-type enzyme and the kinetics of bicarbonate inhibition, that this data might be more likely explained by the sequential ionization of two pairs of two cooperative protons with “crossed” pKa values, i.e., pKa2 < pKa1, with the average pKa near the observed value of 8.2 in Figure 5A. For HICA-W39F, it appears that the pKa values have “uncrossed” to experimentally distinguishable values of approximately 8.1 and 8.9 in the R1 profile. This result gives additional support to the original interpretation of the wild type HICA kinetic data that suggests that the fundamental allosteric unit is a dimer. The pH profiles of RH2O for wild type HICA and HICA-W39F are very similar, with apparent 4-proton ionizations controlling the low pH limb. The high pH limb of both R1 and RH2O appears to be consistent with the cooperative ionization of two protons from either zinc-bound water (R1) or a proton transfer group (RH2O), respectively; in each case the active form of the enzyme must be protonated for the HCO3− dehydration reaction measured by 18O exchange. In the case of HICA-W39F, the pKa of the zinc-bound water may have been perturbed upward slightly (from 8.6 in the wild type enzyme to 8.9 in HICA-W39F), but the pKa of the catalytic proton transfer group remains unchanged with a pKa of approximately 8.5 (Figure 5). The principal kinetic difference between wild type HICA and HICA-W39F is the pH profile of kcat/Km at steady state, where the wild type enzyme appears to be controlled by cooperative 4-proton ionization, and the variant enzyme appears to be better modeled by two sequential one-proton ionizations. This seems somewhat inconsistent with the remainder of the kinetic data; the origin of this inconsistency is not clear.
Structural homology of HICA-D44N and PSCA
Residues 39-50 of HICA-D44N and the homologous residues 157-168 of P. sativum CA (PSCA, PDB 1EKJ) can be nearly perfectly superimposed (Figure 7), to within 0.209 Å rms for main chain atoms. The degree of overlap of the main chain and side chain atoms in this loop, which encompasses the active and allosteric sites of HICA, is remarkable. Other than the sequence variation that distinguishes these two enzymes, the only substantial structural or conformational difference between HICA-D44N and P. sativum CA is the position of the side chain of Arg46. In PSCA, Arg46 interacts with Asp44 to orient it as a hydrogen bond acceptor for the water/hydroxide ion bound to the zinc ion: this ligand is an acetic acid or acetate in the crystal structure of PSCA (10) and is presumably a water molecule in the active enzyme. In HICA-D44N, Asn44 and 46 understandably do not interact, as the negative charge of Asp44 has been neutralized in this enzyme variant. Otherwise, HICA-D44N is a doppelganger for PSCA in the region depicted by Figure 7, including the interaction between Arg64 and the carbonyl oxygen of residue 44. This suggests that the observed conformation of HICA-D44N is not coincidental, but probably a reasonably stable one that can be populated by wild type HICA. We suggest that the structure of HICA-D44N is likely to be representative of the proposed R-state of the native enzyme.
Figure 7.
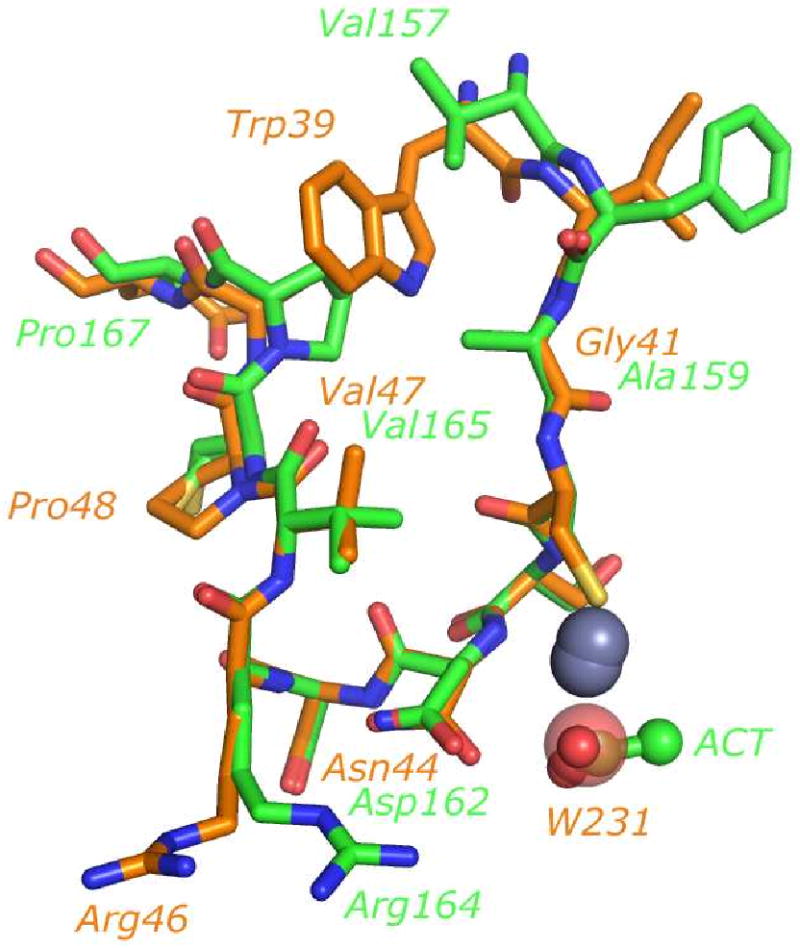
Alignment of allosteric and active site residues of PSCA (PDB 1EKJ) and HICA-D44N (PDB 3E1V). Chain G of PSCA and chain A of HICA-D44N were used in the alignment. Carbon atoms of PSCA are colored green; HICA-D44N carbon atoms are colored orange. Zinc ions are depicted as grey spheres (HICA zinc ion is lower one). An acetate ion (ACT) bound to PSCA is depicted as a ball and stick model. Water-231, bound near the zinc ion in HICA-D44N, is depicted as a transparent sphere. Some key residues are labeled; green labels are for PSCA, orange labels are for HICA-D44N.
To better understand the structural differences between wild type HICA and HICA-D44N structures an animated morph of these two conformations was created (48), and is available in Supporting Information. The principal motions depicted in the morph suggest a possible transition between the R-state (represented by HICA-D44N) and T-state (represented by wild type HICA) of the enzyme.
Figure 7 suggests three key structural elements that may distinguish between allosteric and non-allosteric β-carbonic anhydrases. The principal structural differences between PSCA and HICA-D44N are (1) the substitution of Ala for Gly41; (2) the substitution of Val for Trp39; and (3) the “slippage” of Pro48 by one residue in the C-terminal direction in PSCA. The substitution of Ala for Gly41 results in the Ala side chain projecting into the space that would be occupied by bicarbonate ion in the allosteric site. In addition, the repositioning of Pro48 in HICA to a position one residue later in the amino acid sequence in PSCA results in the reorientation of the proline ring so that it now also protrudes into the allosteric site, rather than pointing away from it as in HICA. The stereoechemical crowding by Ala159, Val165 and Pro167 appears to preclude bicarbonate binding in this pocket in PSCA. In addition, the substitution of Val for Trp39 eliminates one of the stabilizing hydrogen bonds for bicarbonate binding in HICA. This Val residue, together with Ala159, Val165, and Pro167 creates a hydrophobic cluster in PSCA which might serve to stabilize the active, R-state conformation of the enzyme.
Activity of HICA-D44N
While HICA-D44N appears to adopt an R-state-like conformation nearly identical to other, non-allosteric β-CAs, it has little or no catalytic activity. This result contrasts with the catalytic activity of a homologous variant (D34A) in M. thermoautotrophicum β-CA. For this enzyme the Ala variant has kcat and kcat/Km values that are ≈15% of the wild type enzyme, and this observation, combined with crystallographic data of the HEPES complex and the observation that kcat for Asp34 variants can be rescued with imidazole suggest that the active site Asp is more likely to act as a proton shuttle group than a general base or zinc-hydroxide orienting residue (49). This explanation seems inadequate for HICA, as the enzyme is minimally active even in the presence of 1-methyl imidazole and 1,2-dimethylimidazole buffers at steady state, and there is minimal CO2-HCO3− exchange activity (as measured by R1) at chemical equilibrium. If Asp44 were principally acting as a proton transfer group in HICA, one would expect RH2O to be markedly decreased, but R1 to be relatively unaffected, but this is not the case. Therefore, in HICA a role for Asp44 as a general base, hydroxide-orienting residue, or “gatekeeper” residue that excludes anions that cannot donate a hydrogen bond to Asp44 (10) must be considered.
Conclusion
The structural and kinetic evidence presented here provide additional support to the hypothesis that HICA is an allosteric enzyme that can adopt one of two conformations: an inactive T-state, and an active R-state, the equilibrium of which can be mediated by the binding of bicarbonate to an allosteric pocket not far from the active site zinc ion. Two results here are of special significance. The first is that the alteration of a key hydrogen bonding donor to the non-catalytic bicarbonate binding site, Trp39, to a non-hydrogen bonding analog (Phe) results in a significant increase in the observed Ki for bicarbonate inhibition of the enzyme at chemical equilibrium. Perhaps more compelling is that the alteration of the fourth zinc ligand, Asp44, to a non-nucleophilic analog results in HICA adopting a conformation that is eerily similar to non-allosteric β-carbonic anhydrases. The structure of HICA-D44N is likely the first β-CA of the Cys2HisAsp zinc ligation class to be crystallized in an R-state-like conformation.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Prof. Gino Cingolani, Upstate Medical University, Syracuse NY, and Dr. David Davies, Section on Molecular Structure, Laboratory of Molecular Biology, National Institutes of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD for assistance in the collection of X-ray diffraction data.
Footnotes
This work was supported in part by grants (to R.S.R.) from the National Science Foundation (MCB-0741396 and CHE-0418761), and upon research conducted at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the National Science Foundation and the National Institutes of Health/National Institute of General Medical Sciences under NSF award DMR-0225180, using the Macromolecular Diffraction at CHESS (MacCHESS) facility, which is supported by award RR-01646 from the National Institutes of Health, through its National Center for Research Resources.
Coordinates and structure factors have been deposited in the Protein Data Bank as entries 3E24, 3E1V, 3E1W, 3E28, and 3E2A
Abbreviations: CA, carbonic anhydrase; ECCA, Escherichia coli β-carbonic anhydrase; HICA, Haemophilus influenzae β-carbonic anhydrase; PSCA, Pisium sativum β-carbonic anhydrase; EXAFS; extended X-ray absorption fine structure; SDS-PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis; PCR, polymerase chain reaction; TB medium, Terrific Broth medium; LB medium, Luria-Bertani medium; IPTG, isopropyl-β-D-thiogalactoside; DEAE, diethylaminoethyl; HEPES, 4–(2-hydroxyethyl)piperazineethanesulfonic acid; ICP-OES; inductively coupled plasma-optical emission spectroscopy; EDTA, N,N,N′,N′-ethylenediaminetetraacetic acid; PCR, polymerase chain reaction; PEG-400, polyethylene glycol 400.
Unless otherwise noted, the numbering and identification of residues is referenced to that H. influenzae β-CA
Mutation site of oligonucleotides are bolded; restriction endonuclease sites for cloning are underlined.
Supporting Information Available: An animated graphic similar to figure 3 which depicts a morph of active- and allosteric site residues of the wild-type HICA structure (PDB 2A8D) and the HICA-D44N structure (PDB 3E1V) is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hewett-Emmett D. Evolution and distribution of the carbonic anhydrase gene families. EXS. 2000;90:29–76. doi: 10.1007/978-3-0348-8446-4_3. [DOI] [PubMed] [Google Scholar]
- 2.Smith KS, Jakubzick C, Whittam TS, Ferry JG. Carbonic anhydrase is an ancient enzyme widespread in prokaryotes. Proc Natl Acad Sci U S A. 1999;96:15184–15189. doi: 10.1073/pnas.96.26.15184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alber BE, Ferry JG. A carbonic anhydrase from the archaeon Methanosarcina thermophila. Proc Natl Acad Sci U S A. 1994;91:6909–6913. doi: 10.1073/pnas.91.15.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts SB, Lane TW, Morel FMM. Carbonic anhydrase in the marine diatom Thalassiosira weissflogii (Bacillariophyceae) J Phycol. 1997;33:845–850. [Google Scholar]
- 5.Lane TW, Saito MA, George GN, Pickering IJ, Prince RC, Morel FM. Biochemistry: a cadmium enzyme from a marine diatom. Nature. 2005;435:42. doi: 10.1038/435042a. [DOI] [PubMed] [Google Scholar]
- 6.Tripp BC, Bell CB, Cruz F, Krebs C, Ferry JG. A role for iron in an ancient carbonic anhydrase. J Biol Chem. 2004;279:6683–6687. doi: 10.1074/jbc.M311648200. [DOI] [PubMed] [Google Scholar]
- 7.MacAuley SR, Zimmerman SA, Apolinario EE, Evilia C, Hou YM, Ferry JG, Sowers KR. The Archetype gamma-Class Carbonic Anhydrase (Cam) Contains Iron When Synthesized in Vivo. Biochemistry. 2009;48:817–819. doi: 10.1021/bi802246s. [DOI] [PubMed] [Google Scholar]
- 8.Xu Y, Feng L, Jeffrey PD, Shi YG, Morel FMM. Structure and metal exchange in the cadmium carbonic anhydrase of marine diatoms. Nature. 2008;452:56–U53. doi: 10.1038/nature06636. [DOI] [PubMed] [Google Scholar]
- 9.Strop P, Smith KS, Iverson TM, Ferry JG, Rees DC. Crystal structure of the \“cab\”-type .beta class carbonic anhydrase from the archaeon Methanobacterium thermoautotrophicum. J Biol Chem. 2001;276:10299–10305. doi: 10.1074/jbc.M009182200. [DOI] [PubMed] [Google Scholar]
- 10.Kimber MS, Pai EF. The active site architecture of Pisum sativum .beta.-carbonic anhydrase is a mirror image of that of .alpha.-carbonic anhydrases. EMBO J. 2000;19:1407–1418. doi: 10.1093/emboj/19.7.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cronk JD, Endrizzi JA, Cronk MR, O'Neill J W, Zhang KY. Crystal structure of E. coli beta-carbonic anhydrase, an enzyme with an unusual pH-dependent activity. Protein Sci. 2001;10:911–922. doi: 10.1110/ps.46301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cronk JD, Rowlett RS, Zhang KYJ, Tu C, Endrizzi JA, Lee J, Gareiss PC, Preiss JR. Identification of a Novel Noncatalytic Bicarbonate Binding Site in Eubacterial beta-Carbonic Anhydrase. Biochemistry. 2006;45:4351–4361. doi: 10.1021/bi052272q. [DOI] [PubMed] [Google Scholar]
- 13.Mitsuhashi S, Mizushima T, Yamashita E, Yamamoto M, Kumasaka T, Moriyama H, Ueki T, Miyachi S, Tsukihara T. X-ray structure of .beta.-carbonic anhydrase from the red alga, Porphyridium purpureum, reveals a novel catalytic site for CO2 hydration. J Biol Chem. 2000;275:5521–5526. doi: 10.1074/jbc.275.8.5521. [DOI] [PubMed] [Google Scholar]
- 14.Covarrubias AS, Larsson AM, Hogbom M, Lindberg J, Bergfors T, Bjorkelid C, Mowbray SL, Unge T, Jones TA. Structure and function of carbonic anhydrases from Mycobacterium tuberculosis. J Biol Chem. 2005;280:18782–18789. doi: 10.1074/jbc.M414348200. [DOI] [PubMed] [Google Scholar]
- 15.Cox EH, McLendon GL, Morel FMM, Lane TW, Prince RC, Pickering IJ, George GN. The active site structure of Thalassiosira weissflogii carbonic anhydrase 1. Biochemistry. 2000;39:12128–12130. doi: 10.1021/bi001416s. [DOI] [PubMed] [Google Scholar]
- 16.Rowlett RS, Chance MR, Wirt MD, Sidelinger DE, Royal JR, Woodroffe M, Wang YFA, Saha RP, Lam MG. Kinetic and Structural Characterization of Spinach Carbonic Anhydrase. Biochemistry. 1994;33:13967–13976. doi: 10.1021/bi00251a003. [DOI] [PubMed] [Google Scholar]
- 17.Rowlett RS, Tu C, McKay MM, Preiss JR, Loomis RJ, Hicks KA, Marchione RJ, Strong JA, Donovan GS, Jr, Chamberlin JE. Kinetic characterization of wild-type and proton transfer-impaired variants of beta-carbonic anhydrase from Arabidopsis thaliana. Arch Biochem Biophys. 2002;404:197–209. doi: 10.1016/s0003-9861(02)00243-6. [DOI] [PubMed] [Google Scholar]
- 18.Rowlett RS, Tu C, Murray PS, Chamberlin JE. Examination of the role of Gln-158 in the mechanism of CO2 hydration catalyzed by beta-carbonic anhydrase from Arabidopsis thaliana. Arch Biochem Biophys. 2004;425:25–32. doi: 10.1016/j.abb.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 19.Covarrubias AS, Bergfors T, Jones TA, Hogbom M. Structural mechanics of the pH-dependent activity of beta-carbonic anhydrase from Mycobacterium tuberculosis. J Biol Chem. 2006;281:4993–4999. doi: 10.1074/jbc.M510756200. [DOI] [PubMed] [Google Scholar]
- 20.Johansson IM, Forsman C. Solvent hydrogen isotope effects and anion inhibition of CO2 hydration catalysed by carbonic anhydrase from Pisum sativum. Eur J Biochem. 1994;224:901–907. doi: 10.1111/j.1432-1033.1994.00901.x. [DOI] [PubMed] [Google Scholar]
- 21.Johansson IM, Forsman C. Kinetic studies of pea carbonic anhydrase. Eur J Biochem. 1993;218:439–446. doi: 10.1111/j.1432-1033.1993.tb18394.x. [DOI] [PubMed] [Google Scholar]
- 22.Smith KS, Cosper NJ, Stalhandske C, Scott RA, Ferry JG. Structural and kinetic characterization of an archaeal .beta.-class carbonic anhydrase. J Bacteriol. 2000;182:6605–6613. doi: 10.1128/jb.182.23.6605-6613.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monod J, Wyman J, Changeux JP. On the Nature of Allosteric Transitions: A Plausible Model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 24.Sarkar G, Sommer SS. The Megaprimer Method of Site-Directed Mutagenesis. BioTechniques. 1990;8:404–407. [PubMed] [Google Scholar]
- 25.Khalifah RG. Carbon dioxide hydration activity of carbonic anhydrase. I Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem. 1971;246:2561–2573. [PubMed] [Google Scholar]
- 26.Pocker Y, Bjorkquist DW. Comparative studies of bovine carbonic anhydrase in water and water-d2. Stopped-flow studies of the kinetics of interconversion of carbon dioxide and bicarbonate(1-) ion. Biochemistry. 1977;16:5698–5707. doi: 10.1021/bi00645a008. [DOI] [PubMed] [Google Scholar]
- 27.Ghannam AF, Tsen W, Rowlett RS. Activation parameters for the carbonic anhydrase II-catalyzed hydration of carbon dioxide. J Biol Chem. 1986;261:1164–1169. [PubMed] [Google Scholar]
- 28.Rowlett RS, Gargiulo NJ, III, Santoli FA, Jackson JM, Corbett AH. Activation and inhibition of bovine carbonic anhydrase III by dianions. J Biol Chem. 1991;266:933–941. [PubMed] [Google Scholar]
- 29.Silverman DN. Carbonic anhydrase: oxygen-18 exchange catalyzed by an enzyme with rate-contributing proton-transfer steps. Methods Enzymol. 1982;87:732–752. doi: 10.1016/s0076-6879(82)87037-7. [DOI] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor M. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 31.Kissinger CR, Gehlhaar DK, Fogel DB. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 2):484–491. doi: 10.1107/s0907444998012517. [DOI] [PubMed] [Google Scholar]
- 32.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 33.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 34.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 35.Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr D. 1999;55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- 36.Pannu NS, Murshudov GN, Dodson EJ, Read RJ. Incorporation of prior phase information strengthens maximum-likelihood structure refinement. Acta Crystallogr D. 1998;54:1285–1294. doi: 10.1107/s0907444998004119. [DOI] [PubMed] [Google Scholar]
- 37.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 38.Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Macromolecular Crystallography, Pt D. 2003:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- 39.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallographica Section A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 40.Leslie AGW. Recent changes to the MOSFLM package for processing film and image data. Joint CCP4 and ESF-EACMB Newsletter on Protein Crystallography 1992 [Google Scholar]
- 41.CCP4 (Collaborative Computational Project, N. The CCP4 Suite: Programs for protein crystallography. Acta Crystallographica Section D- Biological Crystallography. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 42.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 44.Harding MM. Small revisions to predicted distances around metal sites in proteins. Acta Crystallogr D. 2006;62:678–682. doi: 10.1107/S0907444906014594. [DOI] [PubMed] [Google Scholar]
- 45.Hsin K, Sheng Y, Harding MM, Taylor P, Walkinshaw MD. MESPEUS: a database of the geometry of metal sites in proteins. J Appl Crystallogr. 2008;41:963–968. [Google Scholar]
- 46.Krebs JF, Fierke CA, Alexander RS, Christianson DW. Conformational mobility of His-64 in the Thr-200 .fwdarw Ser mutant of human carbonic anhydrase II. Biochemistry. 1991;30:9153–9160. doi: 10.1021/bi00102a005. [DOI] [PubMed] [Google Scholar]
- 47.Alexander RS, Nair SK, Christianson DW. Engineering the hydrophobic pocket of carbonic anhydrase II. Biochemistry. 1991;30:11064–11072. doi: 10.1021/bi00110a008. [DOI] [PubMed] [Google Scholar]
- 48.Krebs WG, Gerstein M. The morph server: a standardized system for analyzing and visualizing macromolecular motions in a database framework. Nucleic Acids Res. 2000;28:1665–1675. doi: 10.1093/nar/28.8.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith KS, Ingram-Smith C, Ferry JG. Roles of the conserved aspartate and arginine in the catalytic mechanism of an archaeal .beta.-class carbonic anhydrase. J Bacteriol. 2002;184:4240–4245. doi: 10.1128/JB.184.15.4240-4245.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Engh RA, Huber R. Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallographica Section A. 1991;47:392–400. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.