

Abstract
A lack of consensus exists regarding the relative rates of NRAS and BRAF mutations in the radial (RGP) and vertical (VGP) growth phases of individual melanoma tumors. This study was conducted to test the hypothesis that mutations are acquired with progression from RGP to VGP. Using laser capture microdissection, pure tumor DNA was obtained from fifteen in-situ melanomas, and from the RGP and VGP of twenty-nine invasive tumors. NRAS exon 2 and BRAF exon 15 DNA were amplified by PCR and sequenced. Mutations were present in six of fifteen in-situ melanomas (40%). Sixteen of twenty-nine invasive tumors exhibited RGP mutations (55.2%); 22 showed VGP mutations (75.9%). Paired RGP/VGP mutation analysis revealed a trend toward discordance in the distribution of mutations, favoring VGP localization (p = 0.07). Twelve of fifteen samples with mutations in both phases had an increased proportion of mutated DNA in the VGP, measured on DNA chromatograms (p = 0.08). Limitations of this study include a relatively small sample cohort selected for technical reasons from a larger population, presenting the risk of selection bias. These concerns notwithstanding, our findings support the hypothesis that NRAS and BRAF mutations increase with tumor progression from superficial to invasive disease.
Keywords: NRAS, BRAF, RGP, VGP, mutation
Introduction
Cancer develops via a succession of genetic alterations involving pathways which regulate key cellular functions such as apoptosis, growth signaling, DNA repair, and angiogenesis (Hanahan and Weinberg, 2000; Hahn and Weinberg, 2002). Clark has delineated clinical and histopathologic stages of progression for cutaneous melanoma, from precursor neoplasm to in-situ melanoma (also known as the radial growth phase or RGP) to invasive or vertical growth phase (VGP) tumor (Clark et al., 1984). As molecular and genetic abnormalities are identified in melanomas, researchers attempt to identify their origins along the clinical and histopathologic progression model. Mutations of RAS and RAF genes, particularly NRAS exon 2 and BRAF exon 15, have been identified with high frequency in nevi, cutaneous melanomas, and melanoma metastases (Davies et al., 2002; Pollock et al., 2003; Uribe et al., 2003; Houben et al., 2004; Kumar et al., 2004; Akslen et al., 2005; Goydos et al., 2005; Edlundh-Rose et al., 2006; Goel et al., 2006). These mutations lead to amino acid substitutions within the NRAS and BRAF proteins, resulting in constitutive activation of growth signaling through the mitogen-activated protein kinase pathway (Beeram et al., 2005).
A number of studies have been conducted to establish the stage of melanoma at which these mutations are acquired. There is general agreement that pure in-situ melanomas carry a low prevalence of NRAS and BRAF mutations compared to rates seen in other progression phases (Dong et al., 2003; Yazdi et al., 2003; Poynter et al., 2006). In contrast, data from more advanced melanomas, i.e. invasive primary tumors and metastases, have been inconclusive, with some studies showing acquisition of mutations from RGP to VGP and beyond, while others report stable genotypes across all progression stages of an individual tumor (Demunter et al., 2001; Omholt et al., 2002; Dong et al., 2003; Omholt et al., 2003). Differences in patient populations, methods of tumor acquisition, as well as variation in mutation identification procedures and types of mutations tested limit comparison between studies. As a result, the question of whether NRAS and BRAF mutations occur at the earliest tumor stages or are acquired with progression remains unanswered.
The present study attempts to clarify the literature by comparing NRAS and BRAF mutation rates in pure in-situ tumors, and the RGP and VGP of primary invasive melanomas. Methods incorporate laser capture microdissection to assure maximum purity of tumor DNA. Our results show an incremental increase in mutation rates between in-situ melanomas, the RGP of invasive melanomas, and the VGP of the same tumors. In addition, we demonstrate, in tumors with mutations in both phases, an increase in the percentage of mutated DNA in the VGP portion of the tumor. We conclude that NRAS exon 2 and BRAF exon 15 mutations are acquired as melanomas progress from an in-situ to an invasive stage.
Results
Study population
The study objective required the identification and microdissection of melanomas with distinct radial and vertical growth phases. To this end, 285 consecutive entries into the Melanoma Tumor Bank through the M. D. Anderson Cancer Center (MDACC) Melanoma SPORE were screened. Within this group, there were 187 tumors in which both the RGP and VGP could be identified. For an invasive melanoma to qualify for this study, it was required that both growth phases be least 1 mm in least dimension (for adequate tumor cell numbers) and that the two phases be distinct from each other, i.e., sites of overlapping RGP and VGP were avoided. In all, 34 cases were identified and microdissected as separate RGP and VGP samples. A complete set of sequence results, consisting of BRAF exon 15 and NRAS exon 2 for both growth phases, was obtained for 29 of the 34 RGP/VGP pairs.
Seventeen in situ melanomas were identified from the same sample set. Two of these were acral lentigenous in situ melanomas and were excluded from the analysis based on evidence in the literature that the development of this subset of cutaneous melanomas follows a different genetic path than that of melanomas which develop in the more common sites. Therefore, fifteen in situ tumors were analyzed, all of which yielded full sequence results.
Overall mutation rates
DNA sequences for NRAS exon 2 and BRAF exon 15 were obtained for the fifteen in-situ melanomas and twenty-nine paired RGP and VGP samples described above (Fig. 1). Clinical characteristics of the study population are given in Table 1.
Figure 1.

Microdissection of melanoma cells from the radial and vertical growth phases of a cutaneous melanoma. The sample represents #26 in Table 2. Microdissected tumor cells from the VGP (left) had invaded the dermis whereas RGP tumor cells (right) were localized within the epidermis. Hematoxylin and eosin staining, modified for laser capture microdissection; scale bar = 100 um.
Table 1.
Clinical features of patients and tumors
| Characteristic | Invasive Melanomas n (%) |
In-situ Melanomas n (%) |
|---|---|---|
| Age | ||
| ≤ 50 | 12 (41.4) | 6 (40.0) |
| > 50 | 17 (58.6) | 9 (60.0) |
| Gender | ||
| Female | 9 (31.0) | 6 (40.0) |
| Male | 20 (69.0) | 9 (60.0) |
| Location | ||
| Trunk | 11 (37.9) | 7 (46.7) |
| Upper extremity | 10 (34.5) | 3 (20.0) |
| Lower extremity | 6 (20.7) | 4 (26.7) |
| Head and neck | 2 (6.9) | 1 (6.6) |
| Melanoma type | ||
| Superficial spreading | 29 (100.0) | 4 (26.7) |
| Lentigo maligna | 0 (0) | 3 (20.0) |
| Not specified | 0 (0) | 8 (53.3) |
| Breslow thickness | ||
| <1.0 mm | 13 (44.8) | |
| 1.1-2.0 mm | 13 (44.8) | |
| 2.1-3.0 mm | 3 (10.4) | |
| >3.0 mm | 0 (0) | |
| Clark level | ||
| I | 0 (0) | |
| II | 2 (6.9) | |
| III | 17 (58.6) | |
| IV | 10 (34.5) | |
| V | 0 (0) | |
| Ulceration | ||
| Yes | 4 (13.8) | |
| No | 25 (86.2) | |
| Metastasis | ||
| Yes | 4 (13.8) | |
| No | 25 (86.2) |
Mutations were identified in six of the fifteen in-situ melanomas (40%). Five of these were BRAF T1799A point mutations. Additionally, one case had both an NRAS A182G and BRAF GT1798AG mutation. Twenty-three of the twenty-nine invasive melanomas (79.3%) carried mutations, a proportion significantly higher than that of the in situ tumors (p = 0.009, Chi-square). Sixteen tumors harbored mutations in the RGP (55.2%) and twenty-two in the VGP (75.9%). There were nine NRAS and sixteen BRAF mutations including two tumors in which both exons carried a mutation.
Paired RGP/VGP mutation rates
A listing of DNA sequence results for the twenty-nine RGP/VGP pairs is shown in Table 2. Nine tumors (31.0%) showed discordance in genotype between the two growth phases. Eight of these carried wild type DNA in the RGP and mutated DNA in the VGP. A single discordant pair had the reverse pattern, i.e., mutated RGP DNA and wild type VGP DNA. The exact McNemar's test was used to determine if the apparent skewing of discordance toward acquisition of mutations in the VGP was significant. (The presence in sample #4 of both NRAS and BRAF mutations in different locations precluded assignment to the categories required for this test; this sample was excluded from the analysis). Results yielded a p-value of 0.07, approaching significance and supporting the hypothesis of a gain in mutations accompanying dermal invasion of the tumor.
Table 2.
Mutational status of paired radial and vertical growth phase tumor samples
| Sample | NRAS exon 2 | BRAF exon 15 | ||
|---|---|---|---|---|
| RGP | VGP | RGP | VGP | |
| Discordant | ||||
| 1 | WT | C181A | WT | WT |
| 2 | WT | C181A | WT | WT |
| 3 | WT | A182G | WT | GT1798AA |
| 4 | WT | C181A | T1799A | T1799A |
| 5 | WT | WT | WT | T1799A |
| 6 | WT | WT | WT | T1799A |
| 7 | WT | WT | WT | T1799A |
| 8 | WT | WT | WT | TG1799AA |
| 9 | WT | WT | T1799A | WT |
| Concordant | ||||
| 10 | C181A | C181A | WT | WT |
| 11 | C181A | C181A | WT | WT |
| 12 | A182G | A182G | WT | WT |
| 13 | AA182TG | AA182TG | WT | WT |
| 14 | A183T | A183T | WT | WT |
| 15 | WT | WT | T1790A | T1790A |
| 16 | WT | WT | T1799A | T1799A |
| 17 | WT | WT | T1799A | T1799A |
| 18 | WT | WT | T1799A | T1799A |
| 19 | WT | WT | T1799A | T1799A |
| 20 | WT | WT | T1799A | T1799A |
| 21 | WT | WT | T1799A | T1799A |
| 22 | WT | WT | T1799A | T1799A |
| 23 | WT | WT | T1799A | T1799A |
| 24 | WT | WT | WT | WT |
| 25 | WT | WT | WT | WT |
| 26 | WT | WT | WT | WT |
| 27 | WT | WT | WT | WT |
| 28 | WT | WT | WT | WT |
| 29 | WT | WT | WT | WT |
WT, wild type
Proportions of mutated DNA in concordant RGP/VGP pairs
The analysis above included only pairs in which one of the growth phases was entirely wild type. The results, supporting the acquisition of mutations with progression, suggested that in cases where mutations existed in both growth phases, the mutation rate would be higher in cells from the VGP. To explore this possibility, we examined the sequencing chromatograms to estimate the relative amounts of mutated and wild type DNA in a given tumor sample. This approach was first validated using DNA from melanoma cells lines that carry only mutated (A375) or wild type (MeWo) BRAF DNA. Mixtures of known concentrations of DNA from the two cell lines were prepared to obtain 0%, 25%, 50%, 75%, and 100% mutated DNA. The mixtures were amplified by PCR and sequenced using the same procedures as for the melanoma tissue samples. As shown in Figure 2A, the proportions of mutated and wild type nucleotide peaks on the DNA sequencing chromatograms correspond to their respective proportions in the cell line DNA mixtures.
Figure 2.

Acquisition of mutations in the VGP as determined by sequencing chromatograms. (A) Predetermined amounts of BRAF T1799A mutated DNA were mixed with wild type DNA. The contribution of the mutated DNA to the chromatogram reflects the percentage in the DNA mixture. The red peak (T) is wild type; the green peak (A) represents the single nucleotide mutation. (B) Three representative cases of DNA isolated from paired RGP/VGP samples, amplified with primers for BRAF exon 15, and sequenced. Measurement of the chromatograms reveals an increased percentage of T1799A mutations in the VGP DNA.
Having validated this approach, DNA chromatogram measurements comparing the percentage of mutated DNA in the RGP /VGP pairs were made for the fifteen cases in which both growth phases were mutated (samples #10-23 and #4 in Table 2). Representative pairs with their respective mutation rates are shown in Fig 2B; results for all fifteen cases are charted in Fig. 3. Twelve of the fifteen tumors (80%) had an increased percentage of mutated DNA in the VGP compared to the RGP, the increment ranging from 4.8% to 52.4%. The mean percentage of mutated DNA was 39.4% for RGP tumors and 52% for VGP tumors. Results of paired t-test analysis of these data approached significance with a p-value of 0.08. Although the actual percentages reported should be viewed as approximations, these findings support the overall conclusion of an increased proportion of mutated cells in the VGP and are in agreement with the hypothesis of acquisition of mutations in the course of melanoma progression.
Figure 3.
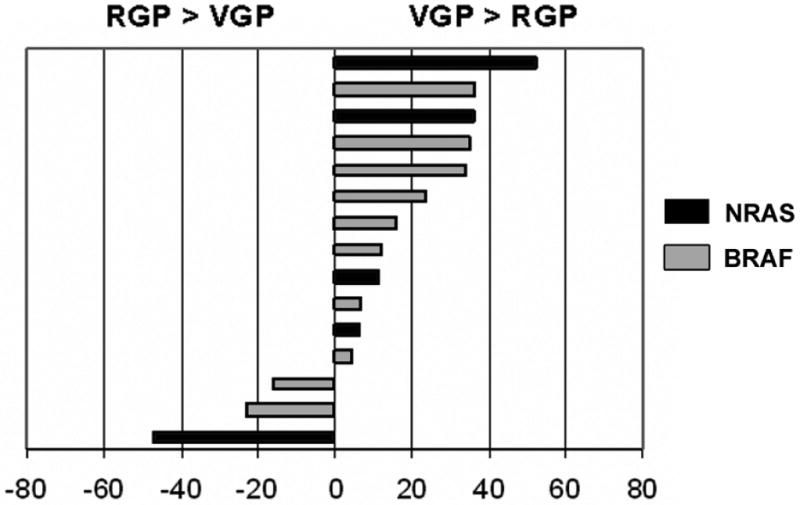
Differences in the percentage of mutated DNA between the RGP and VGP of individual melanomas. Included in this chart are fifteen tumors with identical mutations in both growth phases, representing samples #4 and #10-23 from Table 2. Entries are calculated as (% mutated VGP DNA) minus (% mutated RGP DNA). An increased proportion of mutated DNA is found in the VGP of twelve of the fifteen tumors.
Discussion
The results of this study demonstrate that mutations of NRAS exon 2 and BRAF exon 15 increase in frequency with progression of primary melanoma. We report an incremental increase in NRAS and BRAF mutation rates along the melanoma progression model from purely in-situ melanomas, to the RGP of invasive melanomas, to the VGP of invasive melanomas. Upon examination of mutations within the RGP and VGP of individual tumors, eight of the nine discordant cases carried a mutation in the VGP that was not identified in the RGP. Finally, 80% of tumors in which both growth phases were mutated showed increased proportions of mutated DNA in the VGP compared to the RGP.
Variations in mutation rates between studies notwithstanding, our results are in general agreement with the work of other investigators who found lower NRAS and BRAF mutation rates in purely in-situ melanomas compared to invasive melanomas (Ball et al., 1994; Dong et al., 2003; Yazdi et al., 2003; Poynter et al., 2006). In contrast, our data describing mutational gain from RGP to VGP of the same tumor diverge from findings of other studies employing similar methodology. Olmholt compared the presence of NRAS and BRAF mutations in the RGP and VGP of invasive melanomas and found that all pairs carried identical mutations (Omholt et al., 2002; Omholt et al., 2003). Notably, the samples were selected for the presence of mutations in the VGP and for a patient history of distant metastasis. Similarly, Demunter reported concordance of NRAS mutations in twenty-five RGP/VGP pairs (Demunter et al., 2001). As in the Olmholt studies, the samples were retrospectively selected for VGP mutations and at least one-third of the patients had Stage IV disease. Thus, the 31% RGP/VGP discordance rate that we report differs substantially from the current literature. While the reason for this discrepancy is not entirely clear, part of the explanation may lie in differences in the clinical features of the melanomas examined. The median Breslow thickness of tumors in the two Olmholt studies was 2.55 mm and 2.60 mm, considerably thicker than the median Breslow thickness of 1.25 mm for our samples. Additionally, only four of the twenty-nine patients (13.8%) in our prospectively followed cohort have developed regional metastasis and currently none have distant disease. It is possible that regional and distant spread is preceded by aggressive early acquisition of mutations in the RGP, accounting for the lack of wild type RGP samples in those predominantly Stage IV patient populations. It appears, then, that case selection may influence the results and conclusions of this type of analysis. Although selected based upon technical rather than clinical criteria, our study population, like those in the previous reports, was chosen from a considerably larger patient group and is therefore possibly subject to selection bias. These data should be interpreted with precaution in mind.
The more important difference between our report and those described above is the interpretation of concordant cases, representing 69% and 100% of subjects, respectively. To our knowledge, our study is the first to attempt formal quantitation of mutated DNA within RGP and VGP pairs. Previous reports, taking a “yes or no” approach, have considered any amount of mutated DNA in the RGP and VGP to be equivalent, leading to the conclusion that mutations are not acquired between the growth phases. In contrast, by our approach of measuring chromatogram peaks, we found evidence of a gain of mutations in the VGP. Moreover, we predict that many of the tumors in our study with wild type DNA in both growth phases actually do carry mutations, albeit at levels below the sensitivity of the currently utilized PCR methods. A more sensitive method would likely reveal these low-level mutation rates and demonstrate the same trend toward acquisition of mutations in the VGP. Taken another way, the previous studies using the “yes or no” approach and reporting mutational identity between the RGP and VGP of the same tumors may simply be showing that an RGP with a mutation rate exceeding the detection threshold will be associated with a VGP also above the threshold. In the absence of a quantitative approach, these studies will fail to detect actual differences in the amount of mutated DNA between the two phases and draw a conclusion of equivalency.
Despite the high quality of microdissection provided by the laser capture methodology (Simone et al., 1998), there remains the possibility of contamination of RGP melanoma cells with keratinocytes during the dissection process, leading to an artificial lowering of the RGP mutation rate. Accordingly, steps were taken to avoid contamination, such as leaving a rim of melanoma cells at the microdissection border when possible, and using the smallest laser spot size. Review of the microdissected RGP samples suggests a contamination rate of less than 5%. Furthermore, there is likely to be a similar rate of sporadic contamination of VGP melanoma cells with lymphocytes and endothelial cells, balancing the contribution of normal cells to the RGP preparation. Thus, after taking into account these variables, we believe that the presence of incidental DNA from normal cells did not significantly alter our data or conclusions.
Based on our findings, two non-mutually-exclusive paradigms exist for the acquisition of mutations in melanoma as cells progress from RGP to VGP. In the first scenario, NRAS and BRAF mutations originate in the RGP. The cells bearing these mutations are phenotypically aggressive and more likely to invade the dermis, thus representing a higher percentage of VGP tumor cells. The second model suggests that mutations are not involved in the transition from RGP to VGP, but that mutational pressures are stronger in the dermis than in the epidermis. Unfortunately, the ability to dissect these processes in individual tumors is hampered by the lack of clearly understood mechanisms driving the development of NRAS and BRAF mutations. Processes such as damage from ultraviolet radiation and reactive oxygen species have been proposed, and it is reasonable to speculate that these stimuli may predominate in one compartment or another along the path of melanoma progression (Gilchrest et al., 1999; Meyskins et al., 2004; Fruehauf et al., 2007). It is also clear from our data and those of others that some BRAF and NRAS wild type cells are capable of invading the dermis. It is likely that the wild type cells present in the VGP are biologically distinct from the wild type cells in the RGP, possessing one or more yet to be identified features that promote invasiveness. It appears, then, that NRAS and BRAF mutations are not the entire story and that other invasion-promoting genotypes remain to be discovered.
In conclusion, we have shown that NRAS and BRAF mutations increase with progression of primary melanoma. These findings, if confirmed, may shed light on important aspects of the biology of this disease. Further acquisition of mutations in advanced stages of melanoma will be a focus of interest as we continue to follow our patient cohort.
Materials and Methods
Study population
The study was approved by the MDACC IRB and conducted in accordance with HIPAA and Helsinki guidelines. Written informed consent was obtained from all participants. Fifteen in-situ and twenty-nine invasive primary cutaneous melanomas were selected from consecutive entries into the MDACC Melanoma Program Informatics, Tissue Resource, and Pathology Core. Selection of invasive tumors was based on the criterion of the presence of radial and vertical growth phases in amounts sufficient for PCR amplification and DNA sequencing (approximately 1,000 cells per growth phase).
DNA extraction and amplification
Three 5 micron-thick, formalin-fixed, paraffin-embedded tissue sections per melanoma case were stained using solutions of xylenes (5 minutes), xylenes (5 minutes), 100% ethanol (30 seconds), 100% ethanol (30 seconds), 95% ethanol (30 seconds), sterile deionized distilled water (15 seconds), Mayer's hematoxylin (15 seconds), 0.5% lithium carbonate (30 seconds), sterile deionized distilled water (30 seconds), 70% ethanol (30 seconds), alcoholic eosin (30 seconds), 95% ethanol (15 seconds), 100% ethanol (30 seconds), 100% ethanol (30 seconds), xylenes (1 minute), xylenes (1 minute). Fresh solutions were prepared for each case. Laser capture microdissection of tumor cells was performed by a board-certified pathologist (VRG). A minimum of 1,000 melanoma cells per RGP and VGP were dissected as separate samples using the PixCell II Laser Capture Microdissection System (Arcturus, Mountain View, Ca) according to manufacturer's instructions. Dissected cells were incubated for 72 hours at 37°C in 50-100 ul of lysis buffer containing 10 mM Tris-HCl (pH 8.0), 1% Tween-20, 1 mM EDTA, and 0.04% Proteinase K. The sample was centrifuged for 5 minutes at 14,000 rpm and then heated at 95°C for 8 minutes.
PCR was performed using the GeneAmp Gold PCR Reagent Kit (Applied Biosystems, Foster City, CA). Primers and conditions for NRAS exon 2 and BRAF exon 15 were obtained from Sigma Genosys (The Woodlands, TX). Primer sequences (14) and conditions are listed below:
| NRAS exon 2: | Forward: 5′-GGTGAAACCTGTTTGTTGGA-3′ |
| Reverse: 5′-AACCTAAAACCAACTCTTCCCA-3 | |
| 95 °C 45 s, 57 °C 45 s, 72°C 60 s; 40 cycles; magnesium chloride 2.5 mM | |
| BRAF exon 15: | Forward: 5′-TCATAATGCTTGCTCTGATAGGA-3′ |
| Reverse: 5′-GGCCAAAAATTTAATCAGTGGA-3′ | |
| 94 °C 30 s, 57 °C 60 s, 72 °C 60 s; 40 cycles; magnesium chloride 1.5 mM |
PCR products were purified using QIAquick PCR Purification Kit (Qiagen, Valencia, CA) according to manufacturer's instructions. The purified products were sequenced in both forward and reverse directions by MDACC DNA Core facility using an ABI Prism 3100 DNA Genetic Analyzer (Applied Biosystems) using Big Dye v.3.1 dye terminator chemistry (Applied Biosystems).
Estimation of the proportion of mutated sample DNA
Proportions of mutated DNA were calculated by physically measuring the height of the mutated and wild type nucleotide peaks on the DNA chromatograms. The mutated nucleotide peak measurement was divided by the sum of the mutated and wild type peak measurements resulting in a percentage value for mutated DNA. In the case of tandem point mutations, the proportion of mutated DNA at each nucleotide was calculated and the two values averaged.
Statistical Analysis
Tumors with complete sequence results for NRAS exon 2 and BRAF exon 15 RGP and VGP DNA were analyzed statistically using the exact McNemar's test of equality of paired proportions. The paired t-test was utilized to compare the proportion of mutated DNA in the RGP and the VGP of individual tumors exhibiting mutations in both growth phases.
Acknowledgments
We sincerely appreciate the technical efforts of Ms. Carolyn Cooke and Ms. Marilyn Johnson. We are also indebted to Drs. Jeffrey Gershenwald and Victor Prieto for their efforts in the establishment and maintenance of the Melanoma Program Informatics, Tissue Resource, and Pathology Core. This work was supported by NIH P50 CA093459 (EAG, VRG, MMJ, JAE) and NIH CA16672 (DNA Analysis Facility). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of Interest: The authors state no conflict of interest.
References
- Akslen LA, Angelini S, Straume O, Bachmann IM, Molven A, Hemminki K, et al. BRAF and NRAS mutations are frequent in nodular melanoma but are not associated with tumor cell proliferation or patient survival. J Invest Dermatol. 2005;125:312–7. doi: 10.1111/j.0022-202X.2005.23788.x. [DOI] [PubMed] [Google Scholar]
- Ball NJ, Yohn JJ, Morelli JG, Norris DA, Golitz LE, Hoeffler JP. RAS mutations in human melanoma: a marker of malignant progression. J Invest Dermatol. 1994;102:285–90. doi: 10.1111/1523-1747.ep12371783. [DOI] [PubMed] [Google Scholar]
- Beeram M, Patnaik A, Rowinsky EK. Raf: a strategic target for therapeutic development against cancer. J Clin Oncol. 2005;23:6771–90. doi: 10.1200/JCO.2005.08.036. [DOI] [PubMed] [Google Scholar]
- Clark WH, Jr, Elder DE, Guerry D, Epstein MN, Greene MH, Van Horn M. A study of tumor progression: the precursor lesions of superficial spreading melanoma. Hum Pathol. 1984;15:1147–65. doi: 10.1016/s0046-8177(84)80310-x. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Demunter A, Stas M, Degreef H, De Wolf-Peters C, van den Oord JJ. Analysis of N- and K-Ras mutations in the distinctive tumor progression phases of melanoma. J Invest Dermatol. 2001;117:1483–9. doi: 10.1046/j.0022-202x.2001.01601.x. [DOI] [PubMed] [Google Scholar]
- Dong J, Phelps RG, Qiao R, Yao S, Bernard O, Ronai Z, et al. BRAF oncogenic mutations correlate with progression rather than initiation of human melanoma. Cancer Res. 2003;63:3883–5. [PubMed] [Google Scholar]
- Edlundh-Rose E, Egyhazi S, Omholt K, Mansson-Brahme E, Platz A, Hansson J, et al. NRAS and BRAF mutations in melanoma tumours in relation to clinical characteristics: a study based on mutation screening by pyrosequencing. Melanoma Res. 2006;16:471–8. doi: 10.1097/01.cmr.0000232300.22032.86. [DOI] [PubMed] [Google Scholar]
- Fruehauf JP, Meyskins FL., Jr Reactive oxygen species: A breath of life or death? Clin Cancer Res. 2007;13:789–94. doi: 10.1158/1078-0432.CCR-06-2082. [DOI] [PubMed] [Google Scholar]
- Gilchrest BA, Eller MS, Geller AC, Yaar M. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 1999;340:1341–8. doi: 10.1056/NEJM199904293401707. [DOI] [PubMed] [Google Scholar]
- Goel VK, Lazar AJF, Warneke CL, Redston MS, Haluska FG. Examination of mutations in BRAF, NRAS, and PTEN in primary cutaneous melanoma. J Invest Dermatol. 2006;126:154–60. doi: 10.1038/sj.jid.5700026. [DOI] [PubMed] [Google Scholar]
- Goydos JS, Mann B, Kim HJ, Gabriel EM, Alsina J, Germino FJ, et al. Detection of BRAF and N-RAS mutations in human melanoma. J Am Coll Surg. 2005;200:362–70. doi: 10.1016/j.jamcollsurg.2004.10.032. [DOI] [PubMed] [Google Scholar]
- Hahn WC, Weinberg RA. Rules for making human tumor cells. N Engl J Med. 2002;347:1593–1603. doi: 10.1056/NEJMra021902. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Houben R, Becker JC, Kappel A, Terheyden P, Brocker EB, Goetz R, et al. Constitutive activation of the Ras-Raf signaling pathway in metastatic melanoma is associated with poor prognosis. J Carcinog. 2004;3:6–18. doi: 10.1186/1477-3163-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Angelini S, Snellman E, Hemminki K. BRAF mutations are common somatic events in melanocytic nevi. J Invest Dermatol. 2004;122:342–8. doi: 10.1046/j.0022-202X.2004.22225.x. [DOI] [PubMed] [Google Scholar]
- Meyskens FL, Jr, Farmer PJ, Anton-Culver H. Etiologic pathogenesis of melanoma: A unifying hypothesis for the missing attributable risk. Clin Cancer Res. 2004;10:2581–3. doi: 10.1158/1078-0432.ccr-03-0638. [DOI] [PubMed] [Google Scholar]
- Omholt K, Karsberg S, Platz A, Kanter L, Ringborg U, Hansson J. Screening of N-ras codon 61 mutations in paired primary and metastatic cutaneous melanomas: Mutations occur early and persist throughout tumor progression. Clin Cancer Res. 2002;8:3468–74. [PubMed] [Google Scholar]
- Omholt K, Platz A, Kante L, Ringborg U, Hansson J. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9:6483–8. [PubMed] [Google Scholar]
- Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- Poynter JN, Elder JT, Fullen DR, Nair RP, Soengas MS, Johnson TM, et al. BRAF and NRAS mutations in melanoma and melanocytic nevi. Melanoma Res. 2006;16:267–273. doi: 10.1097/01.cmr.0000222600.73179.f3. [DOI] [PubMed] [Google Scholar]
- Simone NL, Bonner RF, Gillespie JW, Emmert-Buck MR, Liotta LA. Laser-capture microdissection: opening the microscopic frontier to molecular analysis. Trends Genet. 1998;14:272–276. doi: 10.1016/s0168-9525(98)01489-9. [DOI] [PubMed] [Google Scholar]
- Uribe P, Wistuba II, Gonzalez S. BRAF mutation: A frequent event in benign, atypical, and malignant melanocytic lesions of the skin. Am J Dermatopathol. 2003;25:365–70. doi: 10.1097/00000372-200310000-00001. [DOI] [PubMed] [Google Scholar]
- Yazdi AS, Palmedo G, Flaig MJ, Puchta U, Reckwerth A, Rutten A, et al. Mutations of the BRAF gene in benign and malignant melanocytic lesions. J Invest Dermatol. 2003;121:1160–2. doi: 10.1046/j.1523-1747.2003.12559.x. [DOI] [PubMed] [Google Scholar]