

Abstract
Rad51 is the core component of the eukaryotic homologous recombination machinery and assembles into elongated nucleoprotein filaments on DNA. We have used total internal reflection fluorescence microscopy (TIRFM) and a DNA curtain assay to investigate the dynamics of individual S. cerevisiae Rad51 nucleoprotein filaments. For these experiments the DNA molecules were end-labeled with single fluorescent semi-conducting nanocrystals. The assembly and disassembly of the Rad51 nucleoprotein filaments was visualized by tracking the location of the labeled DNA end in real time. Using this approach, we have analyzed yeast Rad51 under a variety of different reaction conditions to assess parameters that impact the stability of the nucleoprotein filament. We show that Rad51 readily dissociates from DNA in the presence of ADP or in the absence of nucleotide cofactor, but that free ATP in solution confers a 5-fold increase in the stability of the nucleoprotein filaments. We also probe how protein dissociation is coupled to ATP binding and hydrolysis by examining the effects of ATP concentration, and by the use of nonhydrolyzable AMP-PNP and ATPase active site mutants. Finally, we demonstrate that the Rad51 gain-of-function mutant I345T dissociates from DNA with nearly identical kinetics to wild-type Rad51, but assembles 30% more rapidly. Taken together these results provide a framework for studying the biochemical behaviors of S. cerevisiae Rad51 nucleoprotein filaments at the single molecule level.
Introduction
To maintain genomic integrity, organisms have evolved repair pathways for repairing damaged DNA1; 2; 3. Double stranded DNA breaks (DSBs) are one of the most deleterious forms of DNA damage and failure to correctly repair them can lead to grave consequences, including cell death or oncogenic transformation4; 5. Homologous recombination (HR) is one evolutionarily conserved mode of DSB repair and is essential for maintaining genomic stability1; 2; 3. When a DSB occurs, the 5′ ends of the break are resected, yielding long 3′ ssDNA overhangs, which serve as the loading site for a DNA recombinase. The resulting nucleoprotein filament aligns the ssDNA with a homologous dsDNA molecule elsewhere in the genome and the ssDNA invades the duplex to form a D-loop structure. The invading ssDNA end then serves as a primer for new DNA synthesis necessary for completion of repair.
The RAD52 epistasis group defines an essential set of eukaryotic proteins required for the repair of DSBs via HR and was discovered initially in the budding yeast S. cerevisiae in a screen for mutants susceptible to DNA-damaging agents6; 7. RAD50, RAD51, RAD52, RAD54, RDH54/TID1, RAD55, RAD57, RAD59, MRE11, and XRS2 genes all belong to the RAD52 epistasis group6. During HR, Rad51 assembles into a nucleoprotein filament on the ssDNA overhangs generated at the DSB. This nucleoprotein filament is responsible for catalyzing the pairing, alignment, and strand invasion steps in the HR reaction8. Homozygous null rad51 mutations in mice result in an embryonic lethal phenotype, demonstrating the central role of Rad51 in HR9. While Rad51 alone is sufficient to catalyze strand exchange in vitro, additional RAD52 epistasis group proteins are required in vivo and their functions range from facilitating Rad51 loading onto ssDNA at the outset of the reaction to promoting removal of Rad51 from the nascent DNA products2; 10; 11; 12; 13; 14.
Rad51 is orthologous to bacterial RecA, which is required for HR in Escherichia coli8. These proteins share a common core with nearly 30% sequence identity within this region, indicating a high degree of evolutionary conservation. This core domain has a 3-dimensional fold resembling the F1-ATPase and also contains the nucleotide-binding Walker A and Walker B motifs, which together are essential for ATP binding and hydrolysis15; 16. The binding of Rad51 to DNA is ATP-dependent, but does not require ATP hydrolysis17; 18; 19. Structures of Rad51 and RecA have been investigated by crystallography and electron microscopy20; 21; 22; 23; 24; 25; 26; 27. Both RecA and Rad51 form right-handed helical filaments that extend the length of the DNA substrate ∼50% relative to B-form DNA. This increase in length is due to a mean rise of ∼5.1 Å per base pair (compared to 3.1 Å for B-DNA) resulting in ∼18.6 bp per turn of DNA. Depending upon ligand binding or reaction conditions, these measurements are variable and can even vary within the same filament. In general, nucleoprotein filaments that are active in a DNA strand exchange assay have a steeper helical pitch (∼90-130 Å) compared to inactive forms (∼65-85 Å)20; 22; 27. The structure of RecA bound to ssDNA reveals that although the overall length of the DNA is extended, the stretching is not uniform28. Rather adjacent bases are organized into groups of three that retain normal B-form helical parameters, but the phosphodiester bonds separating these triplets are highly elongated, accounting for the overall extension of the DNA. It seems likely that this structural organization of the DNA in the extended RecA recombinase filaments is necessary to promote recombination.
Rad51 binds ssDNA and dsDNA with near equal affinity. This presents a potential problem for targeting Rad51 to damage because the amount of ssDNA generated at a break site is much smaller than the total amount of dsDNA in the nucleus 16; 29; 30; 31; 32. Emerging evidence suggests that Rad51 must be continually and actively removed from undamaged chromatin to ensure that a pool of free protein is available for rapid mobilization during DSB repair29. Failure to dissociate Rad51 from the dsDNA product of strand invasion may also prevent subsequent steps in the repair pathway, leading to the accumulation of toxic DNA intermediates and eventual cell death33. To help further evaluate the functional properties of Rad51 nucleoprotein filaments we have established a TIRFM-based single molecule assay that allows us to probe the biochemical properties of individual Rad51 filaments during the assembly and disassembly of Rad51 nucleoprotein filaments on dsDNA34. Here we use this assay to examine the disassembly of S. cerevisiae Rad51-dsDNA nucleoprotein filaments under a variety of reaction conditions to delineate the parameters affecting dissociation of this protein from DNA.
Results
TIRFM based assay to visualize Rad51 nucleoprotein filament assembly and disassembly
We have previously published TIRFM experiments to monitor the assembly of human Rad51 nucleoprotein filaments using DNA curtains that were labeled with the intercalating dye YOYO134. One complication of this assay was that Rad51 displaced YOYO1 from DNA as the nucleoprotein filaments were assembled, making it impossible to accurately track the length of the DNA at higher concentrations of protein34. Moreover, the use of YOYO1 required an oxygen scavenging system to avoid damaging the DNA molecules upon laser illumination35; 36. Preliminary studies from our laboratory have shown that the presence of this oxygen scavenging system prevents the normal dissociation of Rad51 from DNA (data not shown) and also inactivates some proteins involved in HR37. To overcome these problems we developed an alternative labeling strategy in which one end of the DNA was tagged with a fluorescent quantum dot (QD; Figure 1A and B). The use of the QD tagged DNA eliminated the need for an oxygen scavenging system because the QDs are extremely photostable and they do not photobleach on timescales relevant to measurements for biological systems. In addition, because the DNA molecules are not fluorescently stained with an intercalating dye they can be illuminated for long periods of time without succumbing to fluorophore-induced damage. Finally, the QDs serve as a point fluorescent source whose position can be readily monitored over time using a particle-tracking algorithm37.
Figure 1. TIRFM assay for disassembly of Rad51 nucleoprotein filaments.
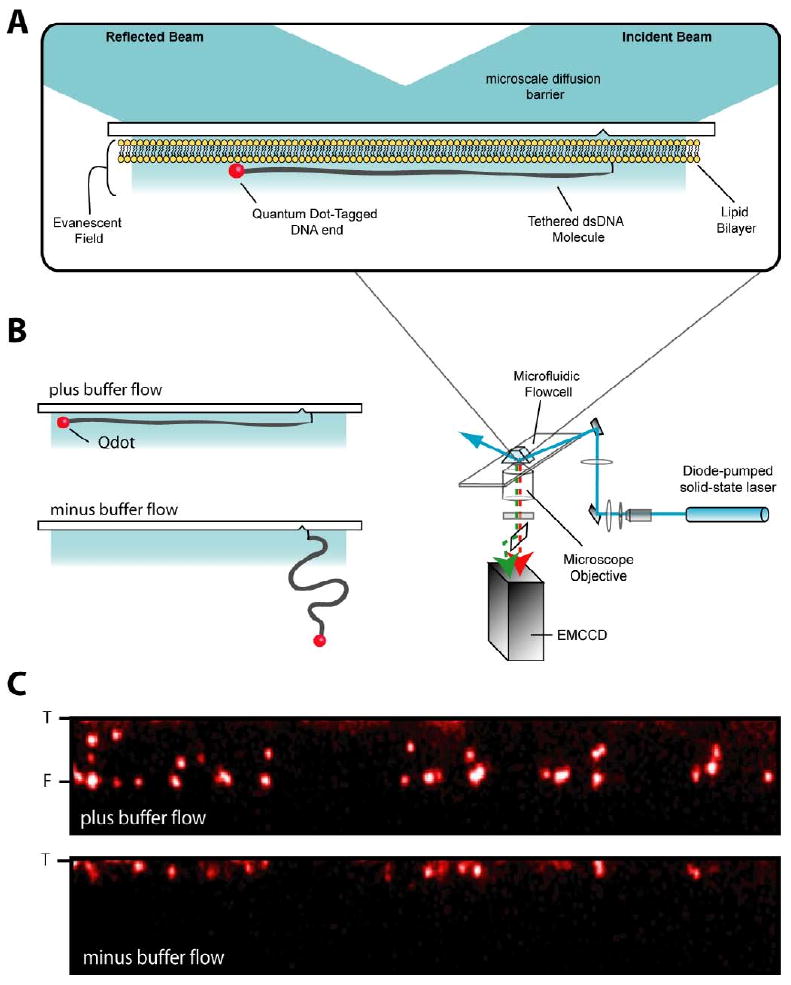
(A) Shows a schematic illustration of the TIRFM system and (B) shows a cartoon image of a QD labeled double-stranded DNA (dsDNA) molecule and its response to variations in buffer flow. The panels in (C) show examples of end-labeled dsDNA curtains in the presence and absence of buffer flow. These and all other images and graphs are oriented such that the direction of buffer flow is from top to bottom. “T” and “F” indicate the tethered and free ends of the dsDNA molecules, respectively, and these designations are used throughout. The red spots in the image are individual quantum dots and those aligned with the “F” in the presence of buffer flow are attached to DNA ends. Note that the shorter DNA fragments arise as a byproduct during the PCR reaction used to make the 23kb DNA fragment. These shorter DNA molecules were excluded from the reported data, but the assembly and disassembly behavior of Rad51 was identical to the behavior observed with the full-length DNA molecules.
DNA curtains were assembled as previously described34; 38. Anti-DIG QDs (see Materials and Methods) were then injected into the sample chamber to label the free ends of the DNA molecules within the curtain. This procedure yielded a molecular curtain comprised of aligned 23 kb DNA molecules and each of the molecules within the curtain was end-labeled with an individual QD (Figure 1). Figure 2 illustrates the assembly and disassembly of Rad51 filaments with the TIRFM assay. After locating a DNA curtain, nucleoprotein filament assembly was initiated by injecting reaction buffer (700 μl) containing 1 μM S. cerevisiae Rad51, along with 40 mM Tris (pH 7.8), 0.2 mg/ml BSA, 10 mM MgCl2, 1 mM DTT, and 1 mM ATP. Unless otherwise stated, these standard conditions were used for all experiments reported below. The injections were done at a constant flow rate of 400 μl/min using a syringe pump system and switch valve, and data acquisition was initiated just before the protein entered the flowcell. Nucleoprotein filament assembly was demarked by an increase in the length of the DNA (Figure 2A). This observed lengthening of the DNA was dependent upon the presence of ATP and confirmed that S. cerevisiae Rad51 assembled into elongated nucleoprotein filaments in our system. While there were modest experiment to experiment variations in apparent DNA length due to variations in buffer flow, the mean length of the unbound DNA based on all of our observations was 5.6 μm and the mean length of the Rad51 bound DNA was 9.4 μm. This corresponds to a 68% increase in the apparent length of the DNA upon formation of the Rad51 nucleoprotein filaments, similar to values we have reported for human Rad5134. This greater than expected increase in the length of the DNA can be attributed to the behavior of the DNA versus the protein-DNA complexes in a shear flow; as the protein binds to the DNA there is an increase in persistence length of the polymer, which makes the molecules easier to stretch at a given flow rate34; 39; 40.
Figure 2. Analysis of Rad51 nucleoprotein filament disassembly.
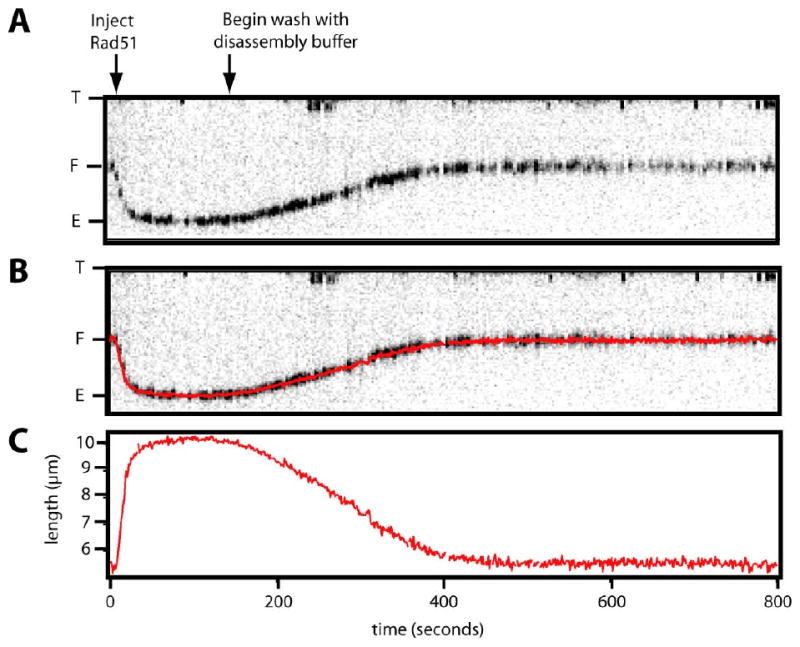
(A) Shows an example of a kymogram extracted from a movie of a DNA curtain illustrating the assembly and disassembly phases of a single Rad51 nucleoprotein filament as a function of time. The assembly phase of the nucleoprotein filament extension was initiated by the injection of Rad51 and ATP, as indicated. Disassembly was initiated by rinsing with buffer that lacked both Rad51 and ATP. As the filament assembles, the DNA lengthens until fully extended. Once there is no more protein entering the flowcell, the filament disassembles and the DNA becomes shorter. The entire reaction can be monitored by visualizing and measuring the location of the QD tagged end. In panel (B) the length of the DNA was automatically tracked by fitting the original data to a 2D-Guassian function to locate the centroid position of the QD and is superimposed onto the kymogram. Panel (C) presents a graphical depiction of the same data, and this is the form the data will be presented in throughout.
Once assembly was complete, the disassembly phase of the reaction was initiated by switching to buffer that lacked free Rad51 (Figure 2). As shown here, the DNA returned to its original length several minutes after initiating disassembly, indicating that Rad51 dissociated from the DNA. For data quantitation, the individual molecules within the DNA curtains were analyzed with a particle-tracking algorithm that used a 2D-Guassian function to locate the centroid position of each individual QD, as previously described37. Figure 2B (middle panel) shows an example of this tracking data superimposed on the corresponding kymogram. The tracking data were then plotted to generate graphs of DNA length versus time as shown in figure 2C (lower panel) and disassembly rates were calculated using a non-linear least squares fitting procedure to find the maximal rate of disassembly (see Materials and Methods). The rates at which the DNA molecules decreased in length were calculated in nanometers per second (nm/sec). Assuming there were ∼3 nucleotides per bound protein monomer, then the 23 kilobase substrates used in this study would be occupied by roughly 7,700 molecules of Rad51 when fully extended. The mean change in length observed for the DNA molecules in the presence of Rad51 was 3.8 μm, corresponding to an apparent increase in length of ∼0.49 nm per molecule of bound protein. This value was used as a conversion factor to estimate the number of Rad51 molecules that dissociate from the DNA based on the observed rates at which the DNA changed length. All disassembly rates determined in this study are reported in table 1.
Table 1.
Disassembly characteristics of wild-type and mutant S. cerevisiae Rad51 nucleoprotein filaments.
| Assembly Conditionsa | Disassembly Conditions b | Disassembly rate (nm/sec) | Disassembly c rate (monomers/sec) |
|---|---|---|---|
| ATP | 0 mM NaCI | 20.55 ± 1.14 | 10.1 ± 0.56 |
| ATP | 10 mM NaCI | 29.05 ± 1.92 | 14.2 ± 0.94 |
| ATP | 25 mM NaCI | 23.57 ± 1.67 | 11.5 ± 0.82 |
| ATP | 50 mM NaCI | 15.17 ± 1.22 | 7.43 ± 0.60 |
| ATP | 100 mM NaCI | 17.47 ± 3.65 | 8.56 ± 1.79 |
| ATP | 250 mM NaCI | 19.35 ± 2.00 | 9.48 ± 0.98 |
| ATP | 500 mM NaCI | 12.26 ± 2.19 | 6.00 ± 1.07 |
| ATP | 0.1 mM MgCl2 | 22.50 ± 2.48 | 11.0 ± 1.21 |
| ATP | 0.5 mM MgCI2 | 19.68 ± 1.67 | 9.64 ± 0.82 |
| ATP | 1.0 mM MgCI2 | 19.71 ± 0.58 | 9.66 ± 0.28 |
| ATP | 2.0 mM MgCI2 | 20.26 ± 1.23 | 9.93 ± 0.60 |
| ATP | 5.0 mM MgCI2 | 20.63 ± 1.07 | 10.1 ± 0.52 |
| ATP | 10 mM MgCI2 | 20.55 ± 1.14 | 10.1 ± 0.56 |
| ATP | 0 mM ATP | 20.55 ± 1.14 | 10.1 ± 0.56 |
| ATP | 0.25 mM ATP | 5.34 ± 0.79 | 2.62 ± 0.39 |
| ATP | 0.5 mM ATP | 5.44 ± 0.91 | 2.67 ± 0.45 |
| ATP | 1.0 mM ATP | 3.24 ± 0.67 | 1.59 ± 0.33 |
| ATP | 2.0 mM ATP | 4.14 ± 0.64 | 2.03 ± 0.31 |
| ATP | 1 mM ADP | 18.94 ± 4.20 | 9.28 ± 2.06 |
| AMP-PNP | 0 mM NaCI | 3.03 ± 0.88 | 1.48 ± 0.43 |
| AMP-PNP | 10 mM NaCI | 2.54 ± 0.39 | 1.24 ± 0.19 |
| AMP-PNP | 25 mM NaCI | 2.39 ± 0.21 | 1.17 ± 0.10 |
| AMP-PNP | 50 mM NaCI | 1.19 ± 0.35 | 0.58 ± 0.17 |
| AMP-PNP | 100 mM NaCI | 2.21 ± 0.32 | 1.08 ± 0.29 |
| AMP-PNP | 250 mM NaCI | 1.27 ± 0.30 | 0.62 ± 0.15 |
| AMP-PNP | 500 mM NaCI | 1.44 ± 0.48 | 0.71 ± 0.24 |
| AMP-PNP | No Nucleotide | 3.03 ± 0.88 | 1.48 ± 0.43 |
| AMP-PNP | 1 mM ADP | 0.92 ± 0.18 | 0.45 ± 0.24 |
| AMP-PNP | 1 mM ATP | 1.49 ± 0.23 | 0.73 ± 0.11 |
| AMP-PNP | 1 mM ATPγS | 1.03 ± 0.28 | 0.50 ± 0.14 |
| AMP-PNP | 1 mM AMP-PNP | 1.01 ± 0.49 | 0.49 ± 0.24 |
| K191R + ATP | 0 mM NaCI | 1.26 ± 0.29 | 0.63 ± 0.14 |
| K191R + ATP | 10 mM NaCI | 2.24 ± 0.71 | 1.10 ± 0.35 |
| K191R + ATP | 25 mM NaCI | 0.99 ± 0.30 | 0.49 ± 0.15 |
| K191R + ATP | 50 mM NaCI | 1.80 ± 0.46 | 0.88 ± 0.23 |
| K191R + ATP | 100 mM NaCI | 1.57 ± 0.22 | 0.77 ± 0.11 |
| K191R + ATP | 250 mM NaCI | 1.96 ± 0.38 | 0.96 ± 0.19 |
| K191R + ATP | 500 mM NaCI | 2.11 ± 0.37 | 1.03 ± 0.18 |
| I345T + ATP | 0 mM NaCI | 20.56 ± 3.19 | 10.1 ± 1.56 |
| I345T + ATP | 10 mM NaCI | 20.70 ± 3.10 | 10.1 ± 1.51 |
| I345T + ATP | 25 mM NaCI | 18.25 ± 3.38 | 8.94 ± 1.66 |
| I345T + ATP | 50 mM NaCI | 16.60 ± 1.82 | 8.13 ± 0.89 |
| I345T + ATP | 100 mM NaCI | 16.45 ± 1.25 | 8.06 ± 0.61 |
| I345T + ATP | 250 mM NaCI | 14.11 ± 1.05 | 6.91 ± 0.51 |
| I345T + ATP | 500 mM NaCI | 15.42 ± 1.38 | 7.56 ± 0.68 |
All assembly reactions contained 1 mM nucleolide. Reactions with wt Rad51 and Rad51 I34ST contained 1 μM protein. Reactions with Rad51 K191R contained 5μM proteinic compensate For its weaker DNA binding activity.
Standard disassembly buffers contained 40 mM Tris-HCI (ph 7.8), 0.2 mg/ml BSA 10 mM MgCI2 and 1 mM DTT.
The observed disassembly rates in nanometers per second were converted to momomers of Rad51 per second using a conversion factor of 0.49 nanometers per monomer See the text for additional discussion of this conversion factor.
Dissociation of ScRad51 from double stranded DNA
To begin dissecting the stability of the nucleoprotein filaments, Rad51 was bound to the DNA in the presence of ATP, and then chased with buffer containing increasing concentrations of NaCl, but lacking ATP and free Rad51 (Figure 3A and 3B). Disassembly of the Rad51 nucleoprotein filaments was not markedly affected by variations in concentration of NaCl. In the absence of NaCl, the disassembly rate was 27.05 ± 3.97 nm/sec and this value decreased to 12.26 ± 2.19 nm/sec at 500 mM NaCl (Figure 3B and Table 1). In addition, the DNA returned to its original length under all concentrations of NaCl that were tested. Magnetic bead pull-down assays were conducted to verify that the changes in DNA length observed with the TIRFM assays correlated with dissociation of the protein from DNA (Figure 3C). For these assays, a biotinylated 60 bp dsDNA oligonucleotide was attached to streptavidin coated magnetic beads and Rad51 was incubated with the beads under the same conditions used for assembly in the TIRFM assays. The beads were then washed with buffer containing increasing amounts of NaCl, but lacking ATP and free Rad51. Finally, the beads were incubated with 2% SDS to remove any proteins remaining bound to the DNA. At each stage of this process samples were removed for analysis by SDS-PAGE. As shown in figure 3C, under buffer conditions identical to those used in the TIRFM assays, Rad51 did not remain bound to the DNA-coupled magnetic beads. This result confirmed that the decreased DNA length observed in the TIRFM assay was correlated with dissociation of the protein from the DNA.
Figure 3. Effects of NaCl on disassembly of Rad51.

(A) Examples are shown of the kinetic reaction between the nucleoprotein filament and dsDNA. The reactions depicted all were initially formed with Rad51 and 1mM ATP. The buffer is then changed for the disassembly phase, each one containing different concentrations of NaCl. The background colors of the assembly phase (blue) and disassembly phase (yellow) are different to emphasize these distinct stages of the reactions, and this color scheme is used throughout subsequent figures. (B) The maximum disassembly rate in nm/sec (+/-standard deviation) was measured during the disassembly phase and graphed as a function of NaCl concentration (see Materials and Methods). (C) The top panel depicts an overview of the magnetic bead-based turnover assay. The lower panel is an SDS-PAGE gel with three reactions containing different concentrations of NaCl in the wash buffer. Each reaction shows supernatant (lane 1), the pooled washes (lane 2), and 2% SDS eluate (lane 3), and these bulk assays confirmed that Rad51 rapidly dissociated from the DNA in the absence of ATP.
Effects of magnesium chloride on the disassembly of yeast Rad51 nucleoprotein filaments
The Rad51 nucleoprotein filament assembly reactions were conducted at 10 mM MgCl2, and we were unable to detect formation of these extended filaments at either 1 or 2 mM MgCl2 (not shown). This MgCl2 dependence during the assembly phase of the reactions was expected based on previously published bulk biochemical experiments19. We next assessed the effect of free MgCl2 concentration on the disassembly of the Rad51 nucleoprotein filaments (Figure 4). Rad51 nucleoprotein filaments were first preassembled at 10 mM MgCl2 as described above. Disassembly was initiated by switching to buffer that contained varying concentrations of MgCl2, but lacked free protein and ATP. These experiments showed that the Rad51 nucleoprotein filaments were insensitive to free MgCl2 concentration during the disassembly phase of the reaction and comparable values were observed over a range spanning 0.1 to 10 mM, despite the fact that 10 mM MgCl2 was required for assembly (Figure 4B). These results were also corroborated with the bead pull down assay, confirming that the protein did in fact dissociate from the DNA (Figure 4C).
Figure 4. MgCl2 effect on the disassembly of Rad51 nucleoprotein filaments.
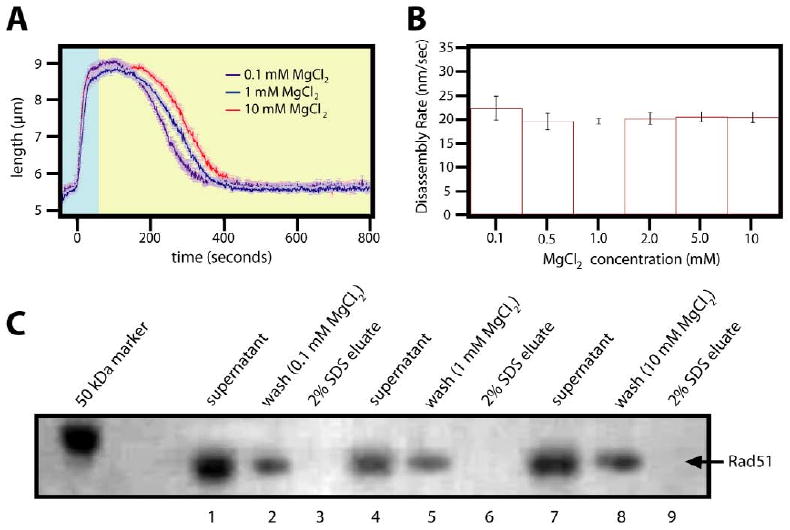
(A) Examples of reaction curves with Rad51 at different concentrations of MgCl2 are shown with blue delineating the assembly phase, yellow the disassembly. (B) A bar graph with the corresponding disassembly rates for all reactions is shown. For these experiments, the Rad51 filaments were assembled in reaction buffer containing 1mM ATP. Disassembly was then initiated by switching to reaction buffer that lacked ATP and contained the indicated concentration of MgCl2. Panel (C) shows an SDS-PAGE gel depicting magnetic bead assay results for three different MgCl2 concentrations.
ATP dramatically increases the stability of S. cerevisiae Rad51 nucleoprotein filaments
To test the influence of ATP on the disassembly rates, nucleoprotein filaments were assembled as described above, and disassembly was initiated with buffer that contained varying concentrations of free ATP. As shown in figure 5A and B, free ATP markedly increased Rad51 nucleoprotein filament stability even at the lowest concentrations of free nucleotide tested. Analysis of this data revealed a disassembly rate of 5.34±0.79 nm/sec measured at 0.25 mM ATP, compared to 20.55±1.14 nm/sec in the absence of ATP, and this value remained essentially unchanged at even the highest concentrations tested (4.14±0.64 nm/sec at 2 mM ATP). These findings were corroborated with the bead pull down assay, which demonstrated that Rad51 remained tightly bound to the DNA when ATP was included in the wash buffers (figure 5C). Our interpretation of the data was that ATP prevented dissociation. An alternative explanation for this outcome with the bulk assay was that the ATP present in the wash buffer simply allowed the protein to rebind to the DNA after it had dissociated. To rule out this possibility, the magnetic bead assay was conducted with a 20-fold excess of free oligonucleotide relative to the bead bound substrate during the wash steps (figure 5C, lanes 1-6), which served as a competitor to prevent rebinding of any Rad51 that may have dissociated. As shown in figure 5C, lanes 1-6, the presence of the competitor DNA during the wash steps did not provoke release of Rad51 from the bead bound DNA when ATP was included in the washes, indicating that the protein did not dissociate from the originally bound substrate. Taken together these bulk and single molecule experiments demonstrated that the presence of free ATP in solution dramatically stabilized the yeast Rad51 nucleoprotein filaments, and suggested that the protein was capable of hydrolyzing multiple molecules of ATP prior to dissociating from the DNA.
Figure 5. ATP stabilizes Rad51 filaments.
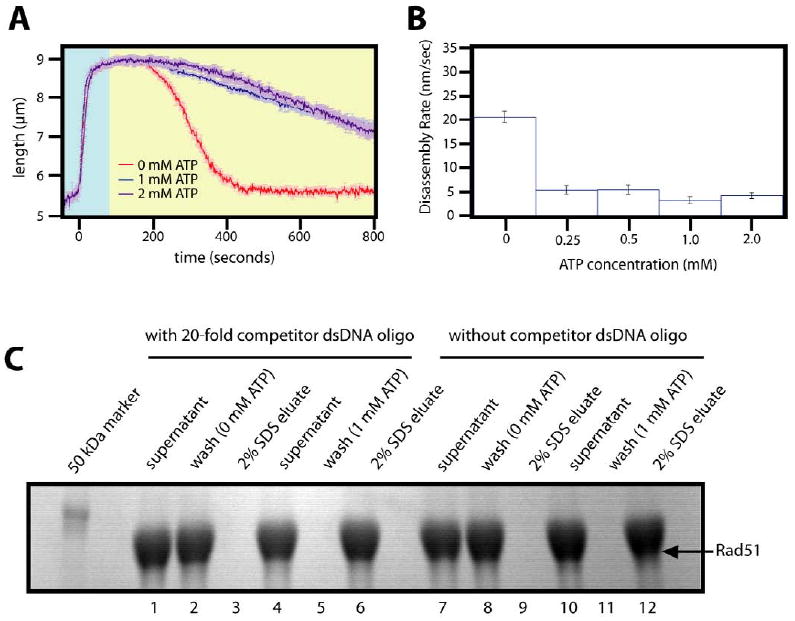
(A) Examples of assembly and disassembly reactions with Rad51 at different concentrations of ATP are shown. (B) The corresponding disassembly rates for all concentrations tested are graphed. For these experiments, the Rad51 filaments were assembled in reaction buffer containing 1mM ATP. Disassembly was then initiated by switching to reaction buffer that contained the indicated concentrations of ATP. The results of the magnetic bead turnover assay with and without ATP in the wash buffer is shown in panel (C) Comparison of lanes 3 and 6, or lanes 9 and 12, shows that free ATP prevents dissociation of Rad51 from the bead coupled DNA substrate. Panel (C), lanes 1-6 included a 20-fold molar excess of a competitor DNA in the wash steps, and lanes 7-12 show results from the same assay when performed without the competitor DNA.
Nucleotide hydrolysis and release is required for disassembly of the Rad51 nucleoprotein filaments
Nonhydrolyzable and slowly hydrolyzable analogs of ATP are often used to study the biochemical role of ATP hydrolysis, and AMP-PNP in particular has been used to aid studies of Rad51 nucleoprotein filaments 15; 16; 17. Therefore we tested the effects of the nonhydrolyzable ATP analog AMP-PNP on the behavior of the Rad51 nucleoprotein filaments. First AMP-PNP and ADP were tested to determine whether they could stabilize Rad51 filaments initially assembled with ATP (Figure 6). As shown in figure 6A and 6C, either ATP or AMP-PNP were sufficient to stabilize filaments preassembled with ATP. However, the nucleoprotein filaments assembled with ATP rapidly disassembled when chased with buffer containing ADP, indicating that ADP alone was insufficient to stabilize the DNA-bound Rad51. In addition, ADP alone did not support assembly of elongated Rad51 filaments under any tested condition (data not shown).
Figure 6. Rad51 nucleoprotein filaments are not stabilized by ADP.
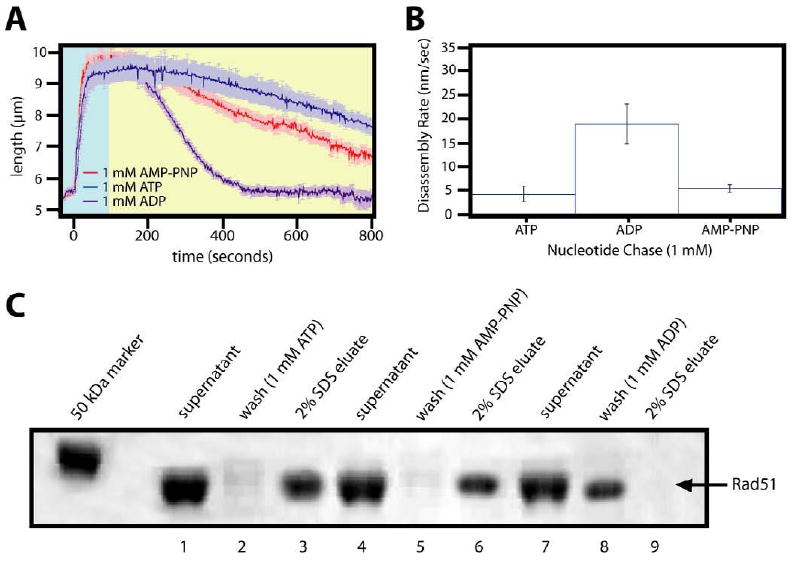
Panel (A) shows examples of assembly and disassembly reactions of Rad51 nucleoprotein filaments formed with ATP and then chased with buffer containing 1 mM ADP, ATP, or AMP-PNP. The disassembly rates for all concentrations tested are graphed in (B). (C) An SDS-PAGE gel depicts results with the magnetic bead turnover assay for the three different nucleotides tested.
We next tested whether nonhydrolyzable nucleotide analogs supported the assembly of elongated Rad51 filaments in our assay by replacing the ATP in the assembly buffer with either ATPγS or AMP-PNP. We were unable to see any DNA extension when ATPγS was used as the cofactor (not shown). This was not surprising, as previous studies have shown that assembly is compromised when ATPγS is used as the nucleotide cofactor19. In contrast to ATPγS, assembly reactions performed with 1 mM AMP-PNP and 1 μM Rad51 resulted in rapid extension of the DNA molecules, consistent with the formation of elongated nucleoprotein filaments. These filaments were highly stable, and disassembled at a rate of 3.03±0.88 nm/sec when washed with buffer lacking free AMP-PNP (figure 7A, B, and C). This disassembly rate was 6.8-fold slower than the observed rates for filaments assembled with ATP and disassembled in buffer lacking nucleotide. In addition, the Rad51 nucleoprotein filaments assembled with AMP-PNP were resistant to dissociation when chased with buffers containing increasing concentrations of NaCl, and there was only a modest increase in the dissociation rate even at 500 mM NaCl.
Figure 7. Rad51 nucleoprotein filaments assembled with AMP-PNP are resistant to disassembly.
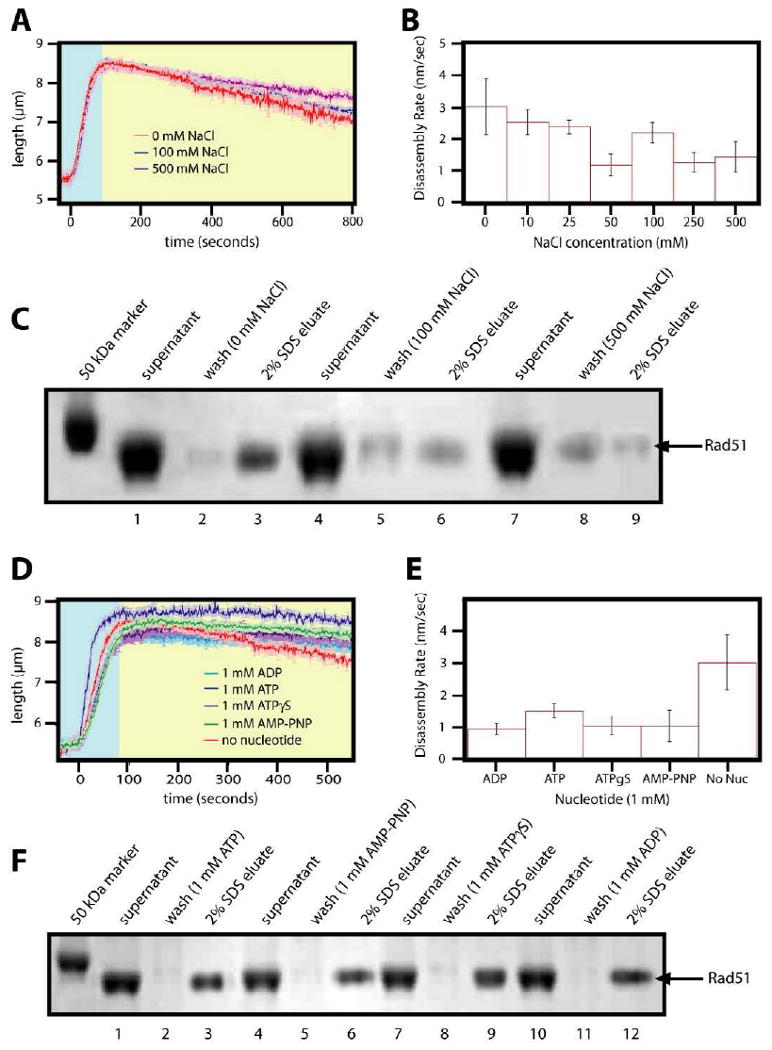
(A) Nucleoprotein filament assembly and disassembly reactions are shown. The filaments were formed with 1mM AMP-PNP and each reaction was challenged with varying concentrations of NaCl. (B) A bar graph with the corresponding disassembly rates for all NaCl concentrations tested. (C) An SDS-PAGE gel depicts the turnover assay results for three different NaCl concentrations where the filament was initially formed with 1mM AMP-PNP. In (D) and (E) the nucleoprotein filaments were formed with 1 μM Rad51 and 1mM AMP-PNP, and then chased with different nucleotides, as indicated. Panel (F) shows an SDS-PAGE gel with the magnetic bead turnover assay results for filaments formed with 1mM AMP-PNP and washed with buffers containing different nucleotides.
Finally we assessed the behavior of Rad51 nucleoprotein filaments initially assembled with AMP-PNP and then chased with disassembly buffer containing various nucleotides (Figure 7D). As shown in figure 7D, 7E and 7F, AMP-PNP assembled nucleoprotein filaments remained stable when chased with buffers containing 1 mM ADP, ATP, ATPγS, AMP-PNP, or even no nucleotide, and there was no appreciable difference in the observed dissociation rates for any of these conditions. The finding that nucleoprotein filaments assembled with AMP-PNP and chased with either no nucleotide or ADP did not provoke more rapid release of Rad51 from the DNA argued that the protein was trapped in the AMP-PNP bound state, was unable to exchange AMP-PNP for other nucleotides, and supports the hypothesis that release of AMP-PNP was the rate-limiting step during dissociation.
The ATPase deficient mutant Rad51 K191R forms highly stable nucleoprotein filaments
To further test the role of ATP hydrolysis in nucleoprotein filament disassembly we assayed Rad51 harboring a mutation known to disrupt ATP hydrolysis. A lysine to arginine mutation in the Walker A box of rad51 K191R disrupts ATP hydrolysis but does not eliminate ATP binding activity15; 16; 18; 41. This mutant protein binds to DNA in a nucleotide-dependent fashion, but does not hydrolyze ATP, and we predicted that the mutant proteins would remain bound to the DNA much longer than the wild-type protein. For experiments with rad51 K191R, we were unable to assemble fully extended nucleoprotein filaments at 1.0 μM protein, therefore the concentration of protein was increased to 5.0 μM in the assembly phase of the reaction to allow full extension of the dsDNA. This requirement for more protein to assemble the nucleoprotein filaments was consistent with the notion that rad51 K191R has a defect in the initial DNA binding during the nucleoprotein filament assembly phase of the reaction18, which was circumvented by increasing the pool of free protein (see below). Despite the defect in initial binding, once assembled, the rad51 K191R nucleoprotein filaments were much more stable than those formed with wild-type Rad51 (Figure 8). In the absence of NaCl the disassembly rate for the K191R mutant protein was just 1.26±0.29 nm/sec, corresponding to approximately a 20-fold decrease in the dissociation rate relative to the wild-type protein. The rad51 K191R nucleoprotein filaments were also resistant to moderately high concentrations of NaCl, and the disassembly rate increased to just 2.11±0.37 nm/sec at 0.5 M NaCl. In fact, the rad51 K191R nucleoprotein filaments assembled with ATP were even more stable than the wild-type Rad51 nucleoprotein filaments formed in the presence of nonhydrolyzable AMP-PNP (compare figure 7A and figure 8A). Importantly, the DNA binding and extension activity of rad51 K191R was entirely dependent upon the presence of ATP, indicating that the protein still required its nucleotide cofactor (data not shown). Taken together, these results show that rad51 K191R formed highly stable and extended nucleoprotein filaments on dsDNA, which were resistant to dissociation.
Figure 8. ATPase-deficient rad51 K191R filaments are resistant to disassembly.

(A) Assembly and disassembly reactions are shown for nucleoprotein filaments formed with ATPase mutant rad51 K191R in the presence of ATP, and then chased with disassembly buffer containing different concentrations of NaCl. The disassembly rates for all tested conditions in are graphed in panel (B). Panel (C) shows the results of the magnetic bead turnover assay using rad51 K191R, and three different NaCl concentrations in the wash steps of the reactions.
Disassembly of Rad51 I345T nucleoprotein filaments occurs at rates comparable to wild-type Rad51
The rad51 I345T gain-of-function mutant was isolated as a suppressor of a null mutation of Rad5742; 43. Rad57 forms an obligate heterodimer with Rad55, and this Rad55/57 complex may assist in loading and/or maintaining Rad51 nucleoprotein filaments on ssDNA ends during homologous recombination. Genetic and biochemical studies have suggested that rad51 I345T overcomes the need for Rad55/57 through an enhancement of its DNA binding activity, thereby eliminating the need for these proteins in vivo42. We tested rad51 I345T in our TIRFM assay and asked whether it could extend dsDNA. As shown in figure 9A, rad51 I345T formed elongated nucleoprotein filaments on dsDNA comparable in length to those observed with wild-type Rad51. When ATP was omitted from the chase buffer, the rad51 I345T nucleoprotein filaments disassembled at rates that were nearly identical to the values observed for the wild-type protein, moreover, these rates did not vary significantly in disassembly buffers containing up to 0.5 M NaCl. The finding that wild-type Rad51 and rad51 I345T had similar properties during nucleoprotein filament disassembly demonstrated that the enhanced binding of rad51 I345T did not impact the dissociation rate of the protein from the DNA and suggested that assembly might be more rapid (see below).
Figure 9. Disassembly of rad51 I345T shows dissociation kinetics identical to wild-type Rad51.
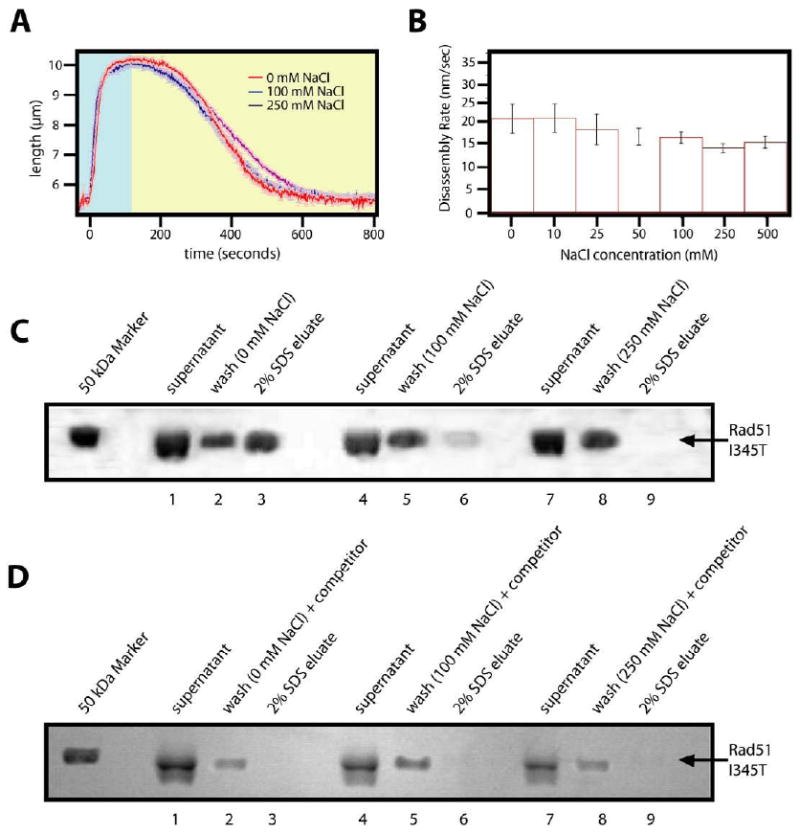
(A) Nucleoprotein filaments that were formed with rad51 I345T were challenged with various concentration of NaCl during the disassembly phase. (B) A bar graph with the corresponding maximum rates of disassembly (nm/s) +/- standard deviation for all reactions is shown. For these experiments, the rad51 I345T filaments were assembled in reaction buffer containing 1mM ATP. Disassembly was then initiated by switching to reaction buffer that contained no nucleotide and included the indicated concentration of NaCl. The results of the turnover assay using rad51 I345T are displayed for three different NaCl concentrations without and with competitor DNA during the wash steps are shown in panels (C) and (D), respectively.
For all other conditions and proteins tested, the results of the magnetic bead pull down assays paralleled those of the TIRFM assays. However, the results obtained with rad51 I345T in the pull down assays were different from the TIRFM assays. In particular, rad51 I345T remained bound to the DNA-coated beads after being washed with buffer lacking ATP and NaCl, despite the fact that the protein appeared to rapidly dissociate under the same conditions in the TIRFM assay (compare 9A and 9C). We reasoned that this apparent discrepancy likely arose from differences between experimental setups. Specifically, in the TIRFM assay, buffer is continuously flowing through the sample chamber. Therefore any protein that dissociates from the DNA is immediately flushed away and can not rebind the DNA molecules. In contrast, the Rad51 in the bead pull-down assays can potentially rebind the DNA substrate because the free protein is not immediately removed from solution. This explanation could only be correct if rad51 I345T had enhanced DNA rebinding activity relative to wild-type Rad51, because the wild-type protein did not remain bound to the beads under these same conditions (see figure 3C). To test this hypothesis we performed the magnetic bead pull down assay, but included a 20-fold excess competitor DNA in the wash buffers to help trap any free rad51 I345T that dissociated from the bead-bound DNA molecules. As shown in figure 9D, when the competitor DNA was included in the wash buffers virtually all of the rad51 I345T dissociated from the bead bound DNA. This result was in good agreement with the TIRFM data, and taken together these assays suggested that rad51 I345T had enhanced activity that increased its assembly on DNA, but did not affect its dissociation rate.
Assembly kinetics for wild-type and mutant Rad51 proteins
The results presented above suggested that the enhanced DNA binding activity of rad51 I345T affected only the assembly phase of the reaction. To test this hypothesis we analyzed the kinetics of nucleoprotein filament assembly by measuring the rates of DNA extension at differing concentrations of protein, and compared the results for wild-type Rad51, rad51 K191R and rad51 I345T (Figure 10). As shown in figure 10A, increasing the concentration of wild-type Rad51 during the assembly reaction led to a corresponding increase in both the rate at which the DNA was extended and the maximum degree of elongation that was achieved during the observation. In contrast to wild-type protein, rad51 K191R displayed a defect during assembly (Figure 10B), whereas rad51 I345T appeared to show more rapid assembly at every tested concentration of protein (Figure 10C). Assembly rates were obtained by fitting the particle tracking data to a sigmoidal function and these rates were then plotted against the concentration of Rad51 (Figure 10D). These results revealed an assembly rate of 0.22 nm/sec•nM for wild-type protein, whereas rad51 I345T had an increased rate of 0.28 nm/sec•nM. In contrast, assembly experiments with rad51 K191R yielded an assembly rate of only 0.015 nm/sec•nM, approximately 15-fold slower than wild-type Rad51.
Figure 10. Assembly kinetics of wild-type and mutant Rad51.
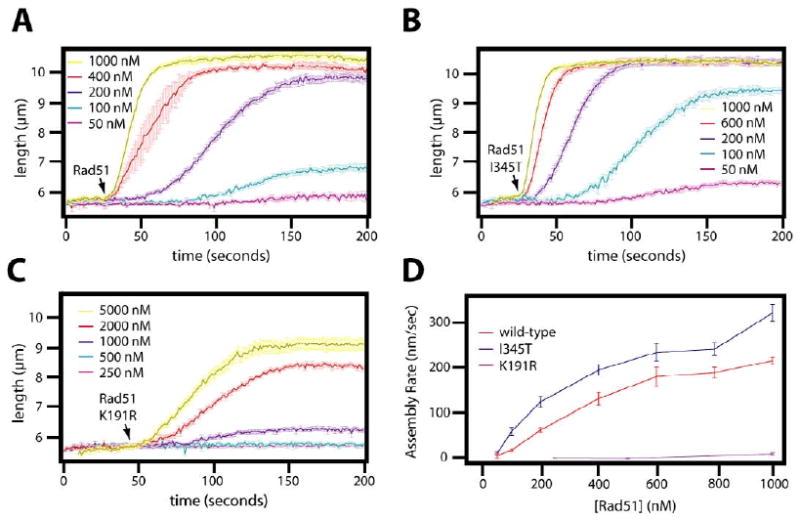
Wild-type Rad51 (A), rad51 I345T (B), or rad51 K191R (C) was injected into flowcells containing DNA curtains, and the length of the DNA was continuously tracked. The assembly phases for the indicated concentrations of protein were then plotted as a function of time. (D) The rate of assembly in nm/sec (+/- standard deviation) were measured from the linear portion of the curves and then plotted as a function of Rad51 concentration. Linear fits to these curves revealed assembly rates of 0.22, 0.28. and 0.015 nm/sec·nM for wild-type Rad51, rad51 I345T, and rad51 K191R, respectively. Please note that the rad51 K191R graph in (D) is truncated so that results from all three proteins can be presented on the same scale.
Discussion
Rad51 filaments represent core elements of the eukaryotic recombination machinery and the dynamics of these filaments must be highly regulated and coordinated with other protein factors to ensure the fidelity of homologous recombination. To begin exploring how these reactions are regulated we have developed a TIRFM-based single-molecule assay that allowed us to probe the assembly and disassembly of Rad51 nucleoprotein filaments on dsDNA, in real time under conditions that do not perturb the biochemical properties of the proteins. Specifically, these TIRFM assays do not require fluorescently tagged proteins, potentially damaging DNA intercalating dyes, or oxygen scavenging systems. These benefits are brought about through the use of DNA molecules that are tagged at one end with a single quantum dot. For this study we have provided an analysis of reaction parameters that influence the disassembly of S. cerevisiae Rad51 nucleoprotein filaments from dsDNA substrates. The importance of this disassembly reaction is that it mimics the later stages of homologous recombination where it is essential for Rad51 to be removed from the DNA in order to ensure that downstream repair proteins can gain access to their substrates. In addition, under normal growth conditions in the absence of DSBs, Rad51 and related recombinases must be actively disassembled from dsDNA in order to maintain a free pool of protein that can be rapidly mobilized when needed for HR. This process is thought to be mediated by the DNA translocases Rad54 and Rdh54, and defects in these proteins can lead to aberrant accumulation of Rad51 on DNA sites that are not damaged. Therefore the disassembly of Rad51 nucleoprotein filaments from dsDNA also has important implications for steps prior to the initiation of homologous recombination.
ATP hydrolysis and disassembly of Rad51 filaments
In our TIRFM assay disassembly of the Rad51 nucleoprotein filaments was rapid when ATP was omitted from buffer, and the proteins completely dissociate from the DNA within ∼3.5 minutes after removal of free nucleotide cofactor. However, this dissociation rate was dramatically slowed when free ATP was included in the disassembly buffer, with concentrations of ATP ranging from 0.25 to 2 mM yielding a 5-fold decrease in the rate at which the protein comes off DNA. This finding indicates that the Rad51 filaments are continually hydrolyzing ATP, releasing ADP + Pi, and then rebinding ATP, all without dissociating from the DNA. Interestingly, inspection of the available recombinase crystal structures suggests that the nucleotide binding pocket is occluded between adjacent protein subunits within the filament20; 28. Therefore the protein must undergo some type of conformational change that is seemingly not reflected in the current crystal structures, but which would be necessary allow the release and exchange of bound nucleotide without concomitant disruption of the nucleoprotein filament.
It should be noted that in vitro DNA topology assays can also be used to monitor the disassembly of Rad51 from circular duplex substrates, and these assays have shown that Rad51 appears to remain tightly bound to DNA for ≥4 hours31. This is in contrast to our experiments, where even in the continued presence of ATP the Rad51 filaments can completely dissociate within tens of minutes. The primary difference between our experimental set up and the bulk topological assays is that the TIRFM assays are conducted within a microfluidic sample chamber. Any protein that dissociates from the DNA in the TIRFM assay is immediately removed from the sample chamber by the flowing buffer and can not rebind to the DNA, therefore these assays give a measure of the dissociation rate in the absence of any rebinding. The bulk assays differ in this respect, because any Rad51 that dissociates from the DNA can potentially rebind the original substrate, even though excess competitor DNA is presented in solution. This is because nucleation on a new DNA molecule is expected to occur more slowly than rebinding to a preexisting filament.
Magnetic tweezer assays with human Rad51 have also examined the effect of nucleotides and nucleotide cofactors on the assembly and stability of these nucleoprotein filaments44. While the properties of human and S. cerevisiae Rad51 appear to be quite similar, there was one notable difference in the disassembly behaviors. With human Rad51, one report indicated that the presence of ATP during the disassembly phase of the reactions had no impact on the kinetics of protein dissociation relative to disassembly reactions conducted in the presence of either ADP or no nucleotide44. This is in contrast to our results with S. cerevisiae Rad51, which demonstrate that the yeast nucleoprotein filaments were much more stable when excess free ATP was present in solution. In addition, our own experiments with human Rad51 also show that free ATP dramatically stabilizes these nucleoprotein filaments (manuscript submitted). We have verified that these seemingly different results arise from the different buffer conditions used in the studies (data not shown).
As expected, AMP-PNP also supported assembly of elongated nucleoprotein filaments in our TIRFM assay, and these filaments do not dissociate rapidly when the free AMP-PNP was flushed from the sample chamber and replaced with buffer containing either ADP or no nucleotide. The stability conferred by AMP-PNP was only moderately greater than that observed with ATP. However, as indicated above, ATP mediated stability required that free ATP be present throughout the disassembly phase of the experiment, whereas nucleoprotein complexes assembled with AMP-PNP remained stable even after the free nucleotide cofactor was flushed from the flowcell and replaced with buffer containing either no nucleotide or ADP. This finding suggested that release of the bound nucleotide was likely to be the rate-limiting step during dissociation of nucleoprotein filaments assembled with AMP-PNP, and also indicates that once assembled, these filaments can not exchange AMP-PNP for ADP present free in solution. However, the observation that free ATP alone could confer a 5-fold decrease in the disassembly rate indicated that the protein monomers within the elongated filaments continually turnover ATP while remaining bound to the DNA.
We also tested the disassembly of ATPase deficient mutants of Rad51. rad51 K191R has a mutation in the Walker A motif that disables ATP hydrolysis activity while only moderately affecting ATP binding15; 16. Genetic and biochemical studies have indicated that the K191R mutation may have several possible consequences for the cell, although the precise mechanism is not entirely clear18. For example, strains expressing rad51 K191R under the control of a native promotor are highly sensitive to ionizing radiation, but this defect can be overcome by the overexpression of Rad5441. This result suggests that Rad54 might be stripping mislocalized rad51 K191R from undamaged chromatin prior to the onset of recombination thereby ensuring a pool of free Rad51 sufficient to load onto ssDNA during repair. Overexpression of rad51 K191R also overcomes the defect in DNA repair, in this case suggesting that the mutation impacts a later step in HR15; 41. More recently, an in vivo study looking at recruitment of rad51 K191R to ssDNA ends revealed that the protein is in fact recruited to ssDNA at DSBs, but much more slowly than wild-type Rad5118. This defective recruitment was overcome by deletion of the SRS2 gene, which encodes a helicase protein with antirecombinase activity that strips Rad51 from ssDNA45; 46. Chromatin immunoprecipitation studies also failed to detect aberrant recruitment of rad51 K191R to random locations in the genome, suggesting that the primary defect was not in mislocalization to chromatin, but rather slowed assembly on ssDNA overhangs18. Finally, it was demonstrated that the rad51 K191R nucleoprotein filaments failed to properly recruit Rad55/57 in vivo, suggesting that the overall filament structure may be altered and/or that ATP hydrolysis is in some way linked to establishing interactions with other recombination proteins 18.
Using our TIRFM assay we confirmed that the rad51 K191R protein is profoundly defective in DNA binding, and displays a 15-fold decrease in the assembly rate on dsDNA compared with wild-type Rad51. This defect could hypothetically arise from a defect in nucleation or reduced cooperativity, but the fact that the protein remains tightly bound after assembly would suggest that cooperative interactions between Rad51 subunits within the polymer are not grossly impaired. These observations are consistent with in vivo results described above indicating that expression of rad51 K191R from a native promotor yields a strain that is sensitive to ionizing radiation and is also defective for mating type switching, but when overexpressed rad51 K191R fully complements DNA repair defects in a rad51 null strain18; 41.
Despite the drastically reduced binding efficiency, this defect can be overcome in vitro at high concentrations of protein, and the K191R mutant is capable of forming elongated nucleoprotein filaments. Our results confirmed that rad51 K191R can form an extended nucleoprotein filament on dsDNA, and these filaments were no different in length from those observed with wild-type Rad51, suggesting that there is not a profound difference in these structures. Once assembled, rad51 K191R nucleoprotein filaments were highly stable. Interestingly, rad51 K191R was even more stable than wild-type Rad51 bound to DNA with nonhydrolyzable AMP-PNP. These findings are in full agreement with a recent biochemical study that demonstrated that the rad51 K191R mutant was defective for initial DNA binding, but once bound formed elongated filaments that were much more stable than wild-type Rad5116. Similar findings have been made for human rad51 K133R, which harbors the equivalent mutation in the Walker A motif17. Taken together, these results suggest that rad51 K191R should be more difficult to remove from dsDNA in vivo and tighter association with DNA might lead to the accumulation of lethal recombination intermediates. In addition, defects may arise from sequestration of the mutant protein at inappropriate sites, which could limit the pool of free Rad51 available for binding to ssDNA at the ends of broken DNA molecules. As indicated above, growth defects in a strain harboring rad51 K191R expressed from the native promotor can be attenuated by overexpression of Rad54, which is a dsDNA translocase thought to remove nonproductively bound Rad51 from dsDNA via its translocation activity33; 41. Based on the tight binding of this mutant protein to dsDNA in our assay, we speculate that overexpression of rad51 K191R in a rad54 null strain may lead to the accumulation of the mutant rad51 protein on undamaged chromatin, which could potentially be lethal for the cell.
Enhanced DNA binding by Rad51 I345T
Rad55 and Rad57 form an obligate heterodimer that assists loading of Rad51 onto ssDNA coated with replication protein A (RPA)6; 42. rad51 I345T was originally identified as gain-of-function mutant that suppressed the requirement for these recombination mediators, and biochemical studies showed that it bound to both dsDNA and ssDNA better than wild-type Rad5142; 43. These findings suggested that the I345T mutation led to enhanced assembly of Rad51 nucleoprotein filaments in vivo, which enabled the mutant protein to bypass the normal requirement for Rad55/57. Indeed, in vitro assays have shown that rad51 I345T does bind better to ssDNA under steady state conditions, and these nucleoproteins are more resistant to disruption by KCl relative to the wild-type protein43. In addition, rad51 I345T is more active than the wild-type protein in a strand exchange assay using oligonucleotide substrates43. Based on these observations, we predicted that rad51 I345T would dissociate from DNA more slowly than the wild-type protein in our assays, and this initially appeared to be true based upon the magnetic bead assays. Yet surprisingly we observed no appreciable change in the disassembly kinetics of rad51 I345T compared to the wild-type protein in the TIRFM assays. This apparent discrepancy could be explained as rapid rebinding of dissociated rad51 I345T to pre-existing nucleoprotein filaments in the bulk magnetic bead assay, which could not occur in the TIRFM assay due to the continuous buffer flow. This conclusion is in agreement with the TIRFM assembly studies which revealed that rad51 I345T assembled onto DNA ∼30% more rapidly than the wild-type protein, yielding a DNA extension rate of 0.28 nm/sec•nM compared to 0.22 nm/sec•nM for the wild-type protein at 1 mM ATP. Since the dissociation kinetics of rad51 I345T from dsDNA were indistinguishable from the wild-type protein in our assays, we attribute the enhanced binding to an increase in the efficiency of nucleoprotein filament assembly.
In summary, we have developed an assay utilizing end labeled DNA substrates, which enables us to monitor the assembly and disassembly of S. cerevisiae Rad51 nucleoprotein filaments. Using this assay we have shown that ATP stabilizes the nucleoprotein filaments 5-fold relative to ADP or the absence of nucleotide cofactor, the ATP hydrolysis defective mutant dissociates from DNA more slowly than wild-type Rad51 assembled with nonhydrolyzable AMP-PNP, and that the gain-of-function mutant rad51 I345T disassembles with kinetics that are indistinguishable from the wild-type protein, but assembles 30% more rapidly. This work provides the foundations for beginning to dissect the interactions between these nucleoprotein filaments and other protein factors involved in their regulation during homologous recombination.
Materials and Methods
Proteins
Wild-type S. cervisiae Rad51 and rad51 K191R were purified from yeast using a combination of ammonium sulfate precipitation, ion exchange, affinity chromatography, and anion exchange as described previously15. rad51 I345T was a generous gift from Dr. Lorraine Symington, and was purified from E. coli as previously described43.
Quantum dots
Quantum dots (QD 705; Invitrogen) coated with primary amines were labeled with polyclonal sheep anti-digoxigenin (anti-DIG) Fab fragments (Roche Applied Sciences). Labeling was performed essentially as recommended by the manufacturer, with a few modifications. The quantum dots (125 μl at 4 μM) were activated by addition of 1 mM SMCC (4-(maleimidomethyl)-1-cyclohexanecarboxylic acid N-hydroxysuccinimide ester) and allowed to react for 1 hour at room temperature. The lyophilized antibodies were resuspended to a final concentration of 1 mg/ml in 300 μl of phosphate buffered saline (PBS), reduced by the addition of DTT to a final concentration of 20 mM, and then incubated for 30 minutes at room temperature. Both the activated quantum dots and the reduced antibodies were then purified on a NAP-5 desalting column (GE Healthcare) to remove unreacted SMCC and excess DTT, respectively. The activated quantum dots and reduced antibodies were then mixed, allowed to incubate for an additional hour at room temperature, and finally quenched with the addition of 10 mM β-mercaptoethanol. The antibody-labeled quantum dots were then concentrated to a final volume of ∼200 μl by ultrafiltration and purified on a Superose 6 or Superdex 200 10/300 GL column (GE Healthcare) equilibrated in PBS. The purified quantum dot conjugates were quantitated by measuring the absorbance at 550 nm and using an extinction coefficient of 1,700,000 M-1cm-1, as suggested by the manufacturer. The antibody-labeled quantum dots were then stored in PBS plus 0.1 mg/ml acetylated BSA at 4°C, and no decrease in performance was observed for 2-4 months under these conditions.
DNA
The DNA substrate was generated using PCR from whole human genomic DNA as the template. This reaction utilized the Expand 20kb PCR kit under the manufacture's recommended standard conditions resulting in a 23 kilobase segment of the human β-globin locus (Roche). PCR was performed according to the manufacturer's recommendation using the following primers: 5′-biotin-TEG-CAC AAG GGC TAC TGG TTG CCG ATT-3′ (forward primer) and 5′-digoxigenin-AGC TTC CCA ACG TGA TCG CCT TTC TCC CAT-3′ (reverse primer). Primers were obtained from Operon and gel purified before use. To remove unreacted primers, the resulting PCR products were purified over a MicroSpin™ S-400 HR column (GE Healthcare) pre-equilibrated in TE. Typical yield from this protocol was ≈50 μl at a final concentration of 1 nM of the 23 kb DNA substrate.
Magnetic Bead-based Turnover Assay
The magnetic bead-based turnover assay has been adapted from a previously described method17. In brief, a 60-mer double stranded oligonucleotide, modified with biotin on one 5′ end was bound to streptavidin-coated magnetic beads. In a total volume of 10μL, Rad51 was bound to the oligonucleotide in reaction buffer containing 40 mM Tris-HCl (pH 7.8), 1 mM DTT, 10 mM MgCl2, and 0.2 mg/mL bovine serum albumin (BSA) plus 1mM KCl. Rad51 was allowed to bind for 2 minutes at 37°C. A magnet was used to pellet the beads, allowing for the removal of the supernatant buffer and the excess, unbound Rad51. The beads were then washed two times, each for 10 minutes at 37°C, under the condition of interest. For each wash, the beads were pelleted as previously and the buffer was exchanged. Once the reaction was complete, remaining Rad51 bound to the oligonucleotide was eluted with reaction buffer plus 2% SDS. Samples of supernatant, the pooled washes, and eluate were then separated on an SDS-PAGE gel and detected with SimplyBlue™ SafeStain (Invirtogen).
Microscopy
The design of the total internal reflection fluorescent (TIRF) microscope used in this study has been previously described34. In brief, the system uses a Nikon TE2000U inverted microscope with a custom-made illumination system to excite the field of view and a back-illuminated EMCCD detector to capture the fluorescent signal. The illumination system uses a 488nm, 200mW, diode-pumped solid-state laser to excite the quantum dots. The laser was attenuated with a neutral density filter and focused through a custom-built fused silica prism to generate an evanescent wave in a microfluidic sample chamber.
Flowcells, sample delivery, and aligned DNA arrays
The details of flowcell construction have been described elsewhere34; 38. Briefly, flowcells were prepared by etching microscale diffusion barriers onto fused silica slides and by boring inlet and outlet ports using a high-speed precision drill press equipped with a diamond tipped bit. The slides were then cleaned extensively with 2% (v/v) Hellmanex, 1M NaOH, and 100% MeOH, dried under nitrogen, and baked at 120°C for at least 1 hour in a vacuum oven. The sample chamber was formed from double-sided sticky tape and borosilicate glass coverslips. Inlet and outlet ports were attached using preformed adhesive rings and cured at 120°C under vacuum for at least 2 hours or with hot glue. The total volume of the sample chamber was ∼4 μL. To control sample buffer and flowrate, a syringe pump and actuated injection valve were used. Temperature regulation at 37°C (±0.1°C) was maintained using computer-controlled feedback with custom-built heaters.
Within the sample chamber, a lipid bilayer was formed on the surface of the fused silica slide as previously described34; 38. A mixture of DOPC, 0.5% biotinylated-DOPE, and 8% PEG(550)-DOPE liposomes within rinse buffer (10 mM Tris-HCl (pH 7.8), 100 mM NaCl) were injected into the sample chamber for 20 minutes, rinsed with Rinse Buffer, and allowed to heal for 20 minutes. The surface was then passivated further with blocking buffer (40 mM Tris-HCl (pH 7.8), 1 mM DTT, 1 mM MgCl2, and 0.2 mg/mL BSA) and incubated for 10 minutes. Neutravidin (330 nM) in blocking buffer was applied to the sample chamber and was allowed to bind for 10 minutes. After thoroughly rinsing the sample chamber with excess blocking buffer, DNA substrate was injected and allowed to incubate for 10 minutes. Application of continuous buffer flow caused the lipid-tethered DNA molecules to align along the diffusion barriers' leading edge, generating a DNA curtain (Figure 1B and 1C). Once the curtain was formed, 1 mL of blocking buffer containing antibody-labeled quantum dots (1nM) was injected into prepared flowcells to bind the digoxigenin-labeled end of the DNA substrate. This allowed for precise measurement of DNA length when the DNA was in a stretched conformation (described below). For all reactions, the flowcells were connected to a syringe pump and injection valve to control the addition of protein to the DNA substrate. All reaction buffers contained reaction buffer (40 mM Tris-HCl (pH 7.8), 1 mM DTT, 10 mM MgCl2, and 0.2 mg/mL BSA) as well as any other additives as indicated in the figure legends.
Reaction conditions
Unless otherwise stated the assembly reactions utilized a 700 μl injection loop containing 1 μM Rad51, a flow rate of 400 μl/min and buffer conditions were optimized for S. cerevisiae Rad51 and the assembly buffer contained 40 mM Tris (pH 7.8), 0.2 mg/ml BSA, 10 mM MgCl2, 1 mM DTT, and 1 mM ATP. The disassembly reactions utilized the same buffer conditions as the assembly reaction with the exception of the indicated variables. The flowcells were mounted in a custom designed heating unit and all reactions were performed at 37°C.
Data collection and analysis
Data collection and particle tracking has been previously described34; 37. In brief, images were collected using either MetaMorph or NI Elements for the entire duration of the reaction, usually 20 minutes. The exposure time for each frame was varied from 50ms to 200ms to generate the best signal to noise ratio. Once the images were collected, ImageJ was used to process the images to further improve the signal to noise ratio. The processed images were then imported into Igor Pro and analyzed using custom-written procedures37.
To measure the rate of the disassembly reaction, the chase phase was fit using a non-linear, least squares fitting method to reduce the chi square value to generate a continuous smooth curve. The specific fitting equation used was of a sigmoid form:
; where Yo is the base value at small X, Ymax is the maximum value at large X, X1/2 is the X value at which Y = (Yo+Ymax)/2 and rise is the rise rate. The derivative of this curve was calculated to generate the instantaneous disassembly rate. The minimum value was then determined from the derivative and reported as the disassembly rate. A similar procedure was used to calculate the assembly rates.
Acknowledgments
This work was supported in part by a grant from Susan G. Komen grant (to E.C.G.) and National Institutes of Health Grants GM074739 (to E.C.G), ES07061 (to P.S.), and GM53738 (to H.K.). R.B.R. was supported by an NIH Biophysics training grant (5T32GM008281). D.N.M. was supported by an NRSA fellowship from the NIH (GM084587). We thank members of this laboratory for comments on the manuscript. We thank Dr. Symington for generously providing purified Rad51 I345T. We thank Luke Kaplan for assistance with data analysis, and we thank members of our laboratories for discussion and comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.West SC. Molecular views of recombination proteins and their control. Nature Reviews. 2003;4:1–11. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 2.Sung P, Krejci L, Van Komen S, Sehorn MG. Rad51 recombinase and recombination mediators. The Journal of Biological Chemistry. 2003;278:42729–42732. doi: 10.1074/jbc.R300027200. [DOI] [PubMed] [Google Scholar]
- 3.Symington LS. Role of RAD52 Epistasis group of genes in homologous recombination and double-strand break repair. Microbiology and Molecular Biology Reviews. 2002;66:630–670. doi: 10.1128/MMBR.66.4.630-670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khanna K, Jackson S. DNA double-strand breaks: signaling, repair and the cancer connection. Nature Genetics. 2001 Mar;27:247–54. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 5.Thompson LH, Schild D. Recombinational DNA repair and human disease. Mutation Research. 2002;509:49–78. doi: 10.1016/s0027-5107(02)00224-5. [DOI] [PubMed] [Google Scholar]
- 6.Krogh BO, Symington LS. Recombination proteins in yeast. Annu Rev Genet. 2004;38:233–71. doi: 10.1146/annurev.genet.38.072902.091500. [DOI] [PubMed] [Google Scholar]
- 7.Symington LS. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol Mol Biol Rev. 2002;66:630–70. doi: 10.1128/MMBR.66.4.630-670.2002. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bianco PR, Tracy RB, Kowalczykowski SC. DNA strand exchange proteins: a biochemical and physical comparison. Frontiers in Bioscience. 1998;3:d530–603. doi: 10.2741/a304. [DOI] [PubMed] [Google Scholar]
- 9.Tsuzuki T, Fujii Y, Sakumi K, Tominaga Y, Nakao K, Sekiguchi M, Yoshimura Y, Morita T. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proceedings of the National Academy of Sciences (USA) 1996;93:6236–6240. doi: 10.1073/pnas.93.13.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sigurdsson S, Trujillo K, Song BW, Stratton S, Sung P. Basis for avid homologous DNA strand exchange by human Rad51 and RPA. The Journal of Biological Chemistry. 2001;276:8798–8806. doi: 10.1074/jbc.M010011200. [DOI] [PubMed] [Google Scholar]
- 11.Mcllwraith MJ, Van Dyck E, Masson JY, Stasiak AZ, Stasiak A, West SC. Reconstitution of the strand invasion step of double-strand break repair using human Rad51, Rad52 and RPA proteins. Journal of Molecular Biology. 2000;304:151–164. doi: 10.1006/jmbi.2000.4180. [DOI] [PubMed] [Google Scholar]
- 12.Baumann P, Benson FE, West SC. Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell. 1996;87:757–766. doi: 10.1016/s0092-8674(00)81394-x. [DOI] [PubMed] [Google Scholar]
- 13.Eggler A, Inman R, Cox M. The Rad51-dependent pairing of long DNA substrates is stabilized by replication protein A. The Journal of Biological Chemistry. 2002 Oct 18;277:39280–8. doi: 10.1074/jbc.M204328200. [DOI] [PubMed] [Google Scholar]
- 14.Mazin A, Zaitseva E, Sung P, Kowalczykowski S. Tailed duplex DNA is the preferred substrate for Rad51 protein-mediated homologous pairing. The EMBO Journal. 2000 Mar 1;19:1148–56. doi: 10.1093/emboj/19.5.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sung P, Stratton SA. Yeast Rad51 recombinase mediates polar DNA strand exchange in the absence of ATP hydrolysis. J Biol Chem. 1996;271:27983–6. doi: 10.1074/jbc.271.45.27983. [DOI] [PubMed] [Google Scholar]
- 16.Li X, Zhang XP, Solinger JA, Kiianitsa K, Yu X, Egelman EH, Heyer WD. Rad51 and Rad54 ATPase activities are both required to modulate Rad51-dsDNA filament dynamics. Nucleic Acids Res. 2007;35:4124–40. doi: 10.1093/nar/gkm412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chi P, Van Komen S, Sehorn MG, Sigurdsson S, Sung P. Roles of ATP binding and ATP hydrolysis in human Rad51 recombinase function. DNA Repair. 2006;5:381–391. doi: 10.1016/j.dnarep.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 18.Fung CW, Fortin GS, Peterson SE, Symington LS. The rad51-K191R ATPase-defective mutant is impaired for presynaptic filament formation. Mol Cell Biol. 2006;26:9544–54. doi: 10.1128/MCB.00599-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zaitseva EM, Zaitsev EN, Kowalczykowski SC. The DNA binding properties of Saccharomyces cerevisiae Rad51 protein. The Journal of Biological Chemistry. 1999;274:2907–2915. doi: 10.1074/jbc.274.5.2907. [DOI] [PubMed] [Google Scholar]
- 20.Conway AB, Lynch TW, Zhang Y, Fortin GS, Fung CW, Symington LS, Rice P. Crystal Structure of a Rad51 filament. Nature Structural and Molecular Biology. 2004;11:791–796. doi: 10.1038/nsmb795. [DOI] [PubMed] [Google Scholar]
- 21.Kinebuchi T, Kagawa W, Enomoto R, Tanaka K, Miyagawa K, Shibata T, Kurumizaka H, Yokoyama S. Structural basis for octomeric ring formation and DNA interaction of the human homologous-pairing protein Dmc1. Molecular Cell. 2004;14:363–374. doi: 10.1016/s1097-2765(04)00218-7. [DOI] [PubMed] [Google Scholar]
- 22.Sehorn MS, Sigurdsson S, Bussen W, Unger VM, Sung P. Human meiotic recombinase Dmc1 promotes ATP-dependent homologous DNA strand exchange. Nature. 2004;429:433–437. doi: 10.1038/nature02563. [DOI] [PubMed] [Google Scholar]
- 23.Shin DS, Pellegrini L, Daniels DS, Yelent B, Craig L, Bates D, Yu DS, Shivji MK, Hitomi C, Arvai AS, Volkmann N, Tsuruta H, Blundell TL, Venkitaraman AR, Tainer JA. Full-length archaeal Rad51 structure and mutants: mechanisms for Rad51 assembly and control by BRCA2. The EMBO Journal. 2003;22:4566–4576. doi: 10.1093/emboj/cdg429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Story RM, Weber IT, Steitz TA. The structure of the E. coli recA protein monomer and polymer. Nature. 1992;355:318–325. doi: 10.1038/355318a0. [DOI] [PubMed] [Google Scholar]
- 25.Wu Y, He Y, Moya IA, Qian X, Luo Y. Crystal structure of archaeal recombinase RadA: a snapshot of its extended conformation. Molecular Cell. 2004;15:423–435. doi: 10.1016/j.molcel.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 26.Yang S, Yu X, Seitz EM, Kowalczykowski SC, Egelman EH. Archeal RadA protein binds DNA as both helical filaments and octomeric rings. Journal of Molecular Biology. 2001;314:1077–1085. doi: 10.1006/jmbi.2000.5213. [DOI] [PubMed] [Google Scholar]
- 27.Yu X, Jacobs SA, West SC, Ogawa T, Egelman EH. Domain structure and dynamics in the helical filaments formed by RecA and Rad51 on DNA. Proceedings of the National Academy of Sciences (USA) 2001;98:8419–8424. doi: 10.1073/pnas.111005398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z, Yang H, Pavletich NP. Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature. 2008;453:489–4. doi: 10.1038/nature06971. [DOI] [PubMed] [Google Scholar]
- 29.Holzen TM, Shah PP, Olivares HA, Bishop DK. Tid1/Rdh54 promotes dissociation of Dmc1 from nonrecombinogenic sites on meiotic chromatin. Genes Dev. 2006;20:2593–604. doi: 10.1101/gad.1447106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kiianitsa K, Solinger JA, Heyer WD. Terminal association of Rad54 protein with the Rad51-dsDNA filament. Proc Natl Acad Sci U S A. 2006;103:9767–72. doi: 10.1073/pnas.0604240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Solinger JA, Kiianitsa K, Heyer WD. Rad54, a Swi2/Snf2-like recombinational repair protein, disassembles Rad51:dsDNA filaments. Mol Cell. 2002;10:1175–88. doi: 10.1016/s1097-2765(02)00743-8. [DOI] [PubMed] [Google Scholar]
- 32.Kiianitsa K, Solinger JA, Heyer WD. Rad54 protein exerts diverse modes of ATPase activity on duplex DNA partially and fully covered with Rad51 protein. J Biol Chem. 2002;277:46205–15. doi: 10.1074/jbc.M207967200. [DOI] [PubMed] [Google Scholar]
- 33.Symington LS, Heyer WD. Some disassembly required: role of DNA translocases in the disruption of recombination intermediates and dead-end complexes. Genes Dev. 2006;20:2479–86. doi: 10.1101/gad.1477106. [DOI] [PubMed] [Google Scholar]
- 34.Prasad TK, Yeykal CC, Greene EC. Visualizing the assembly of human Rad51 filaments on double-stranded DNA. J Mol Biol. 2006;363:713–28. doi: 10.1016/j.jmb.2006.08.046. [DOI] [PubMed] [Google Scholar]
- 35.Gurrieri S, Wells KS, Johnson ID, Bustamante C. Direct visualization of individual DNA molecules by fluorescence microscopy: characterization of the factors affecting signal/background and optimization of imaging conditions using YOYO. Analytical Biochemistry. 1997;249:44–53. doi: 10.1006/abio.1997.2102. [DOI] [PubMed] [Google Scholar]
- 36.Akerman B, Tuite E. Single- and double-strand photocleavage of DNA by YO, YOYO and TOTO. Nucleic Acids Research. 1996;24:1080–90. doi: 10.1093/nar/24.6.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prasad TK, Robertson RB, Visnapuu ML, Chi P, Sung P, Greene EC. A DNA-translocating Snf2 molecular motor: Saccharomyces cerevisiae Rdh54 displays processive translocation and extrudes DNA loops. J Mol Biol. 2007;369:940–53. doi: 10.1016/j.jmb.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Graneli A, Yeykal CC, Prasad TK, Greene EC. Organized arrays of individual DNA molecules tethered to supported lipid bilayers. Langmuir. 2006;22:292–299. doi: 10.1021/la051944a. [DOI] [PubMed] [Google Scholar]
- 39.Doyle PS, Ladoux B, Viovy JL. Dynamics of a tethered polymer in shear flow. Physical Review Letters. 2000;84:4769–4772. doi: 10.1103/PhysRevLett.84.4769. [DOI] [PubMed] [Google Scholar]
- 40.Ladoux B, Doyle PS. Stretching tethered DNA chains in shear flow. Europhysics Letters. 2000;52:511–517. [Google Scholar]
- 41.Morgan EA, Shah N, Symington LS. The requirement for ATP hydrolysis by Saccharomyces cerevisiae Rad51 is bypassed by mating-type heterozygosity or RAD54 in high copy. Molecular and Cellular Biology. 2002;22:6336–6343. doi: 10.1128/MCB.22.18.6336-6343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fortin GS, Symington LS. Mutations in yeast Rad51 that partially bypass the requirement for Rad55 and Rad57 in DNA repair by increasing the stability of Rad51-DNA complexes. EMBO J. 2002;21:3160–70. doi: 10.1093/emboj/cdf293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Malik PS, Symington LS. Rad51 gain-of-function mutants that exhibit high affinity DNA binding cause DNA damage sensitivity in the absence of Srs2. Nucleic Acids Res. 2008 doi: 10.1093/nar/gkn720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van der Heijden T, Seidel R, Modesti M, Kanaar R, Wyman C, Dekker C. Real-time assembly and disassembly of human RAD51 filaments on individual DNA molecules. Nucleic Acids Res. 2007;35:5646–57. doi: 10.1093/nar/gkm629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Veaute X, Jeusset J, Soustelle C, Kowalczykowski SC, Le Cam E, Fabre F. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature. 2003;423:309–12. doi: 10.1038/nature01585. [DOI] [PubMed] [Google Scholar]
- 46.Krejci L, Van Komen S, Li Y, Villemain J, Reddy MS, Klein H, Ellenberger T, Sung P. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature. 2003;423:305–9. doi: 10.1038/nature01577. [DOI] [PubMed] [Google Scholar]