

Abstract
We demonstrate that virus-like particles carrying conformationally-complex membrane proteins (“lipoparticles”) can be used as soluble probes of membrane protein interactions. In order to demonstrate the utility of this approach, we use lipoparticles to rapidly differentiate the relative kinetics of membrane protein interactions using optical biosensor technology. The technique is applied to diverse membrane proteins, including G protein-coupled receptors (GPCRs), and used to rank the relative kinetics of nearly all the commercially available monoclonal antibodies against the chemokine receptor CCR5. These particles serve as versatile probes to screen crude and purified antibody preparations for receptor specificity, epitope reactivity, and relative binding kinetics.
Complex membrane proteins, such as G protein-coupled receptors (GPCRs), ion channels, and oligomeric single-transmembrane proteins, form the interactive interface between the cell and its environment. However, because these proteins rely on the cellular lipid bilayer to maintain their correct structure, it is often difficult or impossible to investigate their molecular interactions using methods that require their liquid suspension or dissolution. We and others have previously described the use of virus-like particles for isolating membrane proteins from cells, a tool that we refer to as lipoparticles (1, 2).
Lipoparticles are created by co-expressing a membrane protein of interest and a retroviral core protein (Gag) in mammalian cells. The Gag core self-assembles and buds from the host cell, resulting in a non-infectious membranous particle, approximately 150 nm in diameter, embedded with non-viral membrane proteins. Unlike other sources of membrane proteins prepared from cells, lipoparticles are homogeneous, physically well-defined, and present high-concentrations of target membrane proteins in their native structure. We have previously used substrate-immobilized lipoparticles to detect membrane protein interaction events (3, 4), but lipoparticles have not previously been used as soluble probes. Immobilization onto a biosensor surface requires direct lipoparticle coupling and the need for receptor regeneration without membrane disruption, which imposes limits on assay sensitivity, speed, and application to diverse receptors. Direct attachment also does not take advantage of the size of lipoparticles, which enables them to be used as solution-phase reagents displaying the incorporated membrane proteins.
Surface plasmon resonance (SPR) biosensors have become a standard method for screening the binding kinetics of monoclonal antibodies (MAbs) from hybridoma supernatants, phage libraries, and following affinity maturation (5–8). Biosensor screening of MAbs is especially valuable for identifying new antibodies of very high (<0.1 nM) or low (>100 nM) affinity, where equilibrium binding assays (e.g. ELISA, flow cytometry, immunofluorescence) are difficult or inaccurate. However, SPR biosensors have not been widely applicable for characterizing the interactions of antibody with integral membrane proteins. While previous approaches, such as capturing receptors from crude membrane preparations, solubilizing receptors in detergent, or immobilizing purified receptors in a reconstituted membrane environment (9–12), have enabled a limited number of readily-manipulated membrane proteins to be studied, they require complex and empirically determined methods for isolating and reconstituting the membrane protein preparations. Because of their size, aqueous suspensions of lipoparticles approximate the behavior of membrane proteins in solution. Here we describe the use of lipoparticles as versatile mobile-phase reagents for the study of membrane protein interactions in optical biosensor analyses. Importantly, this lipoparticle-based method is a modular platform that can readily be adapted to study a broad range of diverse receptors in any format where soluble membrane protein probes are needed.
We reasoned that lipoparticles in aqueous suspension could be flowed across antibodies immobilized on a biosensor flow cell to rapidly screen antibody specificity and the relative kinetics of antibody-membrane protein interactions (Fig. 1a). Antibodies are more readily immobilized to substrates and are more amenable to regeneration than are more fragile membrane structures. Various lipoparticles, each containing a single type of enriched human membrane protein, were produced and purified as described previously (3). Incorporated receptors included seven-transmembrane GPCRs (CXCR3, CXCR4, CCR10, CCR5, 5HT1a, μ-opioid receptor, and CB1), a type I single-transmembrane protein (CD4), and a type II single-transmembrane protein (DC-SIGN, which exists in the lipid membrane naturally as a homotetramer). Where specific MAbs against a receptor were unavailable, receptors were expressed with an extracellular FLAG epitope tag for ease of detection. Lipoparticles were characterized for size and purity by dynamic light scattering, and the incorporation and integrity of each membrane protein was verified by Western blot (data not shown). Biacore biosensor C1 chips were prepared by covalently attaching goat anti-mouse IgG antibodies to all four flow cells using standard NHS/EDC chemistry. Receptor- or FLAG-specific murine antibodies were then sequentially captured onto each flow cell in PBS containing 1 mg/ml BSA. Lipoparticles containing a particular receptor were then flowed over all four flow cells. Each experiment was designed so that one flow cell contained a receptor-specific antibody, while the other three contained antibodies not expected to recognize the incorporated receptor. After each injection of lipoparticles, flow cells were regenerated back to the capture (goat) antibody layer using 0.2% TX-100 followed by 100 mM phosphoric acid. We found that the derivatized chip could be used for at least 100 cycles of antibody capture, lipoparticle binding, and regeneration without significant loss of signal.
Figure 1.

(a) Schematic of the experimental design. (b) A complete capture, binding, and regeneration cycle using lipoparticles on a Biacore 2000 biosensor. (c) Antibodies specific for the membrane receptors CXCR4 (12G5), CCR10 (M2 anti-FLAG), mu-opioid (M2 anti-FLAG), 5HT1a (M2 anti-FLAG), CB1 (368302), CD4 (#19), and DC-SIGN (DC11) were captured onto different flow cells of a Biacore C1 chip. Antibodies 9E10 (anti-myc), and antibodies H36 and H37 (against influenza HA) were used as negative controls. Binding was measured by flowing lipoparticles containing the specific receptor over the immobilized antibodies.
A typical sensorgram showing the complete cycle of murine antibody capture, lipoparticle binding, and chip regeneration, is shown in Figure 1b. In each case, lipoparticles bound only within flow cells containing the antibody specific for the expressed receptor (Fig. 1c, solid lines), but not to the three flow cells containing antibodies reactive against other receptor types (Fig. 1c, dashed lines). Lipoparticles produced without any specific membrane protein (Fig. 1c, Null lipoparticles) did not bind any of the tested antibodies. Because SPR signals are proportional to mass, the signal intensities of the membrane protein interaction are magnified by the large mass of the lipoparticle binding in close proximity to the SPR surface. Assay sensitivity could be readily increased by lengthening the time of interaction or by using increasing amounts of lipoparticles (Fig. S1, Supporting Information). We were also able to detect binding to unpurified monoclonal antibodies in hybridoma supernatant and unpurified polyclonal antibodies in serum (Fig. S2, Supporting Information). The ability to detect dilute antibodies in such complex mixtures is attributed to an antibody capture step coupled with high assay sensitivity due to the large mass change of lipoparticle binding. These data demonstrate that lipoparticles containing diverse GPCRs and other membrane proteins can be readily used as mobile-phase reagents in typical SPR biosensor binding experiments.
To demonstrate that the flowed lipoparticle assay can yield quantitative information for comparing a receptor's interactions with diverse antibodies, we next screened nearly all commercially available monoclonal antibodies against the chemokine receptor CCR5 and ranked them according to the relative strength of their interactions. These twelve antibodies include well-characterized and broadly-inhibitory MAbs with diverse epitope specificities, including both linear and conformation-dependent epitopes (13, 14). An unreactive MAb (9E10) was included as a control. MAbs were sequentially captured to a C1 chip to approximately 50 resonance units (RU) in order to minimize rate differences due to antibody surface density. Lipoparticles incorporating CCR5 were flowed across the chip for two minutes (representative sensorgrams are shown in Figure 2a), and the MAbs were ranked on the basis of the strength of the biosensor signal (Fig. 2b).
Figure 2.
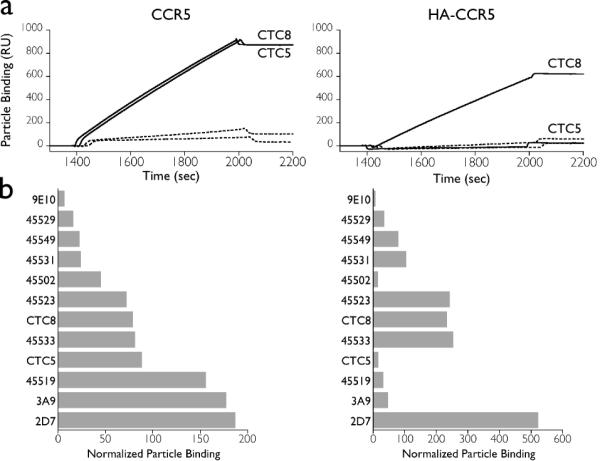
(a) Representative sensorgrams of CCR5 lipoparticle binding experiments on the Biacore. (b) Screening of a panel of CCR5 antibodies. Antibodies reactive against diverse epitopes of CCR5 were captured on a biosensor chip. The binding of CCR5- or HA-CCR5-containing lipoparticles to each of the antibodies was measured in duplicate experiments on a Biacore biosensor. Total lipoparticle binding was normalized by the antibody capture level to correct for small differences in antibody capture. Duplicate injections varied by an average of 4 RU.
The tested antibodies varied considerably in their ability to bind CCR5-containing lipoparticles (Fig. 2b). Three antibodies (45529, 45549, and 45531) bound lipoparticles weakly as compared to the nonspecific binding of lipoparticles to 9E10. Conversely, several antibodies that have not been well described previously, including 3A9 and 45519, showed unexpectedly strong associations, rivaling that of 2D7, the tightest binding and most well-characterized of the known CCR5 antibodies. In order to interpret this variation, we compared our results to published results obtained using some of the same antibodies (13, 14). While no prior studies have analyzed the detailed binding kinetics of these antibodies, the comparisons suggest that the forward rate constant (on-rate) of the receptor-antibody interaction may dominate the binding interactions measured in our experiments. For example, theoretical and experimental analyses of virus binding to cells suggest that the binding interaction is generally irreversible and limited by the on-rate (15–17). Additionally, flow cytometry time course experiments showed semi-quantitative variation in the rate of antibody attachment to the cell surface, from slow (for MAbs 45529, 45531) to fast (for MAb 2D7) (13), that correlate well with our results for these same MAbs. While further detailed kinetic analysis of the on- and off-rates of binding for each MAb is desired, these data support the interpretation that the on-rate of antibody attachment is the strongest determinant of lipoparticle binding in our experiments. This interpretation is not unexpected given the multivalent nature of the interaction; once a single antibody-receptor tether is formed, rapid formation of additional antibody-receptor interactions makes lipoparticle binding essentially irreversible on the timescale of these experiments. By contrast, the off-rate would play an increasingly important role if it exceeds the rate of formation of additional antibody-receptor tethers or if the number of tethers is low, either due to a slow on-rate constant or a low antibody/receptor surface density. Thus, under many conditions, even low affinity antibody-receptor interactions should result in detectable lipoparticle binding. Our results thus suggest that the assay described here is capable of measuring a kinetic component (on-rate) of the interaction, rather than simply measuring equilibrium binding.
The CCR5 N-terminus contains epitopes for several MAbs, such as CTC5 and CTC8 (13). To demonstrate that our methodology can accurately differentiate specific epitopes, we screened the panel of CCR5 MAbs using lipoparticles incorporating either CCR5 or an extracellular N-terminal HA-tagged CCR5 construct (HA-CCR5). As expected, binding to CTC5, which has previously been shown to require an intact N-terminus and not bind to HACCR5 on cells, was substantially reduced (Fig. 2a). In contrast, binding to CTC8, which binds to an internal epitope in the N-terminus and has previously been shown to bind HA-CCR5 on cells, was unaffected by the presence of the tag. Several other MAbs (45502, 45519, and 3A9) that are known to bind to sites on the distal N-terminus were similarly blocked by the HA tag (Fig. 2b). These results demonstrate that epitope specificity is maintained in this assay.
This study demonstrates the use of receptor-containing lipoparticles as mobile phase reagents capable of measuring the interaction of antibodies with diverse membrane proteins. The binding of lipoparticles to captured antibodies is receptor-specific, epitope-specific, and can yield quantitative measurements related to the on-rate of the interaction. Other factors may also contribute to the kinetics of lipoparticle binding, including epitope accessibility, particle diffusion, receptor heterogeneity, and binding stoichiometry. Compared with alternative means of quantifying membrane protein interactions using the Biacore biosensor, the use of lipoparticles as mobile-phase reagents enables the rapid screening of a modest panel of MAbs arrayed in microplates within hours, rather than days, using either purified antibodies or unpurified hybridoma supernatants. Because SPR biosensor signals are proportional to mass, the multivalency and large mass of lipoparticles provides for a highly sensitive assay that is ideal for detecting weakly interacting antibodies or for detecting low abundance antibodies present in polyclonal sera or other complex analytes. Assay sensitivity is also increased by the antibody capture step and, as desired, by increasing reaction time. The dependence of the lipoparticle binding assay on the on-rate allows for the detection of transient receptor-antibody interactions that are specific but do not lead to the formation of a stable complex. Detection of these transient complexes may be useful for understanding immune responses and viral and cellular interactions. The technique developed here should also be applicable to live viruses and to other types of detection technologies where membrane proteins are limited by instrumentation, cell size, or the ability to be solubilized.
Supplementary Material
Acknowledgements
We thank Riley Payne for help with lipoparticle production, Bridget Puffer for molecular biology expertise and helpful discussions, Christopher Laing and Soma Banik for assistance in writing the manuscript, and Ann Rux and Gabriela Canziani for useful discussions and suggestions about the biosensor method. Jim Hoxie kindly provided unpurified 12G5 hybridoma supernatant.
Funding for this work was provided by the National Institutes of Health (GM64924 and RR16832).
Footnotes
Supporting Information Available: Discussion of lipoparticle binding assay sensitivity. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Balliet JW, Bates P. J Virol. 1998;72:671–676. doi: 10.1128/jvi.72.1.671-676.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Endres MJ, Jaffer S, Haggarty B, Turner JD, Doranz BJ, O'Brien PJ, Kolson DL, Hoxie JA. Science. 1997;278:1462–1464. doi: 10.1126/science.278.5342.1462. [DOI] [PubMed] [Google Scholar]
- (3).Hoffman TL, Canziani G, Jia L, Rucker J, Doms RW. Proc Natl Acad Sci U S A. 2000;97:11215–11220. doi: 10.1073/pnas.190274097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Rucker J. Methods Mol Biol. 2003;228:317–328. doi: 10.1385/1-59259-400-X:317. [DOI] [PubMed] [Google Scholar]
- (5).Rich RL, Myszka DG. J Mol Recognit. 2006;19:478–534. doi: 10.1002/jmr.808. [DOI] [PubMed] [Google Scholar]
- (6).Papalia GA, Baer M, Luehrsen K, Nordin H, Flynn P, Myszka DG. Anal Biochem. 2006;359:112–119. doi: 10.1016/j.ab.2006.08.032. [DOI] [PubMed] [Google Scholar]
- (7).Safsten P, Klakamp SL, Drake AW, Karlsson R, Myszka DG. Anal Biochem. 2006;353:181–190. doi: 10.1016/j.ab.2006.01.041. [DOI] [PubMed] [Google Scholar]
- (8).Wassaf D, Kuang G, Kopacz K, Wu QL, Nguyen Q, Toews M, Cosic J, Jacques J, Wiltshire S, Lambert J, Pazmany CC, Hogan S, Ladner RC, Nixon AE, Sexton DJ. Anal Biochem. 2006;351:241–253. doi: 10.1016/j.ab.2006.01.043. [DOI] [PubMed] [Google Scholar]
- (9).Navratilova I, Sodroski J, Myszka DG. Anal Biochem. 2005;339:271–281. doi: 10.1016/j.ab.2004.12.017. [DOI] [PubMed] [Google Scholar]
- (10).Navratilova I, Dioszegi M, Myszka DG. Anal Biochem. 2006;355:132–139. doi: 10.1016/j.ab.2006.04.021. [DOI] [PubMed] [Google Scholar]
- (11).Rice PJ, Kelley JL, Kogan G, Ensley HE, Kalbfleisch JH, Browder IW, Williams DL. J Leukoc Biol. 2002;72:140–146. [PubMed] [Google Scholar]
- (12).Stenlund P, Babcock GJ, Sodroski J, Myszka DG. Anal Biochem. 2003;316:243–250. doi: 10.1016/s0003-2697(03)00046-0. [DOI] [PubMed] [Google Scholar]
- (13).Lee B, Sharron M, Blanpain C, Doranz BJ, Vakili J, Setoh P, Berg E, Liu G, Guy HR, Durell SR, Parmentier M, Chang CN, Price K, Tsang M, Doms RW. J Biol Chem. 1999;274:9617–9626. doi: 10.1074/jbc.274.14.9617. [DOI] [PubMed] [Google Scholar]
- (14).Wu L, LaRosa G, Kassam N, Gordon CJ, Heath H, Ruffing N, Chen H, Humblias J, Samson M, Parmentier M, Moore JP, Mackay CR. J Exp Med. 1997;186:1373–1381. doi: 10.1084/jem.186.8.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Moldovan R, Chapman-McQuiston E, Wu XL. Biophys J. 2007;93:303–315. doi: 10.1529/biophysj.106.102962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Schwartz M. J Mol Biol. 1976;103:521–536. doi: 10.1016/0022-2836(76)90215-1. [DOI] [PubMed] [Google Scholar]
- (17).English TJ, Hammer DA. Biophys J. 2005;88:1666–1675. doi: 10.1529/biophysj.104.047043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.