

Abstract
We formulate a two-dimensional lattice model to study the equilibrium phase behavior of a ternary mixture composed of two enantiomeric forms of a chiral molecule and a nonchiral liquid solvent. Numerical solution of the model invoking the mean-field approximation of statistical mechanics allows the calculation of a ternary phase diagram. A prominent feature of the phase diagram is the appearance of a mirror-image pair of triple points involving coexistence of a liquid phase enriched in one of the enantiomers with two solid phases: a racemic and an enantiopure crystal. Thus, over broad ranges of initial composition, including liquid mixtures containing almost equal amounts of the two enantiomers, the equilibrium state of the system produces liquid-phase chiral amplification. The calculations predict that chiral amplification is favored at low temperatures, and by strengthening those molecular interactions that stabilize the racemic crystal. The phase behavior that we obtain is qualitatively identical to that reported in a recent experimental study of solutions of amino acids, aimed at probing the liquid-phase control of asymmetric catalysis. Those results, and the present calculations that provide molecular-level insight into their underlying causes, suggest a possible thermodynamic scenario for the liquid-phase emergence of chiral imbalance in a prebiotic and presumably nearly racemic world.
Keywords: chirality, asymmetric, behavior
Chiral asymmetry choices exhibited by molecules that are present in living organisms constitute a vivid and scientifically challenging set of observations. Such geometric preferences favoring one enantiomer over its mirror image are obvious in the observed structures of amino acids, sugars, and the biopolymers that they form. The DNA double helix constitutes another mesoscopic example of unvarying chirality. These facts automatically generate fundamental questions about how those chiral asymmetries in the terrestrial biosphere spontaneously arose, and to what extent liquid phases were involved in that scenario. It is also important to know whether the exclusion of the alternate mirror-image enantiomorphs amounts to a basic prerequisite for the origination, survival, propagation, and evolution of living organisms, or whether it is an incidental by-product of the appearance of life.
Not surprisingly, these issues have generated an historically lengthy and diverse literature (e.g., (1, 2)). Several mechanisms have been proposed to explain the spontaneous appearance of chiral bias from a presumably unbiased prebiotic terrestrial environment. One intriguing scenario involves chemical reactions subject to asymmetric autocatalysis, a phenomenon realized experimentally by Soai and collaborators (3). Another invokes chiral crystallization and Ostwald ripening (4). A third possibility focuses on the equilibrium phase behavior of enantiomorphs with a nonchiral third component that acts as a relatively poor solvent (5, 6). In addition to experimental studies, a number of computational examinations of chirality have been published (e.g., (7, 8)). In this study we focus on liquid-phase phenomena and adopt a theoretical approach to calculate the phase behavior.
The theoretical investigation reported in this article was stimulated by the experimental study reported in ref. 5 concerning asymmetric amplification in liquid solutions by amino acid catalysis. Specifically, that investigation quantitatively probed the liquid-phase control of the enantiomorphic excess of a chiral aldol reaction product by the catalyzing role of amino acid enantiomorphic excess present in the solution. A basic conclusion of that study was that the presence of a symmetrical pair of eutectic points† in the ternary phase diagram for the enantiomorphic pair of amino acids and their non-chiral solvent played a key role in the amplification process. In accord with that conclusion, this article introduces and analyzes a simple lattice model that, as shown below, is capable of reproducing, at least qualitatively, such ternary phase diagrams. Although schematic phase diagrams consistent with the type of solid–liquid equilibrium discussed in this work can be found in the stereochemistry literature (9, 10), our focus is on the microscopic origin of such behavior, which has not received comparable attention.
The following section defines the lattice model chosen for study. Although it is conceptually simple, its large-system-limit thermodynamic behavior is currently not amenable to a mathematically exact solution. Consequently, for its analysis we have invoked a standard mean-field approximation, described in the third section. We believe that this approximation preserves the overall essential behavior of the model's thermodynamics, which can be extracted from straightforward numerical calculations. The results of those calculations are then presented. Finally, a summary of our conclusions and remarks about extensions of our model that we believe are feasible are discussed. The technical details are included in supporting information (SI) Appendix.
Lattice Model
To demonstrate that an enantiomorph-solution ternary phase diagram of the type required for liquid-phase chiral amplification can arise from simple molecular interactions, we have constructed and analyzed an elementary two-dimensional lattice model. In particular, this involves a simple square lattice on the sites of which three species can exist: right- and left-handed enantiomorphs (to be denoted “D” and “L” for simplicity), and a nonchiral solvent species “S”. Fig. 1 A illustrates the elements of this model, showing in minimalist fashion how the three species are accommodated on the lattice. The two distinct enantiomorphs, D and L, appear as bent arrows that conform to the square-lattice bond directions, whereas the nonchiral solvent is represented by small open circles at the lattice sites. One and only one of these species resides at each lattice site. Although this model postulates that molecular occupancy patterns can reconfigure freely on the lattice, the individual enantiomorphic molecules are not capable of transforming to their mirror-image forms. Thus, an initially established molecular composition remains invariant thereafter.
Fig. 1.

Schematic of two-dimensional lattice model. (A) The 9 possible occupancy states for each lattice site. Each of the enantiomers can take on 4 different orientations, which are designated by the direction of the backbone: “southeast” (SE), “southwest” (SW), “northwest” (NW), and “northeast” (NE). Rotations are only permitted in the two-dimensional plane, thus preventing a switch of chirality. (B) The interaction between molecules of the same chirality and orientation (v1). (C) The interaction between molecules of opposite chirality and identical orientation (v2).
Our objective is to calculate the thermal equilibrium properties of the model as a function of the molecular composition, and to highlight the resulting ternary phase diagram at any chosen temperature. Each of the two enantiomorphs can reside at any of the N lattice sites in any of 4 orientations. Consequently, there are 9 possible occupancy states for each lattice site after including the occupancy possibility for the structureless solvent molecule. These 9 occupancy states for any lattice site 1 ≤ i ≤ N can formally be denoted by “Ising spin” parameter ξi. Complete specification of the model requires defining the potential energy function Φ(ξ1…ξN) for all possible configurations of the Ising spins.
Three requirements need to be imposed implicitly on the form of Φ. (i) The nonchiral solvent (DMSO, water, or N,N-dimethylformamide in ref. 5) should only produce sparing solubility for the chiral molecules. (ii) At high enantiomorphic excess (EE) and ambient temperature, stable crystals composed only of the excess enantiomer should form. (iii) If the system has a composition close to racemic (1:1), the favored crystal structure should instead be an equimolar “alloy” of D and L, exhibiting periodic spatial order. These requirements evidently can be met by choosing Φ to contain a sum of three terms that each depend on the pattern of parameters ξ1…ξN residing on the lattice:
Here, the quantities v0, v1, and v2 are negative coupling strengths. The function N0 enumerates the number of nearest-neighbor pairs of enantiomorphic molecules, regardless of their chiral type or their orientation. By contrast, N1 enumerates a subset of those in N0, namely, those nearest-neighbor pairs of enantiomorphs of identical chirality and identical orientation (see Fig. 1 B). Finally, N2 represents the number of nearest-neighbor quadruplets of enantiomorphs arranged around an elementary square, with chiral character alternating around the square, and with the bent backbones of all four molecules exhibiting the same orientation (see Fig. 1 C). We choose the interaction parameters to be v0 = −1, v1 = −2, and v2 = −5, in accord with Eq. 2.
The postulated form, Eq. 1, vanishes for pure solvent, as well as for any solution configurations in which all D and L molecules present are fully solvated (i.e., surrounded by 4 nearest-neighbor solvents). Choosing the three coupling parameters v0, v1, and v2 to be negative and sufficiently large in magnitude will ensure that at any temperature the solvent will only be able thermodynamically to support dilute solutions of the enantiomorphs. If the lattice hosts only solvent molecules and just one of the enantiomorphs, the ground state (i.e., the global Φ minimum) will exhibit coexisting phases, respectively, that are pure solvent and a perfect crystal of identically oriented D or L molecules. When equal numbers of D and L molecules are present, with or without solvent, we require that the global Φ minimum exhibit a stable crystal whose structure involves DL alternation around every nearest-neighbor square it occupies, as illustrated in Fig. 1 C. Because of the 4-site nature of the DL interaction, this requirement translates into the following inequality:
It should be mentioned in passing that invoking a 4-site interaction to stabilize a 1:1 racemic crystal, instead of a 2-site interaction favoring contact of chiral opposites, is necessary to suppress high concentrations of chiral-impurity point defects in the otherwise pure enantiomorph crystals.
The model's thermodynamic properties, including ternary phase diagrams, can be extracted from the Helmholtz free energy F. This in turn follows from the classical canonical partition function Q(β) (β = 1/kB T, where kB is Boltzmann's constant and T is the temperature).
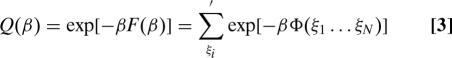 |
The primed summation in this expression includes all distinguishable assignments of solvent, D, and L to the N lattice sites, subject to fixed overall composition. Current interest focuses on intensive thermodynamic properties of the model in its large-system (N → ∞) limit.
Without affecting the large-system-limit results that are of interest, we shall suppose that the square lattice contains N = Nx Ny sites, and is subject to periodic boundary conditions in both the x and y directions. We shall assume that Nx and Ny are large even integers, so that the square lattice consists of two interpenetrating equivalent sublattices “a” and “b.” As a result the ground-state racemic DL crystal can exist with all D's on one of the two equivalent interpenetrating sublattices, and all L's on the other sublattice.
Methods
Mean Field Approximation.
No general analytical method is known to solve rigorously for the thermodynamic properties of our lattice model, which is equivalent to a spin-4 Ising model subject to spatially uniform external fields. However, we propose that the widely used “mean-field approximation” is applicable (11), and should suffice to replicate the desired phase behavior at least qualitatively. In the historical development of Ising model statistical mechanics for order–disorder phenomena, this approach was known as the Bragg–Williams approximation (12).
The long-range-ordered structures of the pure-enantiomer crystals and the stoichiometric racemic crystal involve intrinsic symmetry breaking on the square lattice. In particular, the former can display any of 4 orientations, whereas the latter can have those 4 orientations as well as either of 2 overall sublattice assignments of D's and L's. But without affecting the intensive thermodynamic properties in the large-system limit, one can assume that just one of the respective 4 or 8 possibilities predominates when the free energy indicates that either of these crystal phases should be present. In particular, we assume that in these crystals the backbones of the D and L molecules preferentially orient in the “southeast” (SE) direction, and for the stoichiometric DL crystal the D's occupy the a sublattice and the L's occupy the b sublattice. Of course, the liquid solution phases are isotropic and uniform, and display no such symmetry breaking.
With these innocuous constraints, there are 10 sublattice occupancy probabilities that need to be recognized. Their definitions appear in Table 1. These probabilities are subject to 4 obvious constraints: they must add to unity for each sublattice (two constraints) and they must conform to the assigned mole fractions for the three species (two constraints). Six parametric degrees of freedom remain to be explored so as to attain free-energy minimization. The combination of these 6 parameters describe the overall state of the model (i.e., solution, enantiopure crystal, racemic crystal, etc.).
Table 1.
Definitions of sublattice occupancy probabilities
| Occupying species | Sublattice a | Sublattice b |
|---|---|---|
| Solvent | P1(a) | P1(b) |
| Non-SE D* | P2(a) | P2(b) |
| Non-SE L* | P3(a) | P3(b) |
| SE D* | P4(a) | P4(b) |
| SE L* | P5(a) | P5(b) |
*SE indicates “southeast” backbone orientation; non-SE includes “northeast” (NE), “northwest” (NW), and “southwest” (SW) backbone orientations.
The mean-field approximation assigns a common value to Φ for all configurations consistent with the sublattice probabilities, and this common value is a Φ average over all of those configurations. The corresponding entropy is an elementary combinatorial expression involving the sublattice probabilities. These assignments straightforwardly generate a simple expression for the partition function Q(β) and thus the Helmholtz free energy F that are explicit functions of the sublattice probabilities.
The free energy is a function of composition, temperature, and the 6 mean-field parameters. For a given temperature and composition, the equilibrium state is found by minimizing the free energy with respect to the mean-field parameters. Additionally, systems that phase separate must also obey the phase equilibrium conditions of temperature equality and the equality of the chemical potential of each species between all phases present. The chemical potential of each species μi is straightforwardly calculated from
where ni is the number of molecules of type i. The conditions of phase equilibrium provide a system of nonlinear equations that can be solved for a specified temperature to determine the coexistence regions and tie lines of various phases. Additional details on the techniques used to minimize the free energy and solve the phase equilibrium equations can be found in SI Appendix.
Ternary Diagrams.
Phase diagrams for 3-component systems can be drawn in two ways: (i) a rectangular form where the concentration of the third component is determined by difference, and (ii) a triangular form that shows all 3 components and their concentrations in a symmetric manner. We have chosen the latter method and illustrate how to read a triangular diagram in Fig. 2 (13, 14). The 3 vertices of the triangle each represent a pure species (i.e., pure A, B, and C in Fig. 2). Each side of the triangle is a binary mixture that lacks the species of the opposite vertex (e.g., the bottom side of Fig. 2 is a binary mixture of components A and C). For a point in the middle of the diagram, the fractional concentration for each species is read by tracing a line through the point and parallel to the side opposite the vertex of that species. The intersection of this line with the appropriate side of the triangle gives the species concentration (e.g., the point in Fig. 2 has the composition xA = 0.30, xB = 0.45, and xC = 0.25).
Fig. 2.

Triangular diagram representing the compositions of a ternary mixture with components A, B, and C. The point on the diagram has the composition xA = 0.30, xB = 0.45, and xC = 0.25.
Phase Rule.
The first step in examining phase behavior is the definition of the phase rule for this model. The thermodynamic state of a single-phase C-component system is fixed by the specification of temperature and C − 1 mole fractions.‡ Thus, for a system of P phases, there are a total of PC variables. However, not all of these variables are independent. The temperature of all phases must be the same, resulting in P − 1 restrictions. In addition, for the phases to be in equilibrium, the chemical potential of each species must be equal for all phases present. This results in an additional C(P − 1) restrictions. Thus, the number of degrees of freedom (F) for a C-component system with P phases is given by:
Results
The ternary phase diagram for the reduced temperature T* = 1 (T* = kB T/|v0|) is shown in Fig. 3 and qualitatively reproduces the phase behavior of experiments measuring D- and L-proline concentrations in DMSO solvent in ref. 5. Because of the low solubility of the enantiomers, the region of interest is near the apex of the ternary diagram (i.e., near pure solvent), this region has been enlarged for clarity. Phase diagrams at other temperatures are similar and are shown in the SI Appendix. As temperature decreases, the solubility of the enantiomers decreases and the phase boundaries shift toward the apex of the ternary diagram. In addition, the diagram is perfectly symmetric because D and L molecules have identical interactions. The ternary diagram in Fig. 3 is dominated by two mirror-image 3-phase equilibrium regions. The phase rule for our model (see Eq. 5) dictates a single degree of freedom for 3-phase equilibrium (F = 1 − 3 + 3 = 1). Thus, for a specified temperature, the compositions of the 3 phases are fixed. The composition of the solution phase is given by the appropriate eutectic point, and the solid phases are a near enantiopure phase and a near racemic phase.§ For T* = 1, the eutectic composition corresponds to an enantiomeric excess of 76.5%. Enantiomeric excess is defined by
where αD and αL are the overall mole fractions of D and L, respectively (i.e., αD + αL + αS = 1).
Fig. 3.

Ternary phase diagram for T* = 1. The apex of the diagram is shown on the Left and Right with an illustration of the full diagram in the Center. Open circles mark the eutectic points (labeled E and E′), the solid red lines designate the coexistence region of a solution phase in equilibrium with an enantiopure solid phase, the solid blue lines mark the coexistence region of a solution phase with a racemic solid phase, and the region labeled with green text consists of 3-phase equilibrium between a solution at the eutectic point, a near enantiopure solid, and a near racemic solid. Lines of constant enantiomeric excess (see Eq. 6 for definition) are shown on the Left as a guide. Tie lines are shown as dashed lines on Right. These connect the compositions of the coexisting solution and solid phases.
In addition to the 3-phase coexistence regions, there are regions of Fig. 3 that show the coexistence of two different phases. The phase rule dictates two degrees of freedom for 2-phase equilibrium (F = 1 − 2 + 3 = 2). After specifying the temperature, this leaves a single degree of freedom. Thus, there is a range of conditions for which 2-phase equilibrium is possible. On the outside of the ternary diagram are two mirror-image regions describing equilibrium between a solution phase and a near enantiopure solid. The tie lines in this region (shown on Fig. 3 Right) connect a solution with composition along the red solid line to a crystal with composition on the bottom edge of the diagram near the appropriate vertex. There is an additional 2-phase coexistence region marked by blue lines on Fig. 3. This region represents equilibrium between a solution phase along the top blue line and a solid phase of near racemic composition located in the middle of the bottom axis.
The phase behavior evident in Fig. 3, in conjunction with certain solution phase chemistry, can act as a mechanism for chiral amplification of the solution phase under the following scenario. Imagine a chiral molecule that serves as an asymmetric catalyst with high enantioselectivity for a solution phase reaction (i.e., the chirality of the product of the reaction is determined by the chirality of the catalyst). If a system is prepared with a slight EE of the catalyst and the overall composition lies within the 3-phase coexistence region, then a solution phase, with a composition given by the eutectic point, will coexist with an enantiopure crystal and a racemic crystal. Provided the EE of the eutectic point is sufficiently high, then this leads to a large EE of the catalyst in the solution phase. Thus, a high EE can be achieved for the chiral product of the chemical reaction. The D and L forms of the amino acid serine in water have qualitatively similar phase behavior to that in Fig. 3 with an EE of the eutectic point >99%. In addition, it is known to act as an enantioselective catalyst in aldol reactions (5).
The EE of the eutectic point is a critical aspect of the mechanism for chiral amplification of the solution phase. The composition of the eutectic was calculated for a range of temperatures to explore its sensitivity. Fig. 4 plots the EE of the eutectic as a function of reduced temperature (red line, bottom axis). The ternary diagram (Inset) shows the composition of the eutectic points as a function of temperature. At a reduced temperature of T* = 2.06 the eutectic points lie just above the bottom axis and move toward the apex as temperature decreases. Cooling results in decreased solubility of the D and L enantiomers and shifts the phase boundaries in Fig. 3 toward the apex of the ternary diagram. Fig. 4 shows that, as a consequence of decreased solubility, the EE of the eutectic points increases. Thus, cooling a system with overall composition inside the 3-phase region results in asymmetric amplification of the solution phase. At the coldest temperature studied the EE of the eutectic point reached 83.5%.
Fig. 4.

Variation of the EE of the eutectic point with temperature and fixed interaction parameters (red line, bottom axis) (i.e., v0 = −1, v1 = −2, and v2 = −5). Inset shows the composition of the eutectic points on a ternary diagram as a function of temperature. At T* = 2.06 the eutectic points are nearly on the bottom axis. Cooling decreases the solubility of the enantiomers causing the eutectic points to shift toward the apex of the ternary diagram. Variation of the EE of the eutectic point with the interaction parameter v2 (blue line, top axis). Temperature and the other interaction strengths, v0 and v1, are fixed at T* = 1, v0 = −1, and v1 = −2.
The sensitivity of the EE of the eutectic point to the choice of interaction parameters was also examined. For these calculations the DL interaction strength, v2, was modified. Temperature was fixed at T* = 1 and the v0 and v1 interactions were unchanged (i.e., v0 = −1 and v1 = −2). With these choices, the range of v2 that is in accord with Eq. 2 is v2 ∈ (−∞, −4). For v2 ≥ −4 = 2v1 a racemic crystal phase does not form and the desired phase behavior is not evident. Fig. 4 plots the EE of the eutectic as a function of v2 (blue line, top axis). Increasing the magnitude of the v2 interaction strength confers greater stability to the racemic crystal and expands the 2-phase coexistence region between a solution phase and the racemic crystal. This pushes the eutectic points toward the outside of the ternary diagram and results in a larger EE. The EE of the eutectic point asymptotically approaches 100% as v2 → −∞. In contrast, decreasing the DL interaction strength destabilizes the racemic crystal phase and shrinks the coexistence region between solution and racemic crystal. This brings the eutectic points closer to the middle of the ternary diagram. Consequently, as v2 → −4 = 2v1 the EE of the eutectic approaches zero.
Concluding Remarks
The liquid-phase chiral amplification observed experimentally and reported in refs. 5 and 6 as due to off-symmetry eutectic pairs in the phase diagram of amino acid solutions has been qualitatively replicated here by an elementary lattice model. The model examined is based on a square lattice each of whose sites hosts any one of three species: a structureless solvent molecule, or either enantiomorph of a chemically stable chiral solute molecule. Interactions occur only between nearest neighbors, and have been chosen so that dilute solutions of the enantiomorphs appear in the equilibrium phase diagram at ambient conditions, as well as stable crystals of either pure enantiomorph, and a stoichiometric racemic crystal with periodically alternating enantiomorphs from site to neighboring site.
The mean-field approximation has been invoked to deduce the main qualitative features of the phase diagram for several temperatures and alternative interaction choices. The numerical results indicate that the equilibrium phase behavior of the ternary system can result in chiral amplification of the solution phase, provided the composition lies within one of the 3-phase coexistence regions. Cooling increases the EE of the eutectic point and permits chiral amplification of the solution phase through temperature control. Furthermore, the results demonstrate that the attainable extent of chiral amplification (i.e., the off-symmetry position of the eutectics) is sensitive to the choice of interaction set.
There is reason to believe that, for lattice models with dimension two or higher, the mean-field approximation correctly represents the qualitative equilibrium behavior with respect to first-order phase transitions. Indeed that has been the primary assumption of the work reported herein, where those first-order transitions determine the key eutectic features of the phase diagram. However, it is desirable to check that presumption by invoking a more accurate approach. The well-established Monte Carlo simulation procedure offers such an alternative. We suggest, therefore, that such a follow-up investigation utilizing that procedure would be a natural and desirable addition to our exploratory analysis.
To hold the technical complexity of our lattice model to a minimum, the treatment of the solvent species has been rudimentary. Specifically the nearest-neighbor pair interactions between a pair of solvents, and between a solvent and an enantiomorph, have been set equal (and equal to zero by choice of the energy scale). More generally it would be instructive to determine the extent to which relaxing this simplifying constraint could produce substantial changes in the phase diagram. One possibility might be the appearance of a pair of coexisting liquid phases, one an enantiomorph melt containing a low concentration of solvent, the other a dilute solution of enantiomorphs as described in Results. This would presumably give rise to an upper consolute point upon raising the temperature.
The two-dimensional lattice model possesses a straightforward extension to three dimensions, based on the simple cubic lattice with 6 nearest neighbors. Solvents can continue to be treated as structureless particles occupying a single-lattice site. The enantiomorphs likewise would occupy single-lattice sites but would be endowed with 3 rather than 2 mutually perpendicular “arms” that would orient toward 3 of the 6 nearest neighbors. These arms would be individually distinguishable (“a,” “b,” and “c”), and would be assigned to each molecule in either of 2 ways that are mirror images of one another. Nearest-neighbor pair interactions could be assigned as in the two-dimensional case, including those around each elementary square, to ensure the stability of a stoichiometric crystal at low temperature.
Supplementary Material
Acknowledgments.
We thank Prof. Donna Blackmond for background information and advice concerning this research project. This work was supported by National Science Foundation Collaborative Research in Chemistry Grant CHE0404699 (to P.G.D.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
In this article the term eutectic point refers to the composition of a solution phase that is in equilibrium with two distinct solid phases. This terminology is adopted from ref. 9.
Since our model is formulated on an incompressible lattice, pressure is undefined, and therefore does not contribute to the specification of thermodynamic equilibrium states for single phases nor to the conditions of equilibrium between coexisting phases.
At finite temperature a very few defects are present in the solid structures resulting in near enantiopure and near racemic crystals.
This article contains supporting information online at www.pnas.org/cgi/content/full/0812867106/DCSupplemental.
References
- 1.Weissbuch I, Leiserowitz L, Lahav M. Stochastic “mirror symmetry breaking” via self-assembly, reactivity and amplification of chirality: Relevance to abiotic conditions. Top Curr Chem. 2005;259:123–165. [Google Scholar]
- 2.Mislow K. Absolute asymmetric synthesis: A commentary. Collect Czech Chem Commun. 2003;68:849–864. [Google Scholar]
- 3.Soai K, Shibata T, Morioka H, Choji K. Asymmetric autocatalysis and amplification of enantiomeric excess of a chiral molecule. Nature. 1995;378:767–768. [Google Scholar]
- 4.Cartwright JHE, Piro O, Tuval I. Ostwald ripening, chiral crystallization, and the common-ancestor effect. Phys Rev Lett. 2007;98:165501-1–165501-4. doi: 10.1103/PhysRevLett.98.165501. [DOI] [PubMed] [Google Scholar]
- 5.Klussmann M, et al. Thermodynamic control of asymmetric amplification in amino acid catalysis. Nature. 441:621–623. doi: 10.1038/nature04780. [DOI] [PubMed] [Google Scholar]
- 6.Blackmond DG, Klussmann M. Spoilt for choice: assessing phase behavior models for the evolution of homochirality. Chem Commun. 2007;2007:3990–3996. doi: 10.1039/b709314b. [DOI] [PubMed] [Google Scholar]
- 7.Cao M, Monson PA. A study of the phase behavior of a simple model of chiral molecules and enantiomeric mixtures. J Chem Phys. 2005;122:054505-1–054505-8. doi: 10.1063/1.1842076. [DOI] [PubMed] [Google Scholar]
- 8.Perusquía RA, Peón J, Quintana J. Two-dimensional model for mixtures of enantiomers, bent hard needles: a Monte Carlo simulation. Physica A. 2005;345:130–142. [Google Scholar]
- 9.Jacques J, Collet A, Wilen SH. Enantiomers, Racemates, and Resolutions. New York: Wiley; 1981. [Google Scholar]
- 10.Eliel EL, Wilen SH, Mander LN. Stereochemistry of Organic Compounds. New York: Wiley; 1994. [Google Scholar]
- 11.Stanley HE. Introduction to Phase Transitions and Critical Phenomena. New York: Oxford Univ Press; 1971. [Google Scholar]
- 12.Williams EJ. The effect of thermal agitation on atomic arrangement in alloys - iii. Proc Roy Soc Lond A. 1935;152:231–252. [Google Scholar]
- 13.Sandler SI. Chemical, Biochemical, and Engineering Thermodynamics. 4th Ed. Hoboken, NJ: Wiley; 2006. [Google Scholar]
- 14.Walas SM. Phase Equilibria in Chemical Engineering. Boston: Butterworth; 1985. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.