

Abstract
High-sensitivity microfluidic calorimeters raise the prospect of achieving high-throughput biochemical measurements with minimal sample consumption. However, it has been challenging to realize microchip-based calorimeters possessing both high sensitivity and precise sample-manipulation capabilities. Here, we report chip-based microfluidic calorimeters capable of characterizing the heat of reaction of 3.5-nL samples with 4.2-nW resolution. Our approach, based on a combination of hard- and soft-polymer microfluidics, provides both exceptional thermal response and the physical strength necessary to construct high-sensitivity calorimeters that can be scaled to automated, highly multiplexed array architectures. Polydimethylsiloxane microfluidic valves and pumps are interfaced to parylene channels and reaction chambers to automate the injection of analyte at 1 nL and below. We attained excellent thermal resolution via on-chip vacuum encapsulation, which provides unprecedented thermal isolation of the minute microfluidic reaction chambers. We demonstrate performance of these calorimeters by resolving measurements of the heat of reaction of urea hydrolysis and the enthalpy of mixing of water with methanol. The device structure can be adapted easily to enable a wide variety of other standard calorimeter operations; one example, a flow calorimeter, is described.
Keywords: calorimetry, lab-on-a-chip, nanocalorimetry, biosensor
Fluidic calorimeters provide the ability to characterize the thermodynamics of chemical processes completely without labeling or analyte immobilization. This flexibility is widely used for the study of biomolecular interactions, intramolecular structural changes, and enzyme kinetics (1–3). However, measurement protocols with existing calorimeters involve relatively large sample volumes, typically on the scale of hundreds of microliters, and long measurement times, typically on the order of tens of minutes.
There is growing need for high-throughput, small-volume fluidic calorimeters for both fundamental scientific research and applications in technology. Recent developments in microfabrication now make it possible to build “chip calorimeters” capable of assaying volumes ranging from the microliter to the tens of picoliter scale (4–14). In the future, scale-up to array-based operations will enable high measurement throughput with the reduced sample volumes necessary to make calorimetric screening of large analyte libraries cost effective. Although existing chip calorimeters suggest the feasibility of such future possibilities, practical development and deployment of this technology have been hampered by low device sensitivity and the lack of reliable sample handling down to picoliter volumes.
Chip calorimeters can be classified into two categories according to the configuration of their measurement/reaction chambers. Open-chamber chip calorimeters are built by using thermally isolated wells or platforms onto which samples are spotted as droplets by micropipette or inkjet printing (4–10). Closed-chamber chip calorimeters, by contrast, use microfluidic channels to access closed measurement chambers, into which the samples are introduced and reactions are monitored (11–14). Although open-chamber chip calorimeters provide fairly good thermal isolation, they generally suffer from critical limitations arising from evaporation and awkwardness in sample handling. These can readily lead to erroneous measurements. On the other hand, closed-chamber chip calorimeters generally have greater thermal conductance to their surroundings compared with open-chamber designs. This can result in significant heat loss, which, in turn, can limit device sensitivity. In addition, their sensitivity typically suffers from a larger device heat capacity than that common to open-chamber devices.
We report here the fabrication and operation of microchip-based, closed-chamber calorimeters based on a novel configuration providing greatly enhanced sensitivity. We embed the calorimeter within a thin-film parylene microfluidic system that is thermally isolated from its surroundings by on-chip vacuum encapsulation. Sample handling at 1 nL and below is provided by interfacing these calorimeters with soft pneumatic microfluidics (valves, pumps, and flow channels) for easy and accurate reaction control. This technology can be readily scaled up to array architectures capable of high-throughput calorimetric assays for a wide range of applications in chemistry, the life sciences, and medicine.
Results and Discussion
The calorimeters we have developed comprise three principal components: microfluidics, thermopiles, and vacuum encapsulation (Figs. 1 and 2). The microfluidics include a measurement chamber, flow channels, and pneumatic flow-control components comprising valves and pumps. Integrated within the microfluidic chamber are thermopiles for the local measurement of temperature. The microfluidic channels and measurement chamber are enclosed within vacuum encapsulation (Fig. 1 B and C).
Fig. 1.

Parylene-polymer-based microfluidic calorimeter chip. (A) Device chip (3 × 3 cm) mounted on a vacuum chuck. The sensor leads (Left) and the PDMS microfluidic control tubing (Right) are displayed. (B) Device schematic (cross-sectional view). The parylene membrane is suspended and thermally isolated by vacuum. (C) Optical-microscopy image of the measurement chamber region of the chip, including the boundary of on-chip vacuum space. The vacuum evacuation is through the purple hole visible at the top left. (D) The parylene microfluidics, the thermopile, and the heater on the parylene membrane (≈1.5-mm square). The thermopile has a meandering shape to increase its longitudinal thermal resistance. The suspended parylene membrane is transparent; its reddish color arises mainly from light reflection. (E) Magnified view of the 3.5-nL parylene measurement chamber (200-μm diameter) and connecting channels (35 μm wide) surrounded by vacuum. The reaction chamber swells slightly because of pressure difference (compared with D) when the vacuum chamber is evacuated.
Fig. 2.

Microfluidic layout. Pneumatically controlled PDMS microfluidics chip mated to parylene microfluidics on the calorimeter chip. The PDMS control layer (red) contains valves and peristaltic pumps. The PDMS flow layer (blue) is connected to the parylene channel (black) through an SU-8 via. Four injection pumps (containing large area valves) are built in the right upper corner.
The principal calorimeter components, including the measurement chamber and thermometer, are built on a thin, transparent parylene-C polymer membrane, as shown in Fig. 1D. Parylene-C, a member of the polyxylylene polymer series, is used as the structural material of this membrane and the principal microfluidic components. In this device, these consist of four channels and the reaction/measurement chamber. The chamber is located at the center of the membrane, and it connects to fluidic channels used to inject and purge the sample. In a typical measurement protocol, two different sample solutions are injected into the chamber from two separate channels. After the measurement, buffer solution from a third channel flushes the chamber, and its contents are evacuated through a fourth channel.
In developing a chip-based calorimeter, it is both critical and challenging to engineer a small device heat capacity relative to that of the sample itself. The unique vapor-phase deposition of parylene allows it to form very thin, conformal layers. The parylene microfluidic structures in our device have ≈2-μm-thick walls that permit a very significant reduction in the device heat capacity compared with that of prior closed-chip calorimeter implementations. Parylene is also an excellent insulating material, which provides substantial additional improvement of the calorimetry chamber's thermal isolation.
Parylene has many additional properties, including its chemical resistance and biocompatibility; both are favorable for microfluidic applications (15–17). However, to date, no simple approach has existed to incorporate valves or pumps into parylene microfluidics (18). In this work, to realize a straightforward solution to this problem, we interfaced soft polydimethylsiloxane (PDMS) microfluidics, incorporating valves and pumps, with the “harder” parylene microfluidic components (Figs. 1B and 2). PDMS is a gas-permeable silicone elastomer with Young's modulus three orders smaller than parylene; components made from it provide fluidic control with facility, accuracy, and the potential for great complexity, whereas the parylene microfluidic components have the requisite attributes to serve as the principal calorimeter structures. Because parylene coatings are conformal, the top surfaces of our completed calorimeters were not flat. Therefore, we planarized the structures by using SU-8, a negative photoresist. We used this material to fabricate a thick overlayer that facilitates optimal mating between the parylene calorimeter and PDMS microfluidic structures. A good fluidic seal between the different microfluidic components is obtained by first aligning partially cured PDMS fluidic channels to the parylene microfluidic inlets and outlets, then further curing the mechanically joined structure to achieve a robust bond.
Our device architecture permits variable control of the sample injection volume by sequentially closing four adjacent PDMS valves to facilitate peristaltic pumping. Each valve permits reproducible injection of a 700-pL fluidic volume with ≈50-pL accuracy. The overall operational protocol of the calorimeter can be modified readily by replacing the PDMS microfluidic system with a variety of alternative designs. The PDMS-to-parylene microfluidic interface enables easy integration of calorimeter functions with other forms of lab-on-a-chip devices.
The low thermal conductivity of parylene significantly reduces the heat loss in our devices down to a level comparable to that of the best open-chamber chip calorimeters demonstrated to date. We obtained further enhancement of the thermal isolation of the reaction chamber through a combination of on- and off-chip vacuum encapsulation (Fig. 1 B and C). This configuration proves crucial for achieving maximum sensitivity in closed-chamber, chip-based devices, given their high surface-to-volume ratio. Indeed, for the parylene devices presented here, >90% of the heat loss at ambient pressure is through air. Parylene has very low gas permeability and high mechanical strength, and these properties permit us to apply vacuum across the thin microfluidic walls. Table 1 provides a comparison of the thermal conductance of our vacuum-encapsulated parylene closed-chamber calorimeters with state-of-the-art open and closed chip-based calorimeters.
Table 1.
Chip-based and commercial calorimeters
| Group (source) | Chamber |
Sample handling | Thermal conductance, μW/K | Resolution* |
|||
|---|---|---|---|---|---|---|---|
| Type | Volume, nL | Power, nW | Theoretical energy, nJ† | Practical energy, nJ‡ | |||
| Caltech (this study) | Closed | 3.5 | Multilevel microfluidics | 16 | 4.2 | 6 | 10 |
| Penn State (11) | Closed | 15§ | Syringe pump | 5,000 | 300 | 30 | 105 |
| Eurotronics (12, 13) | Closed | 6,000§ | Syringe pump | 10,000 | 30 | 100 | 1,000 |
| Columbia (14) | Closed | 800§ | Syringe pump | 1,500 | 50 | 30 | 5,000 |
| Scripps-Palo Alto Research Center (4, 5) | Open | 500 | Electrostatic merging | 1,000 | 50 | 100 | 750 |
| Katholieke Universiteit Leuven (6) | Open | 105 to 107 | Micropipette | 5,000 | 5,000 | 105 | 106 |
| University of Glasgow (7) | Open | 0.75 | Micropipette | 100 | 13 | 0.16 | 100 |
| Technische Universität Bergakademie Freiberg (8) | Open | 6,000 | Micropipette | 30,000 | 50 | 500 | 5,000 |
| Vanderbilt (9) | Open | 5–50 | Micropipette | 170 | 22 | 24 | 132 |
| Vanderbilt (10) | Open | 0.05 | Inkjet head | 90 | 150 | 1 | 500 |
| Commercial | Open | 106 | Micropipette | N/A | 10 | N/A | 100 |
N/A, not available.
*Power and energy resolution are based on a 3:1 signal-to-noise ratio.
†Theoretical energy resolution is the product of (noise-limited) power resolution and thermal relaxation time.
‡Practical energy resolution also includes measurement irreproducibility from fluidic volume uncertainties in injection, mixing, and evaporation. Evaporation is particularly deleterious for small, open-chamber calorimeters.
§Flow calorimeters are typically operated at flow rates of 0.5 mL/h.
The vacuum encapsulation comprises two regions. The region above the parylene membrane (the on-chip vacuum space) is defined by SU-8 sidewalls and a top glass coverslip sealed to the calorimeter with UV-curable epoxy. The glass cover facilitates optical imaging of the calorimeter chamber and its contents (Fig. 1B). The SU-8 structure is patterned to achieve both planarization and the vacuum encapsulation in a single step. The region below the parylene membrane (the off-chip vacuum space) is sealed conventionally with a small o-ring within a custom vacuum chuck. The two vacuum spaces are linked by a pumping port etched through the wafer, as depicted in Fig. 1C.
A thin, metal film thermopile, fabricated directly on the parylene membrane, enables measurement of temperature changes within the calorimeter chamber during reactions (Fig. 1D). Thermopower-based sensors provide the most common form of thermometry used in calorimeters. Their principal advantage is that they provide zero-power sensing; that is, sensing uncompromised by additional heat dissipation from the measurement process itself, as encountered with electrically biased resistive sensors. A further advantage over resistive sensing is the circumvention of sensor 1/f noise; however, straightforward thermopower measurements can be degraded by 1/f noise in the readout amplifier. In this work, we connected five thermocouple junctions in series to yield an overall temperature responsivity (total Seebeck coefficient) of 110 μV/K. In the completed devices, the electrical components were sealed between two parylene layers to isolate them from the liquid sample. We measured the thermoelectric voltage from this thermopile by amplifying it with a low-noise, dc-coupled preamplifier (CS 3001; Cirrus Logic). In the output (voltage) domain, the total sensor noise referred to input was ≈10 nV in a 1-Hz bandwidth (0.01–1 Hz). Given the aforementioned temperature responsivity, this indicates the overall temperature resolution for 3:1 signal-to-noise ratio (SNR) is δT = 270 μK in a 1-Hz bandwidth.
The individual thermocouple elements are formed from Au–Ni microjunctions. This choice of materials provides both fabrication convenience and very low 1/f electrical noise. We anticipate that substitution of our Au/Ni thermopiles with previously demonstrated thermopiles providing a higher total Seebeck coefficient should readily provide more than an order of magnitude sensitivity improvement beyond that achieved here (19).
We calibrated the calorimeter by measuring the thermoelectric voltage induced in response to an electric power applied to a gold-resistive heater inside the chamber (Fig. 3A). Thermometer signal response to a step function power is predicted by V(t) = S (P/G) (1 − e−t/τ), where G is thermal conductance, P is amplitude of applied power, and τ is the thermal time constant. From the steady-state response, we found that the device has a heat responsivity, defined as voltage output (V) over applied heating power, of 7.1 V/W. The measured thermal conductance of the device, G = S · P/V, is 16 μW/K under vacuum. This high vacuum-enabled thermal isolation exceeds the performance of prior chip-based calorimeters, which generally range from 100 μW/K to 10 mW/K (Table 1). When a chemical reaction occurs in the chamber, the energy of reaction is obtained as E = tm(t)/Sdt. The time of measurement, tm, is determined by the longer of either the time of the chemical reaction or the thermal relaxation time of the calorimeter. The power sensitivity of the device is G δT ≈ 4.2 nW. During these calibration runs, the measurement chamber is filled with deionized (DI) water. Together, the thermal conductance and the thermal time constant allow us to determine the device heat capacity, C = Gτ. The thermal time constant, τ = 1.3 s, is extracted from the measured rate of exponential growth in response to electrically induced heat steps. These data indicate the heat capacity of the water-filled device is C = 21 μJ/K. By using tabulated values, we estimate that 3.5 nL of water contributes ≈15 μJ/K to the total heat capacity; hence, we deduce the intrinsic (empty) device heat capacity to be ≈6 μJ/K.
Fig. 3.

Thermal response of device. (A) Thermopile response to local electrical heater. A step function of electrical power (4.0 μW) is applied beginning at t = 0. A vacuum of 2 mtorr is maintained, and the device reaction chamber is filled with water. (B) Simulation of thermal response under heat generation inside the chamber at 1 μW. The measurement chamber is the circle. The six U-turn lines are the metallic sensors, and the four rectangles are fluidic channels. (C) Experimental data of thermal conductance, G, of the device under vacuum at different pressures. The fitting formula is G(p) = Gres + Gair/(1 + ), where p is the vacuum chamber pressure (in torr). The fitting parameters are residual thermal conductance (caused by parylene, etc.) Gres = 15.5 μW/K, the thermal conductance of air at 1 atm Gair = 214 μW/K, and a geometric factor d = 170 μm. The geometric factor d is related to the thickness of the vacuum jacket and the size of the measurement chamber.
During these calibrations and all other measurements performed, the enclosure box was maintained at 20 °C, with long-term temperature stability of 0.5 °C (during the ≈1-h measurement sessions). Environmental temperature fluctuations have negligible effect on the accuracy of our measurements because of the close proximity of the two ends of the thermocouple (≈1 mm); the thermopiles only sense the temperature gradient between their two ends. Furthermore, the ends of the thermopiles are isolated from their environment by the vacuum space. To establish a specified reaction temperature the calorimeter and its fluidic contents are maintained within a temperature-regulated enclosure under proportional-integral-derivative control.
Fig. 3B shows results from finite element simulation (FEMLAB; COMSOL) of the calorimeter's thermal conductance. The color scale indicates the temperature change caused by the uniform power dissipated within the chamber. The device is designed in such a way that the temperature gradient inside the measurement chamber (Fig. 3B, circle at the center) is relatively small compared with the gradient over the parylene membrane (Fig. 3B, overall view). The simulation indicates that the maximum temperature gradient in the measurement chamber is <5% of the chamber-averaged temperature change caused by heating. Thus, the temperature inside the measurement chamber can be considered uniform, and the measurement error arising from the position of thermopile junctions will not exceed 5% of total energy. The average temperature change at the thermocouple junction positions, with 1-μW heating, was 0.15 K. The resulting thermal conductance of the device was 6.7 μW/K.
Fig. 3C demonstrates the performance enhancement obtained from on-chip vacuum isolation. Heat loss through air convection and conduction contributes ≈93% of total heat loss at atmospheric pressure. Below 2 mtorr, however, thermal transport through air becomes negligible, and only the parylene microfluidic structure, the electrical leads, and the liquid within the channel provide appreciable pathways for thermal conduction. Thus, vacuum encapsulation provides a 14-fold net increase in signal sensitivity. The solid line in Fig. 3C represents the results of an analytical formula for the thermal conductivity of air between two infinite planes scaled to represent our device geometry but without edge corrections (20). This slip-flow theory provides a reasonably accurate representation for our purposes here. However, to be precise at low pressures, it should be replaced with free-molecule theory. The fitting parameter, d, depends on geometric factors of the device, which include the size of the measurement chamber (200-μm radius) and the distances between the vacuum walls (50 μm, top, and 500 μm, bottom). Fits to the data indicate d ≈ 170 μm, which is comparable with the physical dimensions of the device.
We demonstrated calorimeter performance by observing two separate exothermic reactions. First, we measured the heat of reaction of urea hydrolysis catalyzed by urease:
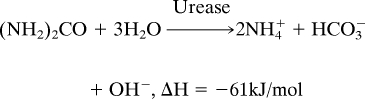 |
To carry out this first measurement, the 3.5-nL reaction chamber was initially filled with an aqueous solution of urease (0.5 mg/mL; Sigma–Aldrich). Subsequently, an aqueous solution of urea (50 mM; USB) was injected into the chamber by using the on-chip PDMS peristaltic pump; this step displaces an equal amount of the urease solution. Both the urease and urea solutions were prepared with the same buffer (0.2 M sodium-phosphate buffer, pH 7.0) to minimize extraneous contributions caused by the heat of mixing. Fig. 4 shows measurements after discrete injections of urea solution, with injection volumes ranging from 700 pL to 2.8 nL, corresponding to 35–140 pmol of urea. The measurement time scale, ≈30 s, was limited by enzyme kinetics and not by the calorimeter's intrinsic thermal response time (τ = 1.33 s). The total energy liberated from hydrolysis can be calculated by integrating the temporal data (Fig. 4 Inset), yielding an enthalpy change of 62 kJ/mol. This finding agrees well with the value of 60.9 ± 2.5 kJ/mol that we measured independently on much larger sample volumes (≈1.5 mL) with a commercial calorimeter (VP-ITC; MicroCal).
Fig. 4.

Calorimetric signature of urea hydrolysis by urease. Calorimetric response after the injection of various volumetric aliquots of 50 mM urea solution into the chamber prefilled with urease solution. Four separate measurements are plotted on the same graph for comparison. (Inset) The total energy of reaction. The red line shows the expected energy of reactions deduced from the heat of reaction, ΔH = 61 kJ/mol.
In these measurements, the total noise at the readout amplifier's output, referred to input (temperature domain), corresponded to ≈500 μK for the present device (1:1 SNR; bandwidth, 0.01–10 Hz). The dominant noise contribution arose from the readout amplifier itself; a secondary contribution arose from the Johnson noise of the thermopile. The contribution from thermal fluctuations of the calorimeter was even smaller; we estimated them to be on an order of 10 μK within the measurement bandwidth. In practice, however, our measurement accuracy does not approach the amplifier-limited value of sensitivity, because of the variance of injection volumes from run to run. To attain full performance from these devices, a more precise sample injection method will be required.
To provide further validation of the capabilities of these calorimeters, we present a second measurement involving the heat of mixing of methanol and water. For this experiment, methanol was prediluted with DI water (14.7% methanol by mole fraction) to reduce the predicted enthalpy change upon mixing to ≈14 μJ (21). The measurement chamber was initially filled with DI water, and 1.4 nL of the 14.7% methanol–water mixture was subsequently injected to attain a final mole fraction of 5.4% methanol. Fig. 5 shows the enthalpy change ensuing from the mixing process. The measured total enthalpy change from the mixing process was on the order of ≈10 μJ. With the current device design, the concentrated sample begins diffusing into the chamber, and thus mixes before the “formal” injection step. The measurement error from this uncontrolled diffusion is rather small for the majority of biochemical reactions because the associated volume of the fluidic channels near the measurement chamber (0.5 pL/μm) is quite small compared with the chamber volume (3.5 nL). Hence, during the short interval between sample loading and injection, only a small amount of reaction/mixing occurs. However, this effect can potentially become more problematic for solutes with large diffusion constants, such as methanol. To circumvent such issues, we have been investigating the separation of samples by immiscible liquids (22). We also note that the temporal resolution of these first-generation devices is limited by the rate of mixing within the chamber (after injection), which occurs primarily by diffusion. Incorporating microfluidic mixers (23) will greatly enhance the rate of mixing.
Fig. 5.

Enthalpy of mixing measurement. (A) Heat of methanol dilution. A total of 1.4 nL of 14.7% methanol–water solution was mixed with 2.1 nL of water. (B) Visualization of the diffusive mixing process, enabled by the injection of red dye into the chamber filled with water.
The vacuum-insulated microfluidic reaction chamber developed in this work is applicable to variety of well-validated approaches in calorimetry, including isothermal titration calorimetry, differential scanning calorimetry, and flow calorimetry. In Fig. 6, we demonstrate one possible configuration we have developed to enable flow calorimetry. Two microfluidic channels, each isolated on a suspended parylene membrane, pass over the two ends of the thermopile. This differential configuration enables suppression of fluidic nonidealities, such as dilution and mixing. Further improvement of the sensitivity should be attainable through use of thermoelectric materials with higher Seebeck coefficients and optimization of parylene suspension geometry to further suppress residual thermal losses under vacuum conditions.
Fig. 6.

Microscale flow calorimeter. (A) Parylene microfluidics and electronic sensor fabricated onto a parylene bridge structure. The measurement chamber is located at the middle of the bridge. (B) Flow calorimeter chip (3 × 3 cm) with two identical measurement chambers (located at the transparent area at the center) for differential measurement.
Summary
Miniaturization brings significant benefits to calorimetry, including the possibility of high-sensitivity, high-throughput analyses with low sample consumption. Past efforts to develop miniaturized calorimetric sensors have led to faster response (better temporal resolution) but generally suffer from low power sensitivity caused by poor thermal isolation. This has proven to be especially deleterious for closed-chamber miniaturized chip calorimeters. In this work, we solved this problem by incorporating vacuum-isolated microfluidics that minimize heat loss and enhance the calorimeter's sensitivity. With these first-generation devices, we demonstrate ≈4.2-nW sensitivity and ≈1.3-s response time with 3.5 nL of total sample volume. This improved performance should enable a new class of cost-effective, high-throughput, automated calorimetric measurements.
Methods
Device fabrication first involves the creation of silicon nitride (SiN) membranes from a SiN wafer polished on both sides (front and back). These SiN membranes subsequently serve as sacrificial layers used to support the polymer devices through the fabrication process. At the end of the fabrication process, they are removed by reactive ion etching (RIE). Fabrication on these membranes proceeds as follows: a first parylene layer (1 μm thick) is deposited on the SiN membrane with the PDS 2010 LABCOTER 2 parylene coater (Specialty Coating Systems). An adhesion promoter, A-174, is applied before the parylene deposition to enhance its adhesion to the substrate. An 80-nm-thick Ni layer and a 60-nm-thick Au layer are sequentially e-beam-evaporated on this basal parylene layer. A 4-nm-thick Ti layer is evaporated as an adhesion layer before both the Ni and Au depositions. These metal layers are subsequently patterned by optical lithography and wet chemical etching to form the thermopiles. Thereafter, a second parylene layer of 1-μm thickness is deposited over the metallic components to isolate them from the fluidic chamber. The parylene microfluidic chamber and its inlet and outlet channels are patterned by conventional methods (15, 16). A 15-μm-thick photoresist is first spun on and patterned to form the inside shapes of the microfluidic structures. A third parylene layer of 2-μm thickness is deposited on these photoresist templates to create the parylene microfluidic structures. An SU-8 device top structure of ≈80-μm thickness is patterned on top of the parylene microfluidic components to planarize the surface and to construct the vacuum encapsulation.
Several etch steps are carried out by using RIE. First, the parylene microfluidic channel opening areas are etched by using an O2 plasma at a pressure of 150 mtorr with a drive power of 140 W. The photoresist inside the channel is removed by using propylene glycol methyl-ether-acetate. After the microfluidic channels are cleared, the parylene covering the electrical components is etched (conditions as above). Finally, the parylene membrane is suspended by etching the sacrificial SiN membrane from its backside; for this step, a CF4 plasma at a pressure of 120 mtorr with a drive power of 140 W is used. For the flow calorimetry devices, an additional parylene etch step is done to pattern the suspended parylene bridges. The final step involves sealing the vacuum encapsulation region with a glass slide by using UV-curable glue.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.Ladbury JE, Chodhry BZ. Biocalorimetry: Application of Calorimetry in the Biological Sciences. Chichester, UK: Wiley; 1998. [Google Scholar]
- 2.Wiseman T, Williston S, Brandts JF, Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 3.Murphy KP, Freire E. Thermodynamics of structural stability and cooperative folding behavior in proteins. Adv Protein Chem. 1992;43:313–361. doi: 10.1016/s0065-3233(08)60556-2. [DOI] [PubMed] [Google Scholar]
- 4.Torres FE, et al. Enthalpy array. Proc Natl Acad Sci USA. 2004;101:9517–9522. doi: 10.1073/pnas.0403573101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Recht MI, et al. Enthalpy array analysis of enzymatic and binding reactions. Anal Biochem. 2008;377:33–39. doi: 10.1016/j.ab.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verhaegen K, Baert K, Simaels J, Driessche WV. A high-throughput silicon microphysiometer. Sens Actuators A Phys. 2000;82:186–190. [Google Scholar]
- 7.Johannessen EA, et al. Micromachined nanocalorimetric sensor for ultra-low-volume cell-based assays. Anal Chem. 2002;74:2190–2197. doi: 10.1021/ac011028b. [DOI] [PubMed] [Google Scholar]
- 8.Lerchner J, Wolf A, Wolf G. Recent developments in integrated circuit calorimetry. J Therm Anal Calorim. 1999;57:241–251. [Google Scholar]
- 9.Xu J, Reiserer R, Tellinghuisen J, Wikswo JP, Baudenbacher FJ. A microfabricated nanocalorimeter: Design, characterization, and chemical calibration. Anal Chem. 2008;80:2728–2733. doi: 10.1021/ac702213d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chancellor EB, Wikswo JP, Baudenbacher F, Radparvar M, Osterman D. Heat conduction calorimeter for massively parallel high-throughput measurements with picoliter sample volumes. Appl Phys Lett. 2004;85:2408–2410. [Google Scholar]
- 11.Zhang Y, Tadigadapa S. Calorimetric biosensors with integrated microfluidic channels. Biosens Bioelectron. 2004;19:1733–1743. doi: 10.1016/j.bios.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 12.Baier V, et al. Highly sensitive thermopile heat power sensor for micro-fluid calorimetry of biochemical processes. Sens Actuators A Phys. 2005;123–124:354–359. [Google Scholar]
- 13.Lerchner J, et al. Nano-calorimetry of small-sized biological samples. Therm Acta. 2008;477:48–53. [Google Scholar]
- 14.Wang L, Sipe DM, Xu Y, Lin Q. A MEMS thermal biosensor for metabolic monitoring applications. J Microelectromech Sys. 2008;17:318–327. [Google Scholar]
- 15.Webster JR, Burns MA, Burke DT, Mastrangelo CH. Monolithic capillary electrophoresis device with integrated fluorescence detector. Anal Chem. 2001;73:1622–1626. doi: 10.1021/ac0004512. [DOI] [PubMed] [Google Scholar]
- 16.Xie J, et al. An electrochemical pumping system for on-chip gradient generation. Anal Chem. 2004;76:3756–3763. doi: 10.1021/ac035188u. [DOI] [PubMed] [Google Scholar]
- 17.Noh HS, Huang Y, Hesketh PJ. Parylene micromolding, a rapid and low-cost fabrication method for parylene microchannel. Sens Actuators B Chem. 2004;102:78–85. [Google Scholar]
- 18.Xie J, Shih J, Lin Q, Yang B, Tai YC. Surface micromachined electrostatically actuated micro peristaltic pump. Lab Chip. 2004;4:495–501. doi: 10.1039/b403906h. [DOI] [PubMed] [Google Scholar]
- 19.Yamashita O, Tomiyoshi S. Bismuth telluride compounds with high thermoelectric figures of merit. J Appl Phys. 2003;93:368–374. [Google Scholar]
- 20.Manglik RM. Heat Transfer and Fluid Flow Data Book. Schenectady, NY: Genium Publishing; 1984. pp. 7–8. section 410.2. [Google Scholar]
- 21.Lama RF, Lu CYB. Excess thermodynamic properties of aqueous alcohol solutions. Chem Eng Data. 1965;10:216–219. [Google Scholar]
- 22.Thorsen T, Roberts WR, Arnold HF, Quake RS. Dynamic pattern formation in a vesicle-generating microfluidic device. Phys Rev Lett. 2001;86:4163–4166. doi: 10.1103/PhysRevLett.86.4163. [DOI] [PubMed] [Google Scholar]
- 23.Ng JM, Gitlin I, Stroock AD, Whitesides GM. Components for integrated poly(dimethylsiloxane) microfluidic systems. Electrophoresis. 2002;23:3461–3478. doi: 10.1002/1522-2683(200210)23:20<3461::AID-ELPS3461>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]