

Abstract
Lysine 5,6-aminomutase (5,6-LAM) catalyzes the interconversions of d- or l-lysine and the corresponding enantiomers of 2,5-diaminohexanoate, as well as the interconversion of l-β-lysine and l-3,5-diaminohexanoate. The reactions of 5,6-LAM are 5′-deoxyadenosylcobalamin- and pyridoxal-5′-phosphate (PLP)-dependent. Like other 5′-deoxyadenosylcobalamin-dependent enzymes, 5,6-LAM is thought to function by a radical mechanism. No free radicals can be detected by electron paramagnetic resonance (EPR) spectroscopy in reactions of 5,6-LAM with d- or l-lysine or with l-β-lysine. However, the substrate analogs 4-thia-l-lysine and 4-thia-d-lysine undergo early steps in the mechanism to form two radical species that are readily detected by EPR spectroscopy. Cob(II)alamin and 5′-deoxyadenosine derived from 5′-deoxyadenosylcobalamin are also detected. The radicals are proximal to and spin-coupled with low-spin Co2+ in cob(II)alamin and appear as radical triplets. The radicals are reversibly formed but do not proceed to stable products, so that 4-thia-d- and l-lysine are suicide inhibitors. Inhibition attains equilibrium between the active Michaelis complex and the inhibited radical triplets. The structure of the transient 4-thia-l-lysine-radical is analogous to that of the first substrate-related radical in the putative isomerization mechanism. The second, persistent radical is more stable than the transient species and is assigned as a tautomer, in which a C6(H) of the transient radical is transferred to the carboxaldehyde carbon (C4′) of PLP. The persistent radical blocks the active site and inhibits the enzyme, but it decomposes very slowly at ≤ 1% of the rate of formation to regenerate the active enzyme. Fundamental differences between reversible suicide inactivation by 4-thia-d- or l-4-lysine and irreversible suicide inactivation by d- or l-lysine are discussed. The observation of the transient radical supports the hypothetical isomerization mechanism.
Lysine 5,6-aminomutase (5,6-LAM1) participates in the fermentation of l- or d-lysine as carbon and nitrogen sources in anaerobic bacteria (1). Anaerobic fermentation of l-lysine proceeds efficiently as in Figure 1, starting with conversion to l-β-lysine by 2,3-LAM, a SAM and PLP-dependent enzyme. 5,6-LAM then converts l-β-lysine into l-3,5-DAH, a molecule poised for dehydrogenation and β-oxidation. Fermentation of d-lysine in Figure 1 begins with conversion to d-2,5-DAH by 5,6-LAM and proceeds to the formation of acetate and butyrate (1).
Figure 1.

Metabolism of lysine in anaerobic bacteria.
5,6-LAM is an adenosylcobalamin- and PLP-dependent enzyme that catalyzes the interconversion of d- or l-lysine with d- or l-2,5-DAH or of l-β-lysine with l-3,5-DAH (1-8). The mechanism of action of 2,3-LAM is well worked out, and the structure of the enzyme is fully compatible with the spectroscopic and chemical evidence supporting the mechanism (9,10). The 2,3-LAM mechanism inspires the hypothetical chemical mechanism for 5,6-LAM shown in Scheme 1 (2,4,9), wherein the 5′-deoxyadenosyl radical from adenosylcobalamin initiates the chemistry by abstracting a C5(H) from lysine to generate the substrate-related radical 2, which is bound as the Nε-aldimine to PLP. Radical isomerization analogous to that in 2,3-LAM leads through the aziridincarbinyl intermediate 3 to the product-related radical 4, which is quenched by hydrogen transfer from 5′-deoxyadenosine. In contrast to 2,3-LAM, little experimental evidence bearing on the mechanism of action of 5,6-LAM is available, apart from the mediation of hydrogen transfer by the 5′-deoxyadenosyl moiety of adenosylcobalamin (7). The X-ray crystal structure of 5,6-LAM raises questions regarding coordination in the actions of PLP and adenosylcobalamin (11).
Scheme 1.

5,6-LAM is a heterotetrameric protein composed of α- and β-subunits (αβ)2. In the available structure, illustrated in Figure 2 with cobalamin, 5′-deoxyadenosine and PLP as ligands, the α-subunit incorporates a TIM barrel and the β-subunit a Rossman domain. Adenosylobalamin binds in a base-off mode, with most interactions to the β-subunit, which projects the 5′-deoxyadenosyl moiety toward the β–barrel of the α–subunit. The major binding contacts of PLP are to the α–subunit, but the β-subunit binds the carboxaldehyde group of PLP as an internal aldimine with Lysβ144 (4,11). The 24 Å separation between 5′-deoxyadenosine and PLP in the structure is too great to represent an active conformation that would allow a substrate to interact chemically with both adenosylcobalamin and PLP.
Figure 2.

Structure of 5,6-LAM and relative locations of adenosylcobalamin and PLP.
The structure is of 5,6-LAM with PLP, 5′-deoxyadenosine and cobalamin as ligands (11). This image was created by H. Adam Steinberg from PDB ID 1XRS.
Spectroscopic experiments show that other adenosylcobalamin-dependent enzymes facilitate the transient and reversible homolytic cleavage of the Co—C5′ bond in adenosylcobalamin to form cob(II)alamin. The resultant 5′-deoxyadenosyl radical initiates catalysis by abstracting a hydrogen atom from the cognate substrate (12-14). Limited evidence for homolytic scission of the Co—C5′ bond is available for 5,6-LAM. Cob(II)alamin is not observable as an intermediate in the steady state with any substrate. The only reported cleavages of the Co—C5′ bond by 5,6-LAM are the formation of cob(III)alamin during suicide inactivation of the enzyme by substrates (2), and the EPR spectroscopic observation of cob(II)alamin in a reaction with the substrate analog 4-thia-l-lysine (15).
EPR spectroscopy is employed in research on the mechanisms of enzymes catalyzing radical reactions, allowing structural assignments to intermediates that are detectable by EPR (16-20). No radical can be detected in the reactions of 5,6-LAM with the natural substrates d-lysine, l-lysine, or l-β-lysine. In this report, we present the results of studies of the reaction of 5,6-LAM with 4-thia-d- and 4-thia-l-lysine. These molecules are structurally similar to d- and l-lysine but have special chemical properties that facilitate the spectroscopic observation of radicals related in structure to possible catalytic intermediates. We also present spectrophotometric evidence for the reaction of 4-thia-d- and 4-thia-l-lysine in cleaving the Co—C5′ bond of adenosylcobalamin to cob(II)alamin and 5′-deoxyadenosine. The results support catalysis of amino group migration by way of the radical mechanism in Scheme 1.
EXPERIMENTAL PROCEDURES
Chemicals
d-Lysine-HCl, adenosylcobalamin, EPPS, EDTA, ME, 4-thia-l-lysine, and PLP were purchased from Sigma. d-[1-14C]Lysine and [U-14C]PLP were obtained from American Radiolabeled Chemicals. l-[3-13C]Cysteine (99% enriched), l-[3-2H2]cysteine (98% enriched), and 1,2-dibromo[2H4]ethane (99% enriched) were purchased from Cambridge Isotope Laboratories. 1,2-Dibromo[13C2]ethane and [15N]phthalimide were from Isotec. Potassium phthalimide and 2-bromoethylamine-HBr were from Aldrich. [adenine-8-14C] Adenosylcobalamin and [5′,5′-2H2]adenosylcobalamin were synthesized enzymatically as described (2). The final specific activity of [adenine-8-14C]adenosylcobalamin was 1.2 × 106 dpm μmol−1. 2-Bromo[1,1,2,2-2H4]ethylamine HBr, 2-bromo[1,2-13C2]ethylamine-HBr, 2-bromo[15N]ethylamine-HBr, and unlabeled 2-bromoethylamine-HBr were prepared from the corresponding potassium phthalimide and 1,2-dibromoethane as described (21). Isotopically labeled 4-thia-d- and l-lysine, and unlabeled 4-thia-d-lysine were prepared from the corresponding isotopically labeled or unlabeled d- or l-cysteine and 2-bromoethylamine-HBr as described (22).
Enzyme
5,6-LAM from Porphyromonas gingivalis was prepared as previously described (4). The specific activity ranged from 9 to 12 IU mg−1. In the preparation of PLP-depleted 5,6-LAM, PLP was omitted from the purification buffer (20 mM triethanolamine-HCl at pH 7.2, 1 mM □□), minimizing PLP to < 2% of sites.
Analytical Methods
UV-visible spectra were measured with a Hewlett-Packard Model 8452A diode array spectrophotometer. Radioactivity was measured by liquid scintillation in a Beckman LS 6500 system. Mass spectra were collected on a Perkin Elmer Sciex API 365 triple quadrupole ESI mass spectrometer (ESI/MS) at the University of Wisconsin Biotechnology Center (Madison, WI) or on an Agilent 110D series HPLC/ESI/MS system equipped with an online diode-array UV detector and a single quadrupole mass detector.
Experimental Conditions
All enzymatic reactions were carried out in a Coy anaerobic chamber shielded from light. Enzyme, coenzymes, unlabeled substrates, and buffers (except d-[14C]lysine) were made anaerobic by repeated evacuation followed by purging with oxygen-free argon before being brought into the anaerobic chamber. d-[14C]Lysine was placed in the anaerobic chamber overnight.
Enzyme Assay
In the radiochemical assay, 5,6-LAM (12 μM) was incubated at 37 °C with 100 mM K+EPPS buffer, 175 μM adenosylcobalamin, 175 μM PLP, and 200 mM d-[1-14C]lysine (total volume 150 μL) for 2 min, quenched with 2N HClO4, removed from the anaerobic chamber and centrifuged. The radio-labeled product was separated from the substrate by paper electrophoresis as described elsewhere (2).
In the HPLC assay, d-lysine was substituted for d-[1-14C]lysine, and the product was assayed by HPLC as its PITC-derivative. After quenching the assay mixture, adenosylcobalamin and 5′-deoxyadenosine were removed from the supernatant fluid with a 500 mg scale Sep-Pak C18 cartridge, the solution was concentrated to ∼50 μL using a SpeedVac apparatus, and d-lysine and d-2,5-DAH were derivatized with PITC (2). PITC-d-lysine and PITC-d-2,5-DAH were separated over a C8 column (Vydac) eluted with a gradient of 65% buffer A (0.05 M ammonium acetate) to 100% buffer B (0.1 M ammonium acetate in 44% H2O, 46% CH3CN, and 10% methanol) at pH 6.8 at a flow rate of 1.0 mL min−1 for 30 min, followed by isocratic elution with 100% buffer B for 5 min, with monitoring at 254 nm. Retention times of PITC-d-lysine and PITC-d-2,5-DAH were 28 and 29.5 min, respectively. d-2,5-DAH formation at various times were plotted to determine initial rates.
Concentrations of 5,6-LAM were determined by measurement of absorbance at 280 nm of the PLP- and adenosylcobalamin-free enzyme and calculation based on the extinction coefficient 1.1 × 105 M−1 cm−1. The extinction coefficient was calculated from the amino acid composition as described (23,24).
Preparation of samples for EPR Measurements
Enzyme, coenzymes, substrates, and buffers were made anaerobic by repeated evacuation followed by purging with oxygen-free argon and placed in the anaerobic chamber. 5,6-LAM (0.2 mM) was incubated at room temperature with 0.25 mM PLP, 0.25 mM adenosylcobalamin, 20 mM 4-thialysine, and 0.1 M K+EPPS buffer at pH 8.5 (total volume 250 μL), with initiation by adenosylcobalamin addition. The solution was transferred to an EPR tube and frozen at selected times by immersion of the tube in an isopentane bath cooled in liquid nitrogen.
EPR Measurements
EPR measurements of 4-thialysyl radical triplets at 77 K were carried out in a Varian E-3 spectrometer equipped with a standard liquid nitrogen immersion Dewar. The EPR spectrometer was interfaced with an UNIX computer for data acquisition. Spin concentrations were estimated by double integration of EPR spectra, using 1 mM CuSO4/10 mM EDTA as standard.
Analysis of EPR Spectra
The resolution of the experimentally observed EPR spectra was enhanced using Fourier filtering methods as described previously [25,26]. The spectra were then analyzed using the following spin Hamiltonian:
| (1) |
The first two terms represent the Zeeman interaction of the low-spin Co2+ of cob(II)alamin and the 4-thia-d-lysine-derived radical(s), respectively. The third and fourth terms represent the magnetic dipole-dipole and isotropic exchange (ZFS) interactions between cob(II)alamin and the 4-thia-D-lysyl radical(s), respectively. Hnuc in equation 2 represents the relevant nuclear hyperfine interactions:
| (2) |
The only nuclear spins that need to be explicitly considered are 59Co (I = 7/2) and its lower axial 14N-ligand (I = 1), which is derived from His133 (3,27). EPR transition energies and probabilities were obtained by diagonalizing the energy matrix, and the field-swept powder spectra were calculated as described previously (27). In this analysis, the hyperfine interactions are treated to first-order, which makes the energy matrix block diagonal with respect to the nuclear quantum number, mI. Initial estimates for the ZFS parameters were taken from measured turning points in the experimental spectra. The g-values and hyperfine parameters of cob(II)alamin were obtained from previously reported values of the species bound to methylmalonyl-CoA mutase (18). These values, along with the g-values of the 4-thialysyl radical and the Euler angles needed to align the D-tensor with the Co2+ g-tensor, were refined by trial and error until reasonable fits were obtained.
Electronic Structure Calculations
To help facilitate characterization of the radical intermediate(s) obtained in reaction mixtures of 5,6-LAM with 4-thialysine, DFT calculations were performed on truncated forms of several putative radical structures (Figure 8). Each of the radicals (except the quinonoid radical) has the pyridine-N1 unprotonated, as it presumably is in 5,6-LAM, where it forms a hydrogen bond with Ser238α (11). The 4-thialysyl moieties of the putative radicals are approximated by a 2-(methylthio)ethylamine group. The geometries of the radicals were optimized using Becke-style 3-parameter density functional theory with the Lee-Yang-Parr correlation functional (B3LYP) and Pople's polarized double-ζ basis set, 6-31G(d), using Gaussian98. Hyperfine and g-tensors tensors were then calculated from the optimized structures using the B3LYP functional in combination with the DFT-optimized valence triple-ζ basis set, TZVP, using Gaussian98 and the ORCA 2.4-41 software package, respectively. The 4′-methyl and 5′-methylene phosphate groups derived from PLP were then replaced with hydrogen and the radical structures were reoptimized at the B3LYP/6−311+G(d,p) level to obtain estimates of the free energy of each species. These calculations were performed at 298 K and 1 atm in the gas phase, using a factor of 0.9877 to scale the vibrational frequencies and zero-point energies.
Figure 8.
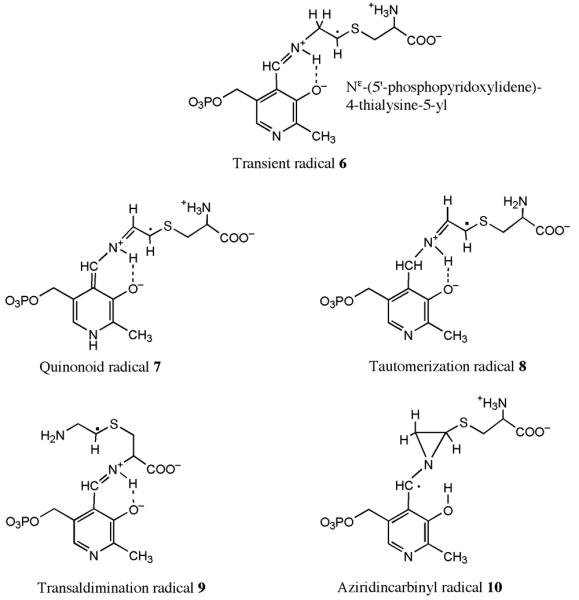
Structures under consideration for the transient and persistent radicals.
At the top is the structure of the 4-thia-analog of radical 2 in Scheme 1, the transient radical 6. The lower four are candidate structures for the persistent radical. The quinonoid radical 7 is the product of proton transfer from C6 of the 4-thialysyl side chain to N1 of PLP. The tautomerization radical 8 is the product of proton transfer from C6 of the 4-thialysyl side chain to C4′ of PLP. The transaldimination radical 9 is the product of aldimine transfer from N6 to N2 of the 4-thialysyl side chain. The aziridincarbinyl radical 10 is the 4-thia-analogue of the intermediate radical 3 in Scheme 1.
Production of 5′-Deoxyadenosine
A solution (250 μL) contained initially 0.1 M NH4EPPS (pH 8.5), 120 μM [adenine-8-14C]adenosylcobalamin, 120 μM PLP, 50 μM 5,6-LAM, and 20 mM 4-thia-d-lysine. After addition of 4-thia-d-lysine, 20 μL-aliquots taken at 0, 10, 25, 35, 45, 60, 75, 120, 360, and 640 s, were quenched with 2 μL 2 N HClO4, removed from the anaerobic chamber, centrifuged, and the supernatant fluids neutralized with KOH. 5′-deoxyadenosine was purified as previously described (2).
LC/MS Analysis of 5′-Deoxyadenosine
5,6-LAM (0.2 mM) was incubated at 37 °C for 20–30 min in the anaerobic chamber with 0.25 mM PLP, 0.25 mM adenosylcobalamin, 20 mM of either 4-thia-d-lysine or 4-thia-d-[5,6-2H4]lysine and 0.1 M NH4EPPS buffer at pH 8.5 in 0.2 mL. The nucleoside product derived from adenosylcobalamin was purified as described and characterized by LC/MS (positive ion mode) (2). 5′-Deoxyadenosine was the only nucleoside product detected.
Reaction of 4-Thia-d-lysine in Suicide Inhibition
5,6-LAM (0.20 mM) was incubated at 37 °C in the anaerobic chamber with 0.25 mM PLP, 0.25 mM adenosylcobalamin, 0.20 mM 4-thia-d-lysine, and 0.1 M NH4EPPS buffer at pH 8.5 in a volume of 0.20 mL. The reaction was initiated by addition of adenosylcobalamin, and timed aliquots (20 μL) were quenched with 2N HClO4, 20 mM DTT, and one equivalent of d-lysine as the internal standard for the quantification of 4-thia-d-lysine. The mixtures were reduced to ∼ 50 μL using a Speedvac apparatus after removal of 5′-deoxyadenosine and adenosylcobalamin. 4-Thia-d-lysine and d-lysine were derivatized with PITC (2,28) and separated as described above. The A254-peak corresponding to authentic PITC-4-thia-d-lysine emerging in 32 min was pooled and characterized by ESI/MS (negative ion mode). The amount of 4-thia-d-lysine was determined by the ratio of the area of PITC-d-lysine versus PITC-4-thia-d-lysine.
RESULTS
As substrates for 5,6-LAM, the values of Km for d- and l-lysine are similar, but the value of kcat is about 12-fold higher for d-lysine (8). The corresponding enantiomers of 4-thialysine also react with 5,6-LAM as suicide inhibitors in PLP- and adenosylcobalamin-dependent processes. Both 4-thia-d- and l-lysine react quickly to form radicals at the active site. The radicals subsequently decay during 20 to 30 min. Neither compound leads to a stable product because 4-thia-2,6-DAH rapidly decomposes. Like the lysyl substrates, 4-thia-d-lysine reacts faster than 4-thia-l-lysine, and for this reason the reaction of the l-enantiomer is the more convenient to study. The two enantiomers react similarly to form carbon-based radicals, cob(II)alamin, and 5′-deoxyadenosine.
Radical Formation in Reaction of 4-Thia-l-lysine with 5,6-LAM
Reaction of 5,6-LAM with 4-thia-d- or l-lysine in the presence of adenosylcobalamin and PLP leads to paramagnetic species that can be detected in freeze-quenched samples. A transient signal appears within 10s with 4-thia-l-lysine, followed by a persistent species within 10 min. 4-Thia-d-lysine reacts faster, and a weak signal for the transient intermediate can be detected only by rapid mix-freeze quench EPR (Supplemental information). The persistent species of the d-enantiomer is formed within 2 min.
The EPR spectrum of the transient species with 4-thia-l-lysine is shown in Figure 3, together with the effects of 2H- or 13C-labels in 4-thia-l-lysine. The signal appears as a broadened spin triplet. Similar triplet signals have been observed for other adenosylcobalamin-dependent mutases (18,19). Whenever 13C is the central atom of a π-radical, the 13C nuclear spin imparts a large axial hyperfine splitting in the spectra. Replacement of 1H by 2H in positions near the radical center narrows the EPR signals owing to the much smaller magnetogyric ratio of 2H. As shown in Figure 3, substitution of 2H or 13C in the side chain at the C5- C6 locus of 4-thia-l-lysine leads to narrowing (2H) or broadening (13C), respectively, in EPR spectra of samples made from the isotopically labeled analogs. No effect is observed in the spectrum of the sample prepared with 4-thia-l-[3-13C]lysine.
Figure 3.

Isotope edited EPR spectra of the transient radical with 4-thia-l-lysine.
EPR spectra at 9.1 GHz of the transient radical with 100 μM activated 5,6-LAM, 100 μM adenosylcobalamin, and 100 μM PLP reacting with 20 mM 4-thia-l-lysine without and with 2H, 13C, or 15N-labeling and freeze quenched at 10 s. In this spectrum, g =2.0 corresponds to 3261 G.
EPR spectra of the persistent species with 4-thia-l-lysine are shown in Figure 4. Spectra of the spin-coupled Co2+-radical pair deviate from those of the transient of 4-thia-l-lysine (Figure 3), indicating a weaker spin-spin interaction, as observed in the signal for a substrate radical intermediate in ethanolamine ammonia-lyase (20). Effects of labeling with 2H or 13C at C5-C6 again indicate the presence of spin in the C5-C6 locus of the 4-thia-l-lysine side chain. Reaction of 4-thia-d-lysine leads to analogous radicals on a faster time scale (Supplemental Information). Deuterium labeling in the carboxaldehyde group of PLP (C4′) narrows the signal (15), clearly showing hyperfine coupling of PLP-C4′–H with the unpaired electron in the persistent species.
Figure 4.

Isotope edited EPR spectra of the persistent radical with 4-thia-l-lysine.
Spectra at 9.1 Ghz were obtained as in Figure 1 but after freeze quenching at 10 min. In this spectrum, g =2.0 corresponds to 3261 G.
Simulations of the transient and persistent cob(II)alamin-radical spectra in the reaction of 4-thia-l-lysine are shown in Figure 5, overlaid on resolution-enhanced experimental spectra. The dipole-dipole tensors obtained from fitting the spectra allow estimates of the distances between the radicals and Co2+ in cob(II)alamin of ∼7 Å for the transient radical and ∼10 Å for the persistent radical. Simulations of both spectra require exchange coupling (J coupling) substantially larger than the dipole-dipole terms. The large exchange coupling gives both spectra triplet character (29).
Figure 5.

EPR spectral simulations of transient and persistent radicals in the reaction of l-4-thialysine.
The upper spectrum is of the transient radical (—) and the simulation (- - -). Parameters used in the calculated spectrum: Co2+ g-tensor, 2.32, 2.22, 2.00; radical g-tensor, 2.009, 2.003, 2.002; ZFS parameters, D = −75 G, E = 0 G; exchange coupling constant, J = 4 × 103 G; 59Co-hyperfine tensor (G), 10, 7, 112; isotropic 14N-hyperfine splitting, 19 G. Euler angles relating the interspin vector to the Co2+ g-axis: ζ = 43°, η = 0°, and ξ = 78°. A uniform line width of 6 G was used in the calculation. The lower spectrum is of the persistent radical (—) and the simulation (- - -). Parameters used in the calculated spectrum: Co2+ g-tensor, 2.32, 2.20, 2.00; radical g-tensor, 2.009, 2.004, 2.002; ZFS parameters, D = −20 G, E = 0 G; exchange coupling constant, J = 525 G; 59Co-hyperfine tensor (G), 10, 7, 112; isotropic 14N-hyperfine splitting, 19 G. Euler angles relating the interspin vector to the Co2+ g-axis: ζ = 60°, η = 0°, and ξ = 35°. A uniform line width of 8 G was used in the calculation. In these spectra, g =2.0 corresponds to 3261 G.
The transient and persistent radicals are much closer to Co2+ of cob(II)alamin than the 24 Å between PLP and adenosylcobalamin in the structure of the free enzyme (Figure 2) (11). The proximities of the radicals and Co2+ confirm the conformational transition that has been postulated based on the crystal structure (11).
Inhibition of 5,6-LAM by 4-Thia-d-lysine
Inhibition of 5,6-LAM accompanies radical formation. The degree of inhibition depends upon the concentration of 4-thialysine and upon which enantiomer is employed. The central observation is that 5,6-LAM is not completely and irreversibly inactivated by either 4-thia-d- or 4-thia-l-lysine under any conditions employed to date. Nor can radical formation be driven to completion, that is, to stoichiometric equivalence with 5,6-LAM.
Figure 6 shows the time course for the appearance of 5′-deoxyadenosine and radical spin for the reaction of 5,6-LAM (and excess PLP and adenosylcobalamin) with 20 mM 4-thia-d-lysine. Radical spin and 5′-deoxyadenosine appeared simultaneously in stoichiometric equivalence to about 64% of enzymatic sites. Enzymatic activity declined at the same rate as the appearance of spin and 5′-deoxyadenosine to the extent of about 23% residual activity (inset, Figure 6). Under the conditions of the experiments in Figure 6, the enzyme was likely nearly completely inhibited before dilution of samples for assay, but upon dilution the non-radical complexes containing 4-thia-d-lysine, which included the Michaelis and external PLP-aldimine complexes, were largely dissociated, leading to the observation of residual activity. The difference between 64% radical formation and 77% inactivation might be explained on the basis that dilution for assay did not lead to complete dissociation of 4-thia-d-lysine from 5,6-LAM. The results in the experiment of Figure 6 and many others clearly showed that 5,6-LAM could not be completely inactivated by either 4-thia-d- or l-lysine, and that radical formation could not be driven to completion with reference to the concentration of 5,6-LAM.
Figure 6.

Time course of inhibition and radical- and 5′-deoxyadenosine-formation.
Reaction of 5,6-LAM (50 μM), 120 μM PLP, 120 μM [adenine-14C]adenosylcobalamin at pH 8.5 (EPPS buffer) was initiated with 20 mM 4-thia-d-lysine. □, Time course for increasing radical spin (μM); ●, time course for the formation of 5′-deoxy[8-14C]adenosine (μM); inset, enzyme activity.
Conditions of equivalent concentrations of 5,6-LAM and 4-thia-d-lysine allow the irreversible decomposition of 4-thia-d-lysine to be observed. At initial concentrations of 100 μM, the concentration of residual 4-thia-d-lysine decreases in a burst to 46 μM within 2 min, and it then declines at a very slow rate (0.25 μM min−1) during the next 30 min. The steady state rate is about 1% of the rate of the burst (Supplemental information, Figure S6). Reaction of 4-thia-l-lysine also leads to very gradual decomposition.2 Decomposition is likely related to the intrinsic instability of the anticipated aminomutation product, 4-thia-2,5-DAH, which spontaneously breaks down to cysteine, ammonia, and acetaldehyde.
Deuterium Transfer to 5′-Deoxyadenosine
Reaction of 4-thia-d-[5,6-2H4]lysine with 5,6-LAM leads to deutero-5′-deoxyadenosine displaying masses (m/z) of 253, 254, and 255, corresponding to the mono-, di-, and tri-deuterated nucleoside. Deuteration of 5′-deoxyadenosine proves the occurrence of deuterium transfer from 4-thia-d-[5,6-2H4]lysine to 5′-deoxyadenosine, and multiple deuteration under all conditions proves reversibility of hydrogen transfer in early steps of the mechanism.
Spectrophotometry in Reactions of 5,6-LAM with 4-Thia-l-lysine
The spectrum of 5,6-LAM in complex with adenosylcobalamin and PLP displays the 525 nm band of adenosylcobalamin and the 420 nm band of the internal PLP-Lys144β-aldimine (Figure 7). Upon addition of excess 4-thia-l-lysine, the spectrum changes to a mixture of adenosylcobalamin and cob(II)alamin, (λmax = 475 nm). The absorbance change represents ∼70%, which is not far from the 64% radical and 5′-deoxyadenosine formation with 4-thia-d-lysine in Figure 6. The band at 420 nm attributed to the PLP-aldimine becomes less prominent after addition of 4-thia-l-lysine.
Figure 7.

Spectrophotometry in the reaction of 5,6-LAM with 4-thia-l-lysine.
The anaerobic solution contained 23 μM 5,6-LAM, 25 μM PLP, and 23 μM adenosylcobalamin in 100 mM K+EPPS buffer at pH 8.5, with 4-thia-l-lysine in a side arm. The solid-line spectra are: (—), 5,6-LAM-adenosylcobalamin-PLP before addition of 4-thia-l-lysine; (—), within 10 s of addition of 4-thia-l-lysine to 20 mM; (—), 30 min after addition of 4-thia-l-lysine. The dashed spectra are: (- - -), 5,6-LAM-adenosylcobalamin and (- - -), 5,6-LAM-cob(II)alamin after anaerobic photolysis of 5,6-LAM adenosylcobalamin. In the cleavage of adenosylcobalamin the 420 nm band is retained as that of the external PLP-4-thia-l-lysine aldimine.
The dashed curves in Figure 7 show the spectra of adenosylcobalamin and cob(II)alamin bound to 5,6-LAM, with the characteristic isosbestic point at 490 nm. Appearance of the same isosbesic point upon addition of 4-thia-l-lysine to the holoenzyme confirms the tranformation of adenosylcobalamin to cob(II)alamin and the absence of other chromophores in this region of the spectra.
All experiments imply that reactions of 4-thia-d- or l-lysine with the complex of 5,6-LAM, PLP, and adenosylcobalamin proceed to an internal equilibrium with the Michaelis complex and adenosylcobalamin-cleavage complexes containing cob(II)alamin, 5′-deoxyadenosine and one of two 4-thialysyl radicals, a transient and a persistent species. Available evidence indicates that the persistent species reacts very slowly to regenerate the active enzyme (equation 3).
| (3) |
This scheme implies that simple dilution of an inhibition reaction mixture should initiate partial reversal of inhibition. During the reaction of 25 μM activated 5,6-LAM with 100 μM 4-thia-l-lysine, activity decreases from 9 to 2 IU mg−1 in 30 min. Upon 25-fold dilution of the reaction mixture with buffer, the activity gradually increases to 2.5 IU mg−1 during 40 min. The increase in activity can be attributed to the slow decomposition of the persistent radical, with regeneration of the active enzyme and reversible binding of 4-thia-l-lysine/4-thia-l-2,5-DAH in the Michaelis complex.
Structures of transient and persistent radicals
The transient and persistent cob(II)alamin-radical triplets are assumed to differ in the structures of the radical components. The transient radical is likely to be the 4-thia-analog of radical 2 in Scheme 1, shown at the top of Figure 8 as Nε-(5′-phosphopyridoxylidene)-4-thialysine-5-yl. This radical should be stabilized by the 4-thia-group (28,30). The effects of the isotopic labels 5,6-2H4 and 5,6-13C2 on the EPR spectra in Figure 3, as well as the reversible transfer of deuterium from 4-thia-[5,6-2H4]lysine to 5′-deoxyadenosine, supports this assignment. The transient radical is regarded as a precursor of the persistent radical.
Figure 8 shows the structures of the transient radical 6 and of candidates for the persistent radical, which are isomeric with the transient radical. The relative energies calculated for models of the radicals are listed in Table 1. For the calculations, the 4′-methyl and 5′-methylene phosphate groups of PLP were replaced by hydrogen. These calculations do not include effects of solvation or the microenvironment of the active site. Therefore, the calculated energies provide estimates of relative stabilities and serve as guidelines that can be considered together with chemical and spectroscopic evidence.
Table 1.
| Radical | Relative energy (kcal mol−1) |
|---|---|
| Transient 6 | Reference |
| Aziridincarbinyl 10 | +17.5 |
| Quinonoid 7 | −10.3 |
| Tautomerization 8 | −6.3 |
DFT-computations are of the structures with the methyl and methylene phosphate groups of PLP replaced with hydrogen and the 4-thialysine moiety approximated by 2-(methylthio)ethylamine, in the gas phase at 298 K and 1 atm.
The persistent radical must be more stable than the transient radical, and it must exhibit appreciable hyperfine coupling of the PLP-C4′(H) with the unpaired electron, as reported elsewhere (15). The electronic structure of the persistent radical must also be consistent with the absorbance spectra in Figure 7. The transaldimination radical 9 in Figure 8 is inspired by the lack of stereospecificity in reactions of 5,6-LAM. The enzyme accepts both d- and l-lysine as substrates (8) and as suicide inactivators (2), and both d- and l-stereoisomers of 4-thialysine are reversible suicide inhibitors. The absence of stereospecificity at C2 implies weakness and potential flexibility in the interactions of the α–amino and carboxylate groups with the active site. Flexibility in binding might allow the transient radical to undergo internal transaldimination with the α-amino group. This possibility is ruled out by the spectra in Figure 7, which show an attenuation of intensity in the band at 420 nm assigned to the external 4-thialysyl-PLP aldimine. The transaldimination radical 9 would display absorption similar to that of the internal aldimine. It also would not display higher extinction near340 nm, nor would it display the hyperfine coupling of the PLP-C4′(H) (15).
The aziridincarbinyl radical 10 in Figure 8 is the 4-thia-analog of the central intermediate 3 in Scheme 1. The electronic properties of the PLP-portion of the aziridincarbinyl radical 10 would be consistent with the absorption spectra of Figure 7, and the structure would account for the magnitude of the PLP-C4′(H) hyperfine coupling (Table 2) (15). Because of its similarity to the aziridincarbinyl radical intermediate 3 of Scheme 1, it is an attractive candidate for the persistent radical. However, this radical is expected to be nearly 18 kcal mol−1 higher in energy than the transient radical (Table 1), which is incompatible with the fact that the persistent radical is more stable. The relative energy of the aziridincarbinyl radical 10 is comparable to that of the intermediate 3 in Scheme 1 (31).
Table 2.
| Radical | g-values | Hyperfine tensors (MHz) |
|
|---|---|---|---|
| 1H (PLP-C4′) | 31P | ||
| Transient 6 | 2.009, 2.003, 2.002 | −1.73, −0.93, 2.58 | −0.13, −0.11, 0.50 |
| Aziridincarbinyl 10 | 2.004, 2.004, 2.003 | −37.6, −14.9, 13.3 | −0.18, 0.18, 1.50 |
| Quinonoid 7 | 2.005, 2.003, 2.002 | −3.58, −3.51, 3.36 | −0.12, −0.04, 0.58 |
| Tautomerization 8 | 2.009, 2.004, 2.002 | 11.7, 12.7, 20.4 | −0.29, −0.22, 0.35 |
DFT-computations are of the structures with the 4-thialysine moiety approximated by 2-(methylthio)ethylamine. Bold numbers refer to the structures in Figure 8.
Chemical isomerizations of the transient radical, wherein a proton is transferred from C6 of the transient radical to C4′ or N1 of PLP, lead to the quinonoid 7 and tautomerization 8 radicals in Figure 8. Both of these isomers are more stable than the transient radical, owing to more extensive delocalization of the unpaired electron (30). The electronic structures of these radicals would account for the decreased intensity in the 420 nm absorption in Figure 7. The quinonoid radical 7 should display a longer wavelength absorption band, in line with the corresponding carbanionic species with its maximum absorbance in the range of 495-505 nm. The absorption spectra of Figure 7 do not present such a band. Moreover, the relative energy of the quinonoid radical, −10.3 kcal mol−1 (Table 1), is so much lower than that of the transient radical that the resulting thermodynamic well should pull all of the enzyme species into the quinonoid form. The lack of complete conversion of the enzyme to this radical (see Figure 6) and the absence of a long wavelength absorbance (see Figure 7) are inconsistent with the quinonoid form. In addition, the magnitude of the PLP-C4′(H) hyperfine coupling calculated for the quinonoid radical 7 is smaller than the observed value (Table 2) (15).
In contrast to problems with the other possible forms of the persistent radical, the expected chemical and electronic properties of the tautomerization radical 8 are fully consistent with the available chemical and spectroscopic data. The electronic structure of the PLP-portion of this radical is consistent with the increased absorbance at 340 nm, the decreased intensity at 420 nm, and the isosbestic point in Figure 7. The electronic structure also accounts for the magnitude of the hyperfine coupling of PLP-C4′(H) (Table 2) (15). The calculated isotropic coupling constant of 44.7 MHz is similar to the experimental value of 40 MHz (15). The DFT calculations for the lowest energy conformation of the tautomerization radical 8 in the gas phase make it −6.3 kcal mol−1 more stable than the corresponding most stable conformation of the transient radical 6 under the same conditions. The actual conformations at the active site are unlikely to be ideal, so the energy difference might be somewhat lower, depending on the true conformations within the active site.
DISCUSSION
Reversible suicide inhibition
The interconversion of the transient radical 6 and the more favored persistent radical 8 establishes the internal equilibrium in equation 3. Because the transformation of the radicals to decomposition products is very slow (≤ 1% of formation rate), inhibition is partial, and its extent is determined by the equilibrium between the radicals and the Michaelis complex. The transient radical 6 is the 4-thia-analog of radical 2 in the mechanism of Scheme 1, the substrate-related radical. The persistent radical is likely the tautomerization radical 8 in Figure 8 and is the species initially observed as a stable radical generated at the active site by 4-thia-l-lysine (15).
The persistent radical probably represents a “blind alley” in that it does not react forward to products. However, reversal of the tautomerization leads to the transient radical 6, which can occasionally form the 4-thia-analog of radical 3 in Scheme 1, the aziridincarbinyl radical 10 in Figure 8, which then breaks down either by reversal to the transient radical 6 or to the active enzyme and decomposition products of 4-thia- l-2,5-DAH. Scheme 2 describes this course of the process. Favored formation of the persistent radical likely accounts for the very slow turnover of 4-thialysine.
Scheme 2.

Alternative modes of isomerization between transient and persistent radical are not ruled out. In any case, formation of the 4-thia-analog of radical 2 in Scheme 1 as a pre-steady state, transient intermediate in reversible suicide inhibition supports the chemical mechanism in Scheme 1.
Formation of the tautomerization radical 8 as the persistent species does not undermine the transformations in Scheme 1 as the mechanism of the reaction of d- and l-lysine. The tautomerization radical 8 is a reversibly formed side product. The structure of this radical constitutes an excellent example of classical radical stabilization by the captodative effect (30). The capto- substituent of C5 (—CH=NH+—) stabilizes the unpaired electron by delocalization to nitrogen, and the dative substituent (—S—) stabilizes the radical by exchange with the doubly-occupied nonbonding p-orbitals of sulfur. These interactions make the tautomerization radical 8 ∼24 kcal mol−1 more stable than the aziridincarbinyl radical 10, the catalytic intermediate.
Regardless of relative stabilities, there must be a mechanism for tautomerization. Given that 5,6-LAM catalyzes the PLP-dependent exchange of C6(H) in d-lysine with solvent protons at a slow rate (6), there must be at least one acid-base group in the near vicinity of C6 in the PLP-Nε-lysine aldimine. At least one acid-base group is also required to bind the Nε-amino group of d- or l-lysine and to catalyze transaldimination to the external aldimine. Such a base could well come into play in facilitating the transfer of a proton from C6 of the lysyl side chain to C4′ of PLP in tautomerization. This process is unlikely to be a normal or compulsory process in the reactions of d- or l-lysine with 5,6-LAM.
Irreversible Suicide Inactivation
Reversible suicide inhibition by 4-thia-d-or l-lysine is unlike irreversible suicide inactivation by d- or l-lysine (2), though both processes take place under anaerobic conditions. Irreversible suicide inactivation by the enantiomers of lysine proceeds by way of the early steps of the mechanism in Scheme 1 to substrate radical intermediates 2 and 4, followed by proton-coupled electron transfer from cob(II)alamin to the lysyl- or DAH-related radicals to generate cob(III)alamin and either lysine or 2,5-DAH (2). No intermediate free radicals can be detected by EPR spectroscopy. The different reactivity of 4-thia-d- or l-lysine relative to d- or l-lysine is most likely due to the inability of cob(II)alamin to reduce the stabilized 4-thialysine-radicals. Delocalization of spin in the 4-thialysyl radicals enhances their stability such that it is possible to observe the radical species by EPR. At the same time, their stability gives them too low a reduction potential to allow electron transfer from cob(II)alamin. Thus, suicide inactivation by d- or l-lysine is irreversible because of the electron transfer step, while suicide inhibition by 4-thia-d- or l-lysine, which involves tautomerization of the radical intermediate as opposed to electron transfer, is reversible.
Ornithine 4,5-aminomutase
In a reaction analogous to that of 5,6-LAM, ornithine 4,5-aminomutase catalyzes the adenosylcobalamin and PLP-dependent interconversion of ornithine and 2,4-diaminopentanoate. Cob(II)alamin and a radical have recently been observed in that enzyme by employing an alternative strategy for stabilizing a radical (32). In the case of ornithine 4,5-aminomutase, the unpaired electron in the substrate-related radical corresponding to 2 in Scheme 1 should reside at C4 of the ornithine-N5-PLP-aldimine, owing to the abstraction of C4(H) by the 5′-deoxyadenosyl radical. The C4-radical was not observed by EPR, presumably because it did not accumulate to a detectable concentration. The substrate analog 2,4-diaminobutyrate, employed in place of ornithine, led to cob(II)alamin and a stable radical. With 2,4-diaminobutyrate, the diaminobutyrate-N4-PLP-aldimine would be formed, and the abstraction of C4(H) by the 5′-deoxyadenosyl radical would lead to a C4-radical in the side chain, as shown in Scheme 3, with the unpaired electron adjacent to the imine and stabilized by delocalization through the imine group and into the pyridine ring of PLP. The resulting highly stabilized radical would be unlikely to react further.3 The results provided support for the basic mechanism of aminomutase action, as presented in Scheme 1 for 5,6-LAM.
Scheme 3.

Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Shyue-Chu Ke for communicating unpublished information on the turnover of 4-thia-l-lysine in the presence of 5,6-LAM, and for useful discussions. We thank Dr. Russell LoBrutto for carrying out preliminary ENDOR measurements.
Footnotes
Supported by Grant No. DK28607 from the National Institute of Diabetes and Digestive and Kidney Diseases (P.A.F), GM35752 from the National Institute of General Medical Sciences (G.H.R), and Fellowship No. GM082085 from the National Institute of General Medical Sciences (S.O.M.).
This paper is dedicated to Dr. Thressa C. Stadtman, who discovered and first characterized lysine 5,6-aminomutase in her research on the metabolism of lysine.
Abbreviations: Adenosylcobalamin, 5′-deoxyadenosylcobalamin; 2,5-DAH, 2,5-diaminohexanoate; 3,5-DAH, 3,5-diaminohexanoate; DFT, density functional theory; DNPH, 2,4-dinitrophenyl hydrazine; DTNB, dithiobis 2-nitrobenzoate; DTT, dithiothreitol; EDTA, ethylenediamine tetraacetate; EPPS, N-(2-hydroxyethyl)piperazine N′-3-propanesulfonic acid; EPR, electron paramagnetic resonance; ESI/MS, electrospray ionization mass spectrometry; HPLC, high performance liquid chromatography; 2,3-LAM, lysine 2,3-aminomutase; 5,6-LAM, lysine 5,6-aminomutase; ME, β-mercaptoethanol; PITC, phenylisothiocyanate; PLP, pyridoxal-5′-phosphate; SAM, S-adenosyl-l-methionine; TIM, triosephosphate isomerase; TLC, thin layer chromatography; ZFS, zero-field splitting.
Personal communication from Dr. Shyue-Chu Ke, National Dong Hwa University, Taiwan.
Two unlikely structures suggested for this radical as isomers in equilibrium are the following (32):  Isomerization of the C4-radical in Scheme 3 to the cyclic radical at left above would require unprecedented transfer of a hydrogen atom from C3 to C4 of the side chain followed by cyclization. In any case, the aziridincarbinyl radical would be far higher in energy than the C4-radical in Scheme 3, owing to strain in the aziridyl ring. The radical at right above is not an isomer of the C4-radical in Scheme 3 or of the cyclic radical but is a hydrogenation product.
Isomerization of the C4-radical in Scheme 3 to the cyclic radical at left above would require unprecedented transfer of a hydrogen atom from C3 to C4 of the side chain followed by cyclization. In any case, the aziridincarbinyl radical would be far higher in energy than the C4-radical in Scheme 3, owing to strain in the aziridyl ring. The radical at right above is not an isomer of the C4-radical in Scheme 3 or of the cyclic radical but is a hydrogenation product.
Supporting Information Available
Figures S1-S6 in the supporting information display additional EPR spectral data, including the spectra of the transient and persistent radicals in the reactions of 4-thia-dl- and d-lysine with 5,6-LAM, as well as data showing biphasic, burst consumption of 4-thia-d-lysine in reaction with 5,6-LAM. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Stadtman TC. Lysine metabolism by Clostridia. Adv. Enzymol. 1973;28:413–448. [Google Scholar]
- 2.Tang K-H, Chang CH, Frey PA. Electron transfer in the substrate-dependent suicide inactivation of lysine 5,6-aminomutase. Biochemistry. 2001;40:5190–5199. doi: 10.1021/bi010157j. [DOI] [PubMed] [Google Scholar]
- 3.Chang CH, Frey PA. Cloning, sequencing, heterologous expression, purification, and characterization of adenosylcobalamin-dependent d-lysine 5, 6-aminomutase from Clostridium sticklandii. J. Biol. Chem. 2000;275:106–114. doi: 10.1074/jbc.275.1.106. [DOI] [PubMed] [Google Scholar]
- 4.Tang K-H, Harms A, Frey PA. Identification of a novel pyridoxal 5′-phosphate binding site in adenosylcobalamin-dependent lysine 5,6-aminomutase from Porphyromonas gingivalis. Biochemistry. 2002;41:8767–8776. doi: 10.1021/bi020255k. [DOI] [PubMed] [Google Scholar]
- 5.Baker JJ, van der Drift C, Stadtman TC. Purification and properties of -lysine mutase, a pyridoxal phosphate and B 12 coenzyme dependent enzyme. Biochemistry. 1973;12:1054–1063. doi: 10.1021/bi00730a006. [DOI] [PubMed] [Google Scholar]
- 6.Morley CD, Stadtman TC. The role of pyridoxal phosphate in the B12 coenzyme-dependent d-α-lysine mutase reaction. Biochemistry. 1972;11:600–605. doi: 10.1021/bi00754a019. [DOI] [PubMed] [Google Scholar]
- 7.Retey J, Kunz F, Stadtman TC, Arigoni D. On the mechanism of the beta-lysine-mutase reaction. Experientia. 1969;25:801–802. doi: 10.1007/BF01897884. [DOI] [PubMed] [Google Scholar]
- 8.Tang KH, Casarez AD, Wu W, Frey PA. Kinetic and biochemical analysis of the mechanism of action of lysine 5,6-aminomutase. Arch. Biochem. Biophys. 2003;418:49–54. doi: 10.1016/s0003-9861(03)00346-1. [DOI] [PubMed] [Google Scholar]
- 9.Frey PA, Magnusson O. Th. S-Adenosylmethionine: a wolf in sheep's clothing, or a rich man's adenosylcobalamin? Chem Rev. 2003;103:2129–2148. doi: 10.1021/cr020422m. [DOI] [PubMed] [Google Scholar]
- 10.Lepore BW, Ruzicka FJ, Frey PA, Ringe D. The x-ray crystal structure of lysine-2,3-aminomutase from Clostridium subterminale. Proc. Natl. Acad. Sci. U. S. A. 2005;102:13819–13824. doi: 10.1073/pnas.0505726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berkovitch F, Behshad E, Tang KH, Enns EA, Frey PA, Drennan CL. A locking mechanism preventing radical damage in the absence of substrate, as revealed by the x-ray structure of lysine 5,6-aminomutase. Proc. Natl. Acad. Sci. U. S. A. 2004;101:15870–15875. doi: 10.1073/pnas.0407074101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frey PA. Importance of organic radicals in enzymatic cleavage of unactivated C–H bonds. Chem. Rev. 1990;90:1343–1357. [Google Scholar]
- 13.Reed GH. Radical mechanisms in adenosylcobalamin-dependent enzymes. Curr. Opin. Chem. Biol. 2004;8:477–483. doi: 10.1016/j.cbpa.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 14.Toraya T. Radical catalysis in coenzyme B12-dependent isomerization (eliminating) reactions. Chem. Rev. 2003;103:2095–2127. doi: 10.1021/cr020428b. [DOI] [PubMed] [Google Scholar]
- 15.Maity AN, Hsieh C-P, Huang M-H, Chen Y-H, Tang K-H, Behshad E, Frey PA, Ke S-C. EPR, ENDOR, and DTF Study of a radical intermediate in the reaction of 4-thia-l-lysine with lysine 5,6-aminomutase: Evidence for conformational movement and radical mechanism. 2009 doi: 10.1021/jp905357a. Submitted. [DOI] [PubMed] [Google Scholar]
- 16.Mansoorabadi SO, Magnusson OT, Poyner RR, Frey PA, Reed GH. Analysis of the Cob(II)alamin-5′-deoxy-3′,4′-anhydroadenosyl radical triplet spin system in the active site of diol dehydrase. Biochemistry. 2006;45:14362–70. doi: 10.1021/bi061586q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerfen GJ, Licht S, Willems J-P, Hoffman BM, Stubbe J. Electron paramagnetic resonance studies of a kinetically competent intermediate in ribonucleotide reduction. J. Am. Chem. Soc. 1996;118:8192–8197. [Google Scholar]
- 18.Mansoorabadi SO, Padmakumar R, Fazliddinova N, Vlasie M, Banerjee R, Reed GH. Characterization of a succinyl-CoA radical-cob(II)alamin spin triplet intermediate in the reaction catalyzed by adenosylcobalamin-dependent methylmalonyl-CoA mutase. Biochemistry. 2005;44:3153–3158. doi: 10.1021/bi0482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bothe H, Darley DJ, Albracht SPJ, Gerfen GJ, Golding BT, Buckel W. Identification of the 4-glutamyl radical as an intermediate in the carbon skeleton rearrangement catalyzed by coenzyme B12-dependent glutamate mutase from Clostridium cochlearium. Biochemistry. 1998;37:4105–4113. doi: 10.1021/bi971393q. [DOI] [PubMed] [Google Scholar]
- 20.Bandarian V, Reed GH. Analysis of the electron paramagnetic resonance spectrum of a radical intermediate in the coenzyme B12-dependent ethanolamine ammonia-lyase catalyzed reaction of S-2-aminopropanol. Biochemistry. 2002;41:8580–8588. doi: 10.1021/bi0201217. [DOI] [PubMed] [Google Scholar]
- 21.Philippossian G, Welti DH, Fumeaux R, Richli U, Anantharaman K. Synthesis and NMR characterization of (15N)taurine [2-(15N)aminoethanesulfonic acid] J. Labelled Comp. Radiopharm. 1989;27:1267–1273. [Google Scholar]
- 22.Cavallini D, De Marco C, Mondovi B, Azzone GF. A new synthetic sulfur-containing amino acid: S-aminoethylcysteine. Experientia. 1955;11:61–62. doi: 10.1007/BF02179026. [DOI] [PubMed] [Google Scholar]
- 23.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 24.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kauppinen JK, Moffatt DJ, Mantsch HH, Cameron DG. Fourier self-deconvolution: A method for resolving intrinsically overlapped bands. Appl. Spectrosc. 1981;35:271–276. [Google Scholar]
- 26.Latwesen DG, Poe M, Leigh JS, Reed GH. Electron paramagnetic resonance studies of a ras p21-MnIIGDP complex in solution. Biochemistry. 1992;31:4946–4950. doi: 10.1021/bi00136a004. [DOI] [PubMed] [Google Scholar]
- 27.Bandarian V, Reed GH. Hydrazine cation radical in the active site of ethanolamine ammonia-lyase: mechanism-based inactivation by hydroxyethylhydrazine. Biochemistry. 1999;38:12394–12402. doi: 10.1021/bi990620g. [DOI] [PubMed] [Google Scholar]
- 28.Miller J, Bandarian V, Reed GH, Frey PA. Inhibition of lysine 2,3-aminomutase by the alternative substrate 4-thialysine and characterization of the 4-thialysyl radical intermediate. Arch. Biochem. Biophys. 2001;387:281–288. doi: 10.1006/abbi.2001.2261. [DOI] [PubMed] [Google Scholar]
- 29.Mansoorabadi SO, Reed GH. Effects of Electron Spin Delocalization and Non-Collinearity of Interaction Terms in EPR Triplet Powder Patterns. In: Telser J, editor. Paramagnetic Resonance of Metallobiomolecules. ACS; Washington D. C: 2003. pp. 82–96. [Google Scholar]
- 30.Sustman R, Korth H-G. The captodative effect. Adv. Phys. Org. Chem. 1990;26:131–178. [Google Scholar]
- 31.Sandala GM, Smith DM, Radom L. In search of radical intermediates in the reactions catalyzed by lysine 2,3-aminomutase and lysine 5,6-aminomutase. J. Am. Chem. Soc. 2006;128:16004–16005. doi: 10.1021/ja0668421. [DOI] [PubMed] [Google Scholar]
- 32.Wolthers KR, Rigby SEJ, Scrutton NS. Mechanism of radical-based catalysis in the reaction catalyzed by adenosylcobalamin-dependent ornithine 4,5-aminomutase. J. Biol. Chem. 2008;283:34615–34625. doi: 10.1074/jbc.M807911200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.