

Synopsis
Gambogic acid (GA) is a polyprenylated xanthone abundant in the resin of Garcinia morella and G. hanburyi with a long history of use as a complementary and alternative medicine. The anti-tumor activity of GA has been well demonstrated and is thought to arise partly from the associated anti-inflammatory activity. Recent studies have indicated that the anti-tumor activity of GA is mediated by its ligation of the transferrin receptor TfR1. Since the cellular expression of TfR1 is down-regulated by lipopolysaccharide (LPS), we hypothesized that an alternative pathway exists in immune cells, such as macrophages, where GA could mitigate the expression of pro-inflammatory genes. Here we demonstrate that GA inhibits the LPS-dependent expression of nuclear factor-κB (NF-κB) target pro-inflammatory genes in macrophages. Western immunoblot, NF-κB luciferase reporter, and gel shift analyses revealed that GA strongly blocked the activation of NF-κB induced by LPS; while 9,10-dihydroGA that lacks the reactive α,β-unsaturated carbonyl group was ineffective. Moreover, GA was able to decrease nuclear p65 levels in RAW264.7 macrophages, where the expression of TfR1 was down-regulated by RNA interference. In-vitro kinase assays coupled with interaction studies using biotinylated GA as well as proteomic analysis demonstrated that IKKβ, a key kinase of the NF-κB signaling axis, was covalently modified by GA at Cys179 causing significant inhibition of its kinase activity. Taken together, these data demonstrate the potent anti-inflammatory activity of GA.
Keywords: Anti-inflammatory, plant-derived bioactives, polyprenylated xanthone, cyclooxygenase-2
INTRODUCTION
Gambogic acid (GA) is a polyprenylated xanthone isolated from the resin of G. hanburyi, commonly used in food preparations in many Asian countries [1]. Recent literature has demonstrated its efficacy as a potent anti-tumor drug in rodent and canine models [2, 3]. Most importantly, GA treatment induced apoptosis of human gastric carcinoma cells [4] and human hepatoma cells in mice at 2-8 mg/kg body weight [5]. Kasibhatla et al have reported that the mechanism of GA-induced apoptosis is mediated through the activation of caspases via the transferrin receptor (TfR1) [6]. Recently, Pandey et al also demonstrated that GA inhibited NF-κB signaling pathway through its interaction with TfR1 [7]. However, the scenario may be different in non-cancerous cells, such as macrophages, where the expression of TfR1 is down-regulated by LPS-treatment [8]. We, therefore, reasoned that GA could possibly inhibit NF-κB activation through an alternate pathway. Given that Michael acceptors like 15-deoxy-Δ12,14-prostaglandin J2 and 4-hydroxynonenal (4-HNE) can interact with proteins of the NF-κB signaling axis [9, 10], we hypothesized that GA could interact similarly and modulate their function. In fact, rearrangement of the olefinic bond at C10 (Fig. 1) to a saturated bond in GA resulted in significantly decreased anti-proliferative activities, suggesting that the α,β-unsaturated group in GA was essential for biological activity [1, 6].
FIGURE 1. Structure of GA.
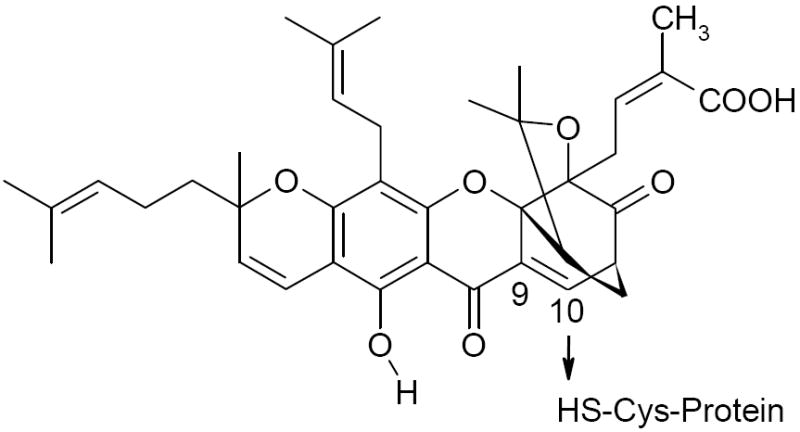
The putative site of addition to Cys thiol is shown. Reduction of the unsaturation (c9-c10) converts GA to DGA.
The NF-κB represents a family of transcription factors that participate in the regulation of diverse biological processes, including immune, inflammatory and apoptotic responses [11-14][15]. Given its ability to regulate expression of inflammatory enzymes, cytokines, chemokines, immunoreceptors, and cell adhesion molecules, NF-κB has often been termed a “central mediator of the immune response” [12, 16]. Macrophages are critical cellular participants in immune regulation and are activated by diverse stimuli (like LPS), to synthesize and secrete cytokines, which initiate and control inflammatory and immune functions [17]. In LPS-treated macrophages, the canonical (classical) pathway of NF-κB is mainly mediated by the IKKβ subunit; [18]. Under normal conditions, the signals mediating NF-κB activation are transient, which are instrumental for programmed cell proliferation and survival. However, activation of NF-κB exacerbates proinflammatory gene expression, inhibits apoptosis, and thus, contributes to inflammation.
Bioactive natural compounds such as sulforaphanes from broccoli [19], curcumin from turmeric [20], caffeic acid phenethylether from the propolis of honeybee [21], zerumbone from ginger [22] and many others cause transcriptional down-regulation of pro-inflammatory genes by inhibiting the pathway of NF-κB activation [23]. In addition, endogenous Michael acceptors with α,β-unsaturated carbonyl moiety such as 15-deoxy-Δ12,14-prostaglandin J2 and 4-HNE [10] inhibit pro-inflammatory gene expression by targeting the NF-κB-dependent transcription via covalent interaction with the IKKβ subunit [24], Cys62 in p50 [25], Cys38 in p65 [26], in addition to its interaction with Cys in c-Jun to promote homodimerization [27]. We hypothesized that GA could inhibit the NF-kB pathway through the α,β-unsaturated carbonyl group to impart potent anti-inflammatory activity. Here we demonstrate that the anti-inflammatory effect of GA occurs via the inhibition of IKKβ activity by covalent modification leading to the consequent inhibition of NF-κB-dependent transcription of pro-inflammatory genes.
EXPERIMENTAL
Cell Culture
The mouse RAW 264.7 macrophage cell line, human embryonic kidney (HEK293) cells, and human monocytic U937 cells, obtained from American Type Culture Collection (Manassas, VA), were cultured in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum (Hyclone), 2 mM L-Glutamine (Invitrogen) and 10 μg/ml ciprofloxacin (Sigma) at 37 °C in a humidified atmosphere of 5% CO2. The cells were sub-cultured either in 6-well or 12-well tissue culture plates.
Flow Cytometric Analysis of TfR1 Expression
The expression of TfR1 (CD71) in RAW264.7 macrophages before and after LPS treatment (1 μg/ml; 12 h)was quantitated by flow cytometry. The cells were incubated with blocking antibody (CD16/CD32; BD Biosciences) for 2 h followed by incubation with anti-CD71-FITC antibody (BD Biosciences). Both anti-CD16/CD32 and anti-CD71-FITC were kindly provided by Dr. Robert Paulson, Penn State University. Results were analyzed using FlowJo® software program (Tree Star, Inc.).
Preparation of Total Cell Lysates and Nuclear Extracts
Gambogic acid and its inactive dihydro-derivative, 9,10-dihydrogambogic acid (DGA), were purchased from Gaia Chemical Corporation, Gaylordsville, CT. To study the effect of GA on the expression of pro-inflammatory genes, RAW264.7 and U937 cells were grown to 80 % confluence and treated with various concentration of GA for 30 min. After pretreatment with GA, cells were stimulated with LPS (1 μg/ml) for 2 h. Cells were washed with PBS and lysed in mammalian protein extraction reagent (M-PER, Pierce) at 4 °C for 20 min. Lysates were centrifuged at 16,000 × g for 10 min and the resulting supernatants were frozen until further analyses. Protein concentrations in the supernatants were determined by BCA protein assay kit (Pierce). Nuclear extracts were isolated using NE-PER kit according to the manufacturer’s instructions (Pierce).
Western Blot Analysis
Nuclear and cytosolic proteins (~10 μg) were resolved by electrophoresis on an SDS-PAGE gel (T = 10 %) and subjected to Western immunoblotting as described previously from our laboratory [9]. Primary antibodies for COX-2 (Cayman Chemicals), iNOS (Cayman Chemicals), p65 (Santa Cruz), pIκBα (Cell Signaling), TfR1 (Cell Signaling), IKKβ (Imgenex) were used. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and RNA polymerase II (Santa Cruz) were used as loading controls. In pull-down experiments, neutravidin-agarose (Pierce) or Anti-HA agarose beads (Santa Cruz) were employed. The bands were visualized by enhanced chemiluminescence (ECL) assay kit (Pierce). The membranes were also stained with Ponceau Red to verify equal loading and uniform transfer of proteins.
TNF-α Secretion Assay
To determine the effect of GA on TNF-α secretion, RAW 264.7 cells were treated with 1μM GA for 30 min and stimulated with LPS (1 μg/ml) for different time points. The culture supernatants were isolated and the accumulation of TNF-α in cultured medium was determined using a commercially available TNF-α ELISA kit (Diaclone Research).
Quantitation of Nitrite Production
Nitric oxide production in culture media supernatant was assayed by measuring the stable degradation product of NO, nitrite, using the Griess reagent (Sigma). RAW 264.7 cells were grown in 12-well plates and incubated with different concentrations of GA (0.1, 0.5, 1.0, and 1.5 μM) for 30 min. The cells, were then stimulated with LPS (1 μg/ml) for 12 h. After 12 h of LPS stimulation, the culture media supernatants were isolated and mixed with an equal volume of Griess reagent and incubated at room temperature for 15 min. The absorbance was measured at 550 nm in a microplate reader. Sodium nitrite (10 μM-100 μM) was used to create a standard calibration curve.
Immunoprecipitation of p65
The nuclear translocation of p65 was examined by immunoprecipitation. Nuclear proteins (50 μg) were immunoprecipitated with 0.5 μg of anti-p65 polyclonal agarose conjugate (Santa Cruz Biotechnology) overnight at 4 °C. The agarose beads were extensively washed with PBS (4 × 400 μl) and subjected to SDS-PAGE (10%) followed by Western blot analysis as described earlier.
Transient Transfection Assays
The effect of GA on NF-κB-dependent reporter gene transcription induced with LPS was analyzed by luciferase assay. Briefly, RAW264.7 cells were seeded at a concentration of 2×105 cells/well in six-well plates. After overnight culture, the cells in each well were transfected with 1 μg of DNA (0.75 μg of COX-2, COX-2 double mutant or 5X multimerized κB-luciferase reporter plasmid (Stratagene) and 0.25 μg pRLTK renilla luciferase control plasmid, Promega) along with 6 μl of lipofectamine-2000 (Invitrogen Life Technologies). in serum-free DMEM media. After a 6 h exposure to the transfection mixture, complete medium with 5 % FBS was added to the cells and incubated at 37 °C for an additional 16 h. The transfected cells were treated with various concentrations of GA (0.1-1.5 μM) for 30 min and the cells were stimulated with LPS (1 μg/ml) for 6 h. The cells were harvested and luciferase activity was measured by dual luciferase assay (Promega). Renilla luciferase activity was used to normalize transfection efficiency.
RNA Interference
RAW264.7 cells were plated in six-well plates and allowed to adhere overnight. Such cells were transfected with TfR1 siRNA (1 μg/well) or si-control (Dharmacon) using the Mirus SiQuest TransIT® reagent (Mirus Bio Corporation). After 48 hours, cells were treated with 1μM GA for 30 min at 37 °C followed by stimulation with 1 μg/ml LPS for 2 h. Cytoplasmic and nuclear fractions were separated from cells using the protocol described earlier. The cytoplasmic and nuclear fractions were used in Western blot analyses using specific antibodies. GAPDH and SP1 were used as controls for cytoplasmic and nuclear fractions, respectively.
Electrophoretic Mobility Shift Assays
NF-κB activation was analyzed by EMSA as described previously [9]. Briefly, nuclear extracts (10 μg) were incubated with 40,000 cpm 32P-labeled NF-κB double stranded oligonucleotide (Promega) and 0.1 μg poly (dI-dC) for 30 min and subjected to electrophoresis under non-denaturing conditions on a 5 % polyacrylamide gel in Tris-borate buffer (BioRad). To confirm specificity of NF-κB binding, unlabeled oligonucleotide (3.5 pmoles) was used as a specific competitor. For super-shift experiments, the nuclear extract from LPS-treated cells was incubated with 2 μg of anti-p50 (Santa Cruz Biotechnology, CA) followed by 32P-labeled oligonucleotides as described above.
Preparation of Biotinylated GA
The carboxyl group of GA was modified by amidation with EZ-link (5-biotinamido)pentylamine (Pierce). Briefly, GA (~2 mg) and EZ-link (5-biotinamido)pentylamine (5 mg) were dissolved in DMSO (300μl) and were allowed to react with 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) in 100 mM MES, pH 5.5, at 37 °C for 3 h. The reaction was stopped by extracting the product with ethylacetate and MES buffer (1:1; v/v). The extracted biotinylated GA was purified using reverse phase HPLC with a linear gradient of acetonitrile (10-100%). The biotinylated GA was dried under nitrogen and dissolved in acetonitrile for further use.
Interaction of Biotinylated GA with IKKβ in HEK Cells
To determine the interaction of biotinylated GA with IKKβ, HEK293 cells were transfected with 5 μg HA-tagged IKKβ wild type mammalian expression construct along with 20 μl of lipofectamine for 16 h. After transfection, the cells were washed with PBS, harvested, and lysed in M-PER. Cell lysates containing 50 μg of protein were treated with 1mM DTT and/or 1.5 μM of biotinylated GA for 2 h at 37 °C and immunoprecipitated with anti-HA agarose beads (Santa Cruz, CA) overnight at 4 °C with constant shaking. The beads were washed three times with TBST buffer by centrifugation at 16,000 × g for 10 min. The proteins were eluted by boiling the beads in SDS sample buffer for 5 min and analyzed by SDS-PAGE. The biotinylated proteins were electrotransfered and immunodetected using the North2South chemiluminescent detection kit (Pierce). The cell lysates, where IKKβ-HA was over-expressed, were incubated with biotinylated-GA mixed with increasing amounts of glutathione (GSH; 0.1-3 mM) to test the effect of GSH on the interaction with IKKβ. To test if Cys179 was being modified by GA, we used the HA-tagged human IKKβ wild-type and IKKβ 179Cys→Ala mutant to compare the reactivity of GA-biotin. The expression constructs were kindly provided by Dr. Michael Karin at University of California at San Diego, CA. The human HA-tagged IKKβ wild type or Cys179→Ala mutant expression constructs were transfected into RAW264.7 macrophages and cell lysates were treated with biotinylated GA as described above.
Mass Spectrometric Analysis of Post-Translational Modification of IKKβ
To further confirm the modification of IKKβ by GA, murine IKKβ peptide-(173-186), LDQGSLCTSFVGTL (synthesized at the Peptide Synthesis Facility, Penn State College of Medicine, Hershey, PA) was incubated with Me2SO4 and purified GA (mol/mol) for 30 min at 37 °C in PBS. The samples were analyzed by M@LDI-TOF-MS for modification at the Mass Spectrometry Facility, Penn State University, University Park, PA.
Inhibition of IKKβ Activity by GA in vitro
To determine whether GA inhibits IKK activation, RAW264.7 cells were transfected with HA-tagged IKKβ wild-type expression vector for 24 h as described earlier. The cells were lysed and the clarified lysates were then incubated with anti-HA prebound to agarose at 4 °C overnight. The beads were washed with TBST containing phosphatase inhibitors and treated with GA (1.0 μM) for 2 h at 37 °C. The kinase reaction was performed on the immunoprecipitates in kinase buffer with GST-IκBα as the substrate for 2 h at 30 °C. The reaction was stopped by adding 10 μl of 1X SDS sample buffer and the reaction mixtures were subjected to SDS-PAGE followed by Western blot analysis. The membrane was probed with monoclonal anti-pIκBα antibodies and reprobed with anti-IKKβ after treating the membrane with the stripping reagent (Pierce).
Cell Viability Assay
Cell viability was determined using CCK-8 kit to count number of living cells (Dojindo Molecular Technologies, Inc., Gaithersburg, MD). CCK-8 utilizes WST-8 (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt), which produces a water-soluble formazan dye upon bio-reduction by cellular dehydrogenases to an orange formazan product that is soluble in the tissue culture medium. The amount of formazan produced is directly proportional to the number of living cells. RAW264.7 cells were plated at a concentration of 5,000 cells in 100 μl of complete medium per well in 96-well plates for 24 h prior to treatment. The cells were treated with the indicated concentrations of GA for different time points. Thereafter, 10 μl of CCK-8 reagent was added to each well and incubated for an additional 3 h. The absorbance was read in a Packard microplate reader at 450 nm and the cell viability was calculated. In addition, RAW264.7 cells were treated with four-log orders of GA (0.01-10 μM) for 24 h and the cell viability was assessed using trypan blue exclusion method. Ratio of live to total cells was calculated using a hemocytometer.
Statistical Analysis
When necessary, data are expressed as mean ± S.D., and the Student’s t test was used in statistical analysis for comparison. P<0.05 was used as the criterion for statistical significance.
RESULTS
Effect of GA on the LPS-induced Expression of COX-2, TNF-α and iNOS
To investigate the anti-inflammatory effects of GA, the expression of three prototypical inflammatory markers, COX-2, TNF-α, and iNOS, were analyzed in RAW macrophages. As shown in Fig. 2A, GA pretreatment markedly reduced the expression of COX-2 in LPS-treated macrophages in a concentration-dependent manner. The EC50 was found to be ~ 0.5 μM. At 1 μM, there was complete inhibition of COX-2 expression. Along the same lines, the production of TNF-α in the extracellular media was analyzed in RAW264.7 macrophages. As shown in Fig. 2B, the production of TNF-α was inhibited in a time- dependent manner in GA pretreated RAW macrophages. The inhibitory effect was apparent as early as 30 min post LPS stimulation; while at later time points (2-4 h), the inhibition in TNFα was even more conspicuous. We also analyzed the expression of iNOS in RAW 264.7 macrophages before and after treatment with GA. As shown in Fig. 3A, pretreatment of RAW 264.7 cells with increasing concentrations of GA promoted a dose-dependent inhibition in the amount of nitrite produced by these cells in response to LPS. The EC50 was calculated to be ~0.5 μM of GA. Western blot analysis of the corresponding cell extracts showed that GA pretreatment inhibited the LPS induced stimulation of iNOS expression. The EC50 in this case was calculated to be ~0.5 μM of GA, which corroborates well with those obtained with activity. The fact that the expression of all three proinflammatory genes was inhibited by GA suggested that the effect of GA was likely mediated at the level of transcription.
FIGURE 2. Effect of GA on expression of COX-2 and TNF-α in LPS activated RAW 264.7 cells.
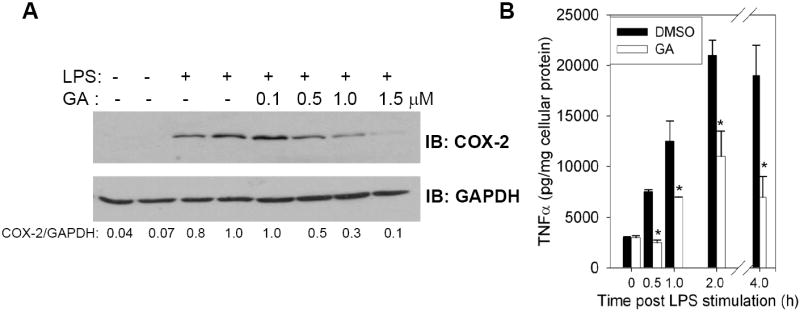
A, The cells were incubated with various concentrations of GA (0.1-1.5 μM) for 30 min followed by stimulation with LPS (1 μg/ml) for 2-4 h at 37 °C. The culture media supernatants and corresponding cell lysates were used for the analyses of COX-2 expression. B, Time-dependent inhibition of the production of TNF-α by GA (1 μM) in LPS-treated RAW264.7 macrophages. The results are expressed as the mean ± S.D. of three independent experiments performed in triplicate. DMSO added was kept constant in all experiments. Representative Western blots of n= 3 are shown.
FIGURE 3. Effect of GA on nitrite production and iNOS expression in LPS-induced RAW 264.7 macrophages.
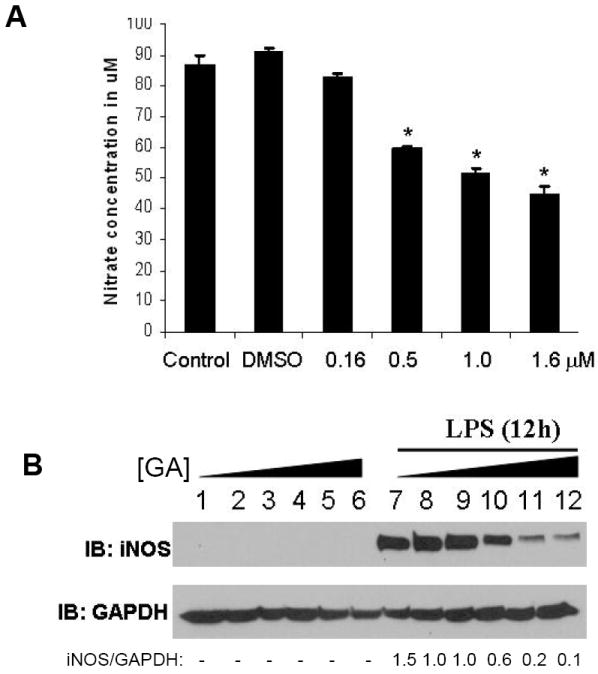
A, Cells were pretreated with GA (0.1-1.5 μM) for 30 min before incubation with 1 μg/ml of LPS for 12 h. Nitrite levels in the cell culture medium are expressed as the mean ± S.D. of three independent experiments performed in triplicate. * p<0.05. B, whole cell extracts were analyzed for iNOS expression by Western blot analysis. Representative of n= 3 shown.
Kasibhatla et al [6] and Pande et al [7] have demonstrated that GA binds to TfR1 to negatively effect transcription of proinflammatory genes in cancer cells. However, flow-cytometric studies indicated a >85 % down regulation of TfR1 (also called CD71) in LPS-treated RAW264.7 cells (Fig. 4A). To address the fact that GA targeted the NF-κB pathway independently of TfR1, we examined the LPS-dependent nuclear translocation of p65 in TfR1 knockdown cells. Results shown in Fig. 4B clearly indicate that even in the absence of TfR1, GA caused a significant decrease in the nuclear levels of p65 in cells treated with LPS. Thus, given the possibility of the existence of alternative cellular targets other than TfR1, we examined the effect of GA on the activation of the NF-κB pathway.
FIGURE 4. TfR1-independent modulation of p65 in GA pretreated macrophages.

A, Flow cytometry histograms of RAW264.7 cells treated with or without LPS (1μg/ml) for 12 h. Cells were blocked with FcR blocking antibody (CD16/CD32), stained for CD71 (TfR1)-FITC and analyzed by flow cytometry. The histograms represent unstained (gray shaded), treated with LPS (dotted line), and untreated with LPS (solid line), respectively. Cell numbers described above are averages of triplicate experiments ± S.D. B, RAW264.7 cells were plated in 6-well plates and were transfected with TfR1 siRNA or si-control at 1 μg/well. After 48 h, cells were treated with 1 μM GA or vehicle (DMSO) for 30 min at 37 °C followed by treatment with 1 μg/ml LPS for 2 h. Cytoplasmic and nuclear extracts were prepared from these cells. Representative Western blot of n = 2 shown.
GA blocks LPS-induced Nuclear Translocation of p65 Subunit of NF-κB
We determined the effect of GA on the LPS-induced nuclear translocation of the p65 subunit of NF-κB by immunoprecipitation of nuclear protein extracts. LPS treatment elicited a rapid nuclear accumulation of p65 in control and DMSO treated cells. Preincubation with GA abolished the LPS-stimulated p65 translocation to the nucleus in RAW264.7 macrophages (Fig. 5). RAW264.7 cells showed enhanced sensitivity towards GA in that the inhibition of p65 translocation was robust at concentrations as low as 0.5 μM; while at higher concentrations (1.0 and 1.5 uM), there was complete inhibition of nuclear translocation of p65. Taken together, these results indicate that GA blocks the nuclear translocation of p65 subunit of NF-κB.
FIGURE 5. GA inhibits LPS-induced nuclear translocation of p65.
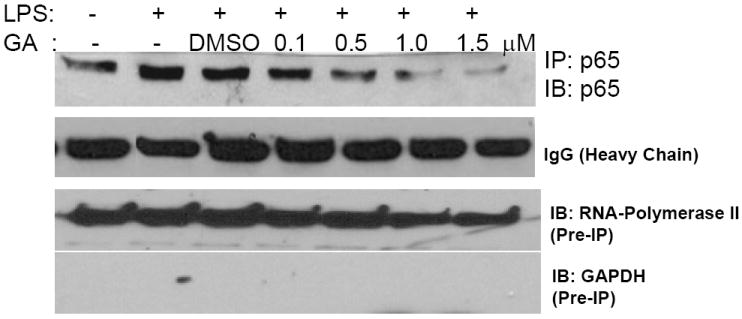
RAW264.7 cells were pretreated with 0.1-1.5 μM GA for 30 min and then stimulated with LPS (1 μg/ml) for 2 h. Nuclear proteins were analyzed for the presence of p65 by immunoprecipitation followed by Western blot analysis. IgG-heavy chain was used to confirm near equal loading of the immunoprecipitate. Cell lysates (pre-IP) were analyzed for Western blotting for GAPDH and RNAP-II as markers of cytosolic and nuclear fractions, respectively. Representative Western blot of n = 4.
Effect of GA on NF-κB DNA Binding
To determine if GA inhibited LPS-induced NF-κB transcriptional activity, transient transfections with a multimerized NF-κB driven luciferase vector, murine COX-2 wildtype (COX-2 WT), and COX-2ΔκB promoter luciferase vectors were carried out (Fig. 6A,B). We tested the modulation of murine COX-2 promoter reporter construct by GA, which has two NF-κB sites that are pivotal to LPS induced transcriptional activation [28, 29]. Preincubation of such COX-2 WT transfected cells with various concentrations of GA clearly indicated that that the LPS-induced NF-κB activation was significantly decreased at ≥0.5 μM of GA and at concentration ≥ 1 μM, there was hardly any activity observed (Fig. 6A). The NF-κB double mutant COX-2 reporter was inactive and exhibited no effect upon LPS and/or GA treatment (Fig. 6A). Furthermore, we studied the effect of GA on the LPS-dependent activation of the multimerized NF-κB luciferase reporter, (κB)5-Luc (Fig. 6B). The results of the inhibition of LPS-dependent luciferase activity by GA corroborate well with those obtained earlier with the COX-2 reporter assays, with the EC50 in the range of 0.3-0.5 μM (Fig. 6B). Interestingly, DGA was unable to repress the NF-κB activity at 1 μM.
FIGURE 6. GA represses NF-κB dependent reporter gene expression induced by LPS.

A, Inhibition of NF-κB dependent COX-2 reporter activity using murine COX-2 wildtype (COX-2WT) and ΔκB-mutant (COX-2-ΔκB) promoter-luciferase reporter constructs by GA (0.1-1.5 μM). B, inhibition of NF-κB-luciferase activity by GA. Cells transfected with multimerized NF-κB-luciferase and pRL-TK vectors were treated with GA (0.1-1.5 μM) followed by stimulation with LPS for 6 h. Data shown are averages ± S.D. of four independent experiments. C and D, EMSA for the binding of NF-κB in the nuclear extracts isolated from RAW264.7 and U937 cells, respectively. Cells were treated with indicated concentrations of GA followed by stimulation with LPS for 2 h. CC corresponds to cold competitive oligonucleotide control. SS represents super-shift with anti-p50 antibody incubated with the LPS-treated nuclear extracts followed by 32P-labeled NF-κB double-strand oligonucleotides. DGA was used at 1 μM in all the experiments as a control. All lanes either had vehicle (DMSO) and/or GA or DGA. The EMSA autoradiogram shown in each case are representative of n= 5.
In support of the reporter studies, we analyzed nuclear extracts from LPS stimulated RAW264.7 macrophages (Fig. 6C) and human U937 monocytic cells (Fig. 6D) before or after GA and DGA treatment for binding to NF-κB oligonucleotide in gel shift assays. LPS at a concentration of 1 μg/ml strongly activated NF-κB activity in both the macrophage cell types. Pretreatment with GA markedly inhibited LPS-dependent NF-κB activation in both cell types. As seen earlier, pretreatment with DGA was ineffective in inhibiting the activation of NF-κB. Taken together, these studies unequivocally indicate that GA inhibits the activation of NF-κB and that the α,β-unsaturated moiety is essential for the inhibitory activity.
In-vitro Kinase Assays
Based on our previous experiments with 15d-PGJ2 [9], bioactive compounds with an α,β-unsaturated moiety have the potential to interact covalently with a key thiol in IKKβ. Therefore, we first examined if IKK activity was affected by GA. To determine the effect of GA on LPS induced IKK activation, we analyzed the cellular levels of pIκBα before and after LPS treatment (Fig. 7). GA-pretreatment significantly decreased pIκBα in a dose dependent manner in RAW264.7 cells (Fig. 7A) at ≤0.5 μM of GA. To further investigate if GA targeted IKKβ, IKKβ-HA was over-expressed in RAW264.7 cells and the immunoprecipitates were examined for the effect of GA (1.0 μM) on kinase activity. As shown in Fig 7B, GA significantly inhibited the IKK activity suggesting the possibility of covalent interaction with IKKβ to inhibit the enzymatic activity.
FIGURE 7. Effect of GA on LPS-induced IKK activity.
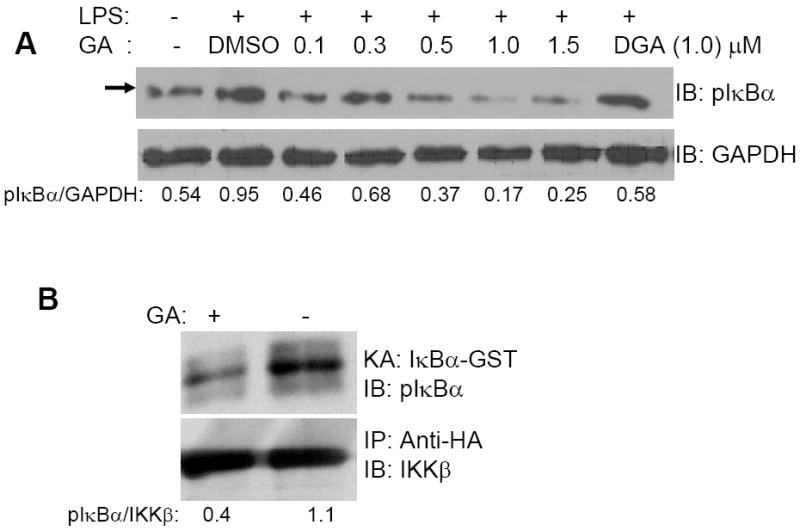
A, RAW264.7 cell lystates from RAW264.7 cells treated with DMSO or GA (0.1-1.5 μM) followed by stimulation with LPS for 2 h. B, IKKβ-HA tagged protein was expressed in RAW264.7 cells and the immunoprecipitate (IKKβ: anti-HA complex) was incubated with DMSO or GA (1.0 μM) for 30 min followed by in-vitro kinase activity assay (KA) using the GST-IκBα(1-55) protein and ATP. In all experiments, the blots were reprobed for GAPDH or IKKβ to normalize for equal protein loading. Representative Western blots of n= 3 shown.
Covalent Binding of GA with IKKβ
To determine whether GA covalently reacted with IKKβ, a biotinylated derivative of GA was added to the lysates from HEK293 cells that were transfected with a pCMV-IKKβ-HA expression construct. The GA-IKKβ adduct was pulled down with anti-HA agarose beads and the biotinylation of IKKβ was examined. It appeared that GA reacted with IKKβ and treatment with 1 mM DTT disrupted the binding of GA with IKKβ (Fig. 8A, upper panel). Alternatively, the biotinylated proteins from the cell lysates were pulled-down with neutravidin-agarose beads. The affinity pull-downs indicated the presence of IKKβ as one of the proteins that interacted with GA (Fig. 8A, lower panel). In addition, we examined if the interaction between IKKβ and GA was modulated by physiological cellular thiols such as GSH. Although, increasing GSH concentration from 0.1 mM to 1.0 mM and 3 mM decreased the ability to IKKβ to bind GA, we were still able to detect interaction even at 3 mM (Fig. 8B). Both these experiments clearly indicated that GA covalently interacted with IKKβ and that the interaction withstood reducing and denaturing conditions during electrophoresis except when DTT or GSH was included during reaction. To further confirm the reaction, GA was incubated with the IKKβ (173-186) peptide and subjected to qualitative M@LDI-TOF-MS analysis. The results clearly demonstrated an increase in the mass of the peptide from 1440.7 to 2066.2, a difference of 625.5 m/z units, indicating that the peptide was covalently modified by GA (Fig. 8C). To address the importance of 179Cys in the interaction with GA, an IKKβ (Cys/Ala) mutant expression construct was transfected into RAW264.7 cells. The cell lysates were incubated with GA as described above. Immunoprecipitation with anti-HA followed by Western immunoblot with streptavidin-HRP clearly showed lack of binding of GA to IKKβ(179Cys/Ala) mutant compared to the wildtype IKKβ (Fig. 8D). Taken together, these studies indicate that GA interacts with the 179Cys residue of IKKβ.
FIGURE 8. Interaction of GA and IKKβ.

A, Total cell lysates (50 μg) from HEK293 cells transfected with IKKβ-HA plasmid were incubated with biotinylated-GA (1.5 μM) in the presence (lane 2) or absence (lane 3) of DTT. The lysates were subjected to immunoprecipitation with anti-HA followed by Western immunoblot with streptavidin-HRP (upper panel) or, a neutravidin-pull down followed by anti-IKKβ Western immunoblot (lower panel). B. Effect of GSH on the interaction of GA with IKKβ. GSH (0.1, 1, and 3 mM) was incubated with biotinylated GA (1 μM) for 2 h at 37 °C. The complex was added to IKKβ over-expressing HEK293 cell lysates (100 μg) for 1 h on ice. Neutravidin affinity chromatography was used to analyze biotinylated GA-modified IKKβ as described above. C, mass spectrometric analysis of the interaction of IKKβ peptide with DMSO and GA. D, interaction of GA with IKKβ-HA wild-type and 179 Cys→Ala mutant proteins expressed in RAW264.7 cells. Total cell lysates (50 μg) from these cells were incubated with biotinylated-GA (1.0 μM) for 30 min followed by immunoprecipitation by anti-HA and Western immunoblot using streptavidin-HRP. The blot was reprobed with anti-IKKβ to confirm equal protein expression and loading. Western blots shown are representative of n= 3.
Cytotoxicity of GA
To further test if GA exhibited any effect on viability of RAW264.7 cells used in this investigation, cytotoxic assays were performed. Results shown in Fig. 9 indicate that in RAW 264.7 cells, no cytotoxic effect was observed even at 24 h of incubation. Furthermore, trypan blue exclusion method also showed that GA did not cause apoptosis RAW264.7 macrophages toxic effects of GA to about 2.0 μM, after which there was increase in cell death (Fig. 9, inset).
FIGURE 9. Cytotoxic effects of GA.
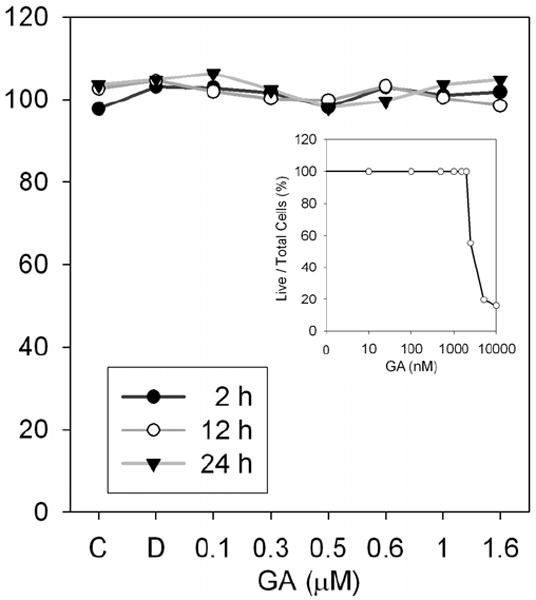
RAW264.7 cells were treated with GA (0.1 μM-1.6 μM) for indicated time points. Thereafter, 10 μl of CCK-8 reagent was added to each well and incubated for an additional 3 h. Cell viability was read in a plate reader at 450 nm. Inset: RAW264.7 cells were treated with GA (0.01-10 μM) for 24 h followed by trypan blue staining.
DISCUSSION
Many signal transduction pathways converge at the multi-subunit IKK complex to mediate activation of NF-κB-dependent gene expression, which is critical to metastatogenesis, tumor promotion and inflammation [30]. Thus, the control of NF-κB activation has significant therapeutic implications. Here, we demonstrate that GA abrogates LPS-activated NF-κB pathway through the inhibition of IKKβ. However, GA has been shown to bind TfR1 that is significantly overexpressed in different types of cancers [6]. Although GA interacted with TfR1 non-covalently; GAO was found to be inactive [6], which suggests that the importance of the α,β-unsaturated group in GA. The use of the dihydro-analog of GA, DGA, in our studies further confirms the requirement of the enone moiety in GA for its NF-κB inhibitory activity. Furthermore, Pandey et al [7] demonstrated that the binding of GA to TfR1 triggered apoptosis in human myeloid leukemia cells via the potentiation of TNFα-induced apoptosis through modulation of the NF-κB pathway. Flow cytometric analysis of TfR1 expression in macrophages indicated a >85 % down-regulation of expression upon LPS-treatment, which agree with the results of Wardrop and Richardson [8]. Studies with siRNA-mediated knockdown of TfR1 confirmed the existence of intracellular targets such as IKKβ to be functional. Therefore, given the down-regulated expression of TfR1 in “inflammatory” macrophages, it is clear that the anti-inflammatory activity of GA arises in part from its interaction with intracellular proteins, particularly components of the NF-κB pathway such as IKKβ.
Based on the results from M@LDI-TOF-MS and site-directed mutagenesis experiments, we strongly believe that 179Cys in IKKβ is an intracellular target. The interaction of GA with IKKβ was inhibited to a significant extent by increasing levels of GSH, a cellular thiol antioxidant. It could be predicted that imbalances in the [GSH]:[GSSG] ratio towards the oxidizing state, as in many inflammatory disease pathologies that activates NF-κB [31], may in fact allow GA to impart its anti-inflammatory activity. Thus, glutathionylation represents a key cellular metabolic event in the regulation of the anti-inflammatory activity of GA.
It is known that electrophilic α,β-unsaturated carbonyl group in bioactive compounds selectively react with protein thiols. Electrophiles such as 15-deoxy-Δ12,14-PGJ2 [24] [9] and 4-HNE [10] interact with IKKβ leading to complete abrogation of its kinase activity. To test if the interactome of GA and 15-deoxy-Δ12,14-PGJ2 with cellular proteins overlapped, we analyzed the ability of GA to modify thioredoxin and p50 that are already established as key protein targets of 15-deoxy-Δ12,14-PGJ2 [25, 32]. Studies showed that GA interacted with both these proteins (data not shown). Molecular modeling studies have provided some insight into the interaction of 15-deoxy-Δ12,14-PGJ2 with Cys thiols in the above proteins to be controlled by molecular recognition by fit of shape and complementarity in addition to accessibility and environment of the Cys thiol [33]. Thus, it is likely that a subset of cellular proteins may meet these requirements to interact with hydrophobic electrophiles such as 15-deoxy-Δ12,14-PGJ2 and GA. Needless to say, the specificity of covalent interaction of GA with Cys thiols in target proteins will need to be further characterized using proteomics, which might provide clues regarding the environment of the Cys thiol and the structural features that are necessary for interaction. Based on these studies, we believe that the spectrum of activities exhibited by GA could be partly attributed to interaction with proteins covalently, as shown here, in addition to the ability to interact non-covalently with TfR1 to effect downstream gene expression.
In summary, our results indicate the existence of an alternate pathway of inhibition of NF-κB activation by GA via specifically modifying 179Cys of IKKβ leading to the down-regulation of expression of COX-2, iNOS, and TNFα, which are implicated in inflammation.
Acknowledgments
We are grateful to the Macromolecular Core Facility, Penn State College of Medicine, Hershey, PA, for the synthesis of the IKKβ peptide, the Penn State Mass Spectrometry Facility and the Center for Quantitative Cell Analysis, for the mass spectral and flow cytometric analyses, respectively. We thank Dr. Michael Karin, UCSD, for the IKK plasmid constructs. This study was funded by a PHS grant (AT-004350) from the National Institutes of Health.
Abbreviations
- NF-κB
Nuclear factor-κB
- GA
gambogic acid
- DGA
dihydrogambogic acid
- LPS
lipopolysaccharide
- TfR1
transferrin receptor-1
Footnotes
This work was supported by a PHS grant (AT 004350) from the National Institutes of Health (to K.S.P.).
References
- 1.Han QB, Cheung S, Tai J, Qiao CF, Song JZ, Xu HX. Stability and cytotoxicity of gambogic acid and its derivative, gambogoic acid. Biol Pharm Bull. 2005;28:2335–2337. doi: 10.1248/bpb.28.2335. [DOI] [PubMed] [Google Scholar]
- 2.Guo QL, Lin SS, You QD, Gu HY, Yu J, Zhao L, Qi Q, Liang F, Tan Z, Wang X. Inhibition of human telomerase reverse transcriptase gene expression by gambogic acid in human hepatoma SMMC-7721 cells. Life Sci. 2006;78:1238–1245. doi: 10.1016/j.lfs.2005.06.046. [DOI] [PubMed] [Google Scholar]
- 3.Wu ZQ, Guo QL, You QD, Zhao L, Gu HY. Gambogic acid inhibits proliferation of human lung carcinoma SPC-A1 cells in vivo and in vitro and represses telomerase activity and telomerase reverse transcriptase mRNA expression in the cells. Biol Pharm Bull. 2004;27:1769–1774. doi: 10.1248/bpb.27.1769. [DOI] [PubMed] [Google Scholar]
- 4.Zhao L, Guo QL, You QD, Wu ZQ, Gu HY. Gambogic acid induces apoptosis and regulates expressions of Bax and Bcl-2 protein in human gastric carcinoma MGC-803 cells. Biol Pharm Bull. 2004;27:998–1003. doi: 10.1248/bpb.27.998. [DOI] [PubMed] [Google Scholar]
- 5.Guo QL, You QD, Wu ZQ, Yuan ST, Zhao L. General gambogic acids inhibited growth of human hepatoma SMMC-7721 cells in vitro and in nude mice. Acta Pharmacol Sin. 2004;25:769–774. [PubMed] [Google Scholar]
- 6.Kasibhatla S, Jessen KA, Maliartchouk S, Wang JY, English NM, Drewe J, Qiu L, Archer SP, Ponce AE, Sirisoma N, Jiang S, Zhang HZ, Gehlsen KR, Cai SX, Green DR, Tseng B. A role for transferrin receptor in triggering apoptosis when targeted with gambogic acid. Proc Natl Acad Sci U S A. 2005;102:12095–12100. doi: 10.1073/pnas.0406731102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pandey MK, Sung B, Ahn KS, Kunnumakkara AB, Chaturvedi MM, Aggarwal BB. Gambogic acid, a novel ligand for transferrin receptor, potentiates TNF-induced apoptosis through modulation of the nuclear factor-{kappa}B signaling pathway. Blood. 2007;110:3517–3525. doi: 10.1182/blood-2007-03-079616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wardrop SL, Richardson DR. Interferon-gamma and lipopolysaccharide regulate the expression of Nramp2 and increase the uptake of iron from low relative molecular mass complexes by macrophages. Eur J Biochem. 2000;267:6586–6593. doi: 10.1046/j.1432-1327.2000.01752.x. [DOI] [PubMed] [Google Scholar]
- 9.Vunta H, Davis F, Palempalli UD, Bhat D, Arner RJ, Thompson JT, Peterson DG, Reddy CC, Prabhu KS. The antiinflammatory effects of selenium are mediated through 15-deoxy-u 12,14-prostaglandin J2 in macrophages. J Biol Chem. 2007;282:17964–17973. doi: 10.1074/jbc.M703075200. [DOI] [PubMed] [Google Scholar]
- 10.Ji C, Kozak KR, Marnett LJ. IkappaB kinase, a molecular target for inhibition by 4-hydroxy-2-nonenal. J Biol Chem. 2001;276:18223–18228. doi: 10.1074/jbc.M101266200. [DOI] [PubMed] [Google Scholar]
- 11.Chen F, Castranova V, Shi X, Demers LM. New insights into the role of nuclear factor-kappaB, a ubiquitous transcription factor in the initiation of diseases. Clin Chem. 1999;45:7–17. [PubMed] [Google Scholar]
- 12.Ghosh S. Regulation of inducible gene expression by the transcription factor NF-kappaB. Immunol Res. 1999;19:183–189. doi: 10.1007/BF02786486. [DOI] [PubMed] [Google Scholar]
- 13.Hatada EN, Krappmann D, Scheidereit C. NF-kappaB and the innate immune response. Curr Opin Immunol. 2000;12:52–58. doi: 10.1016/s0952-7915(99)00050-3. [DOI] [PubMed] [Google Scholar]
- 14.Sun SC, Xiao G. Deregulation of NF-kappaB and its upstream kinases in cancer. Cancer Metastasis Rev. 2003;22:405–422. doi: 10.1023/a:1023733231406. [DOI] [PubMed] [Google Scholar]
- 15.Saitoh T, Nakayama M, Nakano H, Yagita H, Yamamoto N, Yamaoka S. TWEAK induces NF-kappaB2 p100 processing and long lasting NF-kappaB activation. J Biol Chem. 2003;278:36005–36012. doi: 10.1074/jbc.M304266200. [DOI] [PubMed] [Google Scholar]
- 16.Abraham E. NF-kappaB activation. Crit Care Med. 2000;28:N100–104. doi: 10.1097/00003246-200004001-00012. [DOI] [PubMed] [Google Scholar]
- 17.Cavaillon JM. Interleukins and inflammation. Pathol Biol (Paris) 1990;38:36–42. [PubMed] [Google Scholar]
- 18.Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death--a new approach to cancer therapy. J Clin Invest. 2005;115:2625–2632. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu C, Shen G, Chen C, Gelinas C, Kong AN. Suppression of NF-kappaB and NF-kappaB-regulated gene expression by sulforaphane and PEITC through IkappaBalpha, IKK pathway in human prostate cancer PC-3 cells. Oncogene. 2005;24:4486–4495. doi: 10.1038/sj.onc.1208656. [DOI] [PubMed] [Google Scholar]
- 20.Brennan P, O’Neill LA. Inhibition of nuclear factor kappaB by direct modification in whole cells--mechanism of action of nordihydroguaiaritic acid, curcumin and thiol modifiers. Biochem Pharmacol. 1998;55:965–973. doi: 10.1016/s0006-2952(97)00535-2. [DOI] [PubMed] [Google Scholar]
- 21.Natarajan K, Singh S, Burke TR, Jr, Grunberger D, Aggarwal BB. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc Natl Acad Sci U S A. 1996;93:9090–9095. doi: 10.1073/pnas.93.17.9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takada Y, Murakami A, Aggarwal BB. Zerumbone abolishes NF-kappaB and IkappaBalpha kinase activation leading to suppression of antiapoptotic and metastatic gene expression, upregulation of apoptosis, and downregulation of invasion. Oncogene. 2005;24:6957–6969. doi: 10.1038/sj.onc.1208845. [DOI] [PubMed] [Google Scholar]
- 23.Na HK, Surh YJ. Transcriptional regulation via cysteine thiol modification: a novel molecular strategy for chemoprevention and cytoprotection. Mol Carcinog. 2006;45:368–380. doi: 10.1002/mc.20225. [DOI] [PubMed] [Google Scholar]
- 24.Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- 25.Cernuda-Morollon E, Pineda-Molina E, Canada FJ, Perez-Sala D. 15-Deoxy-Delta 12,14-prostaglandin J2 inhibition of NF-kappaB-DNA binding through covalent modification of the p50 subunit. J Biol Chem. 2001;276:35530–35536. doi: 10.1074/jbc.M104518200. [DOI] [PubMed] [Google Scholar]
- 26.Perez-Sala D, Cernuda-Morollon E, Pineda-Molina E, Canada FJ. Contribution of covalent protein modification to the antiinflammatory effects of cyclopentenone prostaglandins. Ann N Y Acad Sci. 2002;973:533–536. doi: 10.1111/j.1749-6632.2002.tb04695.x. [DOI] [PubMed] [Google Scholar]
- 27.Perez-Sala D, Cernuda-Morollon E, Canada FJ. Molecular basis for the direct inhibition of AP-1 DNA binding by 15-deoxy-Delta 12,14-prostaglandin J2. J Biol Chem. 2003;278:51251–51260. doi: 10.1074/jbc.M309409200. [DOI] [PubMed] [Google Scholar]
- 28.Wadleigh DJ, Reddy ST, Kopp E, Ghosh S, Herschman HR. Transcriptional activation of the cyclooxygenase-2 gene in endotoxin-treated RAW 264.7 macrophages. J Biol Chem. 2000;275:6259–6266. doi: 10.1074/jbc.275.9.6259. [DOI] [PubMed] [Google Scholar]
- 29.Zamamiri-Davis F, Lu Y, Thompson JT, Prabhu KS, Reddy PV, Sordillo LM, Reddy CC. Nuclear factor-kappaB mediates over-expression of cyclooxygenase-2 during activation of RAW 264.7 macrophages in selenium deficiency. Free Radic Biol Med. 2002;32:890–897. doi: 10.1016/s0891-5849(02)00775-x. [DOI] [PubMed] [Google Scholar]
- 30.Karin M. The IkappaB kinase - a bridge between inflammation and cancer. Cell Res. 2008;18:334–342. doi: 10.1038/cr.2008.30. [DOI] [PubMed] [Google Scholar]
- 31.Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 32.Shibata T, Yamada T, Ishii T, Kumazawa S, Nakamura H, Masutani H, Yodoi J, Uchida K. Thioredoxin as a molecular target of cyclopentenone prostaglandins. J Biol Chem. 2003;278:26046–26054. doi: 10.1074/jbc.M303690200. [DOI] [PubMed] [Google Scholar]
- 33.Pande V, Ramos MJ. Molecular recognition of 15-deoxy-delta(12,14)-prostaglandin J2 by nuclear factor-kappa B and other cellular proteins. Bioorg Med Chem Lett. 2005;15:4057–4063. doi: 10.1016/j.bmcl.2005.06.025. [DOI] [PubMed] [Google Scholar]