

Abstract
Background
The tick-borne pathogen Anaplasma marginale, which is endemic worldwide, is the type species of the genus Anaplasma (Rickettsiales: Anaplasmataceae). Rhipicephalus (Boophilus) microplus is the most important tick vector of A. marginale in tropical and subtropical regions of the world. Despite extensive characterization of the genetic diversity in A. marginale geographic strains using major surface protein sequences, little is known about the biogeography and evolution of A. marginale and other Anaplasma species. For A. marginale, MSP1a was shown to be involved in vector-pathogen and host-pathogen interactions and to have evolved under positive selection pressure. The MSP1a of A. marginale strains differs in molecular weight because of a variable number of tandem 23-31 amino acid repeats and has proven to be a stable marker of strain identity. While phylogenetic studies of MSP1a repeat sequences have shown evidence of A. marginale-tick co-evolution, these studies have not provided phylogeographic information on a global scale because of the high level of MSP1a genetic diversity among geographic strains.
Results
In this study we showed that the phylogeography of A. marginale MSP1a sequences is associated with world ecological regions (ecoregions) resulting in different evolutionary pressures and thence MSP1a sequences. The results demonstrated that the MSP1a first (R1) and last (RL) repeats and microsatellite sequences were associated with world ecoregion clusters with specific and different environmental envelopes. The evolution of R1 repeat sequences was found to be under positive selection. It is hypothesized that the driving environmental factors regulating tick populations could act on the selection of different A. marginale MSP1a sequence lineages, associated to each ecoregion.
Conclusion
The results reported herein provided the first evidence that the evolution of A. marginale was linked to ecological traits affecting tick vector performance. These results suggested that some A. marginale strains have evolved under conditions that support pathogen biological transmission by R. microplus, under different ecological traits which affect performance of R. microplus populations. The evolution of other A. marginale strains may be linked to transmission by other tick species or to mechanical transmission in regions where R. microplus is currently eradicated. The information derived from this study is fundamental toward understanding the evolution of other vector-borne pathogens.
Background
The genus Anaplasma (Rickettsiales: Anaplasmataceae) contains obligate intracellular organisms found exclusively within membrane-bound inclusions or parasitophorous vacuoles in the cytoplasm of both vertebrate and invertebrate host cells [1,2]. The genus Anaplasma includes pathogens of ruminants, A. marginale, A. centrale, A. bovis, and A. ovis. Also included in this genus are A. phagocytophilum, which infects a wide range of hosts including humans and wild and domesticated animals, and A. platys that infects dogs.
To date, most research has been reported for A. marginale, the type species for the genus Anaplasma [3]. Both cattle and ticks develop persistent infections with A. marginale and therefore can serve as reservoirs of infection. A. marginale is transmitted horizontally by ixodid ticks including Rhipicephalus spp. and Demacentor spp. Rhipicephalus (Boophilus) microplus is considered the most important biological vector in tropical and subtropical regions of the world [4]. Transfer of infected blood by biting flies or blood-contaminated fomites effects mechanical transmission of A. marginale. The complex developmental cycle of A. marginale has been described and shown to be coordinated with the tick feeding cycle [3]. The midgut is the first site of infection, where membrane-bound vacuoles or colonies initially contain reticulated forms that divide by binary fission and subsequently transform into dense forms. Infection of salivary glands and other tissues then occurs which completes the developmental cycle and allows for transmission to susceptible hosts during tick feeding.
Vector-pathogen interactions involve traits from both the vector and the pathogen [5]. Several major surface proteins (MSPs) have been identified and characterized in A. marginale [3,5]. MSPs are involved in interactions with both vertebrate and invertebrate hosts [2,3,6-9], and therefore are likely to evolve more rapidly than other genes because they are subjected to selective pressures exerted by host immune systems. The MSP1a of A. marginale geographic strains differs in molecular weight due to a variable number of tandem 23-31 amino acid repeats, and the sequence of MSP1a has been shown to be a stable marker for identification of geographic strains [10,11]. Functionally, MSP1a was shown to be an adhesin for bovine erythrocytes and tick cells [12-14]. Tick molecules involved in vector-A. marginale interactions were recently identified and functionally characterized [15].
The geographic strains of A. marginale are highly variable, as demonstrated by the analysis of MSP1a sequences [7,11]. Such genetic heterogeneity observed among A. marginale strains in endemic regions could be explained by cattle movement and maintenance of different genotypes by independent transmission events, due to infection exclusion of A. marginale in cattle and ticks which commonly results in the establishment of only one genotype per animal [16-18]. Due to the high degree of sequence variation within most endemic areas, MSP1a sequences have failed to provide phylogeographic information on a global scale [7]. These studies also suggested that multiple introductions of A. marginale strains from different geographic locations had occurred in many regions.
The evolutionary history of vector-pathogen interactions can be reflected in the sequence variation of Anaplasma MSPs. Previous studies demonstrated that A. marginale MSP1a evolved under positive selection [19]. Analysis of A. marginale MSP1a repeats provided evidence of tick-pathogen co-evolution [5,6,20], a result that is consistent with the biological function of MSP1a in pathogen transmission by ticks [14]. However, the study of A. marginale evolutionary history and tick-pathogen co-evolution has remained elusive because of the extensive genetic diversity of MSP1a sequences.
In this study we analyzed MSP1a repeat and microsatellite sequences in order to provide information on the evolution of A. marginale strains by determining their phylogeographic association with world ecological regions (ecoregions) and by testing the effect of different evolutionary pressures associated with the tick vector, mainly R. microplus, ecology in these ecoregions.
Methods
Anaplasma marginale MSP1a repeat sequences
The MSP1a sequences included in this study were obtained from A. marginale strains collected worldwide from infected cattle and recently reviewed by de la Fuente et al. [11]. The data on MSP1a sequences was updated by searching the Genbank sequence database [21]. The amino acid sequence of the first (R1) and last (RL) MSP1a repeats of 111 A. marginale strains were used in this study, from which 39 and 28 unique R1 and RL sequences, respectively, were obtained. For comparison, the sequences of Rn MSP1a repeats, located between R1 and RL, were included in some analyses. MSP1a sequences not included in these studies were from A. marginale strains that were not adequately geo-referenced.
Anaplasma marginale MSP1a microsatellite sequences
A microsatellite was located in the MSP1a 5'UTR between the putative Shine-Dalgarno sequence (GTAGG; [6]) and the translation initiation codon (ATG). The structure of the microsatellite (bold) was GTAGG (G/A TTT)m (GT)n T ATG (Table 1). The microsatellite was sequenced in 115 A. marginale strains collected from infected cattle in the USA, Canada, Mexico, Puerto Rico, Argentina, Brazil, Italy, Israel, South Africa and China [11].
Table 1.
Structure and ecoregion cluster distribution of the A. marginale MSP1a microsatellites.
| Genotype | Number of strains | m | n | SD-ATG distance (nucleotides) | Genotype frequency per ecoregion cluster | |||
| 1 | 2 | 3 | 4 | |||||
| A | 2 | 1 | 7 | 19 | 0.00 | 1.00 | 0.00 | 0.00 |
| B | 5 | 1 | 9 | 23 | 1.00 | 0.00 | 0.00 | 0.00 |
| C | 7 | 2 | 5 | 19 | 0.57 | 0.14 | 0.29 | 0.00 |
| D | 3 | 2 | 6 | 21 | 0.33 | 0.67 | 0.00 | 0.00 |
| E | 12 | 2 | 7 | 23 | 0.75 | 0.00 | 0.25 | 0.00 |
| F | 3 | 3 | 4 | 21 | 0.00 | 0.00 | 1.00 | 0.00 |
| G | 78 | 3 | 5 | 23 | 0.15 | 0.14 | 0.56 | 0.14 |
| H | 3 | 3 | 6 | 25 | 0.00 | 0.67 | 0.33 | 0.00 |
| I | NI | 4 | 6 | 29 | --- | --- | --- | --- |
The MSP1a microsatellite sequences were analyzed in 115 A. marginale strains. The microsatellite (sequence in bold) was located between the Shine-Dalgarno (SD; sequence in brackets) and the translation initiation codon (ATG) with the structure: GTAGG (G/A TTT)m (GT)n T ATG. The SD-ATG distance was calculated in nucleotides as (4 × m) + (2 × n) + 1. Abbreviation: NI, not included in the study because the A. marginale strain was not adequately geo-referenced.
World ecoregions and association with A. marginale strains
Ecoregions are used herein to classify the world across dynamic environmental factors. We assumed that i) ecoregions could be delineated using quantitative abiotic characters based on well-recognized and repeatable attributes and ii) A. marginale strains are associated with each ecoregion and subjected to different environmental conditions that could be analyzed by multivariate geographic clustering [22]. Multivariate geographic clustering involves the use of standardized values for selected environmental conditions in a set of raster maps. Those values serve as coordinates in the environmental data space, in which environmental conditions are further clustered according to their similarities. The feature selected to put together the clusters was the monthly Normalized Difference Vegetation Index (NDVI). NDVI is a variable that reflects vegetation stress, a feature that summarizes information about the ecological background for tick populations [23]. We obtained a 0.1° resolution series of monthly NDVI data for the period 1986-2006. The 12 averaged monthly images were subjected to Principal Components Analysis (PCA) to obtain decomposition into the main axes representing the most significant, non-redundant information. The strongest principal axes were chosen using Cattell's Scree Test [22]. It has been found that the first principal component derived from NDVI typically represents the greenness of the surveyed area [24]. Component 2 is interpreted as a change component, taken to represent a winter/summer seasonality effect. Components 3 and 4 are also essentially seasonal, but represent areas where the timing of green-up is different from that in component 2. Our PCA analysis retained three principal axes, explaining the 92% of total variance. These three axes were related to the mean NDVI values, annual amplitude, and NDVI values in the period May to August, respectively. We then used a hierarchical agglomerative clustering on PCA values to classify multiple geographical areas into a single common set of discrete regions. Mahalanobis distance was used as a measure of dissimilarity and the weighted pair-group average was used as the amalgamation method. A value of 0.05 was used as the cut-off probability for assignment to a given ecoregion. All the procedures adhered to methods previously described [25].
The decision about the number of ecoregions to retain without any prior detail about the information they contain is a problem to which a solution has not yet been found. The main goal is to define unambiguously the A. marginale strains recorded mostly in a single ecoregion cluster and present in the highest number of geographical sites belonging to that cluster. The result is to refine the degree of clustering that gives the optimal degree of association between A. marginale strains ('species') and ecoregions ('sites'). This analysis was done using the 'indicator species' method [26], a previously published multivariate statistics procedure to define 'sites' as a function of their faunal composition ('species'). We began the agglomerative process described above with an unrealistic high number of ecoregions. At every step of the agglomerative process, pathogen strains and ecoregions were ordered by a correspondence analysis, and then analyzed using the 'indicator species' method. The procedure runs iteratively, trying to improve the association with further clustering of ecoregions. However, the method does not force a cluster if specifications of cut-off probabilities for ecoregions are violated, and does not assume any a priori condition about the geographical range of any cluster. The procedure is only an indicator that stops when an optimum degree of ecoregion clustering collectively explaining the association of A. marginale strains with the environment is reached. Such association may be low or high (in the range 0-1) but is the optimal according to the ecoregion features. After the use of these methods we retained a total of four ecoregion clusters (Figure 1) to which A. marginale sequences were unambiguously associated, and the rest were discarded because A. marginale strains were not isolated there.
Figure 1.
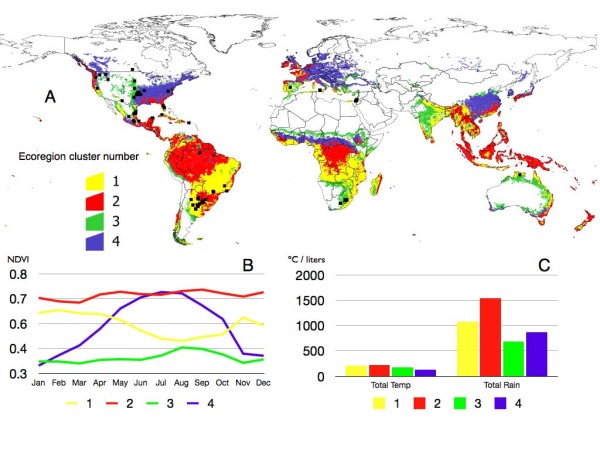
World ecoregions. (A) Clusters of vegetation features of the world computed through an unsupervised classification on Principal Components Analysis decomposition of monthly normalized difference vegetation index (NDVI) values at a resolution of 0.1°. Small islands were removed to improve the presentation. Only the ecoregions providing most information about A. marginale distribution according to a Discriminant Analysis were included, grouped in four main clusters and arbitrarily numbered. Overlying records of A. marginale are in black. (B) Monthly NDVI values detected for each ecoregion clusters (plotted with the same colors as in A). (C) Monthly accumulated temperature and rainfall recorded for each ecoregion cluster.
Analysis of A. marginale MSP1a repeat sequences
The conserved amino acid sequences of MSP1a R1 and RL repeats reported in A. marginale strains from each ecoregion cluster were used to determine the consensus sequence in the cluster. The distances between groups of sequences ascribed to an ecoregion cluster were then computed, both within and between clusters. The hypothesis herein is that highest affinities between R1 and RL isolates are observed when grouped according to the ecoregion to which they are related. An alternative hypothesis is that isolates cluster according to a geographical background. To check for that alternative hypothesis we examined genetic distances with strains arranged into geographical groups. We used only American strains to overcome the undersampling in Africa. They were grouped as Western USA, Eastern USA, Mesoamerica and the Caribbean, and South America, and then distances within and between geographical clusters computed again.
Other analyses involved calculation of the percentage of changes between the consensus MSP1a repeat sequences among different ecoregion clusters as a measure of the genetic distance between A. marginale strains in these clusters. Some R1 and RL sequences were found associated with more than one ecoregion cluster. Therefore, the percentage of changes in amino acid composition for every sequence and the consensus sequence in each cluster were computed as a measure of similarity of each sequence and consensus sequence for each cluster. MSP1a repeat sequences were aligned for pairwise comparison and determination of non-synonymous (dN) and synonymous (dS) substitutions using Mega 4 [27]. The dN and dS were determined among all pairwise comparisons of MSP1a repeat sequences within each ecoregion cluster, estimated by the method of Nei and Gojobori [28] with the correction for multiple substitutions [29]. The ratio of the mean dN/dS was used as an indicator of the level of selection acting on MSP1a repeat sequences.
As an additional test to verify our hypothesis, we performed a Mantel's test. The explanation of the distribution of the strains in terms of environmental variables may be confounded because the variables are intercorrelated among themselves, and so it may be difficult to ascribe causal mechanisms to the environmental variables. Mantel's test is a regression in which the variables are dissimilarity matrices. The operative question is 'do strains that have similar sequences also tend to be similar in terms of the environmental variables?' Therefore we performed a Mantel's test both between a dependent distance matrix (genetic similarities of R1 and RL) and the predictor matrix of geographical distances among strains. A second Mantel's test was done with the same dependent matrix and a predictor dissimilarity matrix based on environmental PCA-derived values. Such correlations will indicate if locations that are closer or locations that are similar environmentally are similar compositionally. Mantel's tests were performed using the Jackard index according to [30].
Analysis of A. marginale MSP1a microsatellite sequences
The extent of genetic differentiation of A. marginale strains at the MSP1a microsatellite was assessed within and among ecoregion clusters using an analysis of molecular variance (AMOVA; [31]) and pairwise population FST significance tests as implemented in ARLEQUIN, version 3.01 [32]. The statistical significance of fixation indices was tested using a non-parametric permutation approach [33] with 20,000 permutations. Ecoregion clusters for which statistically significant subdivision was not detected were pooled to define groups.
The effect of microsatellite size on MSP1a expression was characterized in A. marginale strains Wetumka (OK; Genbank accession number AY010247), Okeechobee (FL; AY010244), Idaho (ID; M32868) and HB-A8 (China; DQ811774) sequences. These strains had microsatellite genotypes G (Wetumka and Okeechobee; distance SD-ATG = 23 nucleotides), C (Idaho; SD-ATG = 19 nucleotides) and I (China; SD-ATG = 29 nucleotides). The msp1alpha gene containing promoter sequences active in Escherichia coli [6] was amplified using oligonucleotide primers MSP1aP: 5'GCATTACAACGCAACGCTTGAG3' and MSP1a3: 5'GCTTTACGCCGCCGCCTGCGCC3' and cloned into pGEM-T vector (Promega, Madison, WI, USA) as reported previously [20]. Three independent clones for each of the MSP1a constructs were transformed in E. coli JM109 cells and grown for 15 to 20 hours at 37°C. Culture volumes of 3 ml were used for RNA and DNA extraction using TriReagent (Sigma, St. Louis, MO, USA) according to manufacturer's instructions. The RNA samples were treated with RNase-free DNase (Invitrogen, Carlsbad, CA, USA) prior to RT-PCR. MSP1a mRNA levels were characterized by real-time RT-PCR using oligonucleotide primers MSP1RT5: 5'ACCAATCGTTGGCAGAAGAG3' and MSP1RT3: 5'ACCTGCTCCCAAAGTAGCAA3' and normalizing against E. coli D-1-deoxyxylulose 5-phosphate synthase gene (dxs) [34] and plasmid DNA copy number by msp1alpha PCR using the oligonucleotide primers and conditions described above. Real-time RT-PCR was conducted using the iScript One-Step RT-PCR Kit with SYBR Green and an iQ5 thermal cycler (Bio-Rad, Hercules, CA, USA). Control reactions were performed using the same procedures but without reverse transcriptase to test for DNA contamination in the RNA preparations and without RNA added to detect contamination of the PCR reaction. The normalized mRNA levels were compared between different MSP1a constructs using an ANOVA test (P = 0.05).
Results
Anaplasma marginale MSP1a repeat sequences show ecoregion-specific signatures
Ecoregion clusters showed different NDVI, temperature and rainfall values (Figure 1). Ecoregion cluster 1 extended over large areas of central Africa and central South America, primarily Argentina and southern Brazil. It involved a region with medium to high NDVI values with a clear seasonal decrease between June and September. This was the ecoregion with the highest recorded temperature and around 1,000 mm of annual rainfall. Ecoregion cluster 2 included vast areas of the Mesoamerican corridor, northern South America and a small territory of eastern South Africa. It consisted of zones with high NDVI along the year without seasonal variability, temperature values similar to those in ecoregion cluster 1 and rainfall around 1,500 mm/year. Ecoregion cluster 3 extended over central South Africa and scattered parts of southern USA and Mexico, with the lowest NDVI values and little change across the year. This ecoregion displayed lower temperature values and the minimum rainfall. Finally, ecoregion cluster 4 extended over large areas of USA and had a clear NDVI signature, very low between November and March and then rising to reach maximum levels around July. This area was the coldest among all the ecoregion clusters and rainfall was around 800 mm/year.
Figures 2 and 3 display the association of the A. marginale strains with the four ecoregion clusters. These figures are plotted according to the values of the first two axes derived from PCA on NDVI time series. Figures plot the 80% confidence ellipses of the annual mean NDVI and the seasonal variation of NDVI for each ecoregion cluster, as well as the plot of the isolates in the NDVI envelope. Analysis showed that 77% of MSP1a R1 unique sequences were associated with only one ecoregion cluster (Figure 2). Ten R1 unique sequences (25.6% of the total number of R1 sequences) were reported exclusively in ecoregion cluster 1 and they shared 16 out of 31 amino acids (51.6% of the total number of amino acids; Table 2). Six R1 unique sequences (12.8%) were reported solely in ecoregion cluster 2 with 64.5% identical amino acids. Twelve R1 unique sequences (30.7%) were found only in ecoregion cluster 3, sharing 64.5% of their amino acids. Only three R1 sequences were exclusively associated with ecoregion cluster 4, with 77.4% identical amino acids. All of the A. marginale MSP1a R1 sequences within each ecoregion cluster appeared to be under positive selection as shown by dN/dS indexes of 1.83, 1.61, 1.54 and 1.21 for ecoregion clusters 1 to 4, respectively.
Figure 2.
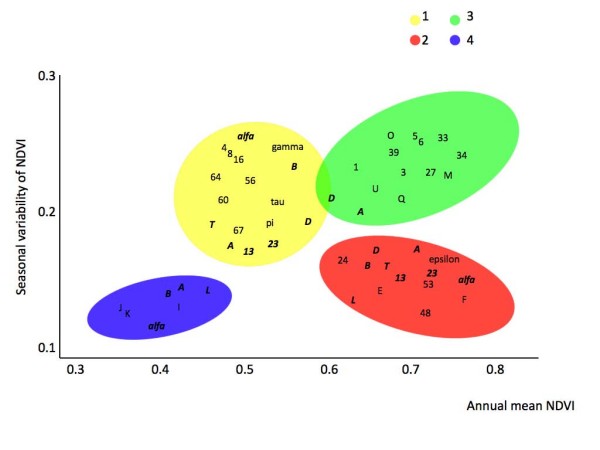
Associations between A. marginale MSP1a R1 repeat sequences and ecoregion clusters, plotted along the values of the first two axes derived from Principal Components Analysis on normalized difference vegetation index (NDVI) time series. Figure plots the 80% confidence ellipses of the annual mean normalized difference vegetation index (NDVI) and the seasonal variation of NDVI for each ecoregion cluster, as well as the plot of the isolates in the NDVI envelope. Each letter displays the mean position of the records for that strain. Unique sequences for each ecoregion are displayed in plain type. Sequences recorded in more than one ecoregion cluster are displayed in italic bold.
Figure 3.
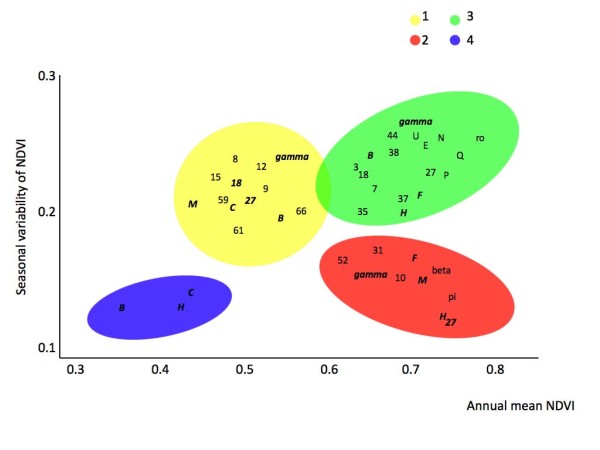
Associations between A. marginale MSP1a RL repeat sequences and ecoregion clusters, plotted along the values of the first two axes derived from Principal Components Analysis on normalized difference vegetation index (NDVI) time series. Figure plots the 80% confidence ellipses of the annual mean normalized difference vegetation index (NDVI) and the seasonal variation of NDVI, for each ecoregion cluster, as well as the plot of the isolates in the NDVI envelope. Each letter displays the mean position of the records for that strain. Unique sequences for each ecoregion are displayed in plain type. Sequences recorded in more than one ecoregion cluster are displayed in italic bold.
Table 2.
MSP1a R1 and RL repeat sequences of unique sequences unambiguously associated to only one ecoregion cluster, including the consensus sequence of the isolates of that cluster.
| MSP1a Repeat | Unique sequences | Consensus sequence | Other strains and number of substitutions |
| R1/Ecoregion 1 | 4, 8, 16, 56, 60, 64, 67, gamma, pi, tau | ***SSA***QQ*SSV*S*S**AS*SSQ*G-- | A(0), B(0), D(1), T(0), 13(1), 23(1), alfa(0) |
| R1/Ecoregion 2 | 28, 48, 53, E, F, epsilon | ***SS**GQQQESSV***S*-ASTSSQLG-- | A(0), B(0), L(1), T(7),13(1), 23(1), alfa(0) |
| R1/Ecoregion 3 | 1, 3, 5, 6, 27, 33, 34, 39, M, O, Q, U | **SSSA*GQQQESSV*****QA*TSSQLG-- | A(0), D(0) |
| R1/Ecoregion 4 | I, J, K | *D*S*A*GQQQESSVSSQS*QASTSSQLG-- | A(0), B(0), L(0), alfa(0) |
| RL/Ecoregion 1 | 8, 9, 12, 15, 59, 61, 66 | ***SSA**QQQES*V*SQS**ASTSSQ*G-- | B(0), C(0), M(0), 18(0), 27(0), gamma(0) |
| RL/Ecoregion 2 | 10, 31, 52, pi, beta | *DSSSA**QQQ*S*V*S*S*-ASTSSQLG-- | F(0), H(0), M(0), 27(0), gamma(0) |
| RL/Ecoregion 3 | 3, 7, 35, 37, 38, 44, E, N, P, Q, U, ro | *DSSSAS*QQQESS**S*S*QA**S*Q*G-- | B(1), F(0), H,18(0), gamma(0) |
| RL/Ecoregion 4 | none | B, C, H | |
Other strains recorded from more than one ecoregion cluster are included in the last column, stating the number of substitutions in their amino acid sequences as compared with the consensus sequence in that ecoregion cluster (in parentheses).
Differences were found among the R1 sequences at each ecoregion cluster. Comparison of consensus sequences between clusters 1 and 2 revealed 22.5% of amino acid differences. Ecoregion clusters 1 and 3 differed by 25% of their consensus sequences while ecoregion clusters 2 and 3 had only 19% of different amino acids in their R1 sequences. Five R1 sequences, T, 13, 23, D and L were found simultaneously in two of the ecoregion clusters (Figure 2, Table 2). Details of their similitude with the consensus sequences of each ecoregion cluster are included in Table 2. Three R1 sequences, A, B and alpha appeared associated with different ecoregions. Pairwise comparisons between A. marginale R1 sequences demonstrated that genetic distances were lower within than between ecoregion clusters (Table 3). However, when strains were compared as clustered geographically, intra-cluster distances were much higher, revealing a clear heterogeneity between geographic strains and rejecting the hypothesis of a pure geographical association. Mantel's test on R1 sequences was 0.82 (P < 0.001) when applied to ecoregion clusters using only unique sequences. Mantel's test on R1 sequences dropped to 0.72 (P < 0.005) when every sequence in the ecoregion clusters was included. The same test provided a value of 0.31 (P = 0.145) for the distances matrix based on geographical association of strains.
Table 3.
Top table shows the genetic distances among and between the MSP1a R1 repeat sequences reported on each ecoregion cluster.
| Ecoregion | Cluster 1 | Cluster 2 | Cluster 3 | Cluster 4 |
| Cluster 1 | 0.212 | 0.311 | 0.319 | 0.337 |
| Cluster 2 | 0.199 | 0.321 | 0.371 | |
| Cluster 3 | 0.175 | 0.214 | ||
| Cluster 4 | 0.156 | |||
| Geographical cluster | Western USA | Eastern USA | Mesoamerican+ Caribbean | South America |
| Western USA | 0.418 | 0.399 | 0.514 | 0.519 |
| Eastern USA | 0.329 | 0.388 | 0.501 | |
| Mesoamerican+ | 0.454 | 0.455 | ||
| Caribbean | ||||
| South America | 0.521 |
Bottom table displays the genetic distances among and between the MSP1a R1 repeat sequences clustered on a geographical basis.
Numbers in bold represent the distance between the MSP1a R1 repeat sequences contained within each ecoregion cluster.
Discriminant analysis showed that 74.8% of MSP1a RL sequences were found associated with one ecoregion cluster only (Figure 3, Table 2). Seven RL sequences (25% of the total number of RL sequences) were reported exclusively in ecoregion cluster 1, with 61.2% identical amino acids. Five RL sequences (17.8%) were reported solely in ecoregion cluster 2 with 64.5% identical amino acids. Twelve RL sequences (32%) were found only in ecoregion cluster 3, sharing 61.2% of their amino acids. No RL unique sequences were found in ecoregion cluster 4. The differences in RL consensus sequences between ecoregion clusters 1 and 2, 2 and 3, and 1 and 3 were 16%, 26% and 32%, respectively. In contrast to A. marginale MSP1a R1 repeat sequences, the RL sequences within each ecoregion cluster did not appear to be under positive selection as shown by dN/dS indexes of 0.89, 0.79 and 0.77 for ecoregion clusters 1 to 3, respectively. Mantel's test on RL sequences was 0.87 (P < 0.001) when applied to ecoregion clusters using only unique sequences. This value dropped to 0.82 (P < 0.001) when every strain in the ecoregion clusters was included in the analysis. The same test provided a value of 0.27 (P = 0.208) for the distances matrix based on geographical association of strains. When the MSP1a Rn repeats, located between R1 and RL were analyzed, a total of 30, 9, 17 and 3 sequences were ascribed to ecoregion clusters 1 to 4, respectively. The Rn consensus sequences contained 14 (45.2%), 17 (54.9%), 17 (54.9%) and 24 (77.4%) identical amino acids in ecoregions clusters 1 to 4, respectively. The number of R1, Rn and RL repeat sequences varied between ecoregion clusters without a clear pattern.
Anaplasma marginale MSP1a microsatellite sequences have ecoregion-specific signatures and affect gene expression in Escherichia coli
The analysis of the MSP1a microsatellite sequences resulted in nine different genotypes among A. marginale strains (Table 1). The different microsatellite sequences produced SD-ATG distances of between 19 and 29 nucleotides, but the predominant distance was 23 nucleotides in all regions (Table 1).
The analysis of MSP1a microsatellite sequences showed phylogeographic clustering of some genotypes (Table 1). However, only three of the eight microsatellite genotypes included in the analysis were exclusive to a particular ecoregion cluster: genotype A to ecoregion cluster 2, genoytpe B to ecoregion cluster 1 and genotype F to ecoregion cluster 3. A significant genetic differentiation was found between ecoregion clusters for MSP1a microsatellite genotype frequencies, with overall FST = 0.16 (P < 0.0001). The ecoregion clusters 1 and 2 were clearly distinct from the other ecoregion clusters, and the largest FST value (0.28) was found between ecoregion clusters 1 and 4 while significant genetic differences were not found between ecoregion clusters 3 and 4 (Table 4). To control for potential confounding effects of geography, we investigated genetic differentiation between broad geographic regions. This showed that genetic differentiation is mostly dependent on ecoregion-based clustering of the strains, with the highest FST value being 0.07 for South America and the Mediterranean (see Table 4). The AMOVA showed that 84% of the variance accounted for within-ecoregion cluster differences, while a geographic-based AMOVA showed that within-population variation explained less (73%) of the variance (Table 5).
Table 4.
Ecoregion and geographic cluster pairwise FST significance tests (bottom half of each section) and P-values (top half of each section) of A. marginale MSP1a microsatellites.
| Ecological | Ecoregion 1 | Ecoregion 2 | Ecoregion 3 | Ecoregion 4 |
| Ecoregion 1 | - | 0.019 ± 0.001 | 0.000 ± 0.000 | 0.001 ± 0.000 |
| Ecoregion 2 | 0.09 | - | 0.027 ± 0.001 | 0.032 ± 0.001 |
| Ecoregion 3 | 0.22 | 0.07 | - | 0.312 ± 0.003 |
| Ecoregion 4 | 0.28 | 0.15 | 0.016 (NS) | - |
| Geographical | North America | South America | Mediterranean | South Africa |
| North America | - | 0.324 ± 0.003 | 0.016 ± 0.001 | 0.232 ± 0.003 |
| South America | 0.004 (NS) | - | 0.036 ± 0.001 | 0.452 ± 0.003 |
| Mediterranean | 0.06 | 0.07 | - | 0.101 ± 0.002 |
| South Africa | 0.01 (NS) | -0.01 (NS) | 0.07 (NS) | - |
Abbreviation: NS, not significant.
Table 5.
Hierarchical analysis of molecular variance (AMOVA) for MSP1a microsatellites.
| Source of variation | d. f. | Sum of squares | Variance components | Percent variation |
| Ecological | ||||
| Among ecoregions | 3 | 3.99 | 0.04 | 16.31 |
| Within ecoregions | 109 | 24.49 | 0.22 | 83.69 |
| Total | 112 | 28.48 | 0.27 | --- |
| Geographic | ||||
| Among continents | 2 | 1.16 | -0.08 | -35.70 |
| Among populations within continents | 6 | 8.76 | 0.15 | 63.18 |
| Within populations | 104 | 18.57 | 0.18 | 72.52 |
| Total | 112 | 28.48 | 0.25 | --- |
Table shows Wright's fixation index, FST. Statistical significance was calculated from 20,000 random permutations. Fixation indices and P-values: (Ecological) FST = 0.16 (P = 0.00 ± 0.00), (Geographic) FST = 0.29 (P = 0.00 ± 0.00).
The effect of MSP1a microsatellite size on gene expression was analyzed using the sequence derived from Wetumka, Okeechobee, Idaho and HB-A8 A. marginale strains (Table 6). The results showed that MSP1a expression was lower in the construct containing the Idaho strain-derived MSP1a sequence with the lowest SD-ATG distance of 19 nucleotides, while differences were not observed between constructs containing the MSP1a sequences from Wetumka, Okeechobee and HB-A8 A. marginale strains with SD-ATG distances of 23 and 29 nucleotides (Table 6).
Table 6.
Effect of microsatellite genotype on the expression of A. marginale MSP1a in E. coli.
| A. marginale strain | Microsatellite genotype | SD-ATG distance (nucleotides) | Normalized MSP1a mRNA levels (arbitrary units) |
| Idaho, ID | C | 19 | 1.57 ± 0.29 (a) |
| Wetumka, OK | G | 23 | 2.31 ± 0.27 (b) |
| Okeechobee, FL | G | 23 | 2.13 ± 0.0 (b) |
| HB-A8, China | I | 29 | 2.39 ± 0.19 (b) |
MSP1a mRNA levels were analyzed by real-time RT-PCR in three independent clones for each of the MSP1a constructs transformed in E. coli JM109. MSP1a mRNA levels were normalized against E. coli dxs gene and plasmid DNA copy number by msp1α PCR. Normalyzed MSP1a mRNA levels were represented in arbitrary units as average ± SD and compared between different constructs using an ANOVA test (different letters denote significant differences; P < 0.02).
Discussion
Vector-borne pathogens have evolved molecular mechanisms of vector-pathogen interactions that involve genetic traits of both the vector and the pathogen [5,35]. For the tick-borne pathogen A. marginale, recent studies have characterized tick and pathogen-derived genes that are involved in tick-pathogen interactions [2,3,6-9,15]. Phylogenetic studies using tick and/or pathogen-derived genetic markers have contributed to our understanding of the evolution of A. marginale strains and tick-pathogen relationships [5,6,20]. However, the impact of these studies has been limited by the genetic diversity of genes involved in tick-pathogen interactions and thus are likely to reflect pathogen evolution and tick-pathogen relationships [5].
In this study we took a different approach to characterize the evolution of A. marginale strains. This is the first study to use remotely sensed vegetation features as a surrogate of an environmental envelope to which genetic variability and structure of a single pathogen is associated. Biogeographic research seeks to identify the processes structuring organism diversity at a variety of geographic and taxonomic scales [36]. Remote sensing is being used increasingly as a tool to discover ecological traits through definite signatures. NDVI is a measure of the vegetation stress, thus a time series of NDVI values over a region reflects the seasonal cycle of vegetation as a surrogate of the seasonal variation in climate. NDVI and other climate features are commonly used to detect ecologically suitable areas for some pathogens and their vectors [37-39]. For example, Randolph and Rogers [40] indicated that climate has directed and constrained the evolution of flaviviruses of the TBE group. Six flaviviruses (SSEV, WTBEV, Russian TBEV, OHFV, and Kyasanur forest virus) fall within a distinct eco-climatic space defined by factors derived from thermal and moisture conditions. Herein we showed that A. marginale MSP1a R1 repeats evolved under positive selection, were associated with specific ecoregion clusters and were not arranged according to geographical features. The different evolutionary pressures operating over different MSP1a repeats was demonstrated previously [6], but the possibility of using the MSP1a R1 repeat as a biogeographical marker has only been suggested [5]. In contrast, MSP1a RL repeat sequences, while still linked to a similar set of ecoregion clusters, did not evolve under positive selection. Consequently, RL repeat sequences were not good genetic markers for the characterization of A. marginale biogeography and evolution.
Analysis of MSP1a microsatellite sequences demonstrated that A. marginale strains are associated with specific ecoregion clusters, thus corroborating the results obtained with repeat sequences. These results may have a functional significance. It has been shown that the SD-ATG distance between the ribosome binding site (Shine-Dalgarno sequence) and the translation initiation codon affects gene expression in prokaryotes [41]. Little is known about the regulation of gene expression in A. marginale [6]. However, as shown here in E. coli, the length of the MSP1a microsatellite could affect the expression of MSP1a, which varies during A. marginale multiplication in both tick cells and bovine erythrocytes, thus affecting pathogen infection and transmission [42]. Since MSP1a repeats and microsatellites are unambiguously associated to ecoregion clusters, these results suggested a new factor that may affect the efficiency by which different A. marginale strains are transmitted under different environmental conditions.
A. marginale is an obligate intracellular parasite, which alternates between the tick vector and the vertebrate host. Our hypothesis was that the link between pathogen strains and definite portions of the environmental envelope could reflect the effects of climate on the tick vector. Temperature and rainfall, which are indirectly captured by the specific signatures of NDVI, are the main factors affecting the ecology and population dynamics of tick species [43] and these operate at critical levels of selection of tick populations, selecting also specific strains of the pathogen. This framework is further obscured by the 'noise' produced by invasive events of the pathogen (by cattle movement or other factors), or by selection of strains transmitted mechanically in areas where ticks are eradicated by acaricide application, contributing to the absence of total consistence between ecoregion clusters and strains.
Adequate reports exist about distribution, seasonal dynamics and abundance of R. microplus populations in the study area, allowing a direct comparison with results presented herein. Ecoregion cluster 1 contained the R1 repeats with the lowest percentage of conserved amino acids and the highest positive selection pressure, in areas with high temperature and medium rainfall. R. microplus ticks are common in these areas and a strict seasonality in tick population dynamics has been reported, allowing for a high selection of tick populations due to winter mortality [44]. Ecoregion cluster 2 contained sites with constant high temperature and rainfall. In these sites, R. microplus ticks are abundant throughout the year without marked seasonality and climate is not a limiting factor in tick mortality [44,45]. In ecoregion cluster 3, tick populations suffer drastic limitations in effectiveness because of low and inadequate rainfall [46], and this high selection pressure on tick populations might be adverse for pathogen transmission and selection. The R1 repeat sequences in ecoregion clusters 2 and 3 had higher number of conserved amino acids and lower positive selection pressure when compared with R1 sequences in ecoregion cluster 1. Finally, the R1 repeat sequences ascribed to ecoregion cluster 4 had the highest percentage of conserved amino acids and the lowest positive selection pressure, recorded only in sites where R. microplus ticks are absent because of the low yearly temperature and thus other tick species act as vectors of A. marginale [18]. The analysis of MSP1a microsatellite sequences also supported differences among all ecoregion clusters, except for ecoregion clusters 3 and 4 where R. microplus has low prevalence or is absent.
Some R1 repeat sequences such as A, B, D and alpha as well as microsatellite genotypes C-D, G and H were present across several ecoregion clusters. These sequences appeared in A. marginale strains collected in zones where R. microplus ticks are common (ecoregion clusters 1 and 2) and in sites, such as central Argentina and southern parts of the USA, where R. microplus has been prevalent in the past but has been eradicated [46]. Additionally, these sequences were also found in sites where other tick vectors such as Dermacentor spp. are prevalent [18]. These R1 repeats and microsatellite sequences could have evolved from ancestor pathogen strains transmitted by R. microplus as the main vector, and then evolved under lower selection pressure, due to pathogen transmission by other tick species or mechanically. The presence of these sequences in sites where R. microplus has been historically absent (that is, north-western USA) and now adapted to transmission by Dermacentor spp. ticks, could be interpreted as invasive events. The results reported here showed that lowest selection pressure exist in sites where Dermacentor spp. ticks are the main biological vectors or where mechanical transmission is predominant because of eradication of R. microplus. Therefore, R1 repeats are evolving under high selection pressure only in sites where R. microplus is the main vector and is subjected to selection because of climate constraints. This hypothesis did not explain the absence of MSP1a genetic diversity in Australia. Analysis of four A. marginale strains in Australia revealed the presence of a single repeat type 8 [11]. We would expect evolution of A. marginale MSP1a towards different repeat sequences, sharing the consensus sequence found in ecoregion cluster 1, into which R1 type 8 is ascribed, even in the case of a single invasive event. Reasons accounting for such a lack of diversity are currently unknown, but the combined pressure exerted by tick population structure [47,48] the A. centrale vaccine, acaricide treatments and cattle movement for pathogen and tick control may have impacted on A. marginale genetic diversity in Australia [48].
A. marginale exclusively infects cattle and wild ruminants [2]. Such high host specificity may results in a relatively low impact of vertebrate host factors on the evolution of A. marginale strains, thus leaving tick-pathogen interactions as the main contributing factor affecting its biogeography and evolutionary history. However, as previously discussed, cattle movement may have contributed to the genetic diversity of A. marginale strains worldwide [11]. Nevertheless, the results reported herein may be relevant in studying the evolution of other vector-borne pathogens. Many vector-borne pathogens, such as some Babesia, Theileria, Rickettsia, Ehrlichia and Plasmodium species, are also highly host-specific [49] and vector-pathogen interactions may play a crucial role in their evolution and biogeography [35].
Conclusion
The results reported herein provided the first evidence that the evolution of A. marginale MSP1a repeat and microsatellite sequences was linked to environmental traits, and that strains are not geographically related. Different evolutionary pressures acting on A. marginale were found associated with zones where climate and rainfall affect presence, abundance and dynamics of vector populations. We hypothesized that some A. marginale strains evolved under conditions of pathogen biological transmission by R. microplus, while others may be linked to transmission by other tick species or to mechanical transmission in regions where R. microplus is currently absent. The procedures outlined herein could be fundamental toward studying the evolution of other vector-borne pathogens.
Authors' contributions
AEP, JF and AJM conceived and designed the study and conducted MSP1a repeat sequence analysis. AEP conducted ecoregion analysis and clustering. VN, KAW and JF conducted MSP1a expression and microsatellite sequence analyses. AEP, KAW KMK, and JF contributed to drafting the manuscript. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
This research was supported by grants from the Ministerio de Ciencia e Innovación, Spain (project BFU2008-01244/BMC), the CSIC intramural project 200830I249 to JF and the Oklahoma Agricultural Experiment Station (project 1669) and was facilitated through the Integrated Consortium on Ticks and Tick-borne Diseases (ICTTD-3), financed by the International Cooperation Program of the European Union, coordination action project No. 510561. VN was funded by the European Social Fund and the Junta de Comunidades de Castilla-La Mancha (Program FSE 2007-2013), Spain.
Contributor Information
Agustín Estrada-Peña, Email: aestrada@unizar.es.
Victoria Naranjo, Email: MVictoria.Naranjo@uclm.es.
Karina Acevedo-Whitehouse, Email: Karina.Acevedo-Whitehouse@ioz.ac.uk.
Atilio J Mangold, Email: AMangold@rafaela.inta.gov.ar.
Katherine M Kocan, Email: Katherine.Kocan@okstate.edu.
José de la Fuente, Email: jose_delafuente@yahoo.com.
References
- Dumler JS, Barbet AC, Bekker CPJ, Dasch GA, Palmer GH, Ray SC, Rikihisa Y, Rurangirwa FR. Reorganization of the genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions subjective synonyms of Ehrlichia phagocytophila. Int J Sys Evol Microbiol. 2001;51:2145–2165. doi: 10.1099/00207713-51-6-2145. [DOI] [PubMed] [Google Scholar]
- Kocan KM, de la Fuente J, Blouin EF, Garcia-Garcia JC. Anaplasma marginale (Rickettsiales: Anaplasmataceae): recent advances in defining host-pathogen adaptations of a tick-borne rickettsia. Parasitol. 2004;129:S285–S300. doi: 10.1017/S0031182003004700. [DOI] [PubMed] [Google Scholar]
- Kocan KM, de la Fuente J, Blouin EF. Advances toward understanding the molecular biology of the Anaplasma-tick interface. Front Biosci. 2008;13:7032–7045. doi: 10.2741/3208. [DOI] [PubMed] [Google Scholar]
- Estrada-Peña A, Bouattour A, Camicas JL, Guglielmone A, Horak I, Jongejan F, Latif A, Pegram R, Walker AR. The known distribution and ecological preferences of the tick subgenus Boophilus (Acari: Ixodidae) in Africa and Latin America. Exp Appl Acarol. 2006;38:219–235. doi: 10.1007/s10493-006-0003-5. [DOI] [PubMed] [Google Scholar]
- de la Fuente J, Kocan KM, Blouin EF, Zivkovic Z, Naranjo V, Almazán C, Esteves E, Jongejan J, Daffre S, Mangold AJ. Functional genomics and evolution of tick-Anaplasma interactions and vaccine development. Vet Parasitol. 2009. [DOI] [PubMed]
- de la Fuente J, Garcia-Garcia JC, Blouin EF, Rodríguez SD, Garcia MA, Kocan KM. Evolution and function of tandem repeats in the major surface protein 1a of the ehrlichial pathogen Anaplasma marginale. Anim Health Res Rev. 2001;2:163–173. [PubMed] [Google Scholar]
- de la Fuente J, Lew A, Lutz H, Meli ML, Hofmann-Lehmann R, Shkap V, Molad T, Mangold AJ, Almazán C, Naranjo V, Gortázar C, Torina A, Caracappa S, García-Pérez AL, Barral M, Oporto B, Ceci L, Carelli G, Blouin EF, Kocan KM. Genetic diversity of Anaplasma species major surface proteins and implications for anaplasmosis serodiagnosis and vaccine development. Anim Health Res Rev. 2005;6:75–89. doi: 10.1079/AHR2005104. [DOI] [PubMed] [Google Scholar]
- Brayton KA, Kappmeyer LS, Herndon DR, Dark MJ, Tibbals DL, Palmer GH, McGuire TC, Knowles DP., Jr Complete genome sequencing of Anaplasma marginale reveals that the surface is skewed to two superfamilies of outer membrane proteins. Proc Natl Acad Sci USA. 2005;102:844–849. doi: 10.1073/pnas.0406656102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning Hotopp JC, Lin M, Madupu R, Crabtree J, Angiuoli SV, Eisen J, Seshadri R, Ren Q, Wu M, Utterback TR, Smith S, Lewis M, Khouri H, Zhang C, Niu H, Lin Q, Ohashi N, Zhi N, Nelson W, Brinkac LM, Dodson RJ, Rosovitz MJ, Sundaram J, Daugherty SC, Davidsen T, Durkin AS, Gwinn M, Haft DH, Selengut JD, Sullivan SA, et al. Comparative genomics of emerging human ehrlichiosis agents. PLoS Genet. 2006;2:e21. doi: 10.1371/journal.pgen.0020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allred DR, McGuire TC, Palmer GH, Leib SR, Harkins TM, McElwain TF, Barbet AF. Molecular basis for surface antigen size polymorphisms and conservation of a neutralization-sensitive epitope in Anaplasma marginale. Proc Natl Acad Sci USA. 1990;87:3220–3224. doi: 10.1073/pnas.87.8.3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente J, Ruybal P, Mtshali MS, Naranjo V, Shuqing L, Mangold AJ, Rodríguez SD, Jiménez R, Vicente J, Moretta R, Torina A, Almazán C, Mbati PM, Torioni de Echaide S, Farber M, Rosario-Cruz R, Gortazar C, Kocan KM. Analysis of world strains of Anaplasma marginale using major surface protein 1a repeat sequences. Vet Microbiol. 2007;119:382–390. doi: 10.1016/j.vetmic.2006.09.015. [DOI] [PubMed] [Google Scholar]
- McGarey DJ, Allred DR. Characterization of hemagglutinating components on the Anaplasma marginale initial body surface and identification of possible adhesins. Infect Immun. 1994;62:4587–4593. doi: 10.1128/iai.62.10.4587-4593.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarey DJ, Barbet AF, Palmer GH, McGuire TC, Allred DR. Putative adhesins of Anaplasma marginale: major surface polypeptides 1a and 1b. Infect Immun. 1994;62:4594–4601. doi: 10.1128/iai.62.10.4594-4601.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente J, Garcia-Garcia JC, Blouin EF, Kocan KM. Differential adhesion of major surface proteins 1a and 1b of the ehrlichial cattle pathogen Anaplasma marginale to bovine erythrocytes and tick cells. Int J Parasitol. 2001;31:145–153. doi: 10.1016/S0020-7519(00)00162-4. [DOI] [PubMed] [Google Scholar]
- de la Fuente J, Blouin EF, Manzano-Roman R, Naranjo V, Almazán C, Pérez de la Lastra JM, Zivkovic Z, Jongejan F, Kocan KM. Functional genomic studies of tick cells in response to infection with the cattle pathogen, Anaplasma marginale. Genomics. 2007;90:712–722. doi: 10.1016/j.ygeno.2007.08.009. [DOI] [PubMed] [Google Scholar]
- de la Fuente J, García-García JC, Blouin EF, Saliki JT, Kocan KM. Infection of tick cells and bovine erythrocytes with one genotype of the intracellular ehrlichia Anaplasma marginale excludes infection with other genotypes. Clin Diagn Lab Immunol. 2002;9:658–668. doi: 10.1128/CDLI.9.3.658-668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente J, Blouin EF, Kocan KM. Infection exclusion of the rickettsial pathogen, Anaplasma marginale, in the tick vector, Dermacentor variabilis. Clin Diagn Lab Immunol. 2003;10:182–184. doi: 10.1128/CDLI.10.1.182-184.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer GH, Knowles DP, Jr, Rodriguez JL, Gnad DP, Hollis LC, Marston T, Brayton KA. Stochastic transmission of multiple genotypically distinct Anaplasma marginale strains in a herd with high prevalence of Anaplasma infection. J Clin Microbiol. 2004;42:5381–5384. doi: 10.1128/JCM.42.11.5381-5384.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente J, Bussche RA Van Den, Prado T, Kocan KM. Anaplasma marginale major surface protein 1α genotypes evolved under positive selection pressure but are not markers for geographic strains. J Clin Microbiol. 2003;41:1609–1616. doi: 10.1128/JCM.41.4.1609-1616.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente J, Bussche RA Van Den, Kocan KM. Molecular phylogeny and biogeography of North American strains of Anaplasma marginale (Rickettsiaceae: Ehrlichieae) Vet Parasitol. 2001;97:65–76. doi: 10.1016/S0304-4017(01)00378-8. [DOI] [PubMed] [Google Scholar]
- Genbank sequence database http://www.ncbi.nlm.nih.gov/
- Hargrove WW, Hoffman FM. Potential of multivariate quantitative methods for delineation and visualization of ecoregions. Environ Manage. 2004;34:S39–S60. doi: 10.1007/s00267-003-1084-0. [DOI] [PubMed] [Google Scholar]
- Estrada-Peña A, Thuiller W. An assessment of the effect of data partitioning on the performance of modelling algorithms for habitat suitability for ticks. Med Vet Entomol. 2008;22:248–257. doi: 10.1111/j.1365-2915.2008.00745.x. [DOI] [PubMed] [Google Scholar]
- Eastman JR, Fulk M. Long sequence time series evaluation using standardized principal components. Photogrammetric Engineer Rem Sens. 1993;59:991–996. [Google Scholar]
- De'Ath G. Multivariate regression trees: a new technique for modeling species-environment relationships. Ecology. 2002;83:1105–1007. [Google Scholar]
- Dufrêne M, Legendre P. Species assemblages and indicator species: the need for a flexible asymmetrical approach. Ecol Monogr. 1997;67:345–366. [Google Scholar]
- Kumar S, Dudley J, Nei M, Tamura K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform. 2008;9:299–306. doi: 10.1093/bib/bbn017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Jukes TH, Cantor CR. Evolution of protein molecules. In: Munro HN, editor. Mammalian protein metabolism. New York, Academic Press, Inc; 1969. pp. 21–132. [Google Scholar]
- Legendre P, Fortin M. Spatial pattern and ecological analysis. Vegetation. 1989;80:107–138. doi: 10.1007/BF00048036. [DOI] [Google Scholar]
- Excoffier L, Smouse P, Quattro J. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Laval G, Schneider S. Arlequin Ver. 3.0: An integrated software package for population genetics data analysis. Evol Bioinform. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- Guo S, Thomson E. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics. 1992;48:361–372. doi: 10.2307/2532296. [DOI] [PubMed] [Google Scholar]
- Lee C, Lee S, Shin SG, Hwang S. Real-time PCR determination of rRNA gene copy number: absolute and relative quantification assays with Escherichia coli. Appl Microbiol Biotechnol. 2008;78:371–376. doi: 10.1007/s00253-007-1300-6. [DOI] [PubMed] [Google Scholar]
- Hume JCC, Lyons EJ, Day KP. Human migration, mosquitoes and the evolution of Plasmodium falciparum. Trends Parasitol. 2003;19:144–149. doi: 10.1016/S1471-4922(03)00008-4. [DOI] [PubMed] [Google Scholar]
- Richards CL, Carstens BC, Knowles LL. Distribution modelling and statistical phylogeography: an integrative framework for generating and testing alternative biogeographical hypotheses. J Biogeogr. 2007;34:1833–1845. doi: 10.1111/j.1365-2699.2007.01814.x. [DOI] [Google Scholar]
- Sumilo D, Bormane A, Asokliene L, Lucenko I, Vasilenko V, Randolph SE. Tick-borne encephalitis in the Baltic States: identifying risk factors in space and time. Int J Med Microbiol. 2006;296:76–79. doi: 10.1016/j.ijmm.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Estrada-Peña A, Vatansever Z, Gargili A, Buzgan T. An early warning system for Crimean-Congo haemorrhagic fever seasonality in Turkey based on remote sensing technology. Geospat Health. 2007;2:127–135. doi: 10.4081/gh.2007.261. [DOI] [PubMed] [Google Scholar]
- Scharlemann JP, Benz D, Hay SI, Purse BV, Tatem AJ, Wint GR, Rogers DJ. Global data for ecology and epidemiology: a novel algorithm for temporal Fourier processing MODIS data. PLoS ONE. 2008;9:e1408. doi: 10.1371/journal.pone.0001408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph SE, Rogers DJ. Tick-borne disease systems: mapping geographic and phylogenetic space. Adv Parasitol. 2006;62:263–291. doi: 10.1016/S0065-308X(05)62008-8. [DOI] [PubMed] [Google Scholar]
- Ma J, Campbell A, Karlin S. Correlations between Shine-Dalgarno sequences and gene features such as predicted expression levels and operon structures. J Bacteriol. 2002;184:5733–5745. doi: 10.1128/JB.184.20.5733-5745.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Garcia JC, de la Fuente J, Blouin EF, Halbur T, Onet VC, Saliki JT, Kocan KM. Differential expression of the msp1α gene of Anaplasma marginale occurs in bovine erythrocytes and tick cells. Vet Microbiol. 2004;98:261–272. doi: 10.1016/j.vetmic.2003.10.021. [DOI] [PubMed] [Google Scholar]
- Estrada-Peña A, Corson M, Venzal JM, Mangold AJ, Guglielmone A. Changes in climate and habitat suitability for the cattle tick Boophilus microplus in its southern Neotropical distribution range. J Vector Ecol. 2006;31:158–167. doi: 10.3376/1081-1710(2006)31[158:CICAHS]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Ivancovich JC, Braithwaite GB, Barnett S. Comportamiento de los estadios no-parasitarios de la garrapata del ganado - Boophilus microplus (Canestrini). IV. Siete diferentes localidades. Bol Ins Nac Tecnol Agropec Est Exp Reg Chaco. 1984;88:71–109. [Google Scholar]
- Lima WS, Ribeiro MF, Guimaraes MP. Seasonal variation of Boophilus microplus (Canestrini, 1887) (Acari: Ixodidae) in cattle in Minas Gerais State, Brazil. Trop Anim Health Prod. 2000;32:375–380. doi: 10.1023/A:1005229602422. [DOI] [PubMed] [Google Scholar]
- Guglielmone AA. The level of infestation with the vector of cattle babesiosis in Argentina. Mem Inst Oswaldo Cruz. 1992;87:133–137. doi: 10.1590/S0074-02761992000700020. [DOI] [PubMed] [Google Scholar]
- Labruna MB, Naranjo V, Mangold AJ, Thompson C, Estrada-Peña A, Guglielmone AA, Jongejan F, de la Fuente J. Allopatric speciation in ticks: genetic and reproductive divergence between geographic strains of Rhipicephalus (Boophilus) microplus. BMC Evol Biol. 2009;9:46. doi: 10.1186/1471-2148-9-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutullé C, Jonsson NN, Seddon J. Population structure of Australian isolates of the cattle tick Rhipicephalus (Boophilus) microplus. Vet Parasitol. 2009;161:283–291. doi: 10.1016/j.vetpar.2009.01.005. [DOI] [PubMed] [Google Scholar]
- de la Fuente J, Estrada-Peña A, Venzal JM, Kocan KM, Sonenshine DE. Overview: Ticks as vectors of pathogens that cause disease in humans and animals. Front Biosci. 2008;13:6938–6946. doi: 10.2741/3200. [DOI] [PubMed] [Google Scholar]