

Summary
Quorum sensing is a process of bacterial communication involving production and detection of secreted molecules called autoinducers. Gram-negative bacteria use acyl-homoserine lactone (AHL) autoinducers, which are detected by one of two receptor types. First, cytoplasmic LuxR-type receptors bind accumulated intracellular AHLs. AHL-LuxR complexes bind DNA and alter gene expression. Second, membrane-bound LuxN-type receptors bind accumulated extracellular AHLs. AHL-LuxN complexes relay information internally by phosphorylation cascades that direct gene-expression changes. Here we show that a small molecule, previously identified as an antagonist of LuxN-type receptors, is also a potent antagonist of the LuxR family, despite differences in receptor structure, localization, AHL specificity, and signaling mechanism. Derivatives were synthesized and optimized for potency, and in each case, we characterized the mode of action of antagonism. The most potent antagonist protects Caenorhabditis elegans from quorum-sensing-mediated killing by Chromobacterium violaceum, validating the notion that targeting quorum sensing has potential for antimicrobial drug development.
Keywords: Quorum Sensing, Receptor, Autoinducer, Antagonist, Agonist, Homoserine Lactone
Introduction
In a process called quorum sensing, bacteria communicate using chemical signal molecules termed autoinducers (Waters and Bassler, 2005). By monitoring fluctuations in autoinducer concentration, quorum-sensing bacteria track changes in cell population density and synchronously transition into and out of group behaviors. Quorum sensing allows bacteria to collectively carry out tasks that would be unsuccessful if carried out by an individual bacterium acting alone. Virulence factor production is controlled by quorum sensing in many clinically relevant bacteria (Fuqua et al., 1996; Waters and Bassler, 2005). Thus, strategies that disrupt quorum sensing are viewed as viable alternatives to traditional antibiotics.
Gram-negative quorum-sensing bacteria typically employ acyl-homoserine lactones (AHLs) as autoinducers and each bacterial species detects the cell-density-dependent accumulation of one (or more) AHL molecule(s) (Fuqua et al., 1996; Fuqua et al., 1994; Waters and Bassler, 2005). AHL detection occurs by one of two mechanisms. First, cytoplasmic LuxR-type proteins detect AHLs that diffuse into the cell (Figure 1, left panel) (Fuqua et al., 1994). In these cases, the ligand-free LuxR-type proteins are insoluble and degraded at low cell density when AHL concentrations are low (Zhu and Winans, 1999, 2001). At high cell density, when AHLs are present, AHL binding to the cognate LuxR-type proteins promotes folding of the LuxR-AHL complexes. The complexes bind DNA and activate quorum-sensing target genes (Zhang et al., 2002). There are a few cases in which the LuxR-type proteins fold, bind DNA, and repress target gene transcription in the absence of AHL (Minogue et al., 2005; Minogue et al., 2002; Sjoblom et al., 2006). In these cases, binding of AHL relieves repression, and this depends on the location of the DNA binding site, rather than on some unique structural feature of the receptor (von Bodman et al., 2003). In the second mechanism, AHLs are detected by membrane-bound two component histidine kinase-type proteins of the LuxN family (Figure 1, right panel) (Freeman et al., 2000; Jung et al., 2007; Swem et al., 2008; Timmen et al., 2006). In these cases, accumulated AHLs are detected outside the cell (e.g. in the periplasm) and AHL binding by the cognate LuxN receptor elicits a change in its auto-phosphorylation and phospho-transfer activities (Swem et al., 2008). This, in turn, causes a change in the phosphorylation state of a downstream DNA binding transcription factor, which alters its activity and promotes a quorum-sensing gene expression response (Lenz et al., 2004; Martin et al., 1989; Showalter et al., 1990; Tu and Bassler, 2007).
Figure 1. The C. violaceum and V. harveyi Quorum-Sensing Circuits.

Left panel: The cytoplasmic quorum-sensing receptor, CviR, from C. violaceum binds to the AHL autoinducer (black ovals) at high cell density (HCD). The CviR-AHL complex binds to DNA and activates expression of the vio genes required for production of the purple pigment, violacein. CviI is the C6-HSL synthase. Right panel: The membrane-bound quorum-sensing receptor, LuxN from V. harveyi binds to the AHL autoinducer (black ovals) at high cell density (HCD) resulting in a phosphorylation cascade that activates expression of the lux genes required for bioluminescence. LuxM is the 3OH-C4-HSL autoinducer synthase.
The defining member of the family of membrane-bound two-component AHL receptor proteins is LuxN from the quorum-sensing bacterium Vibrio harveyi (Henke and Bassler, 2004). LuxN detects the strongest of three V. harveyi autoinducer signals, the AHL 3OH-C4-HSL (Figure 2) (Bassler et al., 1993; Cao and Meighen, 1989). In earlier work, we screened a chemical library and identified 15 small molecule antagonists of V. harveyi LuxN, one of which we studied in detail (Supplemental Figure 1A) (Swem et al., 2008). Our characterization revealed that the antagonist competes for the AHL binding site of LuxN. Even though LuxN-type and LuxR-type AHL receptors have no obvious sequence homology and they employ distinct mechanisms for signal transduction, we reasoned that because each type of receptor must bind an AHL, and AHLs share common structural features, LuxN-type and LuxR-type receptors could have structurally similar AHL binding pockets. We further reasoned that, since we had identified 15 molecules that antagonized a LuxN-type receptor and these molecules appeared to compete for the AHL binding site, some of these antagonists might also antagonize a LuxR-type AHL receptor. Here we test this idea and find that indeed, one of the 15 molecules identified in the screen for LuxN antagonists also strongly antagonizes a canonical LuxR-type protein called CviR from Chromobacterium violaceum (McClean et al., 1997). We synthesized a set of molecules based on the core structure of this potent LuxN and CviR antagonist and thereby identified and characterized additional antagonists, some with increased potency. We determined two mechanisms by which the antagonists function. One class of antagonists prevents CviR from binding DNA. A second set of antagonists allows DNA binding but reduces or eliminates transcriptional activation suggesting that the CviR-antagonist complex cannot productively interact with RNA polymerase. Consistent with our original hypothesis regarding AHL-binding pockets being similar in these diverse receptors, our most potent new CviR antagonist also functions as the strongest antagonist of LuxN. Finally, we show that this antagonist prevents C. violaceum killing of the nematode C. elegans by specifically inhibiting the CviR-dependent quorum-sensing virulence pathway. In total, the antagonists identified here could serve as broad spectrum lead compounds for disrupting acylated homoserine lactone quorum sensing in pathogenic Gram-negative bacteria.
Figure 2. Structures of the Quorum-Sensing Autoinducers and Synthetic Antagonists.

Structures and designations of the quorum-sensing autoinducers and synthetic antagonists for C. violaceum and V. harveyi.
Results
Analyses of CviR Autoinducer Responses
To assess LuxR-type AHL sensitivity and selectivity, we employed C. violaceum ATCC31532. C. violaceum synthesizes and responds to C6-HSL (Figure 2) (McClean et al., 1997). The quorum-sensing readout is the easily quantifiable pigment, violacein. Violacein production requires CviR binding to C6-HSL, which accumulates at high cell density, and subsequent activation of transcription of the vioABCDE biosynthetic gene cluster (Throup, 1995). C. violaceum also possesses a repressor of vioABCDE (McClean et al., 1997). We identified and inactivated this repressor in our violacein assay strain (see supplemental text). We also developed a second, independent assay for CviR activity using an engineered E. coli strain that expresses CviR and contains a plasmid harboring the vioA promoter fused to gfp. CviR-dependent transcription can be assayed in the presence of various molecules.
We first tested the CviR response to a variety of AHL molecules with varying acyl-tail lengths (Figure 3A). CviR activates significant gfp expression in response to C4-HSL, C6-HSL (the native signal), and C8-HSL with EC50 values of 12 μM, 75 nM, and 30 nM, respectively (Figure 3A). While C4-HSL can induce maximal gfp expression, it takes nearly three orders of magnitude more C4-HSL than C6-HSL to do so. The longer acyl-tail length AHL molecules, C10-HSL, C12-HSL, and C14-HSL do not induce gfp expression through CviR even at 100 μM. A curious finding regarding CviR is that it has a similar sensitivity for C6-HSL (EC50 = 75 nM) and C8-HSL (EC50 = 30 nM), yet the two molecules do not activate gfp expression to the same maximal level (Figure 3A). Specifically, C8-HSL induces only half of the maximal gfp expression induced by C6-HSL. We interpret this result to mean that the C6-HSL and C8-HSL autoinducers bind to CviR with similar affinities but C8-HSL induces a conformational change in the CviR protein that, while obviously not completely negating its interaction with RNA polymerase, attenuates its transcriptional activating potential with respect to that of C6-HSL bound CviR. We return to this point later.
Figure 3. CviR Dose-Response Curves.

(A) CviR-dependent vioA-gfp expression in E. coli is plotted as a function of concentration of the specified homoserine lactone (HSL) molecules. (B) Inhibition of CviR-dependent vioA-gfp expression in E. coli is plotted as a function of the concentration of the specified molecule in the presence of a 500 nM C6-HSL. (C) CviR-dependent violacein production in wild type C. violaceum is plotted as a function of specified antagonist molecule. (D) Inhibition of CviR-dependent vioA-gfp expression in E. coli is plotted as a function of the specified antagonist molecule. In all panels, data were fit with a variable-slope sigmoidal dose-response curve to determine EC50 or IC50 values. Error bars represent the standard error of the mean for three independent trials.
Consistent with the inability of long chain AHL molecules to induce CviR transcriptional activation, C10-HSL, C12-HSL, and C14-HSL have been reported to antagonize the CviR protein (McClean et al., 1997). Indeed, we determined that the IC50 values for C10-HSL, C12-HSL, and C14-HSL in the presence of constant (500 nM) C6-HSL are 208 nM, 494 nM, and 268 nM, respectively (Figure 3B). Interestingly, the IC50 values for the long chain AHL antagonists are quite low, suggesting that the CviR receptor binds these molecules with roughly the same affinity with which it binds the native C6-HSL autoinducer. However, binding of the longer acyl-tail length AHL molecules disrupts transcriptional activation by CviR.
Identification of Synthetic CviR Antagonists
As mentioned above, 15 molecules that inhibit AHL detection by the membrane-bound two-component AHL receptor LuxN have been identified (Supplemental Figure 1A) (Swem et al., 2008). We wondered if any of the LuxN antagonist molecules could also interfere with AHL binding to CviR. To examine this, the 15 molecules were tested for inhibition of in vivo violacein production in C. violaceum. For this set of experiments, we used a C. violaceum cviI mutant that produces no endogenous AHL signal. Because of the cviI mutation, this strain does not produce violacein in the absence of exogenously supplied AHL (Supplemental Figure 1B, bar denoted No HSL) (McClean et al., 1997). We added the native C6-HSL signal at a constant concentration of 5 μM and measured violacein production in the presence of 50 μM of each potential antagonist (Supplemental Figure 1B). One of the LuxN antagonists (denoted 4606-4237) completely abolished violacein production without any effect on bacterial growth (Supplemental Figure 1B and Figure 2).
Characterization of the CviR Antagonist 4606-4237
To quantify the in vivo potency of 4606-4237, we characterized violacein production in wild type C. violaceum (i.e., carrying a functional cviI gene that produces native levels of C6-HSL) in response to 4606-4237 at concentrations ranging from 45 nM to 100 μM (Figure 3C). 4606-4237 inhibits violacein production in C. violaceum with an IC50 value of 1.2 μM (Figure 3C). To determine if 4606-4237 inhibits violacein production specifically by interfering with CviR, we tested 4606-4237 for antagonism of CviR in our recombinant E. coli vioA-gfp assay. C6-HSL was held constant at 500 nM, while 4606-4237 was titrated from 1.3 nM to 100 μM (Figure 3D). We chose this concentration of native C6-HSL signal because it induces maximal GFP production in this heterologous assay system. Importantly, no C. violaceum components other than CviR are present in the recombinant E. coli assay strain. Thus, we reasoned that if inhibition occurred, it must be due to a specific interaction between our antagonist molecule 4606-4237 and CviR rather than an interaction of 4606-4237 with some unknown component required for violacein production in C. violaceum. Indeed, the 4606-4237 molecule strongly inhibited gfp expression with an IC50 value of 2.2 μM, similar to what we found for the in vivo inhibition activity (Figure 3C). Thus, 4606-4237 functions by disabling CviR-mediated activation of gene expression in both the C. violaceum in vivo violacein assay and the recombinant E. coli vioA-gfp assay.
Structure-Function Analysis of 4606-4237 and Derivatives
To explore the structural features of 4606-4237 critical for antagonism of CviR, we synthesized a library of molecules based on the core structure of 4606-4237. 4606-4237, while resembling natural AHL signals, differs in two key features: a substituted phenoxy group at the terminus of the acyl chain and a homocysteine thiolactone ring instead of a homoserine lactone ring. Our focused library explored these two features (the syntheses, structures, and antagonist activities of all of the derivatives are supplied in Supplemental Figure 2). Analogous to the parent molecule 4606-4237, most of the derivatives are antagonists of CviR and none are agonists (Supplemental Figure 2). In general, halogenation of the phenoxy ring is important, with modest differences with respect to the position or identity of the halogen. Electron donating groups reduce potency and large para substituents (phenyl, t-butyl) result in loss of activity. Cumulative analysis of the activities of this set of synthesized molecules revealed that maintaining the chlorine atom at the para position of the phenyoxy ring along with elimination of the methyl group from the ortho position of 4606-4237 increased antagonism efficacy (IC50 decreases from 2.2 μM to 1.1 μM; Figures 2 and Figure 3D denoted CTL). Replacing the sulfur with an oxygen atom in the thiolactone head group also increased the potency of the antagonist nearly 10-fold over that of the original 4606-4237 molecule giving an IC50 value of 377 nM (Figures 2 and Figure 3D denoted CL). Thus, CL is the strongest synthetic antagonist identified in this study.
We tested the potency of CTL and CL in vivo against C. violaceum at concentrations ranging from 31 nM to 100 μM. Both CTL and CL inhibited in vivo violacein production in a dose-dependent manner with IC50 values of 723 nM and 295 nM, respectively (Figure 3C) Importantly, the efficacy of 4606-4237, CTL, and CL follow identical patterns in both the E. coli vioA-gfp assay and the in vivo C. violaceum violacein assay (Figure 3C and 3D). This result shows that, similar to the parent molecule 4606-4237, both CTL and CL traverse the E. coli and C. violaceum membranes and likely function by directly interfering with CviR-dependent transcriptional activation.
CviR Protein-Antagonist Binding Analysis
Similar to the paradigmatic LuxR family member TraR from Agrobacterium tumefaciens, we found that CviR is only soluble, able to bind DNA, and activate transcription when bound to its cognate AHL ligand (Figure 4A, DMSO control lanes) (Zhang et al., 2002; Zhu and Winans, 1999, 2001). We analyzed the solubility characteristics of CviR in the presence of the various molecules under study. To do this, CviR was expressed in E. coli in the presence of exogenously supplied C4-HSL, C6-HSL, C8-HSL, C10-HSL, C12-HSL, and C14-HSL, 4606-4237, CTL, and CL. The protein was also expressed in the absence of any AHL or antagonist. In each case, whole cell and soluble fractions of cell lysates were analyzed by SDS-PAGE to assess whether the CviR protein in question had been stabilized.
Figure 4. Solubility Analysis of CviR Bound to Ligands.

(A) SDS-PAGE analysis of E. coli whole cell (W) and soluble (S) extracts of cell cultures expressing CviR in the presence of dimethyl sulfoxide (DMSO) (Lanes 2 and 3), C6-HSL (Lanes 4 and 5), and C10-HSL (Lanes 6 and 7). (B) SDS-PAGE analysis of E. coli whole cell (W) and soluble (S) extracts of cell cultures expressing CviR in the presence of DMSO (Lanes 2 and 3), 4606-4237 (Lanes 4 and 5), Chloro-thiolactone (CTL) (Lanes 6 and 7) and Chlorolactone (CL) (Lanes 8 and 9). The L above the first lane designates the molecular weight ladder.
In contrast to when no ligand was added (Figure 4A, DMSO control lanes), CviR became soluble when expressed in the presence of all the tested AHLs (we show C6-HSL and C10-HSL as representatives in Figure 4A, all others are shown in Supplemental Figure 3). The three synthetic antagonists, 4606-4237, CTL, and CL also promoted CviR folding and solubility (Figure 4B). Thus CviR is agonized by C4-HSL and C6-HSL, partially agonized by C8-HSL, and antagonized by C10-HSL, C12-HSL, C14-HSL, 4606-4237, CTL and CL (Figure 3A-D). Therefore, we come to the conclusion that all of the CviR antagonist molecules (C10-HSL, C12-HSL, C14-HSL, 4606-4237, CTL, and CL) are bound by CviR, and thus do not antagonize the receptor by inhibiting protein folding and solubility.
To verify that the antagonists compete for the CviR C6-HSL binding site, we performed a competition assay. In this experiment, C6-HSL was titrated from 1.3 nM to 100 μM, and 4606-4237 was added exogenously at concentrations of either 0, 1, 10 or 100 μM. We monitored vioA-gfp expression (Supplemental Figure S4). As expected with a competitive binding event, each titration curve exhibited a similar shape but the EC50 value for C6-HSL increased 10-fold with each 10-fold increase in 4606-4237 concentration.
DNA Binding Analysis of CviR
To further investigate the mechanism underlying antagonism of the CviR receptor, we examined whether the soluble CviR antagonist complexes were incapable of binding DNA, and thus unable to activate transcription. To do this, we purified the CviR protein in complex with the native C6-HSL ligand, and in complex with C8-HSL, C10-HSL, C12-HSL and C14-HSL. The CviR protein was also purified in complex with each of the synthetic antagonist molecules, 4606-4237, CTL, and CL. We performed DNA gel mobility shift analyses to assess binding of each of these purified protein complexes to the vioA promoter. We were unable to purify CviR in the presence of C4-HSL, which is consistent with the high EC50 value and, therefore, low affinity CviR likely has for this molecule (Figure 3A).
CviR loaded with its cognate C6-HSL signal molecule binds DNA with high affinity at even the lowest ligand concentration of 100 nM (Figure 5A). CviR loaded with C6-HSL did not bind to a control DNA probe consisting of a 300 nt region of the vioB open region frame (Supplemental Figure S5). CviR protein loaded with C8-HSL bound DNA with similar affinity to CviR loaded with C6-HSL (Figure 5A). However, we showed above that C8-HSL is incapable of activating maximal gfp expression (Figure 3A) suggesting that CviR loaded with C8-HSL can bind DNA properly but the complex is in a conformation that is not fully competent as a transcriptional activator. We found that C10-HSL is an antagonist of the CviR protein (Figure 3B), but surprisingly C10-HSL also does not abolish DNA binding of the CviR protein (Figure 5A). This result suggests that C10-HSL locks CviR into a conformation incapable of productive interaction with RNA polymerase. Purified CviR bound to the remaining antagonist molecules C12-HSL, C14-HSL, as well as the synthetic antagonists, 4606-4237, CTL, and CL displayed no ability to bind DNA (Figure 5A). Therefore, this final set of molecules antagonize the CviR receptor by inhibiting DNA binding, rather than by inhibiting interaction with RNA polymerase.
Figure 5. Gel Mobility Shift Analysis of the CviR Protein Bound to Agonist and Antagonist Molecules.

(A) CviR binding to the vioA promoter at concentrations of 0 nM (Lane 1), 100 nM (Lane 2), 200 nM (Lane 3), 300 nM (Lane 4), 400 nM (Lane 5), and 500 nM (Lane 6). Each panel corresponds to CviR loaded with a different molecule. (B) CviR proteins loaded with C6-HSL and loaded with CL at concentrations of 500 nM were incubated for 20 min with 0, 0.5, 1, 3, 5 or 10 μM CL and C6-HSL, respectively. The vioA probe was added and allowed to incubate at room temperature for 20 additional min prior to being subjected to electrophoresis. No protein (Lane 1), 0 μM (Lane 2), 0.5 μM (Lane 3), 1 μM (Lane 4), 3 μM (Lane 5), 5 μM (Lane 6), 10 μM (Lane 7) CL or C6-HSL.
To determine if CviR could reversibly bind C6-HSL, gel mobility shift analyses of CviR loaded with C6-HSLwere performed in the presence of increasing concentrations of the most potent synthetic antagonist, CL (Figure 5B). C6-HSL loaded CviR protein was incubated with the vioA probe at a concentration of 500 nM and the CL molecule was added at final concentrations of 0, 0.5, 1, 3, 5 and 10 μM. DNA binding decreased in a dose dependent manner with increasing concentrations of the CL antagonist, suggesting that the CL antagonist could exchange with the C6-HSL bound to the CviR protein. In the reciprocal experiment, we tested whether C6-HSL could replace CL bound to CviR. In this experiment, CviR loaded with CL at a concentration of 500 nM was incubated in the presence of increasing concentrations of C6-HSL (from 0 to 10 μM). At a concentration of 1 μM C6-HSL, we could detect a modest shift of the DNA probe (Figure 5B). This shift was dose dependent, with the strongest gel mobility shift observed at a concentration of 10 μM C6-HSL. When CL is exchanged for C6-HSL, the complex transitions from fully capable of DNA binding to fully incapable of DNA binding. However, the gel shifts show that C6-HSL is not likewise capable of replacing CL and to generate fully competent DNA-bound complex. In this case, the protein must exchange the ligand and convert from a DNA-binding incompetent form to a DNA-binding competent form. Most likely, some of the protein misfolds during this process. Nonetheless, these analyses show that ligand binding is a reversible event because exchange can occur after CviR has folded around the native ligand or the synthetic antagonist molecule.
Inhibition of the Membrane-Bound Quorum-Sensing Receptor, LuxN
Our above experiments show that 4606-4237, CTL, and CL bind to the cytoplasmic CviR AHL receptor and act as potent antagonists. However, as mentioned, 4606-4237 was initially identified for its ability to inhibit signaling by the V. harveyi trans-membrane receptor LuxN (Figure 1B) (Swem et al., 2008). We wondered whether the dual inhibitory characteristic of 4606-4237 was a property exclusive to that molecule, or whether the other inhibitors, namely CTL and CL, might also be potent inhibitors of the LuxN receptor. If so, it would indicate that: (1) a rather generic AHL binding site exists in Gram-negative quorum-sensing receptors that recognize medium-chain length AHLs. (2) This binding site occurs irrespective of the receptor’s overall domain architecture. (3) The binding site is also independent of whether signal recognition occurs in the cytoplasm or in the periplasm.
We quantified the in vivo activity of 4606-4237, CTL, and CL for V. harveyi LuxN using dose-response analyses of the endogenous quorum-sensing-activated response, bioluminescence. We examined three different V. harveyi strains: (1) The wild type V. harveyi strain BB120 which has multiple quorum-sensing circuits, and thus responds to multiple autoinducers, only one of which is the 3OH-C4-HSL acting through LuxN (Henke and Bassler, 2004); (2) V. harveyi BB960 which lacks the receptor for the second autoinducer making the AHL-LuxN circuit the dominant input into bioluminescence expression (Bassler et al., 1994); (3) V. harveyi JMH624 which, in addition to lacking the receptor for the second autoinducer lacks the AHL synthase LuxM (Swem et al., 2008). Thus, V. harveyi JMH624 induces bioluminescence exclusively in response to the AHL, 3OH-C4-HSL signal but only when it is supplied exogenously (Cao and Meighen, 1989; Swem et al., 2008). Exogenous addition of 20 nM 3OH-C4-HSL is sufficient to induce maximal bioluminescence in all of these strains. Importantly, however, endogenously produced AHL accumulates to low micromolar levels in culture fluids of V. harveyi strains possessing the AHL synthase, LuxM (Cao and Meighen, 1989).
Figure 6A shows that 4606-4237 does not inhibit bioluminescence in wild type V. harveyi (BB120; squares). However, modest inhibition occurs in the absence of input from the second quorum-sensing system, with an IC50 value of 302 μM (BB960; triangles). Inhibition increases 100-fold in V. harveyi JMH624 in the presence of 20 nM 3OH-C4-HSL giving an IC50 value of 3.0 μM (JMH624; circles). These results show that 4606-4237 is an antagonist of LuxN and can indeed inhibit LuxN-directed quorum sensing in V. harveyi but only if input from the second quorum-sensing circuit has first been eliminated and especially when the level of competing autoinducer signal is also reduced to a concentration significantly below that produced endogenously.
Figure 6. V. harveyi Bioluminescence in Response to Antagonists.
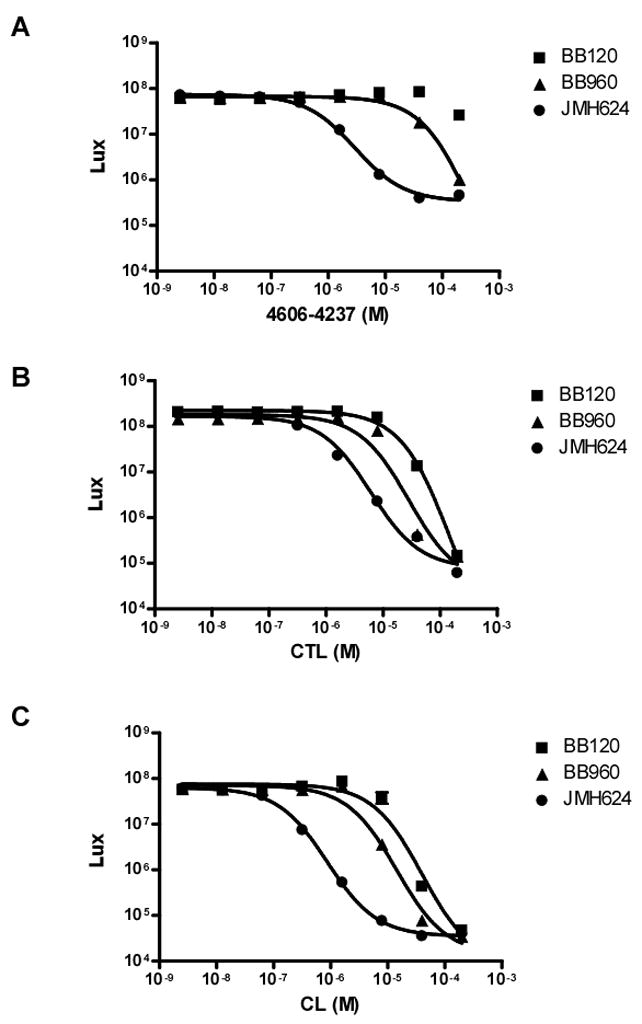
Light production from wild type V. harveyi (BB120), a luxPQ mutant (BB960), and a luxPQ, luxM double mutant (JMH624) was measured in the presence of the specified concentrations of 4606-4237 (A), CTL (B), or (CL) (C). Data were fit with a variable-slope sigmoidal dose-response curve to determine IC50 values. Error bars although small are included and represent the standard error of the mean for three independent trials.
Unlike 4606-4237, the CTL molecule is capable of inhibiting bioluminescence in all three V. harveyi strains; BB120, BB960, and JMH624 (+ 20 nM AHL) with IC50 values of 150 μM, 28 μM, and 6 μM, respectively (Figure 6B). The CL molecule is the most potent LuxN antagonist, with IC50 values for BB120, BB960, and JMH624 (+20 nM AHL) of 40 μM, 14 μM, and 873 nM, respectively (Figure 6C). Thus, the ability of these antagonist molecules to target LuxN parallels what we found for the CviR receptor in terms of potency: CL > CTL > 4606-4237. Strikingly, the more potent antagonists of LuxN are able to interfere with AHL signaling even in the face of a competing quorum-sensing system and in the presence of high levels of endogenously produced AHL signal.
Chromobacterium violaceum Infects Caenorhabditis elegans in a Quorum-Sensing-Dependent Manner
Quorum sensing in both Gram-negative and Gram-positive bacteria often controls processes critical for pathogenesis of eukaryotic hosts (Waters and Bassler, 2005). C. violaceum is a human pathogen frequently infecting by means of introduction through lacerated skin (Richard, 1993). To test whether C. violaceum could infect a model host organism, we employed the broadly utilized animal infection model organism, C. elegans (Aballay and Ausubel, 2002; Dhakal et al., 2006; Vaitkevicius et al., 2006). Lifespan assays reveal that C. violaceum rapidly kills C. elegans. The nematode has a median survival of two days in the presence of C. violaceum compared to a median survival of fifteen days when grown in the presence of the non-pathogenic E. coli strain, OP50 (data not shown). A C. violaceum vioA mutant that is incapable of producing the violacein pigment remains fully capable of shortening C. elegans lifespan, showing that violacein is not responsible for C. violaceum-mediated worm killing (data not shown).
To test if quorum sensing is required for C. violaecum to kill C. elegans, we performed lifespan assays with the C. violaceum cviI mutant that makes no AHL signal and thus does not express genes that are controlled by quorum sensing. In the presence of the mutant strain, the median C. elegans lifespan is twelve days (Figure 7A; DMSO control). Exogenous addition of the native autoinducer molecule, C6-HSL (2 μM) restored quorum sensing and complemented the cviI mutant’s ability to kill C. elegans (median lifespan = 4 days, Figure 7A). This result provides strong evidence that a factor or factors other than violacein that is under quorum-sensing control is responsible for C. violaceum pathogenicity of C. elegans. We tested whether the 4606-4237 antagonist could inhibit quorum-sensing-mediated killing of the nematode by C violaceum. Indeed, the simultaneous addition of C6-HSL (2 μM) and 4606-4237 (50 μM) to the cviI mutant restored C. elegans median survival to twelve days (Figure 7A). The 4606-4237 molecule had little effect on C. elegans when added to the cviI C. violaceum mutant in the absence of the C6-HSL signal (Figure 7A), indicating that the antagonist’s effect is specifically mediated through inhibition of quorum sensing.
Figure 7. C. elegans Survival Following C. violaceum Infection and Treatment.

(A) Kaplan-Meier survival curve of a C. elegans population infected with C. violaceum cviI mutant in the presence of the control solution of dimethyl sulfoxide (DMSO) or the specified molecules. (B) Kaplan-Meier survival curve of a C. elegans population infected with wild type C. violaceum in the absence of any quorum-sensing antagonist or in the presence of CL.
Our ultimate goal is to control virulent processes of fully infective wild type bacteria. Therefore, we tested the most potent antagonist CL, on wild type C. violaceum that is fully capable of producing and responding to native levels of C6-HSL signal. At a concentration of only 20 μM, CL protected C. elegans from killing by wild type C. violaceum (Figure 7B). In this context, C. elegans median lifespan increased from two days to seven days. Thus, the lead quorum-sensing antagonist discovered in this work is capable of specifically disrupting quorum-sensing-directed pathogenicity in a wild type bacterium and, in so doing, dramatically improve the outcome in a nematode animal host.
Discussion
In this study we identify a class of antagonists of AHL signaling that have the unique ability to antagonize two different quorum-sensing receptor types. Specifically, the antagonists inhibit both the membrane bound sensor kinase, LuxN and the cytoplasmic transcriptional regulator, CviR. Remarkably, the chemical features of the antagonists that provide increased potency are identical for both receptor types. This finding is particularly surprising because LuxN and CviR are not evolutionarily related proteins. Both receptors do however, bind and respond to similar ligands; LuxN binds 3OH-C4-HSL and CviR binds C6-HSL (Figure 2) (Cao and Meighen, 1989; McClean et al., 1997).
In the case of CviR, molecules that possess antagonist activity function by two distinct mechanisms: prevention of transcriptional activation or prevention of DNA binding. First we consider the case of C8-HSL and C10-HSL which function by preventing activation of transcription. CviR is highly sensitive to C8-HSL, but only half-maximal GFP production is induced by this molecule. Gel mobility shift analyses inform us that decreased gfp expression is not due to disruption of DNA binding. Therefore, we suggest that C8-HSL places the CviR receptor into a conformation that binds DNA but does not engage in all of the productive interactions with RNA polymerase as does the CviR receptor bound to the native ligand, C6-HSL. Extending the acyl-chain by two additional carbons (C10-HSL) further exacerbates the problem. While CviR binds DNA equally well whether it is in complex with C6-HSL, C8-HSL, or C10-HSL, the CviR-C10-HSL complex apparently cannot make the contacts with RNA polymerase necessary to activate transcription. The second mechanism of CviR antagonism; disruption of DNA binding, is typified by C12-HSL, C14-HSL, 4606-4237, CTL and CL. These antagonists bind and solubilize the CviR receptor, but do not allow the protein to adopt a conformation suitable for interaction with DNA. These findings suggest that at least two distinct events must occur for full activation of LuxR-type proteins: DNA binding and assembly of a transcriptionally competent complex with RNA polymerase. Remarkably, the antagonist molecules described here can affect either process. Furthermore, we have identified ligands, such as C8-HSL, that form semi-active CviR complexes suggesting that molecules designed to interfere with quorum sensing need not function by all-or-none activity mechanisms. Rather, subtle pharmacological control modulated by partial antagonists that function like C8-HSL can be used to fine-tune bacterial quorum-sensing outputs. In scenarios where complete abrogation of quorum-sensing behaviors is desired, more potent antagonists (i.e., C12-HSL, 4606-4237, etc.) can be employed.
Because our strong antagonists also eliminate the LuxN response to its cognate AHL signal, it is not a prerequisite that the particular targeted-quorum-sensing receptor be cytoplasmic, bind DNA, or interact with RNA polymerase for successful interference. In the case of LuxN, the antagonists presumably function by binding to the periplasmic domain. While we have not unequivocally proven that this is the case, our analysis suggests the antagonists act as competitive inhibitors of LuxN binding to the native signal, which we have shown previously binds in the periplasm (Swem et al., 2008).
Irrespective of where the antagonist molecules bind to LuxN, what is clear is that they act to prevent LuxN from switching from kinase-mode to phosphatase-mode, and in so doing, they prevent autoinducer signaling. Therefore, the strong antagonist molecules discovered here have the remarkable capability of inhibiting receptors that are located in different cellular compartments and that function by drastically different mechanisms. Our findings suggest that antagonists can be discovered for any quorum-sensing receptor irrespective of its cellular location and mechanism of signal relay, and furthermore, that such molecules could act ubiquitously against a group of diverse receptors so long as the receptors naturally bind molecules with similar structures.
Typically, bacterial infections are treated with bactericidal or bacteriostatic molecules that impede DNA replication, RNA synthesis, protein production, peptidoglycan biosynthesis, or tetrahydrofolate synthesis (Gale, 1981). Existing methods for treating bacterial infections unfortunately exacerbate the growing antibiotic resistance problem because they inherently select for growth of bacteria that can resist the drug. In this regard, targeting quorum sensing (or other virulence pathways) provides the attractive possibility that since these putative therapies interfere only with signaling and not growth, they could potentially minimize the development of resistance and give such drugs a long functional lifetime (National Research Council (U.S.). Committee on New Directions in the Study of Antimicrobial Therapeutics: New Classes of Antimicrobials. et al., 2006). Anti-quorum-sensing strategies could presumably prevent pathogenic bacteria from initiating the gene expression cascade required for successful establishment in the host, and in so doing, provide the host’s immune system the needed time to eliminate the pathogen. Consistent with this idea, mutant analyses have convincingly demonstrated that clinically relevant pathogens lacking quorum sensing are attenuated for virulence (Jayaraman and Wood, 2008; Kong et al., 2006; Novick and Geisinger, 2008). Such bacteria do not grow unchecked in model host organisms even though their repertoire of canonical virulence factors remain intact and only quorum-sensing-mediated communication is eliminated.
In one elegant study, Wright et al. successfully disrupted the Agr peptide quorum-sensing system in Staphylococcus aureus using the antagonistic peptide, AIP-II which prevented the onset of a murine abscess post infection (Wright et al., 2005). This study also revealed that the proper timing of quorum-sensing-controlled events is critical for successful bacterial infection. Attempts to inhibit AHL-mediated quorum sensing in Gram-negative bacteria have also provided promising results. In studies of Pseudomonas aeruginosa; the major pathogen associated with cystic fibrosis lung infection, keratitis eye infection, and third-degree burn associated skin infections, several antagonists have been identified that inhibit the AHL receptor, LasR (Muh et al., 2006; Willcox et al., 2008). Similar to the antagonists described in this work, many of the identified antagonists contain homoserine lactone head groups with substituted acyl chains (Geske et al., 2008; Geske et al., 2007; Geske et al., 2005; Muh et al., 2006). The P. aeruginosa antagonists await in vivo testing on wild type bacteria. To date, they have only been examined on P. aeruginosa mutant strains lacking the AHL synthase that do not produce endogenous signal that could compete with the putative antagonist for receptor binding. In addition, attenuation of P. aeruginosa virulence has been reported in a pulmonary mouse model by targeting the quorum-sensing signaling pathway with halogenated furanones (Hentzer et al., 2002; Hentzer et al., 2003). However, the mechanism by which these furanones inhibit bacterial quorum sensing is unclear, as mutational analyses suggest that they do not compete for AHL binding to the LuxR-type receptors (Koch et al., 2005).
Very importantly, antagonists of quorum-sensing signals should be especially difficult for bacteria to bypass by mutation, and thus such strategies should be unusually robust to the development of bacterial resistance. Our rationale for making this strong statement is as follows: First, any mutation in the binding pocket of an autoinducer receptor that renders a bacterium immune to an autoinducer antagonist must not interfere with signaling by the endogenous autoinducer. Such mutations will be extremely rare. Second, even if such a mutation could occur, if collective action is required for a quorum-sensing-controlled behavior (e.g., virulence) to be advantageous, then a single mutant bacterium that is blind to an antagonist does not gain an advantage through the mutation. Although the “resistant” bacterium will indeed switch into quorum-sensing mode at the appropriate cell density, other nearby bacteria who are susceptible to the antagonist will not. Thus, the quorum-sensing-controlled behavior will not take place. Third, when an individual bacterium develops resistance to an anti-quorum-sensing drug, it does not gain a growth advantage. Rather, in all likelihood, such a mutant will have decreased fitness because it will undertake the energy-expensive quorum-sensing behaviors but it will not reap the benefits of them because it will be carrying out these tasks in isolation. This quorum-sensing “resistance” scenario is absolutely unlike when resistance to a traditional antibiotic develops and the resistant mutant and its offspring immediately receive a growth advantage. The latter fuels the growth and spread of antibiotic resistant bacteria whereas development of immunity to anti-quorum-sensing therapies could in fact function to limit resistance.
Our results, most notably with the CL molecule, make a strong case and provide compelling in vivo evidence that an anti-quorum-sensing strategy is a valid alternative to traditional antibiotics for Gram-negative bacteria, and that there is merit to pursuing the clinical relevance of such strategies. Specifically, we successfully administered our strongest quorum-sensing antagonists in a model setting and showed that they improve the outcome of a wild type bacterial infection in an animal host, in this case, in the nematode C. elegans. The C. elegans animal model does not allow one to test whether anti-quorum-sensing molecules can be administered subsequent to the establishment of infection. Nonetheless, the results in Figure 5B showing that our strongest antagonist can exchange with the native autoinducer, provide evidence that quorum-sensing controlled virulent processes can indeed be reversed. Together our biochemical and animal studies, coupled with the earlier successful results with AIP-II, suggest that anti-quorum-sensing strategies hold promise as novel non-bactericidal, non-bacteriostatic drug therapies to treat both Gram-negative and Gram-positive infections. Our finding that the CL antagonist molecule is potent enough to protect an animal from wild type virulent bacteria, suggests that quorum-sensing autoinducer-receptor interactions are promising targets for drug development and furthermore, that small molecule antagonists can be identified and synthesized with relative ease.
Experimental Procedures
Strains and Media
Wild type C. violaceum strain ATCC31532 was purchased from the ATCC. The cviI mutant strain, CV026 was kindly supplied by Clay Fuqua and Paul Williams (McClean et al., 1997). C. violaceum strains were grown aerobically at 30°C in either Luria-Bertani broth or Nutrient Broth supplemented with 1mM trypotophan. V. harveyi strains were grown aerobically at 30°C in autoinducer bioassay (AB) broth. AI-1 dose-response curves were generated in V. harveyi strains BB120, BB960 and JMH624 as described. E. coli was grown at 37°C in Luria Bertani broth. The plasmid pET23b (Novagen) was used for expressing cviR in E. coli strain BL21-Gold (DE3) or BL21(DE3) pLysS (Stratagene) and maintained by inclusion of Ampicillin at 100 μg/ml. Plasmid pEVS141 was used for the vioA–gfp promoter fusion and maintained with 50 μg/ml of Kanamycin. A list of strains and plasmids used in this study is provided in the supplemental material (Table S1). C. elegans wild type strain, N2 was used for all of the nematode infection studies. Worms were propagated on Nematode Growth Media (NGM) medium with an E. coli OP50 lawn as the food source and allowed to multiply at 20°C. Primer sequences are available by request.
vioA-gfp Construction
The vioA-gfp promoter fusion was constructed by amplifying the vioA promoter and cloning this region upstream of gfp in pEVS141 at the SphI and SalI sites. Cells containing the vioA-gfp construct were electroporated with the pET23 carrying cviR and selected on LB medium supplemented with 75 μg/mL ampicillin and 50 μg/mL kanamycin. The E. coli strain containing cviR and the vio:gfp reporter is named ELS1257.
GFP and Violacein Dose-Response Analyses
E. coli strains were grown overnight in LB medium with antibiotics at 37°C and sub-cultured into fresh medium at a 1:100 dilution. Various AHL and antagonists were added at constant concentrations or titrated as stated. GFP was measured on an Envision plate reader after 5 hours of growth at 37°C. C. violaceum strains were grown aerobically in 1 mL Nutrient Broth plus 1mM tryptophan at 30°C. Cells were subcultured into 1 mL fresh medium at 1:1000 dilution and the antagonist 4606-4237 or CL was tested at concentrations from 100μM to 0.045 μM and CTL was titrated from 67 μM to 0.03 μM in cultures grown aerobically to steady state at 30°C. Cells were collected via centrifugation at 13000 rpm for 15 minutes. Pellets were resuspended in 1 mL of methanol and incubated at 42 °C for 1 hour. Samples were again collected via centrifugation at 13000 rpm for 5 minutes and analyzed for violacein by measuring optical density at 568 nm on a Beckman Coulter DU-800 spectrophotometer. For the dose-response analyses, all assays were performed in triplicate.
CviR Overexpression and Purification
CviR was overexpresssed in BL21(DE3) pLysS by growing an overnight culture in LB supplemented with 100 μg/mL ampicillin and 10 μg/mL chloramphenicol. The culture was diluted 1:50 into fresh LB supplemented with 100 μg/mL ampicillin and 10 μg/mL chloramphenicol and grown shaking at 37°C to an OD600 of 0.4. Various AHL or antagonist molecules were added at 50μM and incubated an additional 30 minutes at 30°C, after which expression was induced by the addition of 100μM IPTG (isopropyl-β-D-thiogalactopyranoside) for 4 hours at 30° C. Cells were harvested by centrifugation and lysed in 20 mM imidazole pH 8.0, 100 mM NaCl, 0.5 mM EDTA and 5% glycerol via a Micro-fluidizer processor (MicroFluidics). Protein was purified via ion-exchange using a Hitrap Heparin HP column (GE Healthcare). Protein was eluted with a 100 mM to 1 M NaCl gradient. Fractions containing CviR were pooled and diluted in lysis buffer devoid of NaCl, and then further purified by cation exchange chromatography using a Hitrap SP HP column (GE Healthcare). CviR was eluted with a 100 mM to 1 M NaCl gradient. Fractions were pooled and 25% glycerol was added and protein was stored at −80°C.
Gel Mobility Shift Assays
DNA probes for gel mobility shift analyses were generated by standard polymerase chain reaction using primers with a 5′ 6-FAM (fluorescein) tag (Integrated DNA Technologies). The target probe contained about 100 nucleotides of the vioA promoter. The control probe contained 300 bases of vioB intergenic DNA. 37 ng of each probe was incubated for 20 minutes at room temperature with the indicated amounts of CviR (0, 0.1, 0.2, 0.3, 0.4 and 0.5 μM) and 1 μg/μL poly-dIdC in 1X Gel shift buffer (40 mM Tris HCl pH 8.0, 100 mM KCL, 2 mM EDTA, 2 mM DTT, 10% glycerol, 200 μg/mL BSA). Gel mobility shifts were performed on 6% TGE-polyacrylimide gels and visualized using a Storm 860 Imaging System (Molecular Dynamics).
Life Span Assays
C. elegans lifespan assays were completed with at least 100 wild type N2 worms for each condition. C. elegans eggs were harvested from a large population of gravid adults using a standard bleaching protocol (30 mL 5% bleach, 15 mL N KOH, 55 mL DH2O). Harvested eggs were placed on lawns of fresh E. coli OP50 and allowed to hatch and grow to the young adult (L4) stage before being moved onto lawns of C. violaceum. Worms were scored for survival each day and transferred to new C. violaceum lawns every two days until all worms had expired. C6-HSL, 4606-4237 and CL were added directly to NGM medium at 5 μM, 50 μM and 20 μM, respectively.
Chemical Synthesis
Derivatives of 4606-4237 were synthesized by a three step sequence. First, the substituted phenol (21 mmol) was combined with ethyl 4-bromo butyrate (21 mmol) and K2CO3 (42 mmol) in DMF (60 mL) and heated to 100°C for 1 hr. The reaction mixture was cooled to RT, diluted with water (150 mL) and extracted 4 times with heptane (50 mL). The heptane extracts were pooled, dried with anhydrous magnesium sulfate, and the solvent removed by rotary evaporation. The crude phenoxy ester was saponified without further purification by adding a 1:1 mixture of 1M NaOH and methanol and stirred overnight and RT. The reaction mixture was acidified with 6M HCl and the thick white precipitate was filtered and generally provided pure carboxylic acid in 60–70% yield (based on starting phenol). The resulting acid was coupled with either homoserine lactone or homocysteine lactone (1 molar equivalent) using DCC (1 molar equivalent) and triethylamine (3 molar equivalents) in dichloromethane (~40 mL) overnight at RT. Reaction mixtures were filtered, washed with 1M HCl, sat. NaHCO3, dried with anhydrous magnesium sulfate, and the solvent removed by rotary evaporation. The final 4606-4237 derivatives were purified by silica gel column chromatography using 2:1 dichloromethane:ethyl acetate as the eluent.
Supplementary Material
Figure S1. Structures of 15 LuxN Antagonist Molecules and Their Effects on Violacein Production (A) Structures and designations of the 15 identified LuxN antagonists. All molecules were purchased from Chemdiv. (B) The C. violaceum cviI-mutant was grown in the presence of no HSL (first bar), 5 μM exogenously supplied C6-HSL (second bar), or 5 μM exogenous C6-HSL and 50 μM of each antagonist (remaining bars). Violacein was methanol extracted and quantified by measuring the absorbance at 568 nm.
Figure S2. Structures and Activities of the 4606-4237 Molecule Derivatives The synthesis scheme and core structure of each molecule is identical and shown at the top with R representing the position of each side group listed below. The IC50 values in the E. coli CviR-dependent vioA-gfp assay in the presence of each molecule were calculated from triplicate variable-slope sigmoidal dose-response curves and are listed below the structures in Molar (M) units.
Figure S3. Solubility Analysis of CviR Protein solubility was assessed by SDS-PAGE analysis of E. coli whole cell (W) and soluble (S) extracts expressing CviR in the presence of C4-HSL (Lanes 2 and 3), C8-HSL (Lanes 4 and 5), C12-HSL (Lanes 6 and 7) and C14-HSL (Lanes 8 and 9). The L designates the molecular weight ladder.
Figure S4. C6-HSL and 4606-4237 Competition Assay Activation of vioA:gfp expression was measured as a function of C6-HSL concentration at 0, 1, 10 and 100 μM concentration of 4606-4237. Error bars represent the standard error of the mean for three independent trials.
Figure S5. Control Gel Mobility Shift Assay Control DNA probe was subjected to electrophoresis in the absence of CviR protein (Lane 1). 500 nM C6-HSL-loaded CviR (Lane 2) was incubated with control probe for 20 min prior to separation by electrophoresis.
Figure S6. Repression of Violacein Production by Gene CV1055 Violacein production was measured in a C. violaceum strain carrying a transposon insertion in gene cv1055 (this strain is called 31532P1). The assays were carried out with the strain carrying the empty vector, pJAK16 (white bar) or the vector gene Cv1055 (black bar) encoding the putative violacein repressor. Cells were grown overnight in triplicate and the violacein pigment was methanol extracted and measured as function of absorbance at 568 nm. Error bars represent the standard error of the mean.
Table S1.
Acknowledgments
This work was supported by HHMI, NIH grant 5R01GM065859, NIH grant 5R01 AI 054442, and NSF grant MCB-0639855 to B.L.B., and an NIH postdoctoral fellowship GM787552 to L.R.S. The antagonist screen was partly funded with federal funds supplied to the National Cancer Institute’s Initiative for Chemical Genetics, National Institutes of Health, under Contract No. N01-CO-12400 and has been performed with the assistance of the Chemical Biology Platform of the Broad Institute of Harvard and MIT. The C. violaceum cviI mutant (strain CV026) was a generous gift of Dr. P. Williams.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aballay A, Ausubel FM. Caenorhabditis elegans as a host for the study of host-pathogen interactions. Curr Opin Microbiol. 2002;5:97–101. doi: 10.1016/s1369-5274(02)00293-x. [DOI] [PubMed] [Google Scholar]
- Bassler BL, Wright M, Showalter RE, Silverman MR. Intercellular signalling in Vibrio harveyi: sequence and function of genes regulating expression of luminescence. Mol Microbiol. 1993;9:773–786. doi: 10.1111/j.1365-2958.1993.tb01737.x. [DOI] [PubMed] [Google Scholar]
- Bassler BL, Wright M, Silverman MR. Multiple signalling systems controlling expression of luminescence in Vibrio harveyi: sequence and function of genes encoding a second sensory pathway. Mol Microbiol. 1994;13:273–286. doi: 10.1111/j.1365-2958.1994.tb00422.x. [DOI] [PubMed] [Google Scholar]
- Cao JG, Meighen EA. Purification and structural identification of an autoinducer for the luminescence system of Vibrio harveyi. J Biol Chem. 1989;264:21670–21676. [PubMed] [Google Scholar]
- Dhakal BK, Lee W, Kim YR, Choy HE, Ahnn J, Rhee JH. Caenorhabditis elegans as a simple model host for Vibrio vulnificus infection. Biochem Biophys Res Commun. 2006;346:751–757. doi: 10.1016/j.bbrc.2006.05.168. [DOI] [PubMed] [Google Scholar]
- Freeman JA, Lilley BN, Bassler BL. A genetic analysis of the functions of LuxN: a two-component hybrid sensor kinase that regulates quorum sensing in Vibrio harveyi. Mol Microbiol. 2000;35:139–149. doi: 10.1046/j.1365-2958.2000.01684.x. [DOI] [PubMed] [Google Scholar]
- Fuqua C, Winans SC, Greenberg EP. Census and consensus in bacterial ecosystems: the LuxR-LuxI family of quorum-sensing transcriptional regulators. Annu Rev Microbiol. 1996;50:727–751. doi: 10.1146/annurev.micro.50.1.727. [DOI] [PubMed] [Google Scholar]
- Fuqua WC, Winans SC, Greenberg EP. Quorum sensing in bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. J Bacteriol. 1994;176:269–275. doi: 10.1128/jb.176.2.269-275.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furste JP, Pansegrau W, Frank R, Blocker H, Scholz P, Bagdasarian M, Lanka E. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene. 1986;48:119–131. doi: 10.1016/0378-1119(86)90358-6. [DOI] [PubMed] [Google Scholar]
- Gale EF. The Molecular basis of antibiotic action. 2. London; New York: Wiley; 1981. [Google Scholar]
- Geske GD, Mattmann ME, Blackwell HE. Evaluation of a focused library of N-aryl L-homoserine lactones reveals a new set of potent quorum sensing modulators. Bioorg Med Chem Lett. 2008;18:5978–5981. doi: 10.1016/j.bmcl.2008.07.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geske GD, O’Neill JC, Miller DM, Mattmann ME, Blackwell HE. Modulation of bacterial quorum sensing with synthetic ligands: systematic evaluation of N-acylated homoserine lactones in multiple species and new insights into their mechanisms of action. Journal of the American Chemical Society. 2007;129:13613–13625. doi: 10.1021/ja074135h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geske GD, Wezeman RJ, Siegel AP, Blackwell HE. Small molecule inhibitors of bacterial quorum sensing and biofilm formation. Journal of the American Chemical Society. 2005;127:12762–12763. doi: 10.1021/ja0530321. [DOI] [PubMed] [Google Scholar]
- Henke JM, Bassler BL. Three Parallel Quorum-Sensing Systems Regulate Gene Expression in Vibrio harveyi. J Bacteriol. 2004;186:6902–6914. doi: 10.1128/JB.186.20.6902-6914.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentzer M, Riedel K, Rasmussen TB, Heydorn A, Andersen JB, Parsek MR, Rice SA, Eberl L, Molin S, Hoiby N, et al. Inhibition of quorum sensing in Pseudomonas aeruginosa biofilm bacteria by a halogenated furanone compound. Microbiology. 2002;148:87–102. doi: 10.1099/00221287-148-1-87. [DOI] [PubMed] [Google Scholar]
- Hentzer M, Wu H, Andersen JB, Riedel K, Rasmussen TB, Bagge N, Kumar N, Schembri MA, Song Z, Kristoffersen P, et al. Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. Embo J. 2003;22:3803–3815. doi: 10.1093/emboj/cdg366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman A, Wood TK. Bacterial quorum sensing: signals, circuits, and implications for biofilms and disease. Annual review of biomedical engineering. 2008;10:145–167. doi: 10.1146/annurev.bioeng.10.061807.160536. [DOI] [PubMed] [Google Scholar]
- Jung K, Odenbach T, Timmen M. The quorum-sensing hybrid histidine kinase LuxN of Vibrio harveyi contains a periplasmically located N terminus. J Bacteriol. 2007;189:2945–2948. doi: 10.1128/JB.01723-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch B, Liljefors T, Persson T, Nielsen J, Kjelleberg S, Givskov M. The LuxR receptor: the sites of interaction with quorum-sensing signals and inhibitors. Microbiology. 2005;151:3589–3602. doi: 10.1099/mic.0.27954-0. [DOI] [PubMed] [Google Scholar]
- Kong KF, Vuong C, Otto M. Staphylococcus quorum sensing in biofilm formation and infection. Int J Med Microbiol. 2006;296:133–139. doi: 10.1016/j.ijmm.2006.01.042. [DOI] [PubMed] [Google Scholar]
- Larsen RA, Wilson MM, Guss AM, Metcalf WW. Genetic analysis of pigment biosynthesis in Xanthobacter autotrophicus Py2 using a new, highly efficient transposon mutagenesis system that is functional in a wide variety of bacteria. Arch Microbiol. 2002;178:193–201. doi: 10.1007/s00203-002-0442-2. [DOI] [PubMed] [Google Scholar]
- Lenz DH, Mok KC, Lilley BN, Kulkarni RV, Wingreen NS, Bassler BL. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell. 2004;118:69–82. doi: 10.1016/j.cell.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Martin M, Showalter R, Silverman M. Identification of a locus controlling expression of luminescence genes in Vibrio harveyi. J Bacteriol. 1989;171:2406–2414. doi: 10.1128/jb.171.5.2406-2414.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClean KH, Winson MK, Fish L, Taylor A, Chhabra SR, Camara M, Daykin M, Lamb JH, Swift S, Bycroft BW, et al. Quorum sensing and Chromobacterium violaceum: exploitation of violacein production and inhibition for the detection of N-acylhomoserine lactones. Microbiology. 1997;143(Pt 12):3703–3711. doi: 10.1099/00221287-143-12-3703. [DOI] [PubMed] [Google Scholar]
- Minogue TD, Carlier AL, Koutsoudis MD, von Bodman SB. The cell density-dependent expression of stewartan exopolysaccharide in Pantoea stewartii ssp. stewartii is a function of EsaR-mediated repression of the rcsA gene. Mol Microbiol. 2005;56:189–203. doi: 10.1111/j.1365-2958.2004.04529.x. [DOI] [PubMed] [Google Scholar]
- Minogue TD, Wehland-von Trebra M, Bernhard F, von Bodman SB. The autoregulatory role of EsaR, a quorum-sensing regulator in Pantoea stewartii ssp. stewartii: evidence for a repressor function. Mol Microbiol. 2002;44:1625–1635. doi: 10.1046/j.1365-2958.2002.02987.x. [DOI] [PubMed] [Google Scholar]
- Muh U, Schuster M, Heim R, Singh A, Olson ER, Greenberg EP. Novel Pseudomonas aeruginosa quorum-sensing inhibitors identified in an ultra-high-throughput screen. Antimicrob Agents Chemother. 2006;50:3674–3679. doi: 10.1128/AAC.00665-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council (U.S.). Committee on New Directions in the Study of Antimicrobial Therapeutics: New Classes of Antimicrobials., National Research Council (U.S.). Committee on New Directions in the Study of Antimicrobial Therapeutics: Immunomodulation., and ebrary Inc. Treating infectious diseases in a microbial world report of two workshops on novel antimicrobial therapeutics. Washington, D.C: National Academies Press; 2006. p. x.p. 92. [PubMed] [Google Scholar]
- Novick RP, Geisinger E. Quorum sensing in staphylococci. Annu Rev Genet. 2008;42:541–564. doi: 10.1146/annurev.genet.42.110807.091640. [DOI] [PubMed] [Google Scholar]
- Richard C. Chromobacterium violaceum, opportunist pathogenic bacteria in tropical and subtropical regions. Bulletin de la Societe de pathologie exotique (1990) 1993;86:169–173. [PubMed] [Google Scholar]
- Showalter RE, Martin MO, Silverman MR. Cloning and nucleotide sequence of luxR, a regulatory gene controlling bioluminescence in Vibrio harveyi. J Bacteriol. 1990;172:2946–2954. doi: 10.1128/jb.172.6.2946-2954.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoblom S, Brader G, Koch G, Palva ET. Cooperation of two distinct ExpR regulators controls quorum sensing specificity and virulence in the plant pathogen Erwinia carotovora. Mol Microbiol. 2006;60:1474–1489. doi: 10.1111/j.1365-2958.2006.05210.x. [DOI] [PubMed] [Google Scholar]
- Swem LR, Swem DL, Wingreen NS, Bassler BL. Deducing receptor signaling parameters from in vivo analysis: LuxN/AI-1 quorum sensing in Vibrio harveyi. Cell. 2008;134:461–473. doi: 10.1016/j.cell.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Throup J, Winson MK, Bainton NJ, Bycroft BW, Williams P, Stewart GSAB. Signalling in bacteria beyond bioluminescence. In: Campbell LKA, Stanley P, editors. Bioluminescence and Chemiluminescence: Fundamentals and Applied Aspects. Chichester: Wiley; 1995. pp. 89–92. [Google Scholar]
- Timmen M, Bassler BL, Jung K. AI-1 influences the kinase activity but not the phosphatase activity of LuxN of Vibrio harveyi. J Biol Chem. 2006;281:24398–24404. doi: 10.1074/jbc.M604108200. [DOI] [PubMed] [Google Scholar]
- Tu KC, Bassler BL. Multiple small RNAs act additively to integrate sensory information and control quorum sensing in Vibrio harveyi. Genes Dev. 2007;21:221–233. doi: 10.1101/gad.1502407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaitkevicius K, Lindmark B, Ou G, Song T, Toma C, Iwanaga M, Zhu J, Andersson A, Hammarstrom ML, Tuck S, et al. A Vibrio cholerae protease needed for killing of Caenorhabditis elegans has a role in protection from natural predator grazing. Proc Natl Acad Sci U S A. 2006;103:9280–9285. doi: 10.1073/pnas.0601754103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bodman SB, Ball JK, Faini MA, Herrera CM, Minogue TD, Urbanowski ML, Stevens AM. The quorum sensing negative regulators EsaR and ExpR(Ecc), homologues within the LuxR family, retain the ability to function as activators of transcription. J Bacteriol. 2003;185:7001–7007. doi: 10.1128/JB.185.23.7001-7007.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters CM, Bassler BL. QUORUM SENSING: Cell-to-Cell Communication in Bacteria. Annu Rev Cell Dev Biol. 2005;21:319–346. doi: 10.1146/annurev.cellbio.21.012704.131001. [DOI] [PubMed] [Google Scholar]
- Willcox MD, Zhu H, Conibear TC, Hume EB, Givskov M, Kjelleberg S, Rice SA. Role of quorum sensing by Pseudomonas aeruginosa in microbial keratitis and cystic fibrosis. Microbiology. 2008;154:2184–2194. doi: 10.1099/mic.0.2008/019281-0. [DOI] [PubMed] [Google Scholar]
- Wright JS, 3rd, Jin R, Novick RP. Transient interference with staphylococcal quorum sensing blocks abscess formation. Proc Natl Acad Sci U S A. 2005;102:1691–1696. doi: 10.1073/pnas.0407661102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang RG, Pappas T, Brace JL, Miller PC, Oulmassov T, Molyneaux JM, Anderson JC, Bashkin JK, Winans SC, Joachimiak A. Structure of a bacterial quorum-sensing transcription factor complexed with pheromone and DNA. Nature. 2002;417:971–974. doi: 10.1038/nature00833. [DOI] [PubMed] [Google Scholar]
- Zhu J, Winans SC. Autoinducer binding by the quorum-sensing regulator TraR increases affinity for target promoters in vitro and decreases TraR turnover rates in whole cells. Proc Natl Acad Sci U S A. 1999;96:4832–4837. doi: 10.1073/pnas.96.9.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Winans SC. The quorum-sensing transcriptional regulator TraR requires its cognate signaling ligand for protein folding, protease resistance, and dimerization. Proc Natl Acad Sci U S A. 2001;98:1507–1512. doi: 10.1073/pnas.98.4.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Structures of 15 LuxN Antagonist Molecules and Their Effects on Violacein Production (A) Structures and designations of the 15 identified LuxN antagonists. All molecules were purchased from Chemdiv. (B) The C. violaceum cviI-mutant was grown in the presence of no HSL (first bar), 5 μM exogenously supplied C6-HSL (second bar), or 5 μM exogenous C6-HSL and 50 μM of each antagonist (remaining bars). Violacein was methanol extracted and quantified by measuring the absorbance at 568 nm.
Figure S2. Structures and Activities of the 4606-4237 Molecule Derivatives The synthesis scheme and core structure of each molecule is identical and shown at the top with R representing the position of each side group listed below. The IC50 values in the E. coli CviR-dependent vioA-gfp assay in the presence of each molecule were calculated from triplicate variable-slope sigmoidal dose-response curves and are listed below the structures in Molar (M) units.
Figure S3. Solubility Analysis of CviR Protein solubility was assessed by SDS-PAGE analysis of E. coli whole cell (W) and soluble (S) extracts expressing CviR in the presence of C4-HSL (Lanes 2 and 3), C8-HSL (Lanes 4 and 5), C12-HSL (Lanes 6 and 7) and C14-HSL (Lanes 8 and 9). The L designates the molecular weight ladder.
Figure S4. C6-HSL and 4606-4237 Competition Assay Activation of vioA:gfp expression was measured as a function of C6-HSL concentration at 0, 1, 10 and 100 μM concentration of 4606-4237. Error bars represent the standard error of the mean for three independent trials.
Figure S5. Control Gel Mobility Shift Assay Control DNA probe was subjected to electrophoresis in the absence of CviR protein (Lane 1). 500 nM C6-HSL-loaded CviR (Lane 2) was incubated with control probe for 20 min prior to separation by electrophoresis.
Figure S6. Repression of Violacein Production by Gene CV1055 Violacein production was measured in a C. violaceum strain carrying a transposon insertion in gene cv1055 (this strain is called 31532P1). The assays were carried out with the strain carrying the empty vector, pJAK16 (white bar) or the vector gene Cv1055 (black bar) encoding the putative violacein repressor. Cells were grown overnight in triplicate and the violacein pigment was methanol extracted and measured as function of absorbance at 568 nm. Error bars represent the standard error of the mean.
Table S1.