

Abstract
A set of single-tryptophan mutants of the Na+/K+-ATPase isolated, large cytoplasmic loop connecting transmembrane helices M4 and M5 (C45) was prepared to monitor effects of the natural cytoplasmic ligands (i.e., Mg2+ and/or ATP) binding. We introduced a novel method for the monitoring of the changes in the electrostatic surface potential (ESP) induced by ligand binding, using the quenching of the intrinsic tryptophan fluorescence by acrylamide or iodide. This approach opens a new way to understanding the interactions within the proteins. Our experiments revealed that the C45 conformation in the presence of the ATP (without magnesium) substantially differed from the conformation in the presence of Mg2+ or MgATP or in the absence of any ligand not only in the sense of geometry but also in the sense of the ESP. Notably, the set of ESP-sensitive residues was different from the set of geometry-sensitive residues. Moreover, our data indicate that the effect of the ligand binding is not restricted only to the close environment of the binding site and that the information is in fact transmitted also to the distal parts of the molecule. This property could be important for the communication between the cytoplasmic headpiece and the cation binding sites located within the transmembrane domain.
Introduction
Most biochemical reactions are facilitated by the assistance of enzymes. The secret of the enzymes consists in their ability to adopt various conformations, and thus, to provide catalytic residues for chemical reactions, to open or close the pathways for solutes or to change affinities for ligands (1,2). However, description of the enzyme conformation is usually limited to the description of the geometrical features (3). Although the geometry analysis can provide lot of useful information, usually the information about molecular forces causing the movements or molecular interactions is missing. This study describes how we can monitor local changes in the electrostatic surface potential. As the electrostatic forces play an important role in the molecular interactions, this approach can deepen our understanding to the causes and consequences of the enzyme conformational changes. We describe the changes induced by the ligand binding to the cytoplasmic part of the Na+/K+-ATPase.
The Na+/K+-ATPase (sodium pump, EC 3.6.3.9) is the main cytoplasmic sodium regulator within all animal cells. During one catalytic cycle, the pump translocates three sodium ions across the plasma membrane out of the cell, and two potassium ions in the reverse direction; the transport is energized by the ATP hydrolysis (4). The steep gradient of sodium ions concentration created by the Na+/K+-ATPase substantially contributes to the plasma membrane potential and could be used for the secondary transport of other solutes. Hence, it is not surprising that inhibition or malfunction of Na+/K+-ATPase can result in variety of serious diseases, e.g., ischemia, diabetes, etc. (5,6).
Na+/K+-ATPase is a member of the superfamily of cation translocating membrane enzymes, designated as P-type ATPases (7). This designation has origin in the finding that the enzyme is transiently autophosphorylated on the conserved aspartyl residue during the catalytic cycle. Another common feature of these pumps is that they need the Mg2+ ions as a nontransported cofactor to their proper function (8). The first high-resolution structural information about the P-type ATPases was obtained in 2000 in the crystallographic studies of the Ca2+-ATPase from sarco(endo)plasmic reticulum (SERCA) (9). The SERCA was later crystallized under various conditions revealing the different conformations of this enzyme (10–16). A crystallographic structure of Na+/K+-ATPase at 3.5 Å resolution has been published recently (17). It confirms the close relationship of these two enzymes.
Two cation-binding sites (common to both sodium and potassium) were localized in the transmembrane domain. They are formed by the residues of the transmembrane helices M4, M5, M6, and M8 (17). The third binding site for the sodium ion has not been identified so far, but recent data revealed that truncation of the C-terminus greatly reduced the affinity to Na+ (17). Three large domains could be identified on the cytoplasmic side of the membrane. Domain A (actuator) is formed by the N-terminus and the loop between the second and third transmembrane helices (C23). The other two domains are formed by C45. The nucleotide-binding site was localized on the N-domain, whereas the conserved phosphorylation site Asp369 is localized on the central P-domain. The first crystal structure of SERCA showed that the three cytoplasmic domains were well separated each from other, and this structure was assigned to the conformation with high affinity to ATP (9). Later, crystallographic structures (including those of Na+/K+-ATPase) obtained in the presence of various ATP or phosphate analogs, showed several slightly different conformations, where these three domains interacted in the closed complex (10,11,13–15,17). Analysis of these structures revealed that N-domain can rotate by ∼90° around the hinge connecting the N- and P-domains, thus, mutually approaching the nucleotide binding and phosphorylation sites. The A-domain displays two kinds of motion during various conformational transitions. Combining the ∼110° rotation around the axis perpendicular to the membrane or ∼30° rotation around axis parallel with membrane, various A-domain residues can participate to the interactions on the cytoplasmic domains interfaces. Previous kinetic studies suggested that the enzyme conformational change could be the rate limiting step of the catalytic cycle (18).
Cytoplasmic C45 containing the N- and P-domains constitutes ∼40% of the α-subunit mass, and experiments showed that it could be isolated from the rest of the enzyme (19–21) retaining its structure, as well as dynamic and some of its functional (e.g., TNP-ATP- or low-affinity ATP-binding) properties (22–25). Recently, we have described the conformational changes of C45 induced by the cytoplasmic ligand(s) binding (from the point of view of geometry), using the intrinsic tryptophan fluorescence quenching by acrylamide, fluorescence anisotropy decay and molecular dynamic (MD) simulations. We showed that in the absence of any ligand, in the presence of Mg2+ alone or MgATP, the C45 adopted closed conformation, whereas addition of the ATP in the absence of Mg2+ induced the C45 opening (26), i.e., dynamic behavior that is expected for the corresponding part of the entire enzyme. In this study, modification of the quenching experiment (quenching by iodide) enabled us to analyze the conformational changes from the point of view of the electrostatic surface potential. Our data revealed that the set of the sensitive residues was different, and thus, this kind of information can be considered as complementary to the traditional geometrical approach. As the electrostatic forces play an important role in the inter- or intramolecular interactions (27,28), our results indicate that changes in the electrostatic surface potential could mediate the propagation of information about ATP binding to the distal parts of the molecule.
Materials and Methods
Site-directed mutagenesis and protein expression
The native sequence of the C45 from the mouse brain Na+/K+-ATPase contains two Trp residues on the position 385 and 411. The cloning of the isolated C45, subsequent replacement of the two native tryptophans by phenylalanines (WT-W385F, WT-W411F, and WT-W385FW411F = TL; tryptophanless), insertion of the mutations TL-F404W, TL-F426W, TL-F571W, TL-I627W, TL-F683W, and TL-S732W into the tryptophanless construct to yield the set of single tryptophan mutants (Fig. 1), the expression and purification of the (His)6-tagged proteins was described in detail previously (26). Additionally, the mutation TL-V648W was carried out using the upstream primer (altered nucleotides are in italic): TL-V648W 5′ggcatgtctggggttccactggttcactgg3′; the downstream primer was reverse complement.
Figure 1.

Locations of the Trp residues within our set of single-tryptophan mutants. The ATP binding site is represented by the residuum F475.
Tryptophan fluorescence quenching
Steady-state tryptophan fluorescence emission was quenched using acrylamide or KI as a quencher under conditions described previously (26). The excitation wavelength was set to 295 nm to avoid excitation of Tyr residues and the inner filter effect caused by ATP or iodide (in the cuvette with the optical path of 3 mm, both in excitation and emission, the absorbance of 15 mM or 250 mM KI is <0.01, and the inner filter effect is therefore negligible). Emission was set to 350 nm because i), the water-exposed Trp residues have typically emission spectra with maxima at wavelengths >340 nm; and ii), we aimed to avoid contamination of the signal by the Raman scattering of water, which appears near 330 nm. Quenching experiments were carried out in four different setups, i.e., without any ligand, with 15 mM MgCl2 (Lach-Ner, Brno, Czech Republic), 15 mM Na2ATP (Sigma, St. Louis, MO), or 15 mM MgATP (Sigma). The experiments in the absence of magnesium (i.e., free C45, or in the presence of Na2ATP) were carried out in the presence of 5 mM EDTA.
The efficiency of quenching was evaluated by the nonlinear least-squares analysis using the Stern-Volmer formula (29):
where F0 and F are the fluorescence intensities in the absence or in the presence of the quencher, respectively, KSV is the Stern-Volmer quenching constant and [Q] is the concentration of the quencher. Both the F0 and KSV were the fitted parameters, and the mean ± SD from three to five independent measurements was calculated. The bimolecular quenching constant was calculated as ratio of the KSV and the mean fluorescence lifetime.
The Stern-Volmer quenching constants obtained for the quenching by acrylamide or iodide can substantially differ. In addition to the fluorophor accessibility and fluorescence lifetime (that are the same in both cases), the quenching efficiency is influenced also by the diffusion coefficients of quenchers (diffusion coefficient of Trp on a bulky protein is negligibly small), and in the case of the negatively charged iodide (in contrast to the neutral acrylamide), also by electrostatic repulsion or attraction between the quencher and protein. The information about the electrostatic surface potential in the environment of the quenched tryptophan can be extracted by defining the charge parameter (CP) as:
The higher is this value, the more is the iodide attracted to the tryptophan by positively charged residues in its environment, and vice versa. Contribution of all the other factors vanish as they are common to both experiments, only the ratio of diffusion coefficients D(iodide)/D(acrylamide) remains as a multiplicative parameter, which is constant within the whole data set, and thus, does not affect the evaluation of the relative changes.
Note, that these assumptions hold true for the tryptophans located on surface of the protein. However, it was shown that the acrylamide is able to diffuse into the protein interior, in contrast to iodide, which is hydrated, and its ability to penetrate under the surface is very limited (30). Therefore, this approach should be used carefully, namely in the case of tryptophanyl residues buried under the protein surface. Fortunately, according to our model (26), all the examined tryptophans are located on the surface of the protein (except for native W385, which is partly buried), and therefore similarly accessible for both the acrylamide and iodide.
The SD of the CP (ΔCP) was calculated according to error propagation theory (31) as
where KKI and KAA represent the Stern-Volmer quenching constants for the iodide and acrylamide quenching, respectively, and ΔKKI and ΔKAA are the corresponding standard deviations. The significance of the differences in CP was evaluated using Student's t-test (Microsoft Excel utility).
Fluorescence decay measurement
Fluorescence decays of the mutant TL-V648W were measured using the time-correlated single photon counting (TCSPC) method on the PicoHarp300 instrument (PicoQuant, Berlin, Germany). The source of excitation pulses was pulsed LED centered at 298 nm (PLS300; PicoQuant) operating at 10 MHz frequency. The emission was sampled under magic-angle conditions through the monochromator set to 350 nm. The data were collected into the histogram spanning the 100 ns time-range with the 32 ps/channel resolution. The experiment proceeded until 10,000 counts in the peak channel were sampled. The instrument response function was measured using the colloid silica (Ludox, Sigma) as a scatterer. The least-squares fitting was carried out by the Levenberg-Marquardt method using the software FluoFit 4.2.1 (PicoQuant). The model used was the sum of exponentials with instrument response function (IRF) deconvolution:
and the intensity-weighted mean fluorescence lifetime was calculated as
All the experiments were carried out at 20°C (bath-controlled).
Electrostatic potential calculations
The electrostatic potential isocontour maps were calculated for the last snapshots of liganded and unliganded C45 MD simulations (C45-Na, C45-Na2ATP, C45-Mg, and C45-MgATP) reported in Grycova et al. (26) using the program APBS (32) as implemented in the PMV software (33). The dielectric constants of the protein and the solvent were set to 2 and 78.5, respectively. The partial atomic charges for amino acid residues were assigned by AMBER force field of the PMV built-in PDB2PQR converter.
Results
The intrinsic tryptophan fluorescence presents a unique probe for examination of molecular processes on proteins. However, the interpretation of the spectroscopic data is straightforward only for the proteins containing no more than a single tryptophanyl residue. Therefore, we have prepared the set of single-tryptophan mutants, where the Trp residues were placed as reporters on the various locations on the C45 surface. The expression levels of all mutants were high, suggesting that the carried out mutations did not influence the correct protein folding.
Fluorescence decay of the TL-V648W mutant
Fluorescence decays of the TL-V648W mutant were measured under various conditions (i.e., without any ligand, in the presence of Mg2+, MgATP, or Na2ATP). In all cases, the satisfactory fit with the reduced χ2 approaching 1.00, random distribution of the first residuals and autocorrelation function, was obtained for the 3-exponential fit (Table 1) and the decay scheme was very similar in all cases.
Table 1.
Estimated parameters for the fluorescence decays and acrylamide quenching of TL-V648W mutant
| Ligand | A1 | A2 | A3 | τ1 (ns) | τ2 (ns) | τ3 (ns) | τM (ns) | χR2 | KSV (M−1) | kq (109 M−1s−1) |
|---|---|---|---|---|---|---|---|---|---|---|
| None | 44.21 | 43.08 | 12.71 | 0.72 | 2.75 | 7.04 | 4.083 | 1.027 | 5.78 ± 1.35 | 1.42 ± 0.33 |
| Mg | 42.81 | 43.35 | 13.84 | 0.74 | 2.69 | 6.88 | 4.074 | 1.033 | 5.55 ± 1.07 | 1.36 ± 0.26 |
| MgATP | 45.68 | 43.48 | 10.84 | 0.75 | 2.87 | 7.45 | 4.108 | 0.966 | 5.64 ± 0.80 | 1.37 ± 0.19 |
| Na2ATP | 47.76 | 42.07 | 10.18 | 0.97 | 3.16 | 7.82 | 4.203 | 1.015 | 5.63 ± 0.97 | 1.34 ± 0.23 |
The τi represent calculated fluorescence lifetimes, Ai their normalized preexponential factors, τM is the intensity-weighted mean fluorescence lifetime, χR2 characterizes the goodness of fit. KSV represents the Stern-Volmer quenching constant for the acrylamide quenching, and kq the bimolecular quenching constant calculated as KSV/τM.
Acrylamide quenching of TL-V648W mutant
Acrylamide quenching can reveal the information about the steric accessibility of the fluorophor. In our previous study (26), we described how the ligand binding changes the protein conformation. In the absence of any ligand, in the presence of Mg2+, MgATP, or Na2ATP we observed the Stern-Volmer quenching constants (KSV) 5.78 ± 1.35 M−1, 5.55 ± 1.07 M−1, 5.64 ± 0.80 M−1, or 5.63 ± 0.97 M−1, respectively. These constants must be corrected for the possible changes of the excited-state lifetime (i.e., the time, during which the quencher can effectively quench the fluorescence). The bimolecular quenching constant (see Table 1) calculated as the ratio of the KSV and the mean fluorescence lifetime describes already the steric accessibility of the fluorophor. However, for the mutant TL-V648W no significant changes were observed on ligand binding. The acrylamide-quenching experiments for the other mutants were presented in our previous study (26). Briefly, in comparison to the unliganded C45, binding of Mg2+ resulted in decreased accessibility of Trp residue in the TL-F571W mutant only, binding of MgATP resulted in decreased accessibility of tryptophan in TL-F571W and WT-W411F mutants, and the most complex changes were observed on Na2ATP binding, where we observed decreased accessibility of tryptophan in WT-W411F, TL-F404W, TL-F571W, and TL-F683W mutants (26).
Iodide quenching
In comparison to the acrylamide quenching (26), the experiments using quenching by iodide ions (Table 2) carry also interesting information about the local electrostatic surface potential in the fluorophor microenvironment (Fig. 2). Within our set of mutants, the charge parameter (see Materials and Methods) ranged between values 1.35 and 3.50, showing that in the absence of any ligand, the F404 have the most negatively charged environment and the I627 and V648 are surrounded by the positively charged residues (Table 3).
Table 2.
Iodide quenching
| Protein | No substrate | Mg2+ | MgATP | Na2ATP |
|---|---|---|---|---|
| WT-W411F | 8.44 ± 0.40 | 9.19 ± 0.94 | 9.10 ± 0.50 | 8.44 ± 0.18 |
| TL-F404W | 9.96 ± 0.50 | 11.92 ± 0.75 | 9.47 ± 0.68 | 9.36 ± 1.02 |
| WT-W385F | 10.77 ± 0.93 | 12.21 ± 0.70 | 10.79 ± 0.20 | 10.36 ± 0.51 |
| TL-F426W | 12.78 ± 0.56 | 13.09 ± 1.00 | 12.53 ± 0.60 | 11.23 ± 1.05 |
| TL-F571W | 14.43 ± 1.23 | 12.55 ± 0.63 | 11.22 ± 0.13 | 9.65 ± 0.27 |
| TL-I627W | 14.27 ± 0.68 | 14.49 ± 0.73 | 15.25 ± 0.82 | 15.93 ± 0.84 |
| TL-V648W | 20.22 ± 2.38 | 18.49 ± 0.41 | 15.80 ± 1.80 | 10.42 ± 3.07 |
| TL-F683W | 10.32 ± 1.00 | 8.83 ± 0.34 | 10.21 ± 2.14 | 12.18 ± 1.12 |
| TL-S732W | 12.99 ± 0.75 | 11.93 ± 0.78 | 10.97 ± 0.66 | 10.84 ± 0.52 |
Stern-Volmer quenching constants for quenching of the Trp fluorescence by iodide given in M−1 are expressed as mean ± SD from three to five independent titrations. The native sequence of C45 contains two Trp residues on the positions 385 and 411. Thus, WT-W411F or WT-W385F designate mutants where only Trp385 or Trp411 was present, respectively, the other Trp residue was mutated to Phe. For all the other mutants, both the Trp385 and Trp411 were mutated to phenylalanines, and the designation of the protein indicates the position of the reporter Trp residue that was inserted into the tryptophanless (TL) mutant.
Figure 2.

Example of the data from the quenching experiments. Quenching of TL-V648W mutant by (A) acrylamide or (B) iodide in the absence of any ligand (squares) or in the Na2ATP presence (circles).
Table 3.
Charge parameters
| Protein | No substrate | Mg2+ | MgATP | Na2ATP |
|---|---|---|---|---|
| WT-W411F | 1.98 ± 0.10 | 2.39 ± 0.30 | 2.83 ± 0.18 | 2.55 ± 0.12 |
| TL-F404W | 1.35 ± 0.18 | 2.09 ± 0.29 | 1.77 ± 0.47 | 2.29 ± 0.47 |
| WT-W385F | 2.04 ± 0.22 | 2.19 ± 0.37 | 2.12 ± 0.42 | 2.02 ± 0.40 |
| TL-F426W | 1.77 ± 0.08 | 1.98 ± 0.19 | 1.82 ± 0.13 | 1.72 ± 0.22 |
| TL-F571W | 1.80 ± 0.24 | 2.73 ± 0.36 | 1.86 ± 0.13 | 1.74 ± 0.13 |
| TL-I627W | 3.35 ± 0.28 | 3.07 ± 0.24 | 3.37 ± 0.72 | 4.13 ± 0.87 |
| TL-V648W | 3.50 ± 0.91 | 3.33 ± 0.64 | 2.80 ± 0.51 | 1.85 ± 0.63 |
| TL-F683W | 1.80 ± 0.21 | 1.45 ± 0.28 | 2.07 ± 0.48 | 3.07 ± 0.36 |
| TL-S732W | 2.40 ± 0.19 | 2.33 ± 0.16 | 2.29 ± 0.34 | 2.28 ± 0.21 |
Mg2+ binding
The binding of magnesium caused changes in the charge distribution in the proximity of two residues located on the N-domain. When compared to the experiments in the absence of any substrate, the charge parameter increased from 1.35 ± 0.18 to 2.09 ± 0.29 (p < 0.05) or from 1.80 ± 0.24 to 2.73 ± 0.36 (p < 0.05) for the TL-F404W and TL-F571W mutants, respectively, suggesting a shift of the charge toward positive values. Contrary, for the residues located on the P-domain, no significant differences were observed.
MgATP binding
Similarly as in acrylamide-quenching experiments described in our previous study (26), binding of MgATP resulted only in little changes in the observed parameters for most of the mutants. Significant shift of the charge parameter from 1.98 ± 0.10 to 2.83 ± 0.18 (p < 0.01) was observed only in the experiments with the mutant containing the native W385 (WT-W411F mutant). It shows that the charge of the residue environment is shifted toward positive values. Experiments with the other residues showed no significant changes.
Na2ATP binding
Like the acrylamide quenching described in our previous study (26), the most complex changes were observed in the experiments carried out in the Na2ATP presence. On the N-domain, the charge parameter for the mutants WT-W411F or TL-F404W increased from 1.98 ± 0.10 to 2.55 ± 0.12 (p < 0.01) or from 1.35 ± 0.18 to 2.28 ± 0.47 (p < 0.02), respectively. For the TL-F571W mutant we observed change in KSV but not in the charge parameter, indicating only steric but not charge changes in the residue environment. For the other mutants (WT-W385F and TL-F426W) remained both KSV and charge parameter essentially unaltered. On the P-domain, the charge in the environment of the residue F683 is shifted toward positive values, as shown by the shift of the charge parameter from 1.80 ± 0.21 to 3.07 ± 0.36 (p < 0.02). Contrary, the charge in the V648 environment is shifted toward negative values, as shown by the charge parameter change from 3.50 ± 0.91 to 1.85 ± 0.63 (p < 0.02). For the mutants TL-I627W and TL-S732W, both the KSV and charge parameter varied only within the experimental error.
Electrostatic potential maps from the MD simulations
We attempted to correlate the above-mentioned spectroscopic results to the information from the MD simulations reported previously (26) and we calculated the electrostatic potential isocontour maps from the last snapshots of the MD simulations (Fig. 3). We observed a good agreement between the spectroscopic- and MD data for all the N-domain sensitive residues.
Figure 3.

Semitransparent electrostatic potential maps from the final snapshots of the MD simulations of the C45 in the absence of any ligand (C45-Na), in the presence of Mg2+ only (C45-Mg), in the presence of ATP only (C45-Na2ATP), and in the simultaneous presence of Mg2+ and ATP (C45-MgATP). Contour levels were set to −4.0 kT/e (red) and +4.0 kT/e (blue).The picture was generated using the PMV software (33). Residues W385 and F404 are displayed as yellow balls, the residue F571 as well as the nucleotide binding site are located on the rear side of the molecule.
The N-domain seems to be rather rigid (except for the region 393–410; see the Supporting Material), and we observe the positively charged environment of the nucleotide-binding pocket, and a large negatively charged cloud on the opposite site of the N-domain, which fully covers the residue W385, and F404 and F571 are located on its edge. This cloud is shifted on the Na2ATP or MgATP binding, and the W385 gets to the boarder of the cloud, which corresponds to the experimental observations that the electrostatic surface potential shifted toward positive values in these cases. The residue F404 seems to be displaced from the negatively charged cloud in all simulations with ligands. However, evaluation of the results for the F404 residue must be done with caution, as it is located within a very flexible region, and (e.g., in the case of MgATP binding) the difference in the experimentally determined charge parameter was not statistically significant on the p < 0.05 level. The previously suggested binding of the Mg2+ cation in the proximity of F571 (see Fig. 5A in Grycova et al. (26)) could explain the observation of the TL-F571W charge parameter change in the presence of Mg2+.
In contrast, we were not able to reliably link the spectroscopic and MD data for the P-domain. First, numerous P-domain regions became very flexible on Na2ATP binding (see the Supporting Material) and almost every snapshot offered substantially different isocontour map. Second, as reported previously, the last 13 C-terminal residues were truncated in our MD simulation to reduce computational demands. Although this truncation should not substantially influence the C45 geometry during the simulation, it can substantially influence the electrostatic potential maps. Hence, the comparison of MD and spectroscopic data requires further investigation in this case.
Discussion
Conformational changes of the proteins are frequently described only as changes in the molecule geometry. For the P-type ATPases, we have recently obtained detailed information from the crystallographic experiments (9–15,17). However, crystallography data suffer from the facts that buffer composition may be substantially different from the physiological conditions or that the natural ligands usually cannot be used (34). Therefore, these data should be complemented by the conventional spectroscopy experiments, which do not have these limitations (26,35,36). Furthermore, the geometrical approach usually describes only the initial and final states, however, providing only indirect idea about forces causing the movement. In this study, we describe an experimental approach allowing us to monitor the changes in the electrostatic surface potential, which can support the theoretical approaches reported previously (37,38).
The experiments describing the dynamic of transmembrane proteins present still a challenge due to numerous practical difficulties. Fortunately, the central cytoplasmic segment (C45) of Na+/K+-ATPase can be isolated as a soluble protein without the rest of the enzyme, retaining its structural and functional properties (19–25), which greatly facilitates the experimental work. In our previous study (26), we described that the C45 has also the dynamic properties that are expected for this part of the molecule within the entire enzyme. We found that the C45 adopted a closed conformation in the absence of any ligand, in the presence of Mg2+ or in the simultaneous presence of Mg2+ and ATP. Presence of ATP in the absence of Mg2+ induced the C45 opening. Data presented in this study confirmed that the C45 conformations are indeed similar in the absence of any ligand, in the presence of Mg2+ and in the presence of MgATP, whereas the conformation in the presence of ATP (without Mg2+) is substantially different. However, the set of sensitive residues is not the same, which was best demonstrated by the mutant TL-V648W that exhibits little changes from the point of view of the geometry, but large changes in the electrostatic surface potential. Other mutants, however (e.g., WT-W385F or TL-F571W), were sensitive to the changes in geometry (26), but showed no significant change in the electrostatic surface potential. Hence, the potential approach can be considered as complementary to the geometry approach. Moreover, the electrostatic interactions are long-range ones, and we propose that local changes in electrostatic surface potential could be important for the communication between distal parts of the molecule.
Communication between the C45 and other parts of the enzyme
The cation-binding sites are formed by the residues located on the helices M4, M5, M6, and M8 (9,17). Crystallographic experiments with SERCA showed completely different arrangement of the transmembrane helices in the E1 or E2 conformations, respectively. The complex changes were described in detail previously (10), and they concern all 10 of the transmembrane helices. In contrast, interaction partner for the external cofactors (ATP and Mg2+) on the cytoplasmic side is only the C45. Thus, it is interesting to see how the information about the cytoplasmic events can be propagated to the various parts of the transmembrane domain. The influence of the M4 and M5 by the processes occurring on the C45 is easily comprehensible, and it was shown that M4 moves toward cytoplasm in the E2 conformation and M5 inclines on nucleotide binding (10). The N-terminal transmembrane helices (M1–M3) are apparently linked to the C45 through the cytoplasmic A-domain, which is formed by the cytoplasmic N-terminus and C23. The crystalline structures showed that A-domain can form a compact assembly with the N- and P-domains (formed by C45) on nucleotide binding. It seems that the presence of the ATP molecule itself can be a sufficient signal for the A-domain to approach, because the C23- and C45-contact sites are located near the nucleotide-binding- or phosphorylation sites (10). In contrast, it is not straightforward to understand how the information about the nucleotide binding is transmitted to the C-terminal transmembrane helices (M6–M10), because they are located on the side that is reverse to the location of the ATP-binding site on the C45. Obviously, the transmembrane helices are tightly packed and could interact each with other. Nevertheless, numerous authors have already speculated that the short cytoplasmic loop C67 can mediate the information between C45 and C-terminal transmembrane helices (4,39–43). Comparison of our C45 MD simulations (26) with the crystal structure of the complete Na+/K+-ATPase suggests that the interaction partner on the C45 could be the flexible Asn642-Asn649 loop (Fig. 4 and Fig. S1). Indeed, when our reporter tryptophan was located on the position 648, we observed that the ESP in its environment shifted toward more negative values when the C45 adopted the open conformation (induced by Na2ATP binding) as compared to the closed conformation. We propose that this change in the local surface charge could serve as a signal for the charged residues on C67. This hypotheses is supported by mutagenesis experiments, where alterations of charged amino acids within C67 resulted in the decrease of Na+/K+-ATPase activity (40). However, further mutagenesis experiments are required to reveal details of this interaction.
Figure 4.
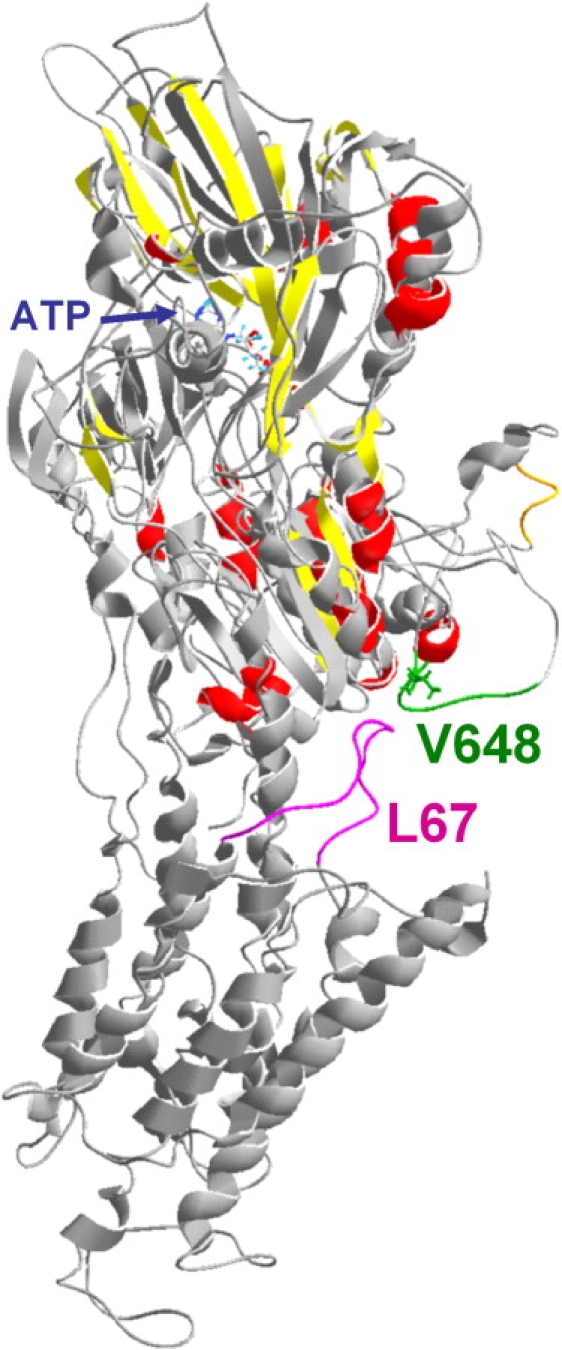
C45 to C67 signaling. The information about the ATP binding to the N-domain of the C45 (the top domain of the colored structure) could be mediated to distal parts of the molecule. Our experiments showed changes in the ESP when the reporter Trp residue was placed on the position 648 within the flexible Asn642-Asn649 turn (green in the final snapshot of the MD simulation of the C45 in the ATP presence described in Grycova et al. (26), or orange in the crystal structure of Na+/K+-ATPase). Comparison with the Na+/K+-ATPase crystal structure (gray; Protein Data Bank entry 3b8e) suggests that this turn could be in the proximity of the C67 (magenta).
Further, we have estimated that the surface charge of four residues located on both N- and P-domains shifted toward positive values. Interestingly, they are all located on the side of the C45 that is reverse to the nucleotide-binding site, on the C-terminal transmembrane helices M7-M10 (Fig. 5). Therefore, it is unlikely that this change could serve as a signal in the communication between the C45 and the A-domain. One may speculate that this could be a signal in the entry/exit pathway of the transported cations, for the cytoplasmic part of the γ-subunit or any other regulatory molecule. However, little structural information is available to support either of these hypotheses.
Figure 5.

Local changes in the ESP on ligand binding. The color code for the local surface charge changes on (A) Mg2+, (B) MgATP, or (C) Na2ATP binding: yellow are the residues with a ESP shift toward positive values, magenta for the residues without change, and cyan for the residue with a ESP shift toward negative values. The orange residue F475 is located within the ATP binding site. (D) View roughly perpendicular to the membrane. The structure of the entire Na+/K+-ATPase is rainbow-colored from the N-terminus (blue) to C-terminus (red), the transmembrane domain of the β-subunit is in gray, transmembrane helix of the γ-subunit is in magenta. Pink spheres represent residues with the positive ESP shift on Na2ATP binding (yellow in C; left to right, positions 404, 385, 683).
Conclusion
This study introduced a novel concept for the evaluation of the enzyme conformational changes. It is based on the monitoring of the local changes of the electrostatic surface potential using the intrinsic tryptophan fluorescence quenching. We found that the effect of the nucleotide binding to the C45 is not restricted only to the close environment of the binding site and that the information is in fact transmitted also to the distal parts of the molecule. We detected changes on the nucleotide binding within the set of residues that are located on the side that is reverse to the nucleotide-binding site. At the moment, however, we are not able to reliably link this observation to any step within the enzyme catalytic cycle. Further, our experiments showed ESP changes in the surrounding of the residue V648 on ATP binding, and we propose that this change could be a signal in the interaction of the Asn642-Asn649 loop and C67. This could be one of the important links in the communication between the cytoplasmic headpiece and the cation binding sites located within the transmembrane domain.
Acknowledgments
Karel Hron from the Department of Mathematical Analysis and Applications of Mathematics, Palacký University in Olomouc, is acknowledged for the expert assistance with the CP error estimation.
This work was supported by the grants of the Czech Science Foundation (GACR 203/07/0564, 203/09/H046, 303/07/0915), and the research projects of Czech Ministry of Schools, Youth and Sports (MSM 6198959215, MSM6198959216).
Supporting Material
References
- 1.Benkovic S.J., Hammes-Schiffer S. A perspective on enzyme catalysis. Science. 2003;301:1196–1202. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- 2.Kantarci-Carsibasi N., Haliloglu T., Doruker P. Conformational transition pathways explored by Monte Carlo simulation integrated with collective modes. Biophys. J. 2008;95:5862–5873. doi: 10.1529/biophysj.107.128447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wen P.C., Tajkhorshid E. Dimer opening of the nucleotide binding domains of ABC transporters after ATP hydrolysis. Biophys. J. 2008;95:5100–5110. doi: 10.1529/biophysj.108.139444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jorgensen P.L., Hakansson K.O., Karlish S.J.D. Structure and mechanism of Na,K-ATPase: Functional sites and their interactions. Annu. Rev. Physiol. 2003;65:817–849. doi: 10.1146/annurev.physiol.65.092101.142558. [DOI] [PubMed] [Google Scholar]
- 5.Therien A.G., Deber C.M. Oligomerization of a peptide derived from the transmembrane region of the sodium pump gamma subunit: effect of the pathological mutation G41R. J. Mol. Biol. 2002;322:583–590. doi: 10.1016/s0022-2836(02)00781-7. [DOI] [PubMed] [Google Scholar]
- 6.Yatime L., Buch-Pedersen M.J., Musgaard M., Morth J.P., Winther A.M.L. P-type ATPases as drug targets: tools for medicine and science. Biochim. Biophys. Acta. 2009;1787:207–220. doi: 10.1016/j.bbabio.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 7.Moller J.V., Juul B., leMaire M. Structural organization, ion transport, and energy transduction of P-type ATPases. Biochim. Biophys. Acta. 1996;1286:1–51. doi: 10.1016/0304-4157(95)00017-8. [DOI] [PubMed] [Google Scholar]
- 8.Skou J.C. Further investigations on a Mg++ + Na+-activated adenosinetriphosphatase, possibly related to the active, linked transport of Na+ and K+ across the nerve membrane. Biochim. Biophys. Acta. 1960;42:6–23. [Google Scholar]
- 9.Toyoshima C., Nakasako M., Nomura H., Ogawa H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 angstrom resolution. Nature. 2000;405:647–655. doi: 10.1038/35015017. [DOI] [PubMed] [Google Scholar]
- 10.Toyoshima C., Nomura H. Structural changes in the calcium pump accompanying the dissociation of calcium. Nature. 2002;418:605–611. doi: 10.1038/nature00944. [DOI] [PubMed] [Google Scholar]
- 11.Toyoshima C., Nomura H., Tsuda T. Lumenal gating mechanism revealed in calcium pump crystal structures with phosphate analogues. Nature. 2004;432:361–368. doi: 10.1038/nature02981. [DOI] [PubMed] [Google Scholar]
- 12.Toyoshima C., Mizutani T. Crystal structure of the calcium pump with a bound ATP analogue. Nature. 2004;430:529–535. doi: 10.1038/nature02680. [DOI] [PubMed] [Google Scholar]
- 13.Sorensen T.L.M., Moller J.V., Nissen P. Phosphoryl transfer and calcium ion occlusion in the calcium pump. Science. 2004;304:1672–1675. doi: 10.1126/science.1099366. [DOI] [PubMed] [Google Scholar]
- 14.Sorensen T.L.M., Olesen C., Jensen A.M.L., Moller J.V., Nissen P. Crystals of sarcoplasmic reticulum Ca2+-ATPase. J. Biotechnol. 2006;124:704–716. doi: 10.1016/j.jbiotec.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 15.Olesen C., Sorensen T.L.M., Nielsen R.C., Moller J.V., Nissen P. Dephosphorylation of the calcium pump coupled to counterion occlusion. Science. 2004;306:2251–2255. doi: 10.1126/science.1106289. [DOI] [PubMed] [Google Scholar]
- 16.Jensen A.M.L., Sorensen T.L.M., Olesen C., Moller J.V., Nissen P. Modulatory and catalytic modes of ATP binding by the calcium pump. EMBO J. 2006;25:2305–2314. doi: 10.1038/sj.emboj.7601135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morth J.P., Pedersen B.P., Toustrup-Jensen M.S., Sorensen T.L.M., Petersen J. Crystal structure of the sodium-potassium pump. Nature. 2007;450:1043–1046. doi: 10.1038/nature06419. [DOI] [PubMed] [Google Scholar]
- 18.Lupfert C., Grell E., Pintschovius V., Apell H.J., Cornelius F. Rate limitation of the Na+,K+-ATPase pump cycle. Biophys. J. 2001;81:2069–2081. doi: 10.1016/S0006-3495(01)75856-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Capieaux E., Rapin C., Thines D., Dupont Y., Goffeau A. Overexpression in Escherichia coli and purification of an ATP-binding peptide from the yeast plasma-membrane H+-ATPase. J. Biol. Chem. 1993;268:21895–21900. [PubMed] [Google Scholar]
- 20.Gatto C., Wang A.X., Kaplan J.H. The M4M5 cytoplasmic loop of the Na,K-ATPase, overexpressed in Escherichia coli, binds nucleoside triphosphates with the same selectivity as the intact native protein. J. Biol. Chem. 1998;273:10578–10585. doi: 10.1074/jbc.273.17.10578. [DOI] [PubMed] [Google Scholar]
- 21.Kubala M., Hofbauerova K., Ettrich R., Kopecky V., Krumscheid R. Phe(475) and Glu(446) but not Ser(445) participate in ATP-binding to the alpha-subunit of Na+/K+-ATPase. Biochem. Biophys. Res. Commun. 2002;297:154–159. doi: 10.1016/s0006-291x(02)02089-2. [DOI] [PubMed] [Google Scholar]
- 22.Kubala M., Plasek J., Amler E. Limitations in linearized analyses of binding equilibria: binding of TNP-ATP to the H-4-H-5 loop of Na/K-ATPase. Eur. Biophys. J. Biophys. Lett. 2003;32:363–369. doi: 10.1007/s00249-003-0278-y. [DOI] [PubMed] [Google Scholar]
- 23.Kubala M., Plasek J., Amler E. Fluorescence competition assay for the assessment of ATP binding to an isolated domain of Na+,K+-ATPase. Physiol. Res. 2004;53:109–113. [PubMed] [Google Scholar]
- 24.Kubala M., Teisinger J., Ettrich R., Hofbauerova K., Kopecky V. Eight amino acids form the ATP recognition site of Na+/K+-ATPase. Biochemistry. 2003;42:6446–6452. doi: 10.1021/bi034162u. [DOI] [PubMed] [Google Scholar]
- 25.Lansky Z., Kubala M., Ettrich R., Kuty M., Plasek J. The hydrogen bonds between Arg(423) and Glu(472) and other key residues, Asp(443), Ser(477), and Pro(489), are responsible for the formation and a different positioning of TNP-ATP and ATP within the nucleotide-binding site of Na+/K+-ATPase. Biochemistry. 2004;43:8303–8311. doi: 10.1021/bi0496485. [DOI] [PubMed] [Google Scholar]
- 26.Grycova L., Sklenovsky P., Lansky Z., Janovska M., Otyepka M. ATP and magnesium drive conformational changes of the Na+/K+-ATPase cytoplasmic headpiece. Biochim. Biophys. Acta. 2009;1788:1081–1091. doi: 10.1016/j.bbamem.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 27.Garciamoreno B., Chen L.X., March K.L., Gurd R.S., Gurd F.R.N. Electrostatic interactions in sperm whale myoglobin—site specificity, roles in structural elements, and external electrostatic potential distributions. J. Biol. Chem. 1985;260:4070–4082. [PubMed] [Google Scholar]
- 28.Warshel A., Sharma P.K., Kato M., Parson W.W. Modeling electrostatic effects in proteins. Biochem. Biophys. Acta. 2006;1764:1647–1676. doi: 10.1016/j.bbapap.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 29.Lakowicz J.R. Kluwer/Plenum; New York: 1999. Principles of Fluorescence Spectroscopy. [Google Scholar]
- 30.Albani J.R. Elsevier; Amsterdam, The Netherlands: 2004. Structure and Dynamics of Macromolecules: Absorption and Fluorescence Studies. [Google Scholar]
- 31.Arras, K. O. 1998. An introduction to error propagation: derivation, meaning and examples of equation Cy=FxCxFxT. In Technical Report of the Autonomous Systems Lab. Swiss Federal Institute of Technology Lausanne, EPFL-ASL-TR-98–01 R3.
- 32.Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanner M.F. Python: a programming language for software integration and development. J. Mol. Graph. 1999;17:57–61. [PubMed] [Google Scholar]
- 34.Kubala M. ATP-binding to P-type ATPases as revealed by biochemical, spectroscopic, and crystallographic experiments. Proteins. 2006;64:1–12. doi: 10.1002/prot.20969. [DOI] [PubMed] [Google Scholar]
- 35.Pratap P.R., Dediu O., Nienhaus G.U. FTIR study of ATP-induced changes in Na+/K+-ATPase from duck supraorbital glands. Biophys. J. 2003;85:3707–3717. doi: 10.1016/S0006-3495(03)74787-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Picard M., Toyoshima C., Champeil P. The average conformation at micromolar [Ca2+] of Ca2+-ATPase with bound nucleotide differs from that adopted with the transition state analog ADP center dot AlFx or with AMPPCP under crystallization conditions at millimolar. J. Biol. Chem. 2005;280:18745–18754. doi: 10.1074/jbc.M501596200. [DOI] [PubMed] [Google Scholar]
- 37.Mead-Savery F.C., Wang R., Tanna-Topan B., Chen S.R.W., Welch W. Changes in negative charge at the luminal mouth of the pore alter ion handling and gating in the cardiac ryanodine-receptor. Biophys. J. 2009;96:1374–1387. doi: 10.1016/j.bpj.2008.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ledvina P.S., Yao N.H., Choudhary A., Quiocho F.A. Negative electrostatic surface potential of protein sites specific for anionic ligands. Proc. Natl. Acad. Sci. USA. 1996;93:6786–6791. doi: 10.1073/pnas.93.13.6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clausen J.D., Andersen J.P. Functional consequences of alterations to Thr(247), Pro(248), Glu(340), Asp(813), Arg(819), and Arg(822) at the interfaces between domain P, M3, and L6–7 of sarcoplasmic reticulum Ca2+-ATPase—roles in Ca2+ interaction and phosphoenzyme processing. J. Biol. Chem. 2004;279:54426–54437. doi: 10.1074/jbc.M410204200. [DOI] [PubMed] [Google Scholar]
- 40.Xu G.Y., Kane D.J., Faller L.D., Farley R.A. The role of loop 6/7 in folding and functional performance of Na,K-ATPase. J. Biol. Chem. 2004;279:45594–45602. doi: 10.1074/jbc.M408147200. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Z.S., Lewis D., Sumbilla C., Inesi G., Toyoshima C. The role of the M6–M7 loop (L67) in stabilization of the phosphorylation and Ca2+ binding domains of the sarcoplasmic reticulum Ca2+-ATPase (SERCA) J. Biol. Chem. 2001;276:15232–15239. doi: 10.1074/jbc.M010813200. [DOI] [PubMed] [Google Scholar]
- 42.Lenoir G., Picard M., Moller J.V., le Maire M., Champeil P. Involvement of the L6–7 loop in SERCA1a Ca2+-ATPase activation by Ca2+ (or Sr2+) and ATP. J. Biol. Chem. 2004;279:32125–32133. doi: 10.1074/jbc.M402934200. [DOI] [PubMed] [Google Scholar]
- 43.Corre F., Jaxel C., Fuentes J., Menguy T., Falson P. Involvement of the cytoplasmic loop L6–7 in the entry mechanism for transport of Ca2+ through the sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 2002;277:13016–13028. doi: 10.1074/jbc.M108899200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.