

Abstract
Efficient synaptic transmission requires the apposition of neurotransmitter release sites opposite clusters of postsynaptic neurotransmitter receptors. Transmitter is released at active zones, which are composed of a large complex of proteins necessary for synaptic development and function. Many active zone proteins have been identified, but little is known of the mechanisms that ensure that each active zone receives the proper complement of proteins. Here we use a genetic analysis in Drosophila to demonstrate that the serine threonine kinase Unc-51 acts in the presynaptic motoneuron to regulate the localization of the active zone protein Bruchpilot opposite to glutamate receptors at each synapse. In the absence of Unc-51, many glutamate receptor clusters are unapposed to Bruchpilot, and ultrastructural analysis demonstrates that fewer active zones contain dense body T-bars. In addition to the presence of these aberrant synapses, there is also a decrease in the density of all synapses. This decrease in synaptic density and abnormal active zone composition is associated with impaired evoked transmitter release. Mechanistically, Unc-51 inhibits the activity of the MAP kinase ERK to promote synaptic development. In the unc-51 mutant, increased ERK activity leads to the decrease in synaptic density and the absence of Bruchpilot from many synapses. Hence, activated ERK negatively regulates synapse formation, resulting in either the absence of active zones or the formation of active zones without their proper complement of proteins. The Unc-51-dependent inhibition of ERK activity provides a potential mechanism for synapse-specific control of active zone protein composition and release probability.
Keywords: autophagy, NMJ, synapse, Drosophila, ATG, active zones, glutamate receptors
Introduction
At the synapse, presynaptic release machinery is directly apposed to neurotransmitter receptors within the postsynaptic density. The active zone is the site of neurotransmitter release, and is composed of a large, macromolecular complex of proteins that are involved in the development and function of the synapse (Fejtova and Gundelfinger, 2006). While many active zone proteins have been identified, little is known of the signaling pathways that regulate the assembly of these proteins and ensure that each active zone receives the proper complement of proteins.
The Drosophila neuromuscular junction is a favorite model system for identifying mechanisms that shape the development and function of the synaptic terminal, but until recently had been little used for the study of active zones (Collins and DiAntonio, 2007). This changed with the identification and characterization of Bruchpilot (Brp), the fly ortholog of the vertebrate active zone protein CAST (Wagh et al., 2006). Bruchpilot is present at every active zone, and is required for the active zone localization of presynaptic calcium channels and T-bars, a dense body specialization thought to promote efficient transmitter release (Kittel et al., 2006). Each Drosophila NMJ comprises hundred of active zones directly apposed to postsynaptic glutamate receptors, and these can be visualized with antibodies against Brp (Wagh et al., 2006) and the essential glutamate receptor subunit DGluRIII (Marrus et al., 2004). While each of these active zones is formed by the same presynaptic motoneuron, they have a heterogeneous release probability that correlates with the abundance of Brp (Marrus and DiAntonio, 2004). This suggests that mechanisms exist to regulate the efficacy of individual release sites, potentially by controlling the protein constituents of the active zone. To identify such mechanisms, we performed a large-scale screen for genes required to ensure that each postsynaptic cluster of glutamate receptors is apposed to an active zone containing Bruchpilot.
Here we report that the serine threonine kinase Unc-51 functions in the presynaptic neuron to ensure that Bruchpilot is apposed to glutamate receptors at each synapse. In the absence of Unc-51, many glutamate receptor puncta are unapposed to Brp, and ultrastructural analysis demonstrates that fewer active zones contain T-bars. In addition to these aberrant synapses, many fewer synapses form in the mutant and there is a large deficit in evoked transmitter release. Mechanistically, Unc-51 promotes synaptic development by inhibiting the activation of the MAP kinase ERK. In the unc-51 mutant, excess ERK activity is responsible for the decreased synaptic density and the absence of Bruchpilot from each synapse. We propose a model in which activated ERK negatively regulates synapse formation, leading to the formation of active zones without Bruchpilot or, in more extreme cases, the complete absence of the synapse. This Unc-51-dependent downregulation of ERK activity is a potential mechanism for synapse-specific control of active zone protein composition and release probability.
Materials and Methods
Fly stocks.
Flies were raised and maintained at 25°C on standard fly media. Wild-Type flies were either Canton S (CS) outcrossed to w−, elav-Gal4 (Yao and White, 1994), or G7-Gal4 (Zhang et al., 2001) based on the experiment. The following fly lines were obtained from the Bloomington Stock Center: P-element collection on the third chromosome (Bellen et al., 2004), deficiency line: Df(3L)BSC10, P-element insertions: P{EPgy2}Atg1EY09216 and P{EPgy2}Atg1EY07351 CG17667EY07351, rolled hypomorph, rl1 (Lim et al., 1999). Excision lines of Unc-51 (unc-513 and unc-5125) that delete the first exon was generated by the excision of the P-element line EP(P) 3348 (obtained from Szeged Drosophila Stock Centre) using Δ2–3 transposase line. Two unc-51 mutant alleles, unc-513 and unc-5125, were generated by imprecise excision, which deleted 804 bp and 1787 bp, respectively, toward downstream from the insertion point, deleting parts of the first exon, including the ATG initiation codon. These mutations caused pupal lethality and failed to complement each other. ΔATG13d line (Scott et al., 2004), another excision mutant was a gift from Dr. Thomas Neufeld (University of Minnesota, Minneapolis, MN). All five of these unc-51 mutant lines and the deficiency in all possible transheterozygote combinations gave qualitatively similar phenotypes. unc-513-rescue construct was generated by cloning the unc-51 cDNA (LD18893, obtained from the Drosophila Genomics Resource Center, Bloomington, IN) into a pUAST vector (Brand and Perrimon, 1993). To test whether varying the amounts of unc-51 would rescue the physiology defect, we used another motoneuron-specific driver OK-6 GAL4 (Aberle et al., 2002) and the C142-Gal4 driver, which expresses weakly in the neurons (de Jong et al., 2005), as well as altering the levels of expression by growing the flies at a lower (18°C) temperature. For the screen, all the lines that were not already balanced with a larval marker were crossed to a TM3, GFP balancer line, and non-GFP larvae were selected under fluorescence dissecting microscope with a GFP filter for dissection.
Immunohistochemistry.
Larvae were dissected and stained as described (Marrus et al., 2004; Wairkar et al., 2008). In brief, wandering third-instar larvae were dissected in cold PBS and fixed for ∼5–10 min in Bouin's fixative and washed at least three times for 5 min each in PBS + 0.1% Triton X-100. Primary antibodies were used in the following dilutions: rabbit anti-GluRIII, 1:2500 (Marrus et al., 2004); mouse anti-BRP, 1:250 (Wagh et al., 2006) [obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the National Institute of Child Health and Human Development and maintained by the Department of Biological Sciences of the University of Iowa (Iowa City, IA)]; rabbit anti-DVGLUT, 1:10,000 (Daniels et al., 2004); mouse anti-Dlg monoclonal antibody (mAb) 4f3, 1:2000 [developed by Corey S. Goodman (Renovis, San Francisco, CA)]; and Cy5-conjugated goat anti-HRP antibody, 1:1000 (Jackson ImmunoResearch). Goat Cy5-, Cy3-, and Alexa 488-conjugated secondary antibodies against mouse and rabbit IgG were used at 1:1000 and were obtained from Jackson ImmunoResearch.
Western blots.
Wandering third-instar larvae were dissected in ice-cold homogenization buffer (67 mm Tris-HCl, pH 8.0, 67 mm NaCl, 2 m urea, 1 mm EDTA, and 1.3% SDS) and stored immediately on dry ice. The samples were run on 10% SDS-PAGE gels according to standard procedures. Mouse anti-diphospho ERK (Sigma, catalog #M8159) and mouse anti-phospho p38 (Cell Signaling Technology) were used at 1:1000. The blot was stripped using standard procedures and then probed for nonphosphorylated ERK-MAP kinase using rabbit anti-rolled antibody (Peverali et al., 1996) at 1:1000. Mouse anti-syntaxin antibody (8C3) and mouse anti-β-tubulin (E7) antibody were obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the National Institute of Child Health and Human Development and maintained by the Department of Biological Sciences of the University of Iowa, were used at 1:100. HRP-conjugated goat anti-rabbit or anti-mouse secondary antibodies (Jackson ImmunoResearch) were used at 1:10,000.
Imaging and analysis.
Imaging and analysis was done as described previously (Viquez et al., 2006) with the following minor differences. The experimenter was blinded to genotypes during both imaging and analysis. Statistical analysis was performed using one-way ANOVA for comparison of samples within an experimental group. n for each condition is described either in the figures or in the results section. All histograms and measurements are shown as mean ± SEM. Bruchpilot (BRP) and DGluRIII puncta were manually counted at MN4b synapses on muscle 4. DGluRIII puncta that were not opposite to a detectable BRP punctum were counted as unapposed DGluRIII puncta. Area of NMJ was calculated by threshold function in MetaMorph for HRP channel. For the analysis of the axon transport phenotype, a box was drawn along a region of axon approximately between segments A3 and A4 and the same size box was used to measure the intensity of BRP in both mutants and wild type. HRP was used to threshold the area within the box. ImageJ was used to analyze the intensity of bands on the Western blots and the measure function in the program was used to quantify the intensity of bands. Ratio of the intensity of diphospho (activated) ERK bands to that of syntaxin bands was calculated and used for calculating the statistical difference between the genotypes. One-way ANOVA was performed to determine significance of the difference between band intensities in an experimental group. For electron microscopy analysis, bouton circumference was measured using the free hand tool in MetaMorph and by measuring around the outer margin of the bouton. T-bars and active zones were readily recognizable due to their electron dense appearance. Both of them were manually counted. An active zone was recognized by electron dense region of a bouton that had the presynaptic and postsynaptic membranes tightly apposed to each other. A T-bar was defined as a membrane structure protruding from the electron-dense active zones and surrounded by vesicles.
Electrophysiology.
Intracellular electrophysiological recordings were performed as previously described (Marrus et al., 2004). Briefly, wandering third-instar larvae were dissected in 0 mm Ca2+ Stewart saline (HL3) (Stewart et al., 1994). Both spontaneous miniature excitatory junction potentials (mEJPs) and evoked potentials (EJPs) were then recorded in either 0.4 or 0.42 mm HL3. Calcium concentration was identical for every cell within an experiment. At least 60 consecutive miniature events were measured per cell using MiniAnalysis (Synaptosoft) and averaged to determine mean mEJP. Events with a slow rise time were rejected as likely artifacts arising from neighboring electrically coupled muscle cells. To record evoked EJPs, segmental nerves were cut and suctioned into a stimulating electrode, in which they received a brief depolarizing pulse. Quantal content was estimated by dividing the mean EJP by the mean mEJP (EJP/mEJP). Cells across all genotypes had similar mean input resistances and resting potentials. Quantal size was not significantly different among any of the genotypes compared with wild-type controls. Statistical analysis was performed using one-way ANOVA to compare samples in an experimental group.
Electron microscopy.
The larval head and tail were pinned and a dorsal slit was made lengthwise, thus filleting the larvae – in phosphate buffer. The larvae were then placed into fixative (2% paraformaldehyde/2.5% glutaraldehyde in cacodylate buffer, pH 7.3) o/n. The larvae were transferred to cacodylate buffer, postfixed in 1% osmium tetroxide for 1 h at 4°C, then washed three times with cacodylate buffer. The larvae were dehydrated through 60 (1×, 7 min), 70, 80, 95 and 100% EtOH (2×, 10 min each step), transferred into propylene oxide (2×, 10 min), then into a 1:1 mixture of propylene oxide and Eponate, and left o/n, capped and at room temp. The larvae were then placed into fresh Eponate and into a mold, oriented and allowed to polymerize at 70°C. Thin sections were made and placed on Superfrost/plus micro slides, heated on a hot plate and stained with Toluidine Blue “O.” Type 1b boutons from NMJ6/7 in segment A2 from wild-type and mutant larval neuromuscular junctions were identified from the thin sections. Sections were cut at 50 nm with a diamond knife, picked up on formvar-coated, copper slot grids, and stained with 5% aqueous uranyl acetate for 15 min followed by Sato's lead stain for 1 min. Samples were observed and photographed in an FEI Tecnai G2 transmission electron microscope.
Results
Drosophila Unc-51 controls synaptic density and apposition
Each Drosophila neuromuscular junction comprises hundreds of individual synapses. At each synapse, the presynaptic machinery for neurotransmitter release is directly apposed to the postsynaptic machinery for neurotransmitter detection. This apposition of presynaptic and postsynaptic specializations is essential for the efficacy of synaptic transmission. To identify molecular pathways that regulate this apposition process, we have undertaken a genetic screen for mutants that disrupt the apposition of presynaptic active zones to postsynaptic glutamate receptor clusters. As an assay for synaptic apposition, we have double-stained for the active zone protein Bruchpilot [Brp, the Drosophila ortholog of the vertebrate active zone protein CAST (Wagh et al., 2006)], and the essential postsynaptic glutamate receptor subunit DGluRIII (Marrus et al., 2004). At a wild-type NMJ, essentially every Brp punctum is aligned with a DGluRIII punctum (Marrus and DiAntonio, 2004) (Fig. 1). We screened a collection of ∼1500 P-element insertion lines on the third chromosome for mutants (Bellen et al., 2004) with abnormalities in the alignment of BRP and DGluRIII. We identified one line, P{EPgy2}Atg1EY09216, in which many DGluRIII clusters are not apposed by a detectable BRP punctum. This P-element is inserted in ATG1 (also called Pegarn), a serine-threonine kinase that is the Drosophila ortholog of Unc-51 from C. elegans and vertebrates (Scott et al., 2004; Ahantarig et al., 2008). Atg1 participates in autophagy in Drosophila, while Unc-51 is required for normal axon outgrowth in mouse and worms (Tomoda et al., 1999; Scott et al., 2004; Tomoda et al., 2004). Due to the confusion in cross-species nomenclature, we will refer to the gene here by the name used in mammals and worms, unc-51. In Drosophila, there is a predicted gene within the intron of unc-51, CG17667, which could also be disrupted by the P-element insertion. Therefore, we obtained additional P-element insertions lines (see Materials and Methods) and generated imprecise excision mutants from a P-element (EP(3)3348), which is inserted within the first exon of the unc-51 gene, 539 bp upstream of the ATG initiation codon. Two independent excision lines (unc-513 and unc-5125) were identified which remove the first exon of unc-51 without disrupting CG17667. Larvae homozygous for each of these alleles also showed unapposed glutamate receptor puncta. To quantify these data, we counted the number of BRP puncta and DGluRIII puncta made by the MN4b motoneuron onto muscle four, although qualitatively similar results were observed at every type I glutamatergic NMJ. In the unc-513 mutant ∼40% of glutamate receptors are not apposed to any detectable BRP puncta (Fig. 1). We see a qualitatively similar apposition phenotype with transheterozygous combinations of the excision mutants with multiple P-insertion lines as well as a deficiency line (Df(3L)BSC10) that deletes the unc-51 gene (supplemental Fig. 1, available at www.jneurosci.org as supplemental material, and data not shown). Since multiple alleles show the same phenotype and, as is shown below, unc-51 transgene expression rescues the phenotype, we conclude that unc-51 is required for proper apposition of active zones and glutamate receptors.
Figure 1.

Unc-51 is required for normal synaptic density and apposition. A, Sample confocal images of larval NMJs from muscle 4 stained with antibodies against BRP (red), DGluRIII (green), and HRP (blue). The genotypes of the larvae are as follows: WT (CS crossed to elav-GAL4), unc-513 deletion mutant, and unc-513-Rescue (elav-GAL4/UAS-unc-51; unc-513). Insets show magnified view of boutons. Note the many DGluRIII puncta (arrowheads) without a detectable BRP punctum in the mutant. B–E, Histograms of NMJ area (B), glutamate receptor density (C), BRP density (D), and ratio of BRP to DGluRIII (E) at WT, unc-513, and unc-513-Rescue NMJs, muscle 4. n = 10 for all the genotypes; ***p < 0.001 and **p < 0.01. Error bars represent SEM. Scale bar, 2 μm.
While we screened for an apposition defect, these mutants display a number of other interesting phenotypes. First, the size of the NMJ is much smaller in the mutant, with an approximately fourfold reduction in the total area (Fig. 1B). However, the change in the size of the NMJ is not commensurate with the change in the number of synapses. When we counted the number of DGluRIII puncta, which are present at every synapse in wild type, we found a more than sixfold decrease (WT: DGluRIII number = 242 ± 22, n = 9; unc-513: DGluRIII number = 39 ± 6, n = 9; p < 0.001). Calculating the density of DGluRIII puncta reveals a >30% reduction in mutant (Fig. 1C). The reduction in the number of Brp puncta is even more striking, showing a nearly 10-fold decrease (WT: BRP number = 242 ± 24, n = 9; unc-513: BRP puncta = 25 ± 8, n = 9; p < 0.001), leading to a >60% reduction in the density of Brp puncta in the mutant (Fig. 1D). These results demonstrate that there is a dramatic decrease in both the absolute number and density of synapses in the mutant. Finally, we observed large accumulations of Brp (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) and the synaptic vesicle protein DVGLUT (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) in mutant axons. Such accumulations are a hallmark of defective axon transport (Duncan and Goldstein, 2006).
To demonstrate that these phenotypes result from the loss of unc-51 and to determine where gene function is required, we performed genetic rescue experiments. We generated transgenic flies carrying the unc-51 gene under the control of UAS promoter (Brand and Perrimon, 1993). Expression of the transgene in the motoneuron using the neuronal driver elav-Gal4 rescues all morphological phenotypes associated with unc-51 mutants. In the rescued mutant the synaptic defects are fully rescued: the density of DGluRIII puncta, the density of BRP puncta, and the apposition of Brp puncta to DGluRIII puncta are all returned to wild-type levels (Fig. 1C–E). In addition, the NMJ area is increased more than twofold compared with the mutant (Fig. 1B), while the axonal accumulations are decreased by about half (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Postsynaptic expression of the unc-51 transgene using G7-Gal4 driver does not rescue any of the defects (data not shown). Hence, unc-51 is required in the presynaptic motoneuron. In addition, selectively inhibiting Unc-51 function in the presynaptic cell via expression of a transgenic RNAi line phenocopies the unc-51 mutant. Neuronal RNAi knockdown leads to unapposed DGluRIII puncta, a reduction in synaptic density, a decrease in NMJ area, and accumulation of synaptic material in the axon including BRP and vesicle associated proteins like DVGLUT (supplemental Fig. 1, available at www.jneurosci.org as supplemental material, and data not shown), suggesting that the axon transport defect is not specific for a particular synaptic protein. Hence, the wild-type function of unc-51 is to act in the motoneuron to promote a high density of synapses, and to ensure that molecular constituents of the release site localize opposite to each postsynaptic density.
In some mutants with axon transport defects such as dynactin mutants, a smaller NMJ is observed and is due in part to retraction of the presynaptic terminal (Eaton et al., 2002). Such retraction could not explain the synaptic apposition defect of the unc-51 mutant, because unapposed DGluRIII puncta are present in a salt-and-pepper pattern throughout the NMJ, rather than only in the distal boutons as would be predicted for the progressive retraction of the synaptic terminal. However the small size of the NMJ in the mutant could be due to such retraction, so we assayed for synaptic retraction in the unc-51 mutant. The hallmark of synaptic retraction is the presence of “synaptic footprints,” in which postsynaptic markers such as the PDZ-protein Dlg are unapposed to synaptic vesicle proteins or presynaptic membrane (Eaton et al., 2002). To test whether unc-51 mutants have a retraction phenotype, we stained wild type and unc-51 mutants for the synaptic vesicle protein DVGLUT and the presynaptic membrane marker HRP, as well as the postsynaptic marker Dlg. We did not observe synaptic footprints, even in severe cases in which the NMJ size was drastically reduced (Fig. 2). Hence, the small size of the NMJ in the unc-51 mutant is not due to a failure to maintain the presynaptic terminal at the NMJ, but instead suggests a defect in the development of the NMJ.
Figure 2.

unc-51 mutants do not show a retraction-like phenotype. Sample NMJs stained with antibodies against postsynaptic membrane marker Discs-Large (Dlg) (red), presynaptic vesicle marker DVGLUT (green), and presynaptic membrane marker HRP (blue). The following genotypes were used: WT (CS crossed to w−) and unc-513 deletion mutant. Even in severe cases (as the example shown in the figure), the presynaptic marker DVGLUT is present opposite the postsynaptic Dlg protein.
The density of active zones and T-bars is decreased in Unc-51 mutants
Our confocal analysis of synaptic structure makes several predictions about synaptic ultrastructure in the unc-51 mutant. First, immunoelectron microscopic studies of glutamate receptors reveal that glutamate receptors are clustered opposite every active zone (Petersen et al., 1997). Since the density of DGluRIII puncta is reduced in the mutant, we predict a decrease in active zone density. Second, Brp is necessary for the localization of the dense body T-bar to the active zone (Kittel et al., 2006). Since many DGluRIII puncta are unapposed to Brp, we predict that there will be fewer T-bars/active zone in the mutant. In addition, since the density of Brp puncta is decreased, we also expect a decrease in the density of T-bars. Electron microscopic analysis of wild-type and unc-51 mutant NMJs from type 1b boutons confirmed our predictions. In wild-type type Ib boutons are filled with vesicles and are surrounded by a subsynaptic reticulum, which is an extensive postsynaptic membrane-infolding (Fig. 3). Active zones are apparent as regions of increased electron density where the presynaptic and postsynaptic membranes are tightly apposed. The T-bar is a dense body specialization thought to promote release of transmitter that is observed as a T-like structure at a subset of active zones. Typically, synaptic vesicles are seen clustered near the T-bars. While the mutant the synaptic ultrastructure is broadly similar to wild type, with active zones, T-bars, and synaptic vesicles, we observed a number of quantitative phenotypes that are consistent with our light-level analysis. First, the density of active zones is decreased by ∼25% in the mutant (Fig. 3B), consistent with the ∼35% decrease in DGluRIII puncta measured at the light level. Second, unc-51 mutants showed a 33% decrease in active zones containing a T-bar (Fig. 3C), which corresponds to the ∼40% of unapposed DGluRIII puncta seen with the confocal. Finally, the mutants also exhibit a >50% decrease in the density of T-bars (Fig. 3D), which is in excellent agreement with the ∼60% reduction in the density of Brp puncta observed with light microscopy. Thus in the absence of unc-51, synaptic density is dramatically decreased and is accompanied by many abnormal active zones which lack T-bar specializations.
Figure 3.
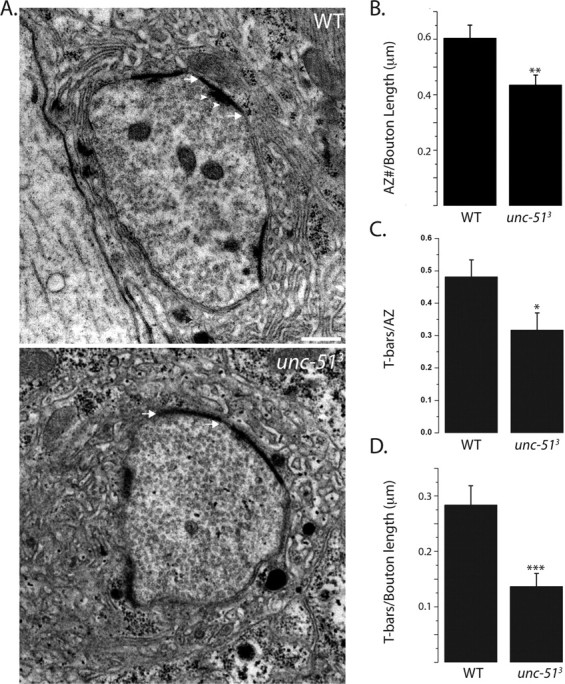
unc-51 mutants have a decreased density of T-bars. A, Sample electron micrographs of 1b boutons from WT and unc-513 mutant larvae. Both the genotypes have an SSR and active zones seen as electron dense regions of presynaptic and postsynaptic membranes (arrows). T-bars are seen in the WT as a T-shaped structure (arrowheads) and typically surrounded by synaptic vesicles. B–D, Histograms show active zone number per bouton circumference (B), T-bars per active zones (C), and T-bars per bouton circumference (D). WT, n = 20 boutons and unc-513, n = 30 boutons; *p < 0.05, **p < 0.01, ***p < 0.001. Error bars represent SEM. Scale bar, 1 μm.
unc-51 mutants display defects in neurotransmitter release
The unc-51 mutant exhibits defects in both the number and structure of active zones, either of which could impair synaptic function. To investigate the strength of synaptic transmission in this mutant, we measured both evoked and spontaneous neurotransmitter release at the NMJ. We performed intracellular electrophysiological recordings from muscle 6, segment A2 or A3 from both wild type and unc-51 mutants (Fig. 4). Consistent with the morphological defects, there is an approximately threefold reduction in the amplitude of evoked excitatory junction potentials (EJPs) in two different allelic combinations of unc-51 mutants compared with wild type (Fig. 4C). A defect in EJP amplitude could be due to the release of fewer vesicles from the nerve, or to a decrease in postsynaptic sensitivity to transmitter. To distinguish between these possibilities, we recorded spontaneous miniature junctional potentials (mEJPs), which are a measure of the postsynaptic response to the fusion of a single synaptic vesicle. In contrast to evoked release, there is no significant difference in the amplitude of mEJPs in these genotypes (WT = 0.88 ± 0.08 mV, unc-513 = 0.73 ± 0.06 mV, unc-513/16816 = 0.80 ± 0.03 mV, n = 10, p = 0.1), suggesting a presynaptic defect in transmitter release. Quantal content is a measure of the number of synaptic vesicles released by the presynaptic terminal following an action potential. Calculating quantal content by the direct method (EJP/mEJP) reveals a three-to-fourfold reduction in the number of synaptic vesicles released in the mutant (Fig. 4D). Consistent with a defect in presynaptic release, we observe a decrease in the frequency of mEJPs in both unc-51 mutants, although the difference is only statistically significant in one genotype (WT = 2.4 ± 0.2, unc-513 = 2.0 ± 0.3 and unc-513/16816 = 1.6 ± 0.2, p < 0.05 for WT: unc-513/16816 and p = 0.3 for WT: unc-513, n = 10). These findings demonstrate that unc-51 is required for normal synaptic transmission, potentially due to the reduced number of morphologically normal synapses in the mutant.
Figure 4.

Evoked transmitter release is impaired in unc-51 mutants. A, Representative spontaneous mEJPs from the following genotypes: WT (CS or eLav crossed to w−), unc-513 deletion mutants, hypomorphic allele unc-513/16816 (unc-513/P{EPgy2}Atg1EY07351 CG17667EY07351), elavGal4/unc-513-rescue, unc-513, and a wild-type larva overexpressing unc-51 presynaptically (elav-Gal4 crossed to UAS-unc-513-Rescue transgene). B, Representative evoked EJPs from the same genotypes as in A. C, D, Histograms show the mean EJP amplitudes (C) and quantal content (D). Quantal content was calculated by dividing the EJP amplitude by the mEJP amplitude for the same cell. ***p < 0.001. Error bars represent SEM and n ≥ 9 for all genotypes.
Transgenic expression of unc-51 rescues the morphological phenotypes of the unc-51 mutant, so we expected that that it would also rescue the physiological phenotypes. Surprisingly, we were unable to rescue the quantal content defect (Fig. 4). Since Unc-51 is a kinase, its overexpression may be deleterious and hence obscure our efforts to observe rescue. To test this possibility, we overexpressed unc-51 in motoneurons in an otherwise wild-type background and observed more than threefold reduction in quantal content (Fig. 4D) as well as a modest reduction in mEJP frequency (WT: 2.4 ± 0.2, unc-513-rescue: 1.3 ± 0.2, unc-513-rescue-overexpression: 1.1 ± 0.1, n ≥ 9 for all genotypes and p < 0.01) (Fig. 4). This demonstrates that the level of unc-51 expression is important for synaptic transmission. Overexpression of unc-51 is unlikely to act as a dominant negative, as its overexpression does not lead to any of the synaptic morphology defects characteristic of the loss-of-function mutant (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). In an effort to express lower levels of unc-51 we tested other Gal4 lines and varied temperature (see Materials and Methods), but we did not find a combination that rescued the physiological phenotype. The failure to rescue this one aspect of the phenotype indicates that either the electrophysiological defect is not due to the loss of unc-51 or that this phenotype is sensitive to the precise levels or timing of unc-51 expression. We suggest that it is likely that the defect in evoked release is due to the loss of unc-51 because the same phenotype is present in all tested unc-51 alleles, the phenotype is consistent with the morphological defects, and both the morphological and physiological defects can be suppressed by inhibiting a downstream target of Unc-51 (see below).
Unc-51 inhibits the activation of the MAP kinase ERK
What might be the mechanism by which Unc-51 regulates the structure and function of synapses? Unc-51 participates in many different pathways in different systems. In yeast, Drosophila, and vertebrates, orthologs of Unc-51 participate in starvation-induced autophagy, a lysosomal process of cytoplasmic degradation that provides nutrients to the cell under starvation conditions (Kamada et al., 2000; Scott et al., 2004; Aladzsity et al., 2007; Cheong and Klionsky, 2008). We tested mutants in other genes required for autophagy and find they do not share the synaptic phenotypes of the unc-51 mutants (Y. Wairkar and A. DiAntonio, unpublished observations). Therefore, the observed synaptic defects are not likely due to a generic block in autophagy. In addition, Unc-51 interacts with microtubule-associated proteins and so could regulate axon transport (Okazaki et al., 2000). Since we see defects consistent with disrupted axon transport, this function is likely conserved in Drosophila. However, as shown below, we have genetically separated the axon transport and synaptic defects, and thus defective transport cannot be the cause of the synaptic defects. Finally, Unc-51 interacts with a number of proteins that function upstream of the ERK-MAP kinase pathway, including the ras-GAP SynGAP and the FGF receptor binding protein FRS 2/3 (Tomoda et al., 2004; Avery et al., 2007). While these interactions are well described, to our knowledge Unc-51-dependent changes in ERK activity in vivo have not been reported. To test the hypothesis that Unc-51 regulates ERK activity, we measured the levels of active diphosphorylated ERK (dp-ERK) from brain and body wall preparations of wild-type and unc-51 mutant larvae. Immunoblot analysis reveals an increase in the levels of dp-ERK in the unc-51 mutants that is rescued by the neuronal expression of unc-51 (dp-ERK: WT = 100% ± 20%, unc-513 = 184% ± 13%, unc-513 + rescue = 101% ± 17%, n = 3, p = 0.02). In these genotypes there is no change in the levels of total ERK (WT = 18.3 ± 2.6 AU, unc-513 = 15.3 ± 5.3 AU, rescue = 13 ± 4.1 AU, n = 3, p > 0.6) or syntaxin (WT = 41.3 ± 9.7 AU, unc-513 = 42.6 ± 6.6 AU, rescue = 45.3 ± 9.1 AU, n = 3, p > 0.9) (Fig. 5). Hence, unc-51 is required in vivo to inhibit ERK activity.
Figure 5.

Unc-51 inhibits ERK activation. Western blot probed using anti-diphospho (activated) ERK, anti-rolled and anti-syntaxin antibody. Activated ERK is increased in unc-51 mutants compared with wild type (n = 3, p < 0.05). Neuronal expression of unc-51 transgene in the mutant rescues the increase in activated ERK levels (n = 3, p < 0.05). Levels of ERK are not significantly different among the three genotypes (n = 3, p = 0.2). Genotypes used are as follows: WT (CS crossed to elav-GAL4), unc-513 deletion mutant, and unc-513-Rescue (elav-GAL4/UAS-unc-51; unc-513).
Mutations in ERK suppress the synaptic defects of unc-51 mutants
If excess ERK activity contributes to the unc-51 synaptic phenotypes, then reducing ERK activity should suppress those defects. ERK null mutants are embryonic lethal (Biggs et al., 1994), so we generated a double mutant between unc-513 and the well characterized ERK (rolled) hypomorph, rl1, that survives past the larval stage (Lim et al., 1999). We stained the NMJs of wild-type, unc-513 mutant, rl1 mutant, and unc-513, rl1 double mutant larvae for the active zone component Brp and the glutamate receptor DGluRIII. Quantitative analysis was performed at the identified NMJ formed by the Mn4b motoneuron onto muscle 4, although results were qualitatively similar at all type 1 glutamatergic NMJs. There is no significant difference in the NMJ area, synapse density, or apposition between Brp and DGluRIII in the ERK hypomorph rl1 when compared with wild type. As shown above, unc-513 mutants display a decrease in synapse area, a decrease in synapse density and a high degree of misapposition between Brp and DGluRIII. In the unc-513, rl1 double mutant there is dramatic suppression of each of these phenotypes (Fig. 6A–D). The double mutant shows a more than twofold increase in the NMJ area, and a complete suppression of the synaptic density and apposition phenotypes to wild-type levels. Hence, increased ERK activity is required for the Unc-51 synaptic defects. There is an additional unc-51 phenotype, the accumulation of synaptic material in the axon that is indicative of an axon transport defect. Of note, this axon transport defect is not suppressed in the double mutant (Fig. 7A,B). Therefore, the axonal and synaptic defects of the unc-51 mutant are genetically separable, and so the transport defects cannot be responsible for the synaptic defects. Instead, these results suggest that Unc-51 regulates at least two pathways. It is required to downregulate ERK activity to ensure proper synaptic development, and works via a second, unknown pathway to promote proper axonal transport.
Figure 6.
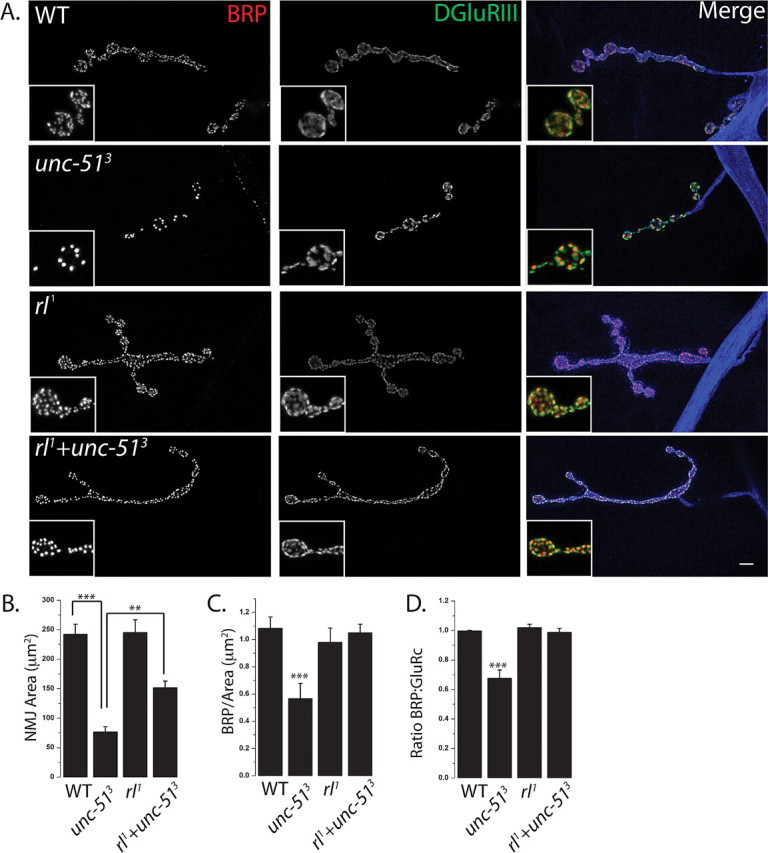
Mutations in rolled suppress the synaptic morphology phenotypes of unc-51 mutants. A, Sample NMJs stained with antibodies against BRP (red), DGluRIII (green), and HRP (blue). The following genotypes were used: WT (CS crossed to w−), unc-513 deletion mutant, homozygous rl1 hypomorphs, and rl1+unc-513 (rl1; unc-513). Inset shows a magnified view of the boutons and highlights the apposition of glutamate receptors and BRP puncta. B–D, Histograms of NMJ area (B), BRP density (C), and ratio of BRP to DGluRIII (D) from the same genotypes as in A. ***p < 0.001, **p < 0.01; n = 9 for all genotypes except rl1+unc-513, where n = 11. Error bars represent SEM. Scale bar, 3 μm.
Figure 7.

Axon transport defects persists in the unc-51, rolled double mutant. A, Sample axons stained with antibodies against BRP (red) and HRP (blue). Dense accumulations of synaptic material are seen in unc-513 axons, which persist in the double mutant of rolled and unc-513. The genotypes are WT (CS crossed to w−), unc-513 deletion mutant, homozygous rl1 hypomorphs, and rl1+unc-513 (rl1; unc-513). B, Histogram showing the intensity of BRP/axon length. ***p < 0.001; n = 9 for all genotypes. Error bars represent SEM. Intensity of HRP was not significantly different among genotypes (n = 9, p = 0.1).
Having demonstrated that rl1 suppresses the synaptic morphology defects of the unc-51 mutant, we next tested whether it also suppresses the defects in synaptic function. Evoked and spontaneous synaptic events were recorded from muscle 6, segments A2/A3 in the same four genotypes described above. While synapse morphology is normal in the rl1 mutant, synaptic transmission is not. There is an approximately fourfold decrease in the amplitude of evoked EJPs in the rl1 mutant, which is very similar to the defect in evoked release observed in the unc-51 mutant. Remarkably, although both single mutants show large decreases in synaptic strength, evoked release returns to wild-type levels in the double mutant. There is no significant difference in the amplitude of mEJPs in any of the four genotypes, so the change in evoked release is likely due to a change in the number of vesicles released from the presynaptic terminal. Calculating quantal content reveals that both the unc-51 mutant and rl1 release at least threefold fewer vesicles following an action potential, while release in the double mutant is indistinguishable from wild type (Fig. 8A–D). These findings have two implications. First, the suppression of the unc-51 phenotype by rl1 is consistent with the morphological findings and demonstrates that synaptic transmission in the unc-51 mutant is impaired due to the failure to inhibit ERK activity. Since increased ERK activity leads to a dramatic decrease in the number of structurally normal synapses, this is likely the cause of the defect in evoked release in the unc-51 mutant. Second, the deficit in evoked release in the rl1 mutant suggests that decreasing levels of ERK also disrupt evoked release, albeit in the absence of synaptic morphology defects, and that this can be suppressed by the loss of Unc-51 inhibition of ERK function. These results suggest that the Unc-51 kinase may bidirectionally regulate ERK activity to control synaptic strength.
Figure 8.

Mutations in rolled suppress the unc-51 defect in evoked transmitter release. A, B, Representative spontaneous mEJPs (A) and EJPs (B) are shown from the same genotypes as in Figure 5. C, D, Histograms show the mean EJP amplitude (C) and quantal content (D) from WT (CS crossed to w−), unc-513 deletion mutant, homozygous rl1 hypomorphs, and rl1+unc-513 (rl1; unc-513). ***p < 0.001; n = 10 for all genotypes; and error bars represent SEM. p < 0.001 when either unc-513 or rl1 is compared with either wild type or double mutant. There is no significant difference between WT and double mutant (p = 0.3). Calibration: mEJP, 2.5 mV, 100 ms; EJP, 5 mV, 5 ms.
Discussion
The close apposition of active zones to postsynaptic neurotransmitter receptors ensures that receptors encounter a high concentration of transmitter following synaptic vesicle fusion. However not every active zone formed by a single presynaptic cell has the same probability of releasing transmitter, implying that synapse-specific mechanisms exist to control active zone function. While many active zone proteins have been identified and characterized, little is known of the mechanisms that ensure that each active zone receives its proper complement of proteins. To identify such mechanisms, we have undertaken a genetic screen to identify genes required to ensure the apposition of such structurally normal active zones opposite each postsynaptic density. We demonstrate that the serine threonine kinase Unc-51 acts presynaptically to regulate the localization of the active zone protein Bruchpilot opposite to glutamate receptors at each synapse. In the absence of Unc-51, there is both a decrease in the density of synapses as well as the presence of abnormal synapses lacking their full complement of active zone proteins, leading to impaired transmitter release. Unc-51 regulates active zone development by inhibiting the activity of the MAP kinase ERK. This Unc-51-dependent inhibition of ERK activity provides a potential mechanism for synapse-specific control of active zone protein composition and release probability.
A screen for active zone mutants
Active zones are the site of synaptic vesicle docking and fusion (Rosenmund et al., 2003). Many active zone proteins have been identified (Zhai and Bellen, 2004; Zhen and Jin, 2004), although mutational analysis has not identified any proteins that are essential for active zone formation. Instead, loss of function analysis reveals defects in the function or morphology of active zones. For example, mutations in syd-2/liprin in both worms and flies lead to larger active zones, demonstrating its central role for organizing the active zone (Dai et al., 2006; Patel et al., 2006). Drosophila contains orthologs of most vertebrate active zone proteins, the best characterized of which is Bruchpilot, the ortholog of CAST (Kittel et al., 2006; Wagh et al., 2006). Bruchpilot is present at the active zone, and promotes the active zone localization of calcium channels and the electron-dense T-bar specialization. As such, Bruchpilot is likely a key determinant of release probability at each active zone. Consistent with this view, synaptic transmission is impaired in bruchpilot mutants (Wagh et al., 2006), and active zones with a higher release probability tend to have more Bruchpilot (Marrus and DiAntonio, 2004).
We performed a large-scale anatomical screen to identify mutants where not every glutamate receptor cluster is apposed to Bruchpilot. We put aside mutants with a global decrease in Brp or DGluRIII across the NMJ, and instead focused on mutants in which Brp was absent from a subset of synapses. Such mutants were identified by the presence of glutamate receptor clusters unapposed to Bruchpilot puncta. In this screen we identified mutants in unc-51.
Unc-51 promotes the localization of Bruchpilot to every active zone
In the unc-51 mutant many DGluRIII clusters are unapposed to Brp. Such misapposition could reflect either DGluRIII clusters unapposed to active zones, or receptor clusters apposed to abnormal active zones that do not contain Brp. The ideal experiment to distinguish between these possibilities would be to stain for other presynaptic active zone proteins. Unfortunately the only other such protein that can be visualized in Drosophila is the calcium channel cacophony, and since its localization depends on Brp this experiment is not be informative. Nonetheless, two results strongly suggest that a subset of glutamate receptors is apposed to abnormal active zones. First, the decreased density of DGluRIII clusters observed via confocal microscopy approximates the decrease in active zone density observed via electron microscopy. If many DGluRIII clusters were unapposed to active zones, then we would have expected a more dramatic decrease in active zone density. Second, ultrastructural analysis demonstrates a decrease in the proportion of active zones containing T-bars. Brp is not necessary for the formation of active zones, but is required for the localization of T-bars to active zones (Kittel et al., 2006; Wagh et al., 2006). If the absence of Brp were due to the absence of the entire active zone, then each active zone would contain Brp and we would have predicted a normal ratio of T-bars/active zones. Instead, the decrease in T-bars/active zone is consistent with the presence of active zones missing Brp and, hence, lacking T-bars. Therefore, we conclude that Unc-51 is required for the high fidelity of active zone assembly, ensuring that Brp is present at every active zone.
In addition to the presence of abnormal synapses in the unc-51 mutant, there is also a decrease in the number and density of synapses. We speculate that the decrease in synaptic density and the presence of abnormal synapses may be related phenotypes that differ in severity. In this view, Unc-51 promotes synapse formation. In its absence, active zone assembly would be less efficient, resulting in either the formation of abnormal active zones missing crucial proteins such as Brp, or in more severe cases leading to complete failure of active zone assembly and, hence, the absence of a synapse. The complete suppression of both the synaptic density and apposition phenotypes by mutation of the downstream target ERK is consistent with these phenotypes sharing an underlying mechanism. As expected, this defect in the number and proper assembly of synapses leads to a dramatic decrease in synaptic efficacy.
In addition to these synaptic defects, the unc-51 mutant also has a smaller NMJ and accumulations of synaptic material in the axons, suggesting defects in axonal transport. One mechanism that could link a small NMJ with defective transport is synaptic retraction, in which entire presynaptic boutons or branches retract leaving a footprint of postsynaptic proteins (Eaton et al., 2002). However, we did not observe such footprints in the unc-51 mutant, so this is not the cause of the small NMJ. Synaptic growth requires the retrograde transport of a BMP signal to the nucleus (Marqués et al., 2003; McCabe et al., 2003), however we found no change in the levels of phosphorylated MAD in motoneuron nuclei (data not shown), suggesting that this is not a likely cause of the growth defect. Finally, in worms and mice Unc-51 is required for axon outgrowth, which may be somewhat analogous to defects in NMJ growth in Drosophila. However, to form an NMJ the axon must navigate out of the ventral nerve cord and cross a wide expanse of muscle before reaching its target and forming a junction. Since we do not observe defects in the pattern of neuromuscular innervation, it is unlikely that a generic defect in axon outgrowth is responsible for the small NMJs. The apparent axonal transport defect is consistent with findings from mammals suggesting a function for Unc-51 in regulating axon transport. We have not investigated the role of Unc-51 for transport, but note that we were able to genetically separate the axonal transport and synapse development phenotypes, so the transport phenotypes may not be primary cause of the synaptic defects.
Unc-51 inhibits ERK signaling to promote synapse formation
Our data support the model that Unc-51 inhibits ERK activation to promote proper active zone development. In the unc-51 mutant we observed a modest increase in the levels of activated ERK, demonstrating that Unc-51 is a negative regulator of ERK activation in vivo. This increased ERK activity is responsible for the defects in active zone formation. Double mutants between unc-51 and the ERK hypomorph rl1 completely suppress the synapse density and apposition phenotypes of the unc-51 mutant, and restore synaptic strength to wild-type levels. Hence, ERK is required for the synaptic phenotypes observed in the unc-51 mutant. The axonal transport defects were not suppressed in the double mutant, so Unc-51 must act through other pathways as well. In mammalian cells Unc-51 can downregulate ERK by inhibiting the binding of a scaffolding protein to the FGF receptor. To date, no receptor tyrosine kinase has been identified that regulates active zone formation in Drosophila. Future studies to characterize the mechanism by which Unc-51 inhibits ERK in Drosophila motoneurons may provide clues toward identification of such a pathway. In addition, it is unclear how ERK regulates active zone formation. A previous study demonstrated that phospho-ERK localizes to the active zone, which would suggest a direct mechanism (Koh et al., 2002). Unfortunately, we have been unable to replicate these localization findings. The same study demonstrated that the transgenic expression of a constitutively active ras or a gain-of-function ERK allele both lead to an increase in the number of synaptic boutons, which is not consistent with our finding of a smaller NMJ. Active zone structure and number were not assessed. We speculate that the global activation of ERK may result in different phenotypes than relief of Unc-51 inhibition of ERK, which could show temporal and spatial specificity.
Regulating release probability at single release sites
In mammalian and Drosophila neurons, release probability varies across release sites formed by a single neuron (Rozov et al., 2001; Marrus and DiAntonio, 2004). One potential mechanism would be the differential localization or activity of core active zone proteins. In Drosophila, Bruchpilot is an excellent candidate for such a protein. It is required for the localization of calcium channels to the active zone, so changes in its localization or function would impact calcium influx and, hence, release probability at an active zone. The unc-51 mutant demonstrates that signaling pathways can differentially regulate the localization of Brp to individual release sites within a single neuron. As such, the Unc-51/Erk signaling pathway is a candidate mechanism to regulate active zone protein composition and release probability in a synapse-specific manner.
Footnotes
This work was supported by a grant from the National Institutes of Health (NS043171) to A.D. We thank Dr. Thomas Neufeld for the gift of ATG1 deletion mutant flies, the fly stock centers at Bloomington and Vienna for a number of fly lines used in this study, Xiaolu Sun for excellent technical help, and John Hardy and Marcia Miller for help with electron microscopy. We are grateful for excellent comments on this manuscript by James Skeath, E. J. Brace, and Sarah Naylor.
References
- Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhães TR, Goodman CS. wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron. 2002;33:545–558. doi: 10.1016/s0896-6273(02)00589-5. [DOI] [PubMed] [Google Scholar]
- Ahantarig A, Chadwell LV, Terrazas IB, Garcia CT, Nazarian JJ, Lee HK, Lundell MJ, Cassill JA. Molecular characterization of Pegarn: a Drosophila homolog of UNC-51 kinase. Mol Biol Rep. 2008 doi: 10.1007/s11033-008-9314-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aladzsity I, Tóth ML, Sigmond T, Szabó E, Bicsák B, Barna J, Regos A, Orosz L, Kovács AL, Vellai T. Autophagy genes unc-51 and bec-1 are required for normal cell size in Caenorhabditis elegans. Genetics. 2007;177:655–660. doi: 10.1534/genetics.107.075762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery AW, Figueroa C, Vojtek AB. UNC-51-like kinase regulation of fibroblast growth factor receptor substrate 2/3. Cell Signal. 2007;19:177–184. doi: 10.1016/j.cellsig.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Bellen HJ, Levis RW, Liao G, He Y, Carlson JW, Tsang G, Evans-Holm M, Hiesinger PR, Schulze KL, Rubin GM, Hoskins RA, Spradling AC. The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics. 2004;167:761–781. doi: 10.1534/genetics.104.026427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs WH, 3rd, Zavitz KH, Dickson B, van der Straten A, Brunner D, Hafen E, Zipursky SL. The Drosophila rolled locus encodes a MAP kinase required in the sevenless signal transduction pathway. EMBO J. 1994;13:1628–1635. doi: 10.1002/j.1460-2075.1994.tb06426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Cheong H, Klionsky DJ. Dual role of Atg1 in regulation of autophagy-specific PAS assembly in Saccharomyces cerevisiae. Autophagy. 2008;4:724–726. doi: 10.4161/auto.6375. [DOI] [PubMed] [Google Scholar]
- Collins CA, DiAntonio A. Synaptic development: insights from Drosophila. Curr Opin Neurobiol. 2007;17:35–42. doi: 10.1016/j.conb.2007.01.001. [DOI] [PubMed] [Google Scholar]
- Dai Y, Taru H, Deken SL, Grill B, Ackley B, Nonet ML, Jin Y. SYD-2 Liprin-alpha organizes presynaptic active zone formation through ELKS. Nat Neurosci. 2006;9:1479–1487. doi: 10.1038/nn1808. [DOI] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Gelfand MV, Dant J, Brooks ES, Krantz DE, DiAntonio A. Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J Neurosci. 2004;24:10466–10474. doi: 10.1523/JNEUROSCI.3001-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong S, Cavallo JA, Rios CD, Dworak HA, Sink H. Target recognition and synaptogenesis by motor axons: responses to the sidestep protein. Int J Dev Neurosci. 2005;23:397–410. doi: 10.1016/j.ijdevneu.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Duncan JE, Goldstein LS. The genetics of axonal transport and axonal transport disorders. PLoS Genet. 2006;2:e124. doi: 10.1371/journal.pgen.0020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton BA, Fetter RD, Davis GW. Dynactin is necessary for synapse stabilization. Neuron. 2002;34:729–741. doi: 10.1016/s0896-6273(02)00721-3. [DOI] [PubMed] [Google Scholar]
- Fejtova A, Gundelfinger ED. Molecular organization and assembly of the presynaptic active zone of neurotransmitter release. Results Probl Cell Differ. 2006;43:49–68. doi: 10.1007/400_012. [DOI] [PubMed] [Google Scholar]
- Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol. 2000;150:1507–1513. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittel RJ, Wichmann C, Rasse TM, Fouquet W, Schmidt M, Schmid A, Wagh DA, Pawlu C, Kellner RR, Willig KI, Hell SW, Buchner E, Heckmann M, Sigrist SJ. Bruchpilot promotes active zone assembly, Ca2+ channel clustering, and vesicle release. Science. 2006;312:1051–1054. doi: 10.1126/science.1126308. [DOI] [PubMed] [Google Scholar]
- Koh YH, Ruiz-Canada C, Gorczyca M, Budnik V. The Ras1-mitogen-activated protein kinase signal transduction pathway regulates synaptic plasticity through fasciclin II-mediated cell adhesion. J Neurosci. 2002;22:2496–2504. doi: 10.1523/JNEUROSCI.22-07-02496.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YM, Nishizawa K, Nishi Y, Tsuda L, Inoue YH, Nishida Y. Genetic analysis of rolled, which encodes a Drosophila mitogen-activated protein kinase. Genetics. 1999;153:763–771. doi: 10.1093/genetics/153.2.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marqués G, Haerry TE, Crotty ML, Xue M, Zhang B, O'Connor MB. Retrograde Gbb signaling through the Bmp type 2 receptor wishful thinking regulates systemic FMRFa expression in Drosophila. Development. 2003;130:5457–5470. doi: 10.1242/dev.00772. [DOI] [PubMed] [Google Scholar]
- Marrus SB, DiAntonio A. Preferential localization of glutamate receptors opposite sites of high presynaptic release. Curr Biol. 2004;14:924–931. doi: 10.1016/j.cub.2004.05.047. [DOI] [PubMed] [Google Scholar]
- Marrus SB, Portman SL, Allen MJ, Moffat KG, DiAntonio A. Differential localization of glutamate receptor subunits at the Drosophila neuromuscular junction. J Neurosci. 2004;24:1406–1415. doi: 10.1523/JNEUROSCI.1575-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe BD, Marqués G, Haghighi AP, Fetter RD, Crotty ML, Haerry TE, Goodman CS, O'Connor MB. The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron. 2003;39:241–254. doi: 10.1016/s0896-6273(03)00426-4. [DOI] [PubMed] [Google Scholar]
- Okazaki N, Yan J, Yuasa S, Ueno T, Kominami E, Masuho Y, Koga H, Muramatsu M. Interaction of the Unc-51-like kinase and microtubule-associated protein light chain 3 related proteins in the brain: possible role of vesicular transport in axonal elongation. Brain Res Mol Brain Res. 2000;85:1–12. doi: 10.1016/s0169-328x(00)00218-7. [DOI] [PubMed] [Google Scholar]
- Patel MR, Lehrman EK, Poon VY, Crump JG, Zhen M, Bargmann CI, Shen K. Hierarchical assembly of presynaptic components in defined C. elegans synapses. Nat Neurosci. 2006;9:1488–1498. doi: 10.1038/nn1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A. Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron. 1997;19:1237–1248. doi: 10.1016/s0896-6273(00)80415-8. [DOI] [PubMed] [Google Scholar]
- Peverali FA, Isaksson A, Papavassiliou AG, Plastina P, Staszewski LM, Mlodzik M, Bohmann D. Phosphorylation of Drosophila Jun by the MAP kinase rolled regulates photoreceptor differentiation. EMBO J. 1996;15:3943–3950. [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Rettig J, Brose N. Molecular mechanisms of active zone function. Curr Opin Neurobiol. 2003;13:509–519. doi: 10.1016/j.conb.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Rozov A, Burnashev N, Sakmann B, Neher E. Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J Physiol. 2001;531:807–826. doi: 10.1111/j.1469-7793.2001.0807h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell. 2004;7:167–178. doi: 10.1016/j.devcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Stewart BA, Atwood HL, Renger JJ, Wang J, Wu CF. Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J Comp Physiol A Neuroethol Sens Neural Behav Physiol. 1994;175:179–191. doi: 10.1007/BF00215114. [DOI] [PubMed] [Google Scholar]
- Tomoda T, Bhatt RS, Kuroyanagi H, Shirasawa T, Hatten ME. A mouse serine/threonine kinase homologous to C. elegans UNC51 functions in parallel fiber formation of cerebellar granule neurons. Neuron. 1999;24:833–846. doi: 10.1016/s0896-6273(00)81031-4. [DOI] [PubMed] [Google Scholar]
- Tomoda T, Kim JH, Zhan C, Hatten ME. Role of Unc51.1 and its binding partners in CNS axon outgrowth. Genes Dev. 2004;18:541–558. doi: 10.1101/gad.1151204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viquez NM, Li CR, Wairkar YP, DiAntonio A. The B' protein phosphatase 2A regulatory subunit well-rounded regulates synaptic growth and cytoskeletal stability at the Drosophila neuromuscular junction. J Neurosci. 2006;26:9293–9303. doi: 10.1523/JNEUROSCI.1740-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagh DA, Rasse TM, Asan E, Hofbauer A, Schwenkert I, Dürrbeck H, Buchner S, Dabauvalle MC, Schmidt M, Qin G, Wichmann C, Kittel R, Sigrist SJ, Buchner E. Bruchpilot, a protein with homology to ELKS/CAST, is required for structural integrity and function of synaptic active zones in Drosophila. Neuron. 2006;49:833–844. doi: 10.1016/j.neuron.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Wairkar YP, Fradkin LG, Noordermeer JN, DiAntonio A. Synaptic defects in a Drosophila model of congenital muscular dystrophy. J Neurosci. 2008;28:3781–3789. doi: 10.1523/JNEUROSCI.0478-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao KM, White K. Neural specificity of elav expression: defining a Drosophila promoter for directing expression to the nervous system. J Neurochem. 1994;63:41–51. doi: 10.1046/j.1471-4159.1994.63010041.x. [DOI] [PubMed] [Google Scholar]
- Zhai RG, Bellen HJ. The architecture of the active zone in the presynaptic nerve terminal. Physiology (Bethesda) 2004;19:262–270. doi: 10.1152/physiol.00014.2004. [DOI] [PubMed] [Google Scholar]
- Zhang YQ, Bailey AM, Matthies HJ, Renden RB, Smith MA, Speese SD, Rubin GM, Broadie K. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell. 2001;107:591–603. doi: 10.1016/s0092-8674(01)00589-x. [DOI] [PubMed] [Google Scholar]
- Zhen M, Jin Y. Presynaptic terminal differentiation: transport and assembly. Curr Opin Neurobiol. 2004;14:280–287. doi: 10.1016/j.conb.2004.05.013. [DOI] [PubMed] [Google Scholar]