

Abstract
Phosphorylation-dependent ubiquitination and degradation of the IFNAR1 chain of Type I interferon (IFN) receptor is regulated by two different pathways one of which is ligand-independent. We report that this pathway is activated by inducers of the endoplasmic reticulum (ER) stress, including viral infection, in a PERK-dependent manner. Upon infection, activation of this pathway promotes phosphorylation-dependent ubiquitination and degradation of IFNAR1, and specifically inhibits Type I IFN signaling and antiviral defenses. Either knock-in of an IFNAR1 mutant insensitive to virus-induced turnover or conditional knockout of PERK prevented ER stress- and virus-induced IFNAR1 degradation while restoring cellular responses to Type I IFN and resistance to viruses. The role of this novel mechanism in pathogenesis of viral infections and therapeutic approaches to their treatment is discussed.
Keywords: interferon, receptor, ubiquitin, ER stress, unfolded protein response, PERK, IFNAR1, knock-in
Introduction
Animal hosts defend themselves against infectious agents by utilizing the mechanisms of innate and adaptive immunity. Importantly, diverse pathways of innate immunity converge on the induction of cytokines that belong to a family of type I interferons (IFN) including various types of IFNα and IFNβ that play a major role in host defenses against the viruses. Unlike IFNγ, which belongs to type II IFN group, all members of the Type I family act on cells via the same cognate receptor that consists of two sub-units: IFNAR1 and IFNAR2c (reviewed in (Pestka, 2000)).
Dimerization of receptor chains in response to the ligands results in the activation of Janus kinase (Jak) family members Jak1 and Tyk2 that phosphorylate each other, the aforementioned receptor subunits and the recruited signal transducers and activators of transcription (Stat1 and Stat2) at specific tyrosines. Phosphorylated Stat proteins translocate to the nucleus, bind to IFN-stimulated regulatory elements (ISRE) and activate transcription of a large number of IFN-stimulated genes (ISGs, reviewed in Stark et al, 1998). ISGs mediate a plethora of IFNα effects that play key roles in anti-viral defense (Brassard et al., 2002; Katze et al., 2002a), inhibition of cell proliferation (Brassard et al., 2002; Kirkwood, 2002; Stark et al., 1998) and modulation of immune responses (Biron, 2001; Brassard et al., 2002). The ability of IFNα to evoke these outcomes makes it an attractive therapeutic agent extensively used for treatment of patients with neoplastic diseases (Kirkwood, 2002), chronic viral infections (Brassard et al., 2002; Katze et al., 2002a), and multiple sclerosis (Karp et al., 2000).
Studies in cell culture revealed that anti-viral effects of IFN are best seen when it is added to cells prior to the infection (Blalock and Baron, 1979; Pfeffer and Colamonici, 1991). While decreased efficacy of IFN added to already infected cells is largely explained by insufficient time to transcribe and translate ISG products (reviewed in (Friedman and Sonnabend, 1970; Pfeffer and Colamonici, 1991)), additional mechanisms such as a negative effect of virus on IFN action have been also postulated (Lockart, 1963; Lockart and Horn, 1963). Indeed, many viruses evolved to employ a multitude of specific mechanisms to protect themselves against Type I IFN. These mechanisms usually involve a rapid synthesis of numerous virus type-specific proteins that impede diverse elements of pathways converging on either IFN production or IFN signaling (reviewed in (Katze et al., 2002b)).
A need for the robust synthesis of viral polypeptides, however, poses additional problems for the virus as it challenges the capacity of the host cell to properly fold and activate proteins. Accumulation of sub-optimally folded proteins in the ER of the host cell induces a series of signaling events known as the ER stress or the unfolded protein response (UPR) (Welihinda et al., 1999). While the ER protein chaperone BiP is central to initiating virtually all branches of the response, subsequent signaling proceeds via a number of defined mechanisms that include other transmembrane sensors including ATF6, IRE1 and PKR-like ER kinase (PERK). The activation of PERK and ensuing phosphorylation of eIF2α restricts translation to alleviate the load of unfolded proteins (reviewed in (Malhotra and Kaufman, 2007; Ron and Walter, 2007)). Viruses are known to both induce UPR and produce the means of inhibiting these responses. The latter is necessary in order to protect the host cells from ER stress-mediated death, to enable translation of viral proteins and to continue virus production (He, 2006; Schroder and Kaufman, 2006; Wang and Weinman, 2006; Waris et al., 2002).
While investigating the mechanisms that govern proteolytic degradation of Type I IFN receptor we found that IFNAR1 undergoes ligand-induced Tyk2 activity-dependent phosphorylation on specific Ser residues (Ser535 in humans and Ser526 in mice). This phosphorylation leads to the recruitment of βTrcp E3 ubiquitin ligase followed by IFNAR1 ubiquitination, internalization, and lysosomal degradation (Kumar et al., 2007a; Kumar et al., 2004; Kumar et al., 2003; Marijanovic et al., 2006). Intriguingly, there is also a ligand- and Jak-independent pathway resulting in phosphorylation and turnover of IFNAR1 in cells that over-expressed this receptor (Liu et al., 2008).
Here we present evidence that UPR triggers including viral infection activate PERK to promote ligand- and Jak-independent phosphorylation of IFNAR1 within its phosphor-degron, leading to IFNAR1 ubiquitination and degradation as well as to suppression of Type I IFN signaling. We propose that this mechanism helps viruses to obviate the IFN system and enable efficient replication. Our data also suggest a potential for therapeutic targeting of PERK in treatment of viral infections.
Results
The UPR induces PERK-dependent phosphorylation of IFNAR1 degron
Overexpressed IFNAR1 undergoes ligand- and Jak-independent degron phosphorylation followed by ubiquitination and degradation of this receptor (Liu et al., 2008). Increasing the amount of transfected IFNAR1 plasmid led to a disproportionate increase in phospho-IFNAR1 signal that cannot be explained solely by higher levels of total IFNAR1 expressed in these cells (Figure 1A). Furthermore, lysates from these transfected cells displayed an elevated ability to phosphorylate bacterially produced GST-IFNAR1 protein on Ser535 (Figure 1B) indicating that forced expression of IFNAR1 activates a signal transduction pathway inducing an unknown protein kinase activity that phosphorylates IFNAR1 within its degron.
Figure 1.
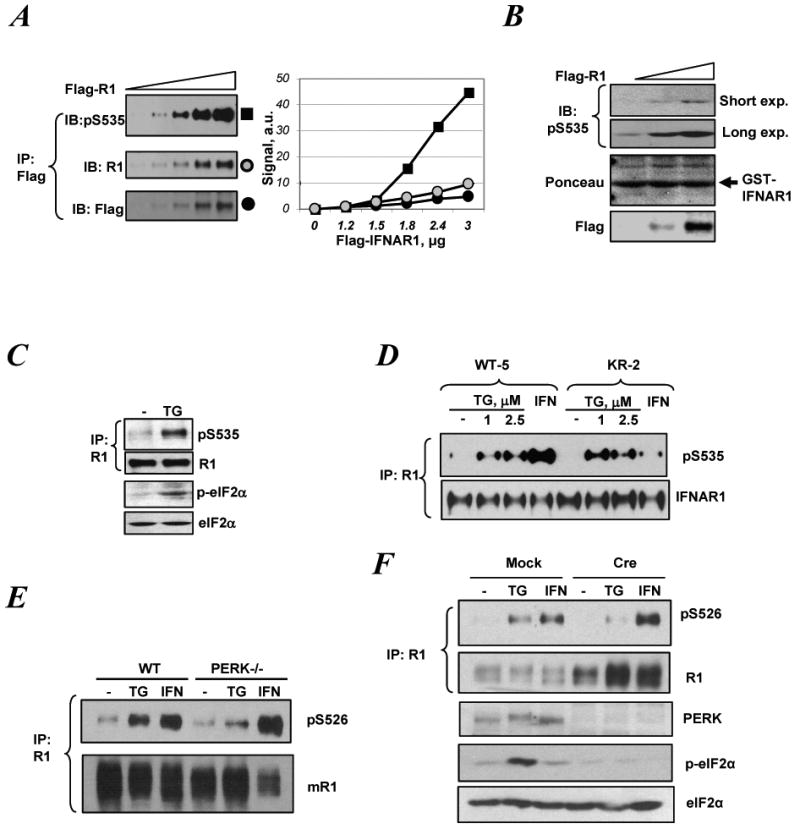
ER stress induces IFNAR1 Ser535 kinase activity and promotes phosphorylation of IFNAR1 within its destruction motif in a manner that does not require Tyk2 activity but relies on activity of PERK.
A. Lysates from KR-2 cells (lacking catalytic activity of Tyk2) transfected with Flag-IFNAR1 plasmid (0 – 3.0μg) were subjected to immunoprecipitation (IP) using anti-Flag antibody followed by immunoblotting (IB) using the indicated antibodies. Relative intensity of bands in IB using either anti-phospho-S535 (black squares) or IFNAR1 (R1, gray squares) or Flag (black squares) was quantified and plotted in the right panel.
B. Lysates from KR-2 cells transfected with Flag-IFNAR1 (1.5-3.0μg) or empty vector were used as a source of kinase activity in an in vitro kinase assay using GST-IFNAR1 as a substrate. The reactions were analyzed by IB using anti-phospho-S535 antibody (upper panel; both shorter and longer exposures shown) and by Ponceau staining to detect the substrate levels (middle panel). Levels of Flag-IFNAR1 in the cell lysates were analyzed by IB using anti-Flag antibody (lower panel).
C. 293T cells were treated with thapsigargin (TG, 1μM) for 30 min. Endogenous IFNAR1 was immunoprecipitated and analyzed by IB using indicated antibodies. Aliquots of the whole cell lysates were analyzed for levels of phospho- and total eIF2α.
D. Cells harboring wild type (WT-5) or the kinase dead Tyk2 (KR-2) were treated with IFNα (1000IU/ml) or TG at indicated concentrations for 30 min. Endogenous IFNAR1 was analyzed by IP-IB as in panel C.
E. WT or PERK-/- MEFs were treated with TG (1μM) or murine IFNβ (1000 U/ml) for 30 min. Mouse endogenous IFNAR1 was analyzed for its phosphorylation and levels using the indicated antibodies.
F. MEFs from PERKfl/fl mice that received an empty vector (Mock) or vector for expression of Cre recombinase (Cre) were treated and analyzed as in panel F. Whole cell lysates from these cells were also analyzed by IB using the indicated antibodies.
Overexpression of secretory and transmembrane proteins (such as IFNAR1) might overpower the ability of a cell to properly fold these proteins in the ER and, therefore initiate the UPR (Welihinda et al., 1999). Indeed, as seen in Supplementary Figures 1 and 4 (SF1, 4), forced expression of IFNAR1 induced the markers of the UPR such as BiP and ATF4 and promoted phosphorylation of eIF2α. Similar results along with phosphorylation of endogenous IFNAR1 on Ser535 were obtained upon overexpression of unrelated IFNγ receptor IFNGR1 (SF2). It appears that eIF2α phosphorylation was dependent on PERK as evident from experiments using RNAi approach to knock down this kinase (SF3-4). Remarkably, thapsigargin, a known inducer of UPR stimulated phosphorylation of endogenous IFNAR1 on Ser535 in the absence of IFN (Figure 1C). Similar results were obtained using other known UPR stimuli such as DTT (SF5) and tunicamycin (not shown).
We next investigated whether activity of Tyk2, which is required for IFNAR1 phosphorylation in response to IFN (Liu et al., 2008; Marijanovic et al., 2006), plays a role in the ligand-independent pathway. To this end, we utilized derivatives of human fibrosarcoma 2fTGH-derived cell lines originally sensitive to Type I IFN (John et al., 1991) but then having lost Tyk2 expression (Velazquez et al., 1992). These cells were reconstituted with either wild type (WT) Tyk2 or its catalytically inactive (KR) mutant (Marijanovic et al., 2006). In line with the latter report, IFNα-stimulated phosphorylation was inhibited in KR cells; however, thapsigargin induced comparable levels of Ser535 phosphorylation of IFNAR1 in both cell lines (Figure 1D). These data suggest that UPR mediates phosphorylation of IFNAR1 in a ligand- and Tyk2 kinase-independent manner.
We then investigated which branch of UPR signaling is involved in regulating IFNAR1 phosphorylation. Embryo fibroblasts derived from PERK-null mice exhibited attenuated IFNAR1 phosphorylation in response to thapsigargin but not to murine IFNβ (Figure 1E). We further corroborated this data using the fibroblasts from mice harboring a conditional knockout allele of PERK (PERKfl/fl) where PERK is acutely excised upon transduction with retrovirus encoding the Cre recombinase (Zhang et al., 2002). The acute deletion of PERK inhibited IFNAR1 phosphorylation induced by thapsigargin without affecting IFN-triggered phosphorylation (Figure 1F). Phosphorylation of IFNAR1 in response to thapsigargin in human cells was not inhibited by either knockdown of IRE1 or the expression of a dominant negative mutant of IRE1 (SF6 and data not shown). Conversely, the knockdown of PERK noticeably decreased the efficacy of IFNAR1 phosphorylation induced by thapsigargin but not by IFNα in human cells (Figures SF7).
Collectively, these data suggest that PERK is required for IFNAR1 degron phosphorylation stimulated by UPR. Given that activated PERK was not capable of phosphorylating IFNAR1 in vitro (data not shown) it is likely that a kinase(s) downstream of PERK is responsible for the direct phosphorylation of IFNAR1 degron.
The UPR promotes IFNAR1 ubiquitination and degradation by inducing degron phosphorylation in a ligand- and Tyk2-independent manner
Phosphorylation within the IFNAR1 degron is expected to promote ubiquitination of this receptor and its degradation in the lysosome (Kumar et al., 2004; Kumar et al., 2003; Marijanovic et al., 2006). Indeed, treatment of cells with thapsigargin decreased the levels of IFNAR1 in human cells within two hours even in the absence of IFN. This decrease was prevented by pre-treating cells with methylamine HCl (MA), an inhibitor of the lysosomal pathway (Figure 2A). Furthermore, thapsigargin treatment induced ubiquitination of IFNAR1 and downregulated this receptor in human fibrosarcoma cells that express either wild type (WT) or catalytically inactive (KR) Tyk2 (Figure 2B) as well as in 293T cells (SF8). Ligand-independent stimulation of IFNAR1 ubiquitination by thapsigargin was also seen in IFNAR1-null mouse fibroblasts reconstituted with IFNAR1WT but not with IFNAR1SA mutant lacking phosphorylation site (SF9). These results indicate that induction of the UPR promotes phosphorylation-dependent ubiquitination of IFNAR1 and downregulates its levels in a manner independent of Tyk2 and of exogenous IFN.
Figure 2.
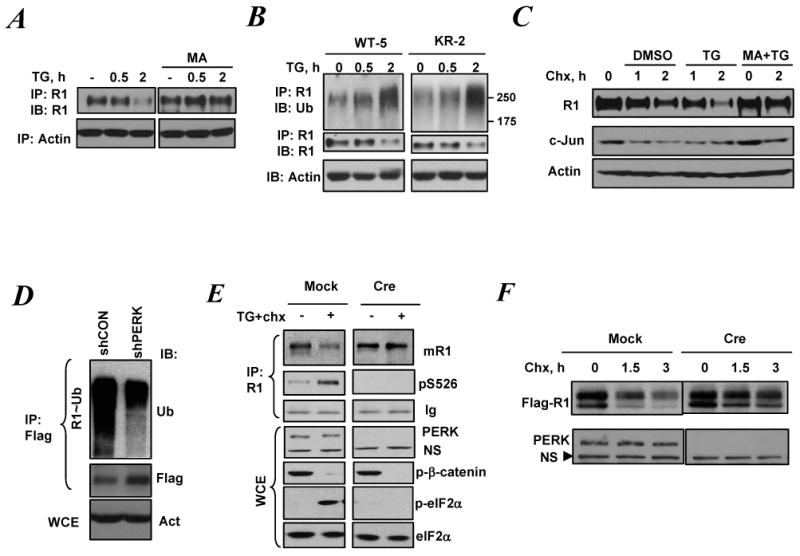
ER stress promotes IFNAR1 ubiquitination and degradation in a ligand/Jak-independent manner
A. Levels of endogenous IFNAR1 in 293T cells pre-treated or not with methylamine HCl (MA, 20mM) for 1 h and then treated with TG (1μM) for indicated time were analyzed by IP-IB. Levels of β-actin in whole cell lysates are also shown.
B. Cells harboring the WT Tyk2 (WT-5) or the kinase dead Tyk2 (KR-2) were treated with TG as indicated and ubiquitination and levels of endogenous IFNAR1 were analyzed by IP-IB. Aliquots of whole cell lysates were also analyzed by IB using anti-β-actin antibody (lower panel).
C. 293T cells were pre-treated or not with MA for 1 h and then treated with cycloheximide (Chx, 50μg/ml) alone or together with TG (1μM) for indicated times. Levels of endogenous IFNAR1 were analyzed by IP-IB. Levels of c-Jun and β-actin in whole cell lysates were also determined by IB using indicated antibodies.
D. Ubiquitination of Flag-tagged IFNAR1 co-expressed with the indicated shRNA constructs in 293T cells was analyzed by IP using anti-Flag antibody followed by IB using anti-ubiquitin and anti-Flag antibodies as indicated. Aliquots of whole cell lysates were also analyzed by IB using anti-β-actin antibody (lower panel).
E. PERKfl/fl MEFs that either underwent acute deletion of PERK (Cre) or not (Mock) were treated with 1μM of TG (together with Chx, 10μg/ml) for 45 min as indicated. Endogenous mouse IFNAR1 was analyzed by IP-IB using the indicated antibodies. Ig: heavy chain immunoglobulins. Whole cell lysates were also subjected to IB analysis to determine levels of phosphorylated β-catenin and eIF2α as well as total levels of PERK and eIF2α using respective antibodies. NS: non-specific band.
F. Mouse Flag-IFNAR1 expressed in PERKfl/fl MEFs that either underwent acute deletion of PERK (Cre) or not (Mock) were analyzed by IB using anti-Flag antibody. Levels of PERK are shown in lower panel. NS: non-specific band that serves as a loading control.
Treatment of cells with thapsigargin decreased the half life of IFNAR1 but not of an unrelated short lived protein, c-Jun, in 293T cells treated with cycloheximide to inhibit translation (Figure 2C). Knockdown of PERK decreased the ubiquitination of exogenously overexpressed IFNAR1 and noticeably increased its level in human cells (Figure 2D). Similarly, thapsigargin-induced ubiquitination of IFNAR1 was alleviated in PERK-deficient cells mouse cells (data not shown). In addition, acute Cre-mediated ablation of PERK slowed down UPR-induced turnover of both endogenous (Figure 2E) and exogenously expressed mouse IFNAR1 (Figure 2F). In contrast to that, degradation of another β-Trcp substrate, phosphorylated β-catenin, was not affected under these conditions (Figure 2E). Collectively, these data suggest that UPR promotes ubiquitination and degradation of IFNAR1 in a PERK-dependent manner.
We next sought to investigate whether UPR-stimulated IFNAR1 degradation is mediated via phosphorylation of serine residues within the IFNAR1 degron. To this end, we generated mouse embryonic stem (ES) cells that harbor one mutant IFNAR1S526A allele introduced via a homologous recombination approach (Figure 3A-B). These cells were grown as embryoid bodies (EB) and differentiated into fibroblast-like cells for analysis. Although treatment with thapsigargin induced a comparable level of eIF2α phosphorylation in both wild type and S526A knock-in cells, the latter displayed a grossly reduced phosphorylation of IFNAR1 on Ser526 (Figure 3C). Furthermore, thapsigargin-stimulated degradation of IFNAR1 was clearly inhibited in the S526A knock-in cells (Figure 3D). These data indicate that UPR-induced acceleration of proteolytic turnover of IFNAR1 depends on its phosphorylation within the specific degron.
Figure 3.
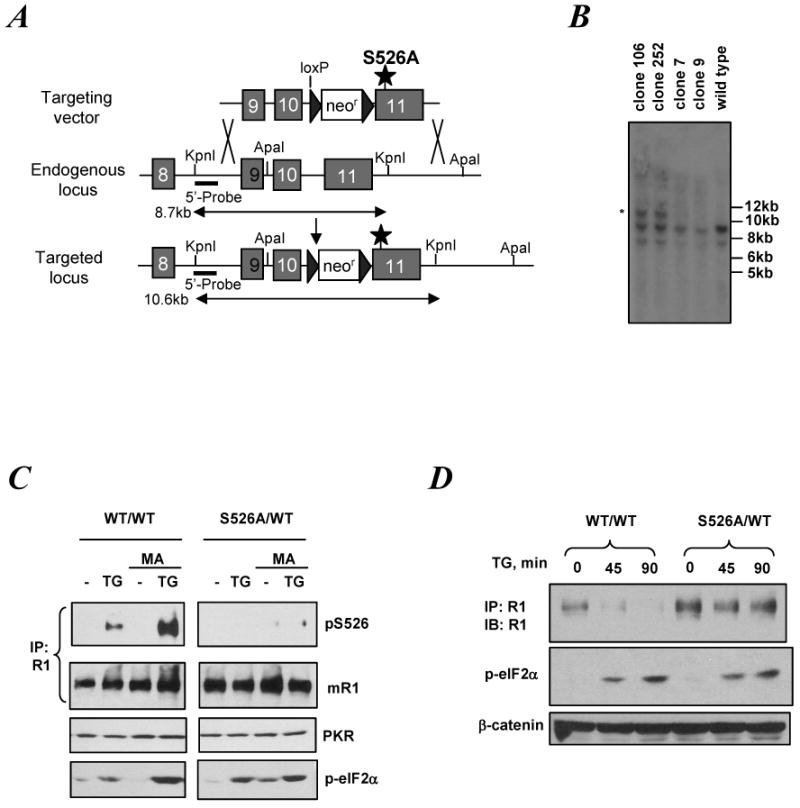
ER stress ER stress promotes IFNAR1 degradation in a manner depending on IFNAR1 phosphorylation within its phospho-degron
A. Targeting strategy to generate an S526A allele in mouse ES cells (C57/BL6). Position of mutated Ser residue, resistance markers, loxP sites as well as restriction sites and the probe used for Southern analysis are also shown.
B. Southern analysis of several selected ES clones that underwent homologous recombination (marked by an asterisk) was performed on genomic DNA digested with KpnI. Correct targeting yielded a 10.6 kb band in clones 106 and 252 (besides the 8.5 kb band indicative of the WT allele).
C. Embryoid bodies (EB) derived from the WT (WT/WT) or the mutant (S526A/WT) ES were trypsinized and the EB-derived cells were propagated on gelatinized plates. Cells were pre-treated or not with MA (20mM for 1 h) and then treated with TG (1μM, 15min) as indicated. Endogenous mouse IFNAR1 was immunoprecipitated and analyzed by IB using the indicated antibodies. Phosphorylation of eIF2α and levels of PKR were also determined in aliquots of whole cell lysates by IB.
D. EB-derived cells were treated with TG (1μM) for indicated times and analyzed for total levels of endogenous IFNAR1 (by IP-IB). Levels of eIF2α phosphorylation and total β-catenin were shown as stress and loading controls, respectively.
VSV and HCV accelerate the degradation of IFNAR1 via induction of PERK-dependent IFNAR1 degron phosphorylation
While IFNα/β play a major role in the defense against viruses, pre-treatment of yet uninfected cells with these cytokines are often required to obtain the protective effect. Numerous viruses including hepatitis C virus (HCV, (Ciccaglione et al., 2007; Wang and Weinman, 2006; Zheng et al., 2005)) are known to massively express their proteins and cause ER stress. Therefore, we sought to investigate whether virus-induced UPR might also affect IFNAR1 phosphorylation and stability that may also lead to inhibiting IFN responsiveness of already infected cells.
Infection of 2fTGH human fibrosarcoma cells with vesicular stomatitis virus (VSV) induced the expression of UPR markers (SF10). Intriguingly, this infection also stimulated IFNAR1 phosphorylation on Ser535 and decreased total levels of IFNAR1 (SF11). Similar results and an increase in the extent of IFNAR1 ubiquitination were observed in the KR derivative cell line (Figure 4A) indicating that VSV infection promotes IFNAR1 phosphorylation, ubiquitination and degradation in a Tyk2-independent manner. Furthermore, given that Tyk2 activity is essential for IFN-induced IFNAR1 phosphorylation (Marijanovic et al., 2006), this result suggests that the effects of VSV on IFNAR1 downregulation could not be attributed solely to induction of endogenous IFN.
Figure 4.
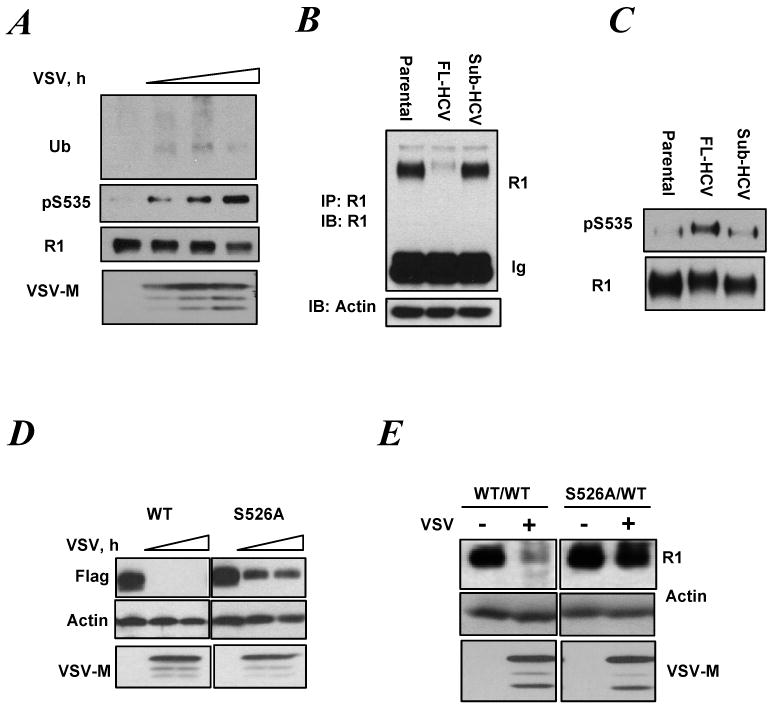
Viral infection promotes phosphorylation-dependent ubiquitination and downregulation of IFNAR1 in a Tyk2-independent and S535/526-dependent manner.
A. Ubiquitination, phosphorylation and total levels of endogenous IFNAR1 from KR-2 cells infected with VSV (for 16, 18 and 20 h) were analyzed by IP using anti-IFNAR1 antibody followed by IB using the indicated antibodies. Viral protein accumulation is shown by the levels of VSV-M.
B. Levels of endogenous IFNAR1 in the lysates from Huh7 cells (parental or harboring either a full length or subgenomic HCV) were analyzed by IP-IB. Levels of β-actin in the lysates aliquots are also shown.
C. Endogenous IFNAR1 proteins were immunopurified from the indicated cells (as in panel C) and loaded onto the gel to yield comparable levels of total IFNAR1 (lower panel). Phosphorylation of IFNAR1 was then analyzed by IB using indicated antibody (upper panel).
D. MEFs from IFNAR1-/- mice were stably reconstituted with murine Flag-IFNAR1 (either wild type or S526A mutant) and then infected with VSV (for 16-18 h.) Levels of IFNAR1, VSV-M and β-actin were analyzed by IB.
E. EB-derived cells of WT (WT/WT) or mutant (S526A/WT) genotype were infected (or not) with VSV for 12 h and lysed. Under these conditions, levels of VSV-M become saturated at 10hr post-infection (data not shown). Levels of endogenous mouse IFNAR1 were determined by IP-IB. Levels of β-actin and VSV-M in the lysates were also determined.
Infection with hepatitis C virus (HCV) promotes the ER stress (Tardif et al., 2005; Waris et al., 2002) that is robustly stimulated by the synthesis of structural proteins (Ciccaglione et al., 2005) known to reside in ER lumen of infected cells (Wu, 2001). In human hepatoblastoma Huh7 cells, total levels of endogenous IFNAR1 were dramatically down regulated by stable transfection of a complete HCV genome (Figure 4B). When the levels of IFNAR1 taken into immunoprecipitation were normalized to yield comparable total levels of IFNAR1, a robust phosphorylation of IFNAR1 degron was detected in Huh7 cells expressing the complete HCV genome (Figure 4C). Thus, it is plausible that the effects of viral infection/expression of viral proteins on IFNAR1 levels could be mediated via IFNAR1 degron phosphorylation and ensuing degradation.
Indeed, while VSV infection dramatically down regulated murine Flag-tagged IFNAR1 (re-expressed in MEFs from IFNAR1-null mice), a noticeably lesser effect was observed on mutant IFNAR1S526A (Figure 4D). This result was further corroborated in IFNAR1S526A knock-in cells that were more resistant in decreasing IFNAR1 levels in response to VSV (Figure 4E). These data indicate that phosphorylation of IFNAR1 degron is implicated in the receptor downregulation stimulated by virus.
Knock-down of PERK in human 2fTGH cells (SF12) attenuated degron phosphorylation and downregulation of IFNAR1 in cells infected with VSV (Figure 5A). Similarly, downregulation of IFNAR1 in VSV-infected 2fTGH cells was prevented by shRNA against PERK but not by a number of irrelevant shRNAs or shRNA against IRE1 (SF13). Accordingly, a much lesser extent of degradation of IFNAR1 promoted by VSV infection (measured in cycloheximide-treated cells) was detected in PERK knockdown cells (Figure 5B). Furthermore, CRE-mediated ablation of PERK decreased the extent of downregulation of cell surface IFNAR1 levels in response to either TG treatment or VSV infection (Figure 5C). In addition, knockdown of PERK partially rescued a decrease in IFNAR1 observed in Huh7 cells expressing the complete HCV genome (Figure 5D). These data suggest that viral infection and expression of structural viral proteins promote downregulation and degradation of IFNAR1 via a PERK-dependent signaling.
Figure 5.
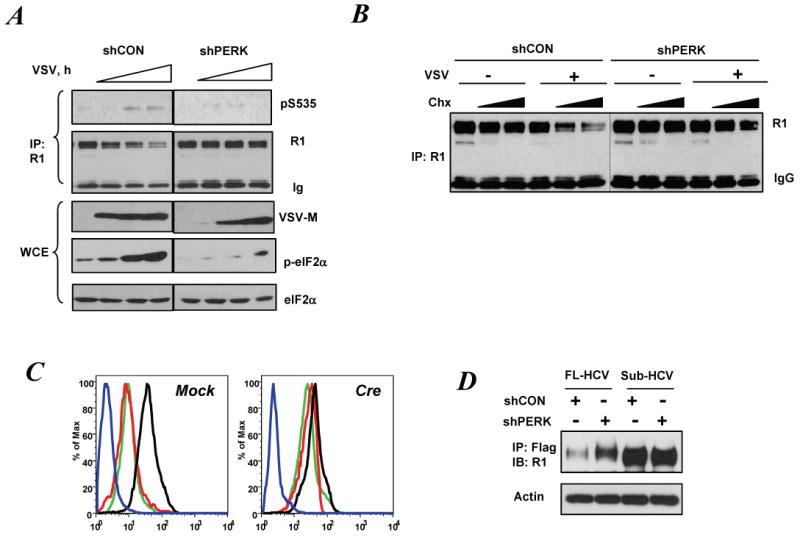
Role of PERK in virus-induced degradation of IFNAR1.
A. Phosphorylation and levels of endogenous IFNAR1 in 2fTGH cells that received indicated shRNA constructs and then were infected with VSV (for 16, 18 and 20 h) was analyzed by IP-IB using the indicated antibodies. Aliquots of IP supernatants were used for analysis of VSV-M, p-eIF2α and eIF2α levels by IB.
B. Control or PERK-depleted 2fTGH cells (as in panel A) were infected with VSV (for 17 h) and then treated with Chx (1 or 10μg/ml for 1.5 h). Total levels of IFNAR1 were determined by IP-IB.
C. Levels of cell surface IFNAR1 analyzed by FACS using monoclonal anti-mIFNAR1 antibody in MEFs from PERKfl/fl mice (transduced with either empty vector (Mock) or construct for expression of Cre) were either left untreated (black line) infected with VSV (for 17 h, red line) or treated with TG (1μM for 4h, green line). Blue line represents the isotype Ig control.
D. Levels of IFNAR1 and actin in Huh7 cells harboring the full-length or subgenomic HCV that were co-transfected with Flag-IFNAR1 and indicated shRNA constructs were analyzed using indicated antibodies.
VSV and HCV attenuate cellular responses to Type I IFN via PERK-dependent phosphorylation and downregulation of IFNAR1
Attenuated anti-viral defense observed in cells from IFNAR1+/- mice suggests that levels of IFNAR1 are important for Type I IFN signaling (Hwang et al., 1995; Muller et al., 1994). Therefore, IFNAR1 downregulation triggered by UPR activation is expected to inhibit cellular responses to IFNα/β. Indeed, either infection of cells with VSV (Figure 6A) or pre-treatment of cells with thapsigargin (SF14) noticeably decreased tyrosine phosphorylation of Stat1 induced by IFNα in human cells. Much lesser inhibition was seen in cells treated with IFNγ that utilizes an entirely different receptor (Pestka, 2000; Schreiber and Farrar, 1993), although yet requiring IFNAR1 for maximal signaling (Takaoka et al., 2000).
Figure 6.
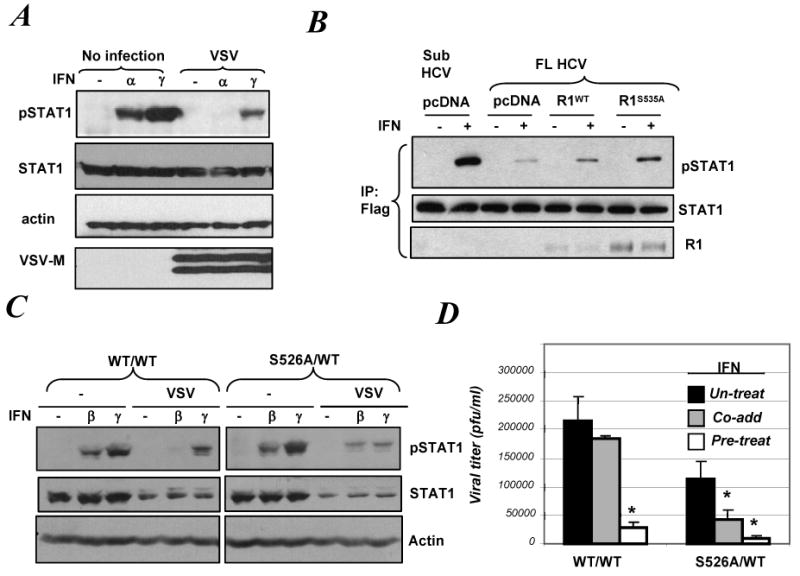
Viral infection inhibits Type I IFN signaling via accelerating Ser526 phosphorylation-dependent degradation of IFNAR1
A. 2fTGH cells infected or not with VSV (for 20 h) were treated with 50 IU/ml of IFNα or IFNγ for 30 min. Phosphorylation of Stat1 and total levels of Stat1, actin and VSV-M were analyzed by IB.
B. Indicated Huh7 cell line derivatives were co-transfected with Flag-STAT1 and either empty vector (pcDNA3) or Flag-IFNAR1 (WT or S535A) as indicated. Lysates from these cells treated or not with IFNα (50 IU/ml) were immunoprecipitated using anti-Flag antibody and these reactions were analyzed by IB using the indicated antibodies.
C. EB-derived cells of wild type (WT/WT) or mutant (S526A/WT) genotype were infected with VSV (for 12 h) and then were treated with murine IFNβ (100 IU/ml) or IFNγ (5ng/ml) for 30 min. Phosphorylation of Stat1 and total levels of Stat1 and actin were analyzed by IB.
D. Titer of VSV produced in EB-derived cells 14 h after infection (an incubation of cells with VSV at MOI 1.0 for 1h) is depicted. The effect of IFNβ (20 IU/ml) added either 16 h prior to the infection (pre-treat) or immediately after infection (co-add) was determined. Data shown (the mean ± SD) are representative of two independent experiments (each in triplicate). Asterisk denotes p<0.01 in comparison with untreated cells.
The expression of the complete HCV genome in Huh7 cells dramatically inhibited Stat1 phosphorylation induced by IFNα while IFNγ signaling was only modestly affected ((Luquin et al., 2007) and SF15). Remarkably, expression of IFNAR1 proteins in these cells partially rescued Type I IFN signaling; this effect was especially pronounced when a stabilized IFNAR1S535A mutant (that lacks Ser responsible for UPR-driven degradation, Figure 4) was used (Figure 6B). Similarly, a lesser inhibitory effect of VSV infection on Type I (but not Type II) IFN signaling was observed in cells derived from the IFNAR1S526A knock-in ES cells (Figure 6C). These results indicate that ER stress and viral infection inhibit Type I IFN signaling via phosphorylation-dependent downregulation of IFNAR1.
We then sought to investigate the role of this regulation in anti-viral defense. While pre-treatment of wild type cells with IFNβ exhibited an anti-viral effect, this cytokine was inefficient when added immediately after the virus. However, under the latter conditions, cells that harbored the knocked-in IFNAR1S526A mutant were capable of utilizing IFNβ to significantly reduce VSV propagation (Figure 6D). These results indicate that viruses at least temporarily benefit from the induced phosphorylation-dependent degradation of IFNAR1 and the ensuing suppression of the anti-viral defenses.
This hypothesis was further corroborated when we investigated the role of PERK in Type I IFN-induced signaling and anti-viral defense. Either knockdown of PERK in human cells (using RNAi approach) or acute genetic ablation of PERK in mouse fibroblasts (using Cre expression) led to the rescue of Stat1 phosphorylation in response to Type I IFNs (Figures 7A-B). Judging from expression of viral VSV-M protein, PERK-deficient cells contained fewer viruses; however, even when exposed to a five-fold higher viral titer to achieve a comparable expression of VSV-M, these cells remained competent in IFNβ-induced activation of Stat1 (Figure 7C). Such protection was not seen in infected MEFs lacking a related kinase, PKR (SF16). Specific role of PERK was further corroborated by data demonstrating that PERK knockdown in Huh7 cells expressing complete HCV genome also restored the ability of these cells to conduct Type I IFN signaling (Figure 7D). These data suggest that PERK plays an important role in virus-mediated inhibition of cellular responses to IFNα/β.
Figure 7.
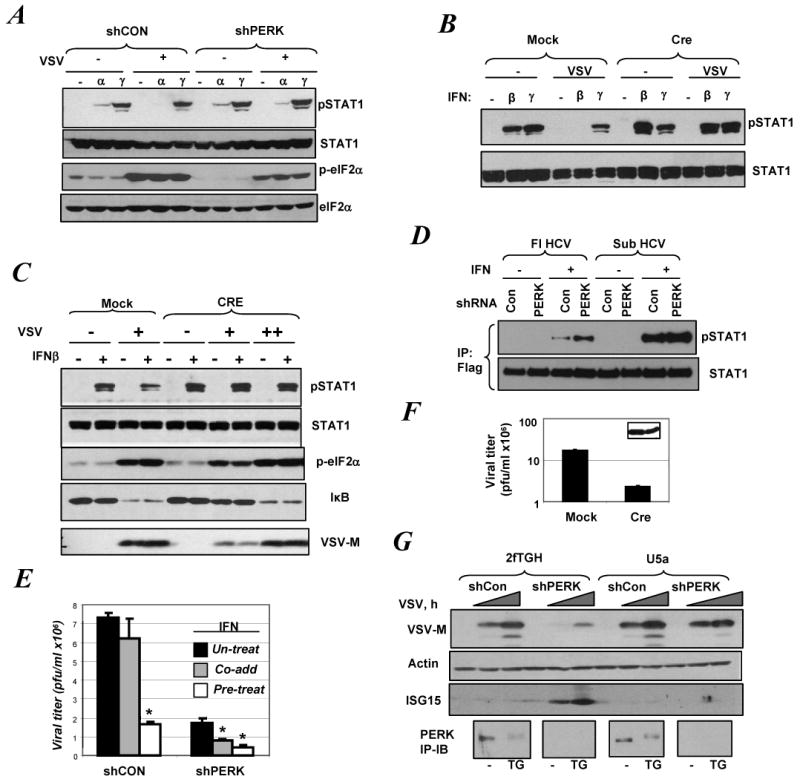
Role of PERK in virus-induced suppression of Type I IFN signaling
A. Control or PERK-depleted derivatives of 2fTGH cells were infected with VSV (for 20 h) and then treated with IFNα or IFNγ (50 IU/ml for 30 min). Phosphorylation and total levels of Stat1 and eIF2α were analyzed by IB.
B. MEFs from PERKfl/fl mice transduced with either empty vector (Mock) or construct for expression of Cre were infected with VSV (for 20 h) and then treated with IFNβ (100 IU/ml) or IFNγ (5ng/ml) for 30 min. Phosphorylation and total levels of Stat1 were analyzed by IB.
C. MEFs from PERKfl/fl mice transduced as indicated were infected with VSV at MOI 0.1 (+) or 0.5 (++) for 20 h and then treated with IFNβ for 30 min. IB analyses using indicated antibodies are shown.
D. Indicated derivatives of Huh7 cells were co-transfected with indicated shRNA constructs and Flag-STAT1 and treated with IFNα (50 IU/ml) for 30 min. Stat1 proteins were immunoprecipitated using anti-Flag antibody and analyzed by IB using anti-phospho-Stat1 and anti-Stat1 antibody.
E. Titer of VSV produced in control or PERK-depleted derivatives of 2fTGH cells 14 h after infection (an incubation of cells with VSV at MOI 1.0 for 1h) is depicted. The effect of IFNα (20 IU/ml) added either 16 h prior to the infection (pre-treat) or immediately after infection (co-add) was determined. Data shown (the mean ± SD) are representative of two independent experiments (each in triplicate). Asterisk denotes p<0.01 in comparison with untreated cells.
F. MEFs from PERKfl/fl mice transduced as indicated were infected with VSV (MOI 1.0). 20 h after infection, viral titer in the culture supernatant was determined. Values represent the mean ± SD of three independent experiments each performed in triplicate. VSV-M protein levels analyzed by IB in cell lysates are also shown in the inset.
G. 2fTGH and isogenic IFNAR2-deficient U5a cells were transduced with indicated shRNA constructs and then infected with VSV (MOI 1.0) for 18-20 h. Levels of VSV-M, ISG15 and β-actin were determined by IB. In a parallel experiment, these cells were treated with TG (1μM for 30 min) and analyzed for PERK levels by IP-IB (lower panel).
PERK knockdown in human cells increased their overall resistance to VSV and promoted the ability of cells to utilize IFNα added after the virus to significantly suppress the replication of VSV in these cells (Figure 7E). This finding is counterintuitive as both previous report (Baltzis et al., 2004) and our own data (SF17) indicated that mouse fibroblasts derived from conventional PERK knockout embryos are somewhat more sensitive to viral infection. Accordingly, conventional PERK-null MEFs displayed a somewhat reduced response to IFN as evident from analysis of the ISRE-driven luciferase reporter (SF18). However, re-expression of PERK in these cells did not stimulate either IFN responses or anti-viral defenses (SF17-18) suggesting that conventional PERK-null cells underwent an additional alteration to decrease IFN signaling. One cannot rule out that this alteration was to compensate an impaired ability to downregulate IFNAR1 via the ligand-independent pathway that has been previously shown to impede cell growth (Liu et al., 2008). Conversely, a compensatory pathway that rescued growth of cells overexpressing a dominant negative mutant of PERK has been also reported (Yamaguchi et al., 2008). Therefore, to investigate the role of PERK in murine cells, we turned to a model of Cre-mediated acute ablation of PERK in MEFs from PERKfl/fl mice that did not display defects in ISRE-dependent transcription (data not shown).
Remarkably, expression of Cre rendered these MEFs more resistant to VSV in the absence of added IFN (as seen from a decreased viral titer and expression of VSV-M, Figure 7F). Furthermore, PERK-deficient 2fTGH cells elevated the expression of interferon stimulated protein ISG15 and became more resistant to VSV infection (as judged by the lower levels of VSV-M protein, Figure 7G). These effects of PERK loss of function appeared to largely depend on the IFN pathway as much lesser changes were observed in isogenic U5a cells lacking IFNAR2. This conclusion was also supported by the fact that expression of VSV-M in infected PERK knockdown cells was noticeably increased by treatment with anti-IFNα/β antibodies (SF19). In all, these results suggest that activation of PERK is utilized by VSV to inhibit cellular responses to IFN and anti-viral defenses; this mechanism at least partially relies on UPR-induced PERK-dependent downregulation of IFN receptor.
Discussion
Ligand-stimulated, Jak-dependent ubiquitination and degradation of Type I IFN receptor plays a key role in the negative regulation of IFNα/β signaling (Kumar et al., 2003). However, recent evidence suggested the existence of a ligand- and Jak-independent pathway that controls stability of IFNAR1 in a phosphorylation-dependent manner. The importance of the latter pathway remained unclear as it was largely observed under the conditions of IFNAR1 overexpression (Liu et al., 2008). Here we report that this pathway is triggered by activation of the ER stress in a manner that requires function of PERK. Among the evidence supporting this conclusion are the following: (i) stimuli that cause UPR induce Ser phosphorylation within the IFNAR1 degron and promote IFNAR1 ubiquitination and degradation in cells that were not treated with IFN and in a Tyk2-indepenent manner; (ii) UPR-induced ubiquitination and degradation of IFNAR1 is inhibited in cells harboring knocked-in IFNAR1 mutant lacking phospho-acceptor site in its degron; and (iii) phosphorylation, ubiquitination and degradation of IFNAR1 induced by UPR are attenuated in PERK-deficient cells.
Furthermore, we found that this pathway, which leads to accelerated degradation of IFNAR1, is utilized by some viruses (including VSV and HCV). Future studies are necessary to identify other viral species that might inhibit IFN signaling by downregulating the receptor. Infection by VSV and expression of HCV genome led to downregulation of IFNAR1 and to inhibition of signaling and anti-viral effects induced by Type I IFN. These effects are at least partially impaired in cells that either lack PERK or contain phospho-degron mutant of IFNAR1 that is insensitive to PERK-induced phosphorylation and degradation. Given that infection with many of human and animal viruses are known to induce the UPR (He, 2006; Schroder and Kaufman, 2006; Wang and Weinman, 2006; Waris et al., 2002), we propose that some rapidly propagating viruses might generally employ the ligand-independent degradation of IFNAR1 to suppress anti-viral defenses in cells that have not yet been exposed to IFN. It is also tempting to speculate that this mechanism might play a role in pathogenesis of some viral infectious diseases.
ER stress has evolved to help the cells to deal with protein overload, which among other scenarios also occurs during acute viral infections. According to a current paradigm, being a cellular protective mechanism, UPR as a whole should help to limit viral infection (He, 2006). Our data, however, strongly suggest that specific activation of the PERK branch of UPR could instead favor viral replication via IFNAR1 degradation and suppression of IFN responses. Such an observation is not entirely unexpected, considering that one major consequence of PERK activation is an inhibition of translation through eIF2α phosphorylation, which, in cells infected by viruses, can also be carried out by PKR. It is plausible that this redundancy in means of translational inhibition might permit a sustained stimulation of PERK to negate IFN signaling and promote the infection. Intriguingly, while viruses often impede PKR-dependent phosphorylation of eIF2α (Bergmann et al., 2000; Gale et al., 1997; Gil et al., 2006; Langland and Jacobs, 2002), the examples of perturbation of PERK activation per se are rare (He, 2006).
During the initial rounds of infection, ligand-independent degradation of IFNAR1 could be of particular importance for a virus that has penetrated a naïve cell and started to produce massive amounts of viral proteins to prepare for replication. At this time, activation of ER-triggered IFNAR1 degron phosphorylation and ensuing degradation is expected to dramatically reduce the sensitivity of an infected cell to either exogenous or endogenously produced and secreted IFNα/β (as seen in Figures 6-7). Such alterations should benefit the offending virus in at least two ways. First, accelerated degradation of IFNAR1 will prevent an efficient induction of expression and activities of diverse anti-viral proteins (including 2′-5′ oligoadenylate synthetases, the Mx proteins, PKR, and the double-stranded-RNA-specific adenosine deaminase) that are known to suppress various steps of viral replication (reviewed in (Guidotti and Chisari, 2001)). Second, and, perhaps equally important, downregulation of IFNAR1 protects the host cell from the proapoptotic effects of IFN (Chawla-Sarkar et al., 2002a; Chawla-Sarkar et al., 2002b), and, therefore, affords the virus a sufficient time for completion of its infectious cycle. Regardless of which of these pathways are more relevant for each specific virus, our data strongly suggests that UPR-mediated downregulation of Type I IFN receptor and its signaling are important for unabated completion of initial rounds of infection when majority of target host cells are yet to encounter IFN.
Although such a mechanism should briefly benefit a virus that has already entered the cell, it cannot be expected to persist for a protracted period of time or to ensure that progeny released from this infected cell will have a better chance of infecting additional host cells. In order to properly synthesize their proteins, viruses have to attenuate the UPR responses, which they are indeed known to do using a plethora of diverse mechanisms (reviewed in (He, 2006; Schroder and Kaufman, 2006). Once ER stress is resolved, the PERK-dependent pathway that facilitate turnover of IFNAR1 will be suspended disabling a described general mechanism for impeding IFN signaling. Under these conditions, viruses will have to resort to individual tricks to maintain a degree of virulence in the environment containing IFNα/β. Such mechanisms (including prevention of microorganism-associated pattern recognition, reduced synthesis and secretion of IFN, inhibition of the activity of regulatory kinases, etc) have been indeed widely reported (reviewed in (Katze et al., 2002b). These mechanisms are of obvious importance for viral replication and subsequent transmission; they contribute to the forces that drive co-evolution of the pathogen and the mammalian host.
However, from the practical point of view of the host, interfering with a non-specific yet important mechanism enabling initial steps of infection would represent an attractive strategy toward either preventing viral infectious diseases or directing the development of these diseases toward an abortive course. Based on presented here data, it is tempting to speculate that inhibitors of PERK-dependent phosphorylation of IFNAR1 might exhibit a potent anti-viral activity. As Type I IFN also plays an important immunomodulatory role (Tompkins, 1999), it is plausible that such effects would be even more pronounced in vivo. Ongoing generation of phospho-degron knock-in animals and the design of the molecular means to inhibit the ligand-independent IFNAR1 phosphorylation are expected to gain an important insight. Given that we did not observe direct phosphorylation of IFNAR1 by PERK (data not shown), future studies aimed at understanding how exactly viruses and UPR mediate phosphorylation-dependent ubiquitination and degradation of IFNAR1 are warranted.
Additional incentive to proceed with further delineation of the mechanisms that confer ligand-independent IFNAR1 degradation is the fact that Type I IFN are widely used in treatment of patients with chronic viral infections (e.g., hepatitis C), multiple sclerosis and some malignancies. Whereas in cancer patients, the rationale for combining IFN with other anti-tumor agents that cause UPR (for example, proteasome inhibitors (Fribley et al., 2004; Nawrocki et al., 2005; Obeng et al., 2006)) might be re-evaluated, design of the means that would impede HCV-mediated ER stress and ensuing degradation of IFNAR1 (e.g., inhibitors of PERK-dependent pathway) is expected to benefit the patients whose therapeutic regiment includes IFNα.
Experimental Procedures (additional details could be found in the Supplemental Information)
Plasmids and Reagents
Vectors for bacterial expression of GST-ctIFNAR1 and mammalian expression of human and mouse Flag-IFNAR1 were described previously (Kumar et al., 2004; Kumar et al., 2007b; Kumar et al., 2003); other plasmids were generous gifts from J. Darnell (Flag-STAT1), R. Bartenschlager (HCV constructs) and K.U. Wagner (Cre). All shRNA constructs used were based on pLKO.1. The specific hairpin sequences are outlined in. Recombinant human IFNα2 was purchased from Roche Diagnostics. Recombinant human and mouse IFNγ and mouse IFNβ were purchased from PBL. Thapsigargin, cycloheximide and methylamine HCl were purchased from Sigma.
Cell culture and Virus
All cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (Hyclone) and various selection antibiotics when indicated. To acutely delete PERK in MEFs, MEFs harboring PERKfl/fl were infected with control retrovirus or retrovirus expressing Cre. The transduced cells were selected by puromycin for 72 h. The surviving clones were pooled and used for further analysis. IFNAR1-null MEFs reconstituted with pBABE-puro-based retroviral vector encoding Flag-tagged mIFNAR1wt and mIFNAR1S526A (Kumar et al., 2003) were generated and cultured in the presence of 4 μg/ml of puromycin. Huh7-derivative cells introduced with a complete HCV genome or a subgenomic genome were described in details in (Luquin et al., 2007) and were cultured in the presence of 500μg/ml of G418.
Mouse ES clone harboring a S526A mutation were obtained by homologous recombination. The targeting vector containing this mutation (Figure 3A) was introduced via electroporation into the C57/BL6 ES cells. The cells were subjected to neomycin selection and DNA samples from survived clones were analyzed by Southern blotting using the indicated probes to identify the homologous recombinants. For experiments, ES cells were differentiated into embryonic bodies according to ATCC recommendations established protocol (Maatman et al., 2003). The embryonic bodies were trypsinized and plated in gelatinized plates using IMDM containing 10% FBS. VSV (Indiana serotype, a gift from R. Harty) was propagated in HeLa cells.
Transfections and lentiviral vector-mediated gene knockdown
Transfection of 293T cells and KR-2 cells using LIPOfectamine Plus and of Huh7-derivatives using LIPOfecatimine-2000 (Invitrogen) was carried out according to manufacturer's recommendations. Replication-deficient lentiviral particles encoding shRNA against GFP (shCON), hPERK and hIRE1α, or the empty virus control were prepared via co-transfecting 293T cells with three other helper vectors as previously described (Dull et al., 1998). Viral supernatant were concentrated by PEG8000 precipitation and were used to infect 2fTGH and U5A lines in the presence of 3μg/ml of polybrene (Sigma). Cells were selected and maintained in the presence of 1.5 μg/ml of puromycin.
Cell treatment and viral infection
For examining the signaling event occurring after initiation of ER stress, cells were treated with vehicle (DMSO) or TG (1μM, unless otherwise indicated) for 0.5–2 h as shown in the figure legends. Unless otherwise specified, cells were inoculated with VSV at an initial MOI of 0.1-1.0 for 1 h. After removing the virus inoculum, cells were then fed with fresh medium. Cells were harvested at different time points afterwards; most of the effects were observed when the cells were uniformly infected and viral markers were at saturation. In some experiments, virus-infected cells were pulsed with IFNs for 30 min and then harvested.
To examine the anti-viral effect of IFN in relation to the time of its addition, 20 IU/ml of IFN was either added overnight prior to VSV infection or was added after the initial virus inoculation/removal. Culture supernatant was generally harvested 20 h after the initial inoculation for analysis of viral titer. VSV titer determination was performed as described elsewhere (Sharma et al., 2003).
Immunotechniques
Antibodies against pSTAT1, p-eIF2α, p-β-catenin, β-catenin, IRE1α (Cell Signaling), STAT1 (Cell Signaling), eIF2α (Biosources), hIFNAR1, PKR, c-Jun, IκBα (Santa Cruz), mIFNAR1 (R&D Systems), Flag tag, β-actin (Sigma) and ubiquitin (clone FK2, Biomol), ISG15 and PERK (Rockland) were used for immunoprecipitation and immunoblotting. Monoclonal antibody 23H12, specific for the M protein of VSV (VSV-M), was kindly provided by D. S. Lyles (Wake Forest University School of Medicine, Winston-Salem, N.C.). Antibody against IFNAR1 phosphorylated on Ser535 (in human receptor) or Ser526 (in murine receptor) were described previously (Kumar et al., 2004). Cells lysis, immunoprecipitation and immunoblotting procedures were described earlier (Kumar et al., 2004). Kinase assay with cell lysates and GST-ctIFNAR1 as a substrate was previously described (Liu et al., 2008).
Supplementary Material
Acknowledgments
We thank Dr. E. Bobrovnikova-Marjon for initial characterization of anti-PERK antibody, Drs. R. Kaufman, R. Harty, G.R. Stark, D. Ron, D.S. Lyles, K.U. Wagner, S. Pellegrini, and J. Darnell for reagents, Drs. Z. Ronai and J. Alwine for critical comments, members of Fuchs and Diehl lab for discussion. This work was supported by the NIH grants CA92900 (to S.Y.F.) and CA104838 (to J.A.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Baltzis D, Qu LK, Papadopoulou S, Blais JD, Bell JC, Sonenberg N, Koromilas AE. Resistance to vesicular stomatitis virus infection requires a functional cross talk between the eukaryotic translation initiation factor 2alpha kinases PERK and PKR. J Virol. 2004;78:12747–12761. doi: 10.1128/JVI.78.23.12747-12761.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann M, Garcia-Sastre A, Carnero E, Pehamberger H, Wolff K, Palese P, Muster T. Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J Virol. 2000;74:6203–6206. doi: 10.1128/jvi.74.13.6203-6206.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron CA. Interferons alpha and beta as immune regulators--a new look. Immunity. 2001;14:661–664. doi: 10.1016/s1074-7613(01)00154-6. [DOI] [PubMed] [Google Scholar]
- Blalock JE, Baron S. Mechanisms of interferon induced transfer of viral resistance between animal cells. J Gen Virol. 1979;42:363–372. doi: 10.1099/0022-1317-42-2-363. [DOI] [PubMed] [Google Scholar]
- Brassard DL, Grace MJ, Bordens RW. Interferon-alpha as an immunotherapeutic protein. J Leukoc Biol. 2002;71:565–581. [PubMed] [Google Scholar]
- Chawla-Sarkar M, Leaman DW, Jacobs BS, Borden EC. IFN-beta pretreatment sensitizes human melanoma cells to TRAIL/Apo2 ligand-induced apoptosis. J Immunol. 2002a;169:847–855. doi: 10.4049/jimmunol.169.2.847. [DOI] [PubMed] [Google Scholar]
- Chawla-Sarkar M, Leaman DW, Jacobs BS, Tuthill RJ, Chatterjee-Kishore M, Stark GR, Borden EC. Resistance to interferons in melanoma cells does not correlate with the expression or activation of signal transducer and activator of transcription 1 (stat1) J Interferon Cytokine Res. 2002b;22:603–613. doi: 10.1089/10799900252982089. [DOI] [PubMed] [Google Scholar]
- Ciccaglione AR, Costantino A, Tritarelli E, Marcantonio C, Equestre M, Marziliano N, Rapicetta M. Activation of endoplasmic reticulum stress response by hepatitis C virus proteins. Arch Virol. 2005;150:1339–1356. doi: 10.1007/s00705-004-0487-4. [DOI] [PubMed] [Google Scholar]
- Ciccaglione AR, Marcantonio C, Tritarelli E, Equestre M, Vendittelli F, Costantino A, Geraci A, Rapicetta M. Activation of the ER stress gene gadd153 by hepatitis C virus sensitizes cells to oxidant injury. Virus Res. 2007;126:128–138. doi: 10.1016/j.virusres.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fribley A, Zeng Q, Wang CY. Proteasome inhibitor PS-341 induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in head and neck squamous cell carcinoma cells. Mol Cell Biol. 2004;24:9695–9704. doi: 10.1128/MCB.24.22.9695-9704.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RM, Sonnabend JA. Mechanism of action of interferon. Arch Intern Med. 1970;126:51–63. [PubMed] [Google Scholar]
- Gale MJ, Jr, Korth MJ, Tang NM, Tan SL, Hopkins DA, Dever TE, Polyak SJ, Gretch DR, Katze MG. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology. 1997;230:217–227. doi: 10.1006/viro.1997.8493. [DOI] [PubMed] [Google Scholar]
- Gil LH, van Olphen AL, Mittal SK, Donis RO. Modulation of PKR activity in cells infected by bovine viral diarrhea virus. Virus Res. 2006;116:69–77. doi: 10.1016/j.virusres.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol. 2001;19:65–91. doi: 10.1146/annurev.immunol.19.1.65. [DOI] [PubMed] [Google Scholar]
- He B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 2006;13:393–403. doi: 10.1038/sj.cdd.4401833. [DOI] [PubMed] [Google Scholar]
- Hwang SY, Hertzog PJ, Holland KA, Sumarsono SH, Tymms MJ, Hamilton JA, Whitty G, Bertoncello I, Kola I. A null mutation in the gene encoding a type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc Natl Acad Sci U S A. 1995;92:11284–11288. doi: 10.1073/pnas.92.24.11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John J, McKendry R, Pellegrini S, Flavell D, Kerr IM, Stark GR. Isolation and characterization of a new mutant human cell line unresponsive to alpha and beta interferons. Mol Cell Biol. 1991;11:4189–4195. doi: 10.1128/mcb.11.8.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp CL, Biron CA, Irani DN. Interferon beta in multiple sclerosis: is IL-12 suppression the key? Immunol Today. 2000;21:24–28. doi: 10.1016/s0167-5699(99)01541-8. [DOI] [PubMed] [Google Scholar]
- Katze MG, He Y, Gale M. Viruses and interferon: A fight for supremacy. Nat Rev Immunol. 2002a;2:675–687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- Katze MG, He Y, Gale M., Jr Viruses and interferon: a fight for supremacy. Nat Rev Immunol. 2002b;2:675–687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- Kirkwood J. Cancer immunotherapy: the interferon-alpha experience. Semin Oncol. 2002;29:18–26. doi: 10.1053/sonc.2002.33078. [DOI] [PubMed] [Google Scholar]
- Kumar KG, Barriere H, Carbone CJ, Liu J, Swaminathan G, Xu P, Li Y, Baker DP, Peng J, Lukacs GL, Fuchs SY. Site-specific ubiquitination exposes a linear motif to promote interferon-alpha receptor endocytosis. J Cell Biol. 2007a;179:935–950. doi: 10.1083/jcb.200706034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar KG, Krolewski JJ, Fuchs SY. Phosphorylation and specific ubiquitin acceptor sites are required for ubiquitination and degradation of the IFNAR1 subunit of type I interferon receptor. J Biol Chem. 2004;279:46614–46620. doi: 10.1074/jbc.M407082200. [DOI] [PubMed] [Google Scholar]
- Kumar KG, Liu J, Li Y, Yu D, Thomas-Tikhonenko A, Herlyn M, Fuchs SY. Raf inhibitor stabilizes receptor for the type I interferon but inhibits its anti-proliferative effects in human malignant melanoma cells. Cancer Biol Ther. 2007b;6:1437–1441. doi: 10.4161/cbt.6.9.4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar KG, Tang W, Ravindranath AK, Clark WA, Croze E, Fuchs SY. SCF(HOS) ubiquitin ligase mediates the ligand-induced down-regulation of the interferon-alpha receptor. Embo J. 2003;22:5480–5490. doi: 10.1093/emboj/cdg524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langland JO, Jacobs BL. The role of the PKR-inhibitory genes, E3L and K3L, in determining vaccinia virus host range. Virology. 2002;299:133–141. doi: 10.1006/viro.2002.1479. [DOI] [PubMed] [Google Scholar]
- Liu J, Plotnikov A, Banerjee A, Kumar Suresh KG, Ragimbeau J, Marijanovic Z, Baker DP, Pellegrini S, Fuchs SY. Ligand-independent pathway that controls stability of interferon alpha receptor. Biochem Biophys Res Commun. 2008;367:388–393. doi: 10.1016/j.bbrc.2007.12.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockart RZ., Jr Production of an Interferon by L Cells Infected with Western Equine Encephalomyelitis Virus. J Bacteriol. 1963;85:556–566. doi: 10.1128/jb.85.3.556-566.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockart RZ, Jr, Horn B. Interaction of an Interferon with L Cells. J Bacteriol. 1963;85:996–1002. doi: 10.1128/jb.85.5.996-1002.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luquin E, Larrea E, Civeira MP, Prieto J, Aldabe R. HCV structural proteins interfere with interferon-alpha Jak/STAT signalling pathway. Antiviral Res. 2007;76:194–197. doi: 10.1016/j.antiviral.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Maatman R, Gertsenstein M, de Meijer E, Nagy A, Vintersten K. Aggregation of embryos and embryonic stem cells. Methods Mol Biol. 2003;209:201–230. doi: 10.1385/1-59259-340-2:201. [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18:716–731. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marijanovic Z, Ragimbeau J, Kumar KG, Fuchs SY, Pellegrini S. TYK2 activity promotes ligand-induced IFNAR1 proteolysis. Biochem J. 2006;397:31–38. doi: 10.1042/BJ20060272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Nawrocki ST, Carew JS, Dunner K, Jr, Boise LH, Chiao PJ, Huang P, Abbruzzese JL, McConkey DJ. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005;65:11510–11519. doi: 10.1158/0008-5472.CAN-05-2394. [DOI] [PubMed] [Google Scholar]
- Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestka S. The human interferon alpha species and receptors. Biopolymers. 2000;55:254–287. doi: 10.1002/1097-0282(2000)55:4<254::AID-BIP1001>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Pfeffer LM, Colamonici OR. Transmembrane signalling by interferon-alpha. Pharmacol Ther. 1991;52:149–157. doi: 10.1016/0163-7258(91)90005-7. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Schreiber RD, Farrar MA. The biology and biochemistry of interferon-gamma and its receptor. Gastroenterol Jpn. 1993;28 Suppl 4:88–94. doi: 10.1007/BF02782897. discussion 95-86. [DOI] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. Divergent roles of IRE1alpha and PERK in the unfolded protein response. Curr Mol Med. 2006;6:5–36. doi: 10.2174/156652406775574569. [DOI] [PubMed] [Google Scholar]
- Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- Takaoka A, Mitani Y, Suemori H, Sato M, Yokochi T, Noguchi S, Tanaka N, Taniguchi T. Cross talk between interferon-gamma and -alpha/beta signaling components in caveolar membrane domains. Science. 2000;288:2357–2360. doi: 10.1126/science.288.5475.2357. [DOI] [PubMed] [Google Scholar]
- Tardif KD, Waris G, Siddiqui A. Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol. 2005;13:159–163. doi: 10.1016/j.tim.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Tompkins WA. Immunomodulation and therapeutic effects of the oral use of interferon-alpha: mechanism of action. J Interferon Cytokine Res. 1999;19:817–828. doi: 10.1089/107999099313325. [DOI] [PubMed] [Google Scholar]
- Velazquez L, Fellous M, Stark GR, Pellegrini S. A protein tyrosine kinase in the interferon alpha/beta signaling pathway. Cell. 1992;70:313–322. doi: 10.1016/0092-8674(92)90105-l. [DOI] [PubMed] [Google Scholar]
- Wang T, Weinman SA. Causes and consequences of mitochondrial reactive oxygen species generation in hepatitis C. J Gastroenterol Hepatol. 2006;21 Suppl 3:S34–37. doi: 10.1111/j.1440-1746.2006.04591.x. [DOI] [PubMed] [Google Scholar]
- Waris G, Tardif KD, Siddiqui A. Endoplasmic reticulum (ER) stress: hepatitis C virus induces an ER-nucleus signal transduction pathway and activates NF-kappaB and STAT-3. Biochem Pharmacol. 2002;64:1425–1430. doi: 10.1016/s0006-2952(02)01300-x. [DOI] [PubMed] [Google Scholar]
- Welihinda AA, Tirasophon W, Kaufman RJ. The cellular response to protein misfolding in the endoplasmic reticulum. Gene Expr. 1999;7:293–300. [PMC free article] [PubMed] [Google Scholar]
- Wu JZ. Internally located signal peptides direct hepatitis C virus polyprotein processing in the ER membrane. IUBMB Life. 2001;51:19–23. doi: 10.1080/15216540119497. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Larkin D, Lara-Lemus R, Ramos-Castaneda J, Liu M, Arvan P. ER chaperone regulation, and survival of cells compensating for deficiency in the ER stress response kinase, PERK. J Biol Chem. 2008 doi: 10.1074/jbc.M802466200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol. 2002;22:3864–3874. doi: 10.1128/MCB.22.11.3864-3874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Gao B, Ye L, Kong L, Jing W, Yang X, Wu Z. Hepatitis C virus non-structural protein NS4B can modulate an unfolded protein response. J Microbiol. 2005;43:529–536. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.