

Abstract
The effects of vasoactive intestinal peptide camptothecin (VIP-CPT) conjugates were investigated on breast cancer cells and cells transfected with VIP receptors (R). (Ala2,8,9,19,24.25.27, Nle17, Lys28)VIP, (A-NL-K)VIP, was synthesized and Lys28 was coupled to a linker, N-methyl-amino-ethyl-glycine, L2, which formed a carbamate bond with CPT. The resulting (A-NL-K)VIP-L2-CPT was cytotoxic for MCF7 breast cancer cells, which have VPAC1-R, with IC50 values of 380 and 90 nM using the MTT and clonogenic assays, respectively. (A-NL-K)VIP, (A-NL-K)VIP-L2 and (A-NL-K)VIP-L2-CPT inhibited specific binding of 125I-VIP to 3T3 cells transfected with VPAC1-R with IC50 values of 1.9, 56 and 126 nM respectively. In contrast, (A-NL-K)VIP, (A-NL-K)VIP-L2 and (A-NL-K)VIP-L2-CPT inhibited specific binding of 125I-Ro25-1553 to 3T3 cells transfected with VPAC2-R with IC50 values of 3.9, 3162 and 2690 nM respectively. (A-NL-K)VIP, (A-NL-K)VIP-L2 and (A-NL-K)VIP-L2-CPT caused increased cAMP after addition to MCF7 cells. 125I-(A-NL-K)VIP-L2-CPT was internalized by MCF7 cells at 37°C but not 4°C. These results indicate that (A-NL-K)VIP-L2-CPT is a VPAC1-R agonist which is cytotoxic for breast cancer cells.
Keywords: camptothecin, VIP receptors, cellular proliferation, breast cancer, receptor-mediated cytotoxicity
1. Introduction
Breast cancer, which kills approximately 40,000 women annually in the United States, utilizes numerous growth factors to stimulate proliferation. Approximately 30% of breast cancer tumors over-express Her2/Neu, a tyrosine kinase [7]. Patients, with amplified Her2/Neu, can be treated with the monoclonal antibody Herceptin [44]. Herceptin delays progression of breast cancer in 90% of the treated patients, allowing time for treatment with other agents such as the chemotherapeutics adriamycin and taxol [47]. Approximately half of the breast cancer tumors express the estrogen receptor, and these patients can be treated with the anti-estrogens tamoxifen or raloxifene [10]. There is a need, however, to develop additional modes of treatment for patients whose tumors are estrogen receptor and Her-2/Neu negative.
The vasoactive intestinal peptide (VIP) receptor (R), in addition to Her-2/Neu and the estrogen receptor, is a molecular target for breast cancer. High densities of VPAC1-R and mRNA are present in all breast cancer cells tested [15,45,48]. VIP, which is a 28 amino acid peptide, binds with high affinity to VPAC1-R and VPAC2-R [18,22,41]. VIP is structurally related to pituitary adenylate cyclase activating polypeptide (PACAP), which binds with high affinity to VPAC1-R, VPAC2-R and and PAC1-R [1,38,42]. VPAC1-R, VPAC2–R and PAC1-R interact with a stimulatory guanine nucleotide binding protein after activation by agonist, resulting in increased adenylyl cyclase activity [28]. The elevated cyclic AMP causes activation of protein kinase A, resulting in the phosphorylation of protein substrates such as CREB [46]. This causes increased expression on nuclear oncogenes such as c-fos or c-jun, leading to elevated levels of vascular endothelial cell growth factor [3]. Using in vitro autoradiographic techniques, VPAC1-R has been detected in numerous tumors including neuroblastoma as well as bladder, breast, colon, liver, lung, stomach, thyroid, uterus and prostate cancer [40]. VPAC2-R predominates in gastric leimyomas, whereas PAC1-R is abundant in glioblastoma, adrenal and pituitary cancer [39].
The overexpression of VIP receptors, as well as other cell surface receptors, is increasingly being used to develop new drugs for the treatment of cancer patients. VIP receptor antagonists such as VIPhybrid or (N-Stearyl1, Nle17)VIP not only inhibit the proliferation of lung and breast cancer cells [17,34], but they potentiate the cytotoxicity of chemotherapeutic agents [14,31]. The overexpression of peptide receptors by the tumor can be used for receptor-targeted delivery of peptide receptor cytotoxic agents [35]. Previously, we reported that VIP-ellipticine (E) conjugates were cytotoxic for lung and breast cancer cells [26]. VIP-LALA-E was internalized by cancer cells containing VPAC1-R, and subsequently metabolized by proteolytic enzymes. The cytotoxic ellipticine (E), which was released after proteolytic metabolism of the VIP-E conjugate, inhibited topoisomerase enzymes leading to cellular cytotoxicity. In the present study we have extended these studies and synthesized VIP-camptothecin (CPT) conjugates which have a novel carbamate linker with a built-in nucleophile associated releasing group, L2. [11,27,33]. After receptor-mediated endocytosis, (Ala2,8,9,19,24.25.27, Nle17, Lys28)VIP-L2-camptothecin ((A-NL-K)VIP-L2-CPT) is metabolized by cytochrome P450 enzymes, releasing CPT. Our results show that (A-NL-K)VIP-L2-CPT is a selective VPAC1-R agonist, which is cytotoxic for breast cancer cells.
2. Materials and Methods
2.1 Materials
The following cells and materials were obtained from the sources indicated: BALB 3T3 (mouse fibroblast), T47D and MCF7 (human breast cancer) and SupT1 cells were from American Type Culture Collection (ATCC), Rockville, MD; Dulbecco’s minimum essential medium (DMEM), phosphate-buffered saline (PBS), Roswell Park Memorial Institute (RPMI-1640), trypsin-EDTA and fetal bovine serum (FBS), from Biofluids, Rockville, MD; G418 sulfate from Life Technologies, Inc., Grand Island, NY; Na125I (2200 Ci/mmol) was from Amersham Pharmacia Biotech; CPT, formic acid, ammonium formate, disodium tetraborate, 4-(dicyanomethylene)-2-methyl-6-(4-dimethylaminostyryl)-4-H-pyran (DCM); 2-dimethylaminoisopropyl chloride hydrochloride (DIC); 1-methyl-2-pyrrolidinone (NMP); dimethylaminopyridine (DMAP); soybean trypsin inhibitor, bacitracin, leupeptin, 4-(2-aminoethyl)-benzenesulfonyl fluoride (AEBSF), poly-L-lysine and triton X-100 were from Sigma-Aldrich, St. Louis, MO; 1,2,4,6-tetrachloro-3α,6α-diphenylglycouril (IODO-GEN) from Pierce Chemical Co., Rockford, IL; VIP and Ro25-1553 were purchased from Bachem, Torrance, CA; AG 1-X8 resin from Bio-Rad, Richmond;[3-(4,5 dimethylthiazol-2-yl)-2.5-diphenyl-2H-tetrazolium bromide] (MTT) and iodo-nitrotetrazolium violet was from Sigma Chemical Co., St. Louis, MO; Agarose was purchased from FMC Corp., Rockford, ME.
2.2 Cell culture
MCF7, SupT1 and T47D cells were cultured in RPMI-1640 medium containing 10% heat-inactivated FBS (Life Technologies, Rockville, MD). Cells were split weekly 1/20 with trypsin-EDTA. Balb 3T3 cells stably expressing human VPAC1-R or VPAC2-R were made as described previously [21] and were grown in DMEM supplemented with 300 mg/liter of G418 sulfate. Cells were mycoplasma free and were used when they were in exponential growth phase after incubation at 37°C in 5% CO2, 95% air.
2.3 Preparation of peptides
(A-NL-K)VIP, which is an analog of the selective VPAC1-R agonist, (Ala2,8,9,11,19,24,25,27,28)VIP, was synthesized using solid phase techniques. The VIP-CPT conjugates were synthesized by coupling the CPT to the Lys28 of (A-NL-K)VIP-L2 as described previously [19]. The product (A-NL-K)VIP-L2-CPT had >97% purity, by preparative HPLC (C18 silica), mass spectrometry and amino acid analysis. 125I-VIP, 125I-(A-NL-K)VIP-L2-CPT and 125I-Ro25-1553 (Ro) had specific activities of 2200 Ci/mmol, were prepared as previously described [19]. The radiolabeled peptides were separated using a C18 Sep-Pak (Waters Associates, Milford, MA) and fractions with the highest radioactivity were pooled, neutralized with 0.2 M Tris buffer (pH 9.5) and stored with 0.5% bovine serum albumin (w/v) at −20°C.
2.4 Binding of 125I-labeled VIP-related peptides
Binding was performed as described previously [21]. The standard binding buffer contained 24.5 mM HEPES (pH 7.4), 98 mM NaCl, 6 mM KCl, 5 mM MgCl2, 2.5 mM NaH2PO4, 5 mM sodium pyruvate, 5 mM sodium fumarate, 0.01% (w/v) soybean trypsin inhibitor, 1% amino acid mixture, 0.2% (w/v) bovine serum albumin (BSA), and 0.05% (w/v) bacitracin. BALB 3T3 cells stably expressing human VPAC1-R (0.3 × 106), VPAC2-R (0.03 × 106), SupT1 or T47D cells were incubated with 50 pM 125I-labeled ligand at 22°C for 60 min. Aliquots (100 μl) were removed and centrifuged through 300 μl of incubation buffer in 400 μl microfuge tubes at 10,000 ×g for 1 min using a Beckman micro-centrifuge B. The pellets were washed twice with buffer and counted for radioactivity in a gamma counter. The nonsaturable binding was the amount of radioactivity associated with cells in incubations containing 50 pM radioligand (2200 Ci/mmol) and 1 μM unlabeled ligand. Nonsaturable binding was <10% of total binding in all the experiments. Inhibition constants (Ki) were determined using a least-square, curve-fitting program (KaleidaGraph) and the Cheng-Prusoff equation [5].
The internalization of 125I-(A-NL-K)VIP-L2-CPT was investigated. 125I-(A-NL-K)VIP-L2-CPT (0.1 nM) was incubated with MCF-7 cells at 4°C for 2 h in PBS containing 2% BSA and 200 μg/ml bacitracin in the presence or absence of 1 μM VIP. One of the plates was incubated at 37°C for 5 min. The plates were washed in cold PBS containing 2% BSA and 200 μg/ml bacitracin three times to remove free 125I-(A-NL-K)VIP-L2-CPT. The cells were treated with 150 mM NaCl/100 mM acetic acid for 5 min at 4°C. The supernatant, which contained radiolabeled peptide bound to the cell surface, was counted. The cells, which contained radiolabeled peptide that was internalized, were dissolved in 100 mM NaOH and counted in a gamma counter.
2.5 Cyclic AMP assays
Cyclic AMP assays were conducted by radioimmunoassay. NCI-H1299 cells in 24 well plates were incubated with VIP-CPT conjugates in the presence of 250 μl of SIT medium containing 50 μM IBMX, 1 mg/ml bacitracin and 1% BSA for 5 min at 37°C. After 5 min, the samples were treated with 250 μl of cold ethanol. The samples were mixed and stored at −80°C until assayed. The samples were assayed in duplicate by radioimmunoassay (New England Nuclear, Boston, MA). The mean value ± S.D. of 4 determinations were calculated.
2.6 Proliferation assays
Growth studies were performed using the [3-(4,5dimethylthiazol-2-yl)-2,5-diphenyl]-2H-tetrazolium bromide (MTT) assay [24]. MCF7 human breast cancer cells (104/well) were placed in SIT medium (100 μl) and various concentrations of (A-NL-K)VIP-L2-CPT, (A-NL-K)VIP or (A-NL-K)VIP-L2 added. After 4 days, 15 μl (1 mg/ml) of MTT was added. After 4 h, 150 μl of DMSO was added. After 16 h, the optical density at 570 nm was determined using an ELISA reader. The proliferation rates were calculated from the OD readings with various concentrations of VIP-CPT conjugate using the untreated cells as 100.
The effects of VIP-CPT conjugates on the growth of MCF7 cells were investigated using a clonogenic assay. The base layer consisted of 3 ml of 0.5% agarose in SIT medium containing 5% fetal bovine serum in 6 well plates. The top layer consisted of 3 ml of SIT medium in 0.3% agarose, VIP-CPT conjugates and 5 × 104 MCF7 cells. Triplicate wells were plated and after 2 weeks, 1 ml of 0.1% p-iodonitrotetrazolium violet was added and after 16 hours at 37°C, the plates were screened for colony formation; the number of colonies larger than 50 μm in diameter were counted using an Omnicon image analysis system.
3. Results
3.1 (A-NL-K)VIP, (A-NL-K)VIP-L2 and (A-NL-K)VIP-L2-CPT bind to cells containing VPAC1-R or VPAC2-R
125I-VIP and 125I-Ro25-1553 (Ro) were examined by radioreceptor assay for their ability to interact with human cancer cells and cells stably transfected with human VIP receptors (Fig. 1). 125I-VIP bound specifically to 3T3 cells stably transfected with VPAC1-R (total binding was 5057 ± 263 cpm and nonspecific binding was 225 ± 27 cpm) and VPAC2-R, whereas 125I-Ro bound specifically to 3T3 cells stably transfected with VPAC2-R (total binding was 2945 ± 176 and nonspecific binding was 220 ± 107 cpm) but not VPAC1-R. Also, 125I-VIP bound specifically to T47D breast carcinoma cells (total binding was 1127 ± 52 cpm whereas nonspecific binding was 68 ± 5 cpm), which have VPAC1-R [19], whereas 125I-Ro bound with high affinity to SupT1 cells (total binding was 764 ± 39 cpm and nonspecific binding was 15 ± 1 cpm), which have VPAC2-R [21].
Figure 1.
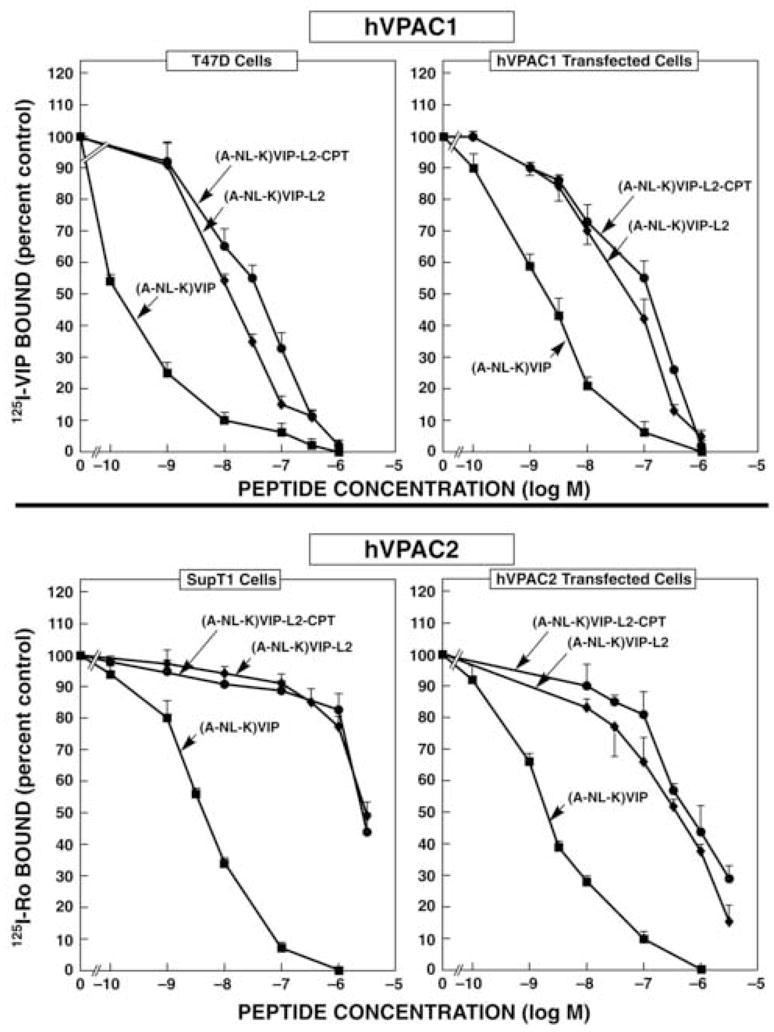
VIP-CPT conjugate binding. (Top) The ability of (A-NL-K)VIP (■), (A-NL-K)VIP-L2-CPT (●) and (A-NL-K)VIP-L2 (◆) to inhibit specific 125I-VIP binding to T47D and 3T3 cells containing VPAC1-R was determined. (Bottom) The ability of (A-NL-K)VIP (■), (A-NL-K)VIP-L2-CPT (●) and (A-NL-K)VIP-L2 (◆) to inhibit specific 125II-Ro25-1553 binding to SupT1 and 3T3 cells containing VPAC2-R was determined. The mean value ± S.D. of 3 determinations is indicated. This experiment is representative of 4 others
(A-NL-K)VIP inhibited specific binding of 125I-Ro with high affinity to cells containing VPAC2-R or 125I-VIP binding to cells containing VPAC1-R. Little specific 125I-Ro binding to 3T3 cells or SupT1 was inhibited by 0.1 nM (A-NL-K)VIP, but almost all specific binding was inhibited by 100 nM (A-NL-K)VIP (Fig. 1, bottom). The IC50 values for (A-NL-K)VIP and 3T3 containing VPAC2-R or SupT1 cells were 1.9 ± 0.1 or 4.3 ± 0.2 nM respectively. Similarly, (A-NL-K)VIP inhibited specific 125I-VIP binding to 3T3 cells containing VPAC1-R with an IC50 value of 1.9 ± 0.1 nM (Fig. 1, top). Surprisingly, (A-NL-K)VIP inhibited specific 125I-VIP binding to T47D cells with an IC50 value of 0.13 ± .01 nM. These results indicate that (A-NL-K)VIP binds with high affinity to cells containing either human VPAC1-R or VPAC2-R.
The ability of (A-NL-K)VIP-L2 or (A-NL-K)VIP-L2-CPT to bind to cells containing VPAC2-R or VPAC1-R was investigated. Specific 125I-Ro binding to 3T3 cells containing VPAC2-R was inhibited by (A-NL-K)VIP-L2 or (A-NL-K)VIP-L2-CPT with IC50 values of 372 ± 15 and 589 ± 23 nM, respectively (Fig. 1, bottom). Similarly, specific 125I-Ro binding to SupT-1 cells was inhibited by (A-NL-K)VIP-L2 or (A-NL-K)VIP-L2-CPT with IC50 values of 3162 ± 126 and 2690 ± 800 nM, respectively. Specific 125I-VIP binding to 3T3 cells transfected with VPAC1-Rs was inhibited by (A-NL-K)VIP-L2 or (A-NL-K)VIP-L2-CPT with IC50 values of 56 ± 2 and 126 ±11 nM, respectively (Fig. 1, top). Specific 125I-VIP binding to T47D cells was inhibited by (A-NL-K)VIP-L2 or (A-NL-K)VIP-L2-CPT with IC50 values of 13.2 ± 0.7 and 39.8 ± 2.3 nM, respectively. Similar results were obtained using MCF7 cells which have VPAC1-R. (data not shown). These results indicate that (A-NL-K)VIP-L2 and (A-NL-K)VIP-L2 CPT bind with 5–30 fold higher affinity to cells containing VPAC1-Rs than cells containing VPAC2-Rs.
125I-(A-NL-K)VIP-L2-CPT bound specifically to MCF7 cells. Table 1 shows that at 4°C, most of the specific 125I-(A-NL-K)VIP-L2-CPT bound to the cell surface and only 22.1% was internalized. In contrast at 37°C, most of the specific 125I(A-NL-K)VIP-L2-CPT bound to MCF7 cells was internalized (61.8%). Similar results were obtained using 125I-(A-NL-K)VIP and MCF7 cells (data not shown).
Table 1.
(A-NL-K) VIP-L2-CPT internalization by MCF7 cells.
| Temperature | % Internalization |
|---|---|
| 4°C | 22.1 ± 2.0 |
| 37°C | 61.8 ± 4.1 |
The % specific binding of 125I-(A-NL-K)VIP-L2-CPT that was internalized was determined at 4°C or 37°C. The mean value ± SD of 3 determinations each repeated in triplicate is indicated.
3.2 (A-NL-K)VIP, (A-NL-K)VIP-L2 and (A-NL-K)VIP-L2-CPT are VPAC1-R agonists
The ability of VIP-CPT conjugates to elevate cAMP was investigated in MCF7 breast cancer cells, which have VPAC1-R [48], by radioimmunoassay. Table 2 shows that addition of 10 nM VIP to MCF7 cells increased the cAMP 7.4-fold from 7 to 52 fmol. Forskolin, which interacts with Gs resulting in maximal adenylyl cyclase activity, increased the cAMP 10.9-fold from 7 to 76.3 fmol. (A-NL-K)VIP, (A-NL-K)VIP-L2 and (A-NL-K)VIP-L2-CPT (1000 nM) increased the cAMP in MCF-7 cells 10.1-fold, 8.3-fold and 8.7-fold respectively. These results indicate that (A-NL-K)VIP, (A-NL-K)VIP-L2 and (A-NL-K)VIP-L2-CPT are VPAC1-R agonists.
Table 2.
Ability of VIP-CPT conjugates to elevate cAMP in MCF7 cells.
| Addition | cAMP, fmol |
|---|---|
| None | 7.0 ± 2.0 |
| VIP, 10 nM | 52.2 ± 6.0 |
| (A-NL-K)VIP, 1000 nM | 70.5 ± 7.2 |
| (A-NL-K)VIP-L2, 1000 nM | 58.3 ± 4.4 |
| (A-NL-K)VIP-L2-CPT, 1000 nM | 60.8 ± 7.3 |
| Forskolin, 50 uM | 76.3 ± 3.1 |
The agents were added to MCF7 cells for 5 min at 37°C. The mean value ± S.D. of 4 determinations each repeated in duplicate is indicated. This experiment is representative of 2 others
3.3 (A-NL-K)VIP-L2-CPT inhibits the growth of breast cancer cells
The ability of VIP-CPT conjugates to inhibit the growth of MCF7 cells was investigated. Using the MTT assay, 30 nM (A-NL-K)VIP-L2-CPT had little effect on proliferation, whereas 1000 nM (A-NL-K)VIP-L2-CPT strongly inhibited MCF7 proliferation (Fig. 2). The IC50 value was 380 nM for (A-NL-K)VIP-L2-CPT, whereas (A-NL-K)VIP had little effect on MCF7 growth at concentrations up to 1000 nM. Similarly, (A-NL-K)VIP-L2 did not affect MCF7 proliferation (data not shown). These results indicate that (A-NL-K)VIP-L2-CPT inhibits the growth of breast cancer cells.
Figure 2.
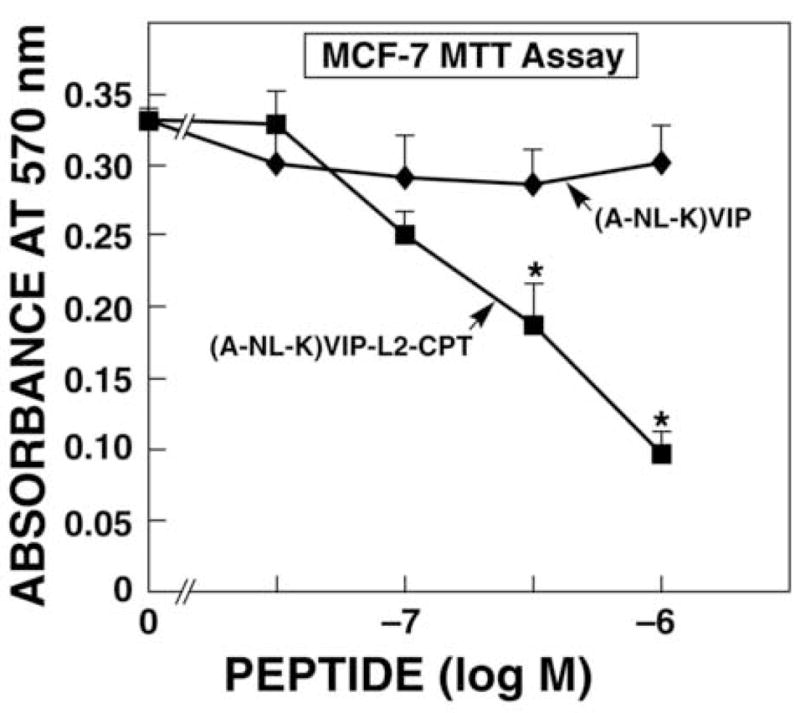
MTT assay. The ability of varying doses of (A-NL-K)VIP-L2-CPT (■) and (A-NL-K)VIP (◆) to inhibit MCF7 proliferation was determined. The mean value ± S.D. of 8 determinations is indicated; p < 0.05; * using Student’s t-test. This experiment is representative of 2 others.
Using the clonogenic assay, (A-NL-K)VIP-L2-CPT reduced MCF7 proliferation with an IC50 value of 90 nM (Fig. 3). In contrast, (A-NL-K)VIP had little effect on MCF7 growth. Similarly, (A-NL-K)VIP-L2 had little effect on MCF7 colonies (data not shown). Figure 4 shows that a large number of robust MCF7 colonies formed in the absence (Fig. 4A) or presence of 30 nM (A-NL-K)VIP-L2-CPT (Fig. 4B). In the presence of 100 or 300 nM (A-NL-K)VIP-L2-CPT, MCF7 colonies were smaller and absent (Fig. 4C and 4D, respectively). These results indicate that (A-NL-K)VIP-L2-CPT but not (A-NL-K)VIP or (A-NL-K)VIP-L2 is cytotoxic for MCF-7 cells.
Figure 3.
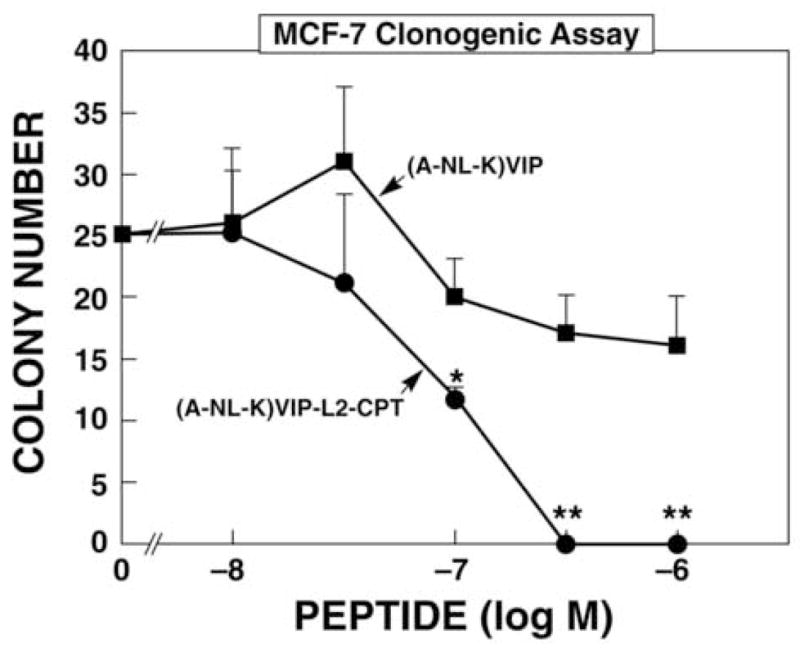
Clonogenic assay. The ability of varying doses of (A-NL-K) VIP-L2-CPT (●) and (A-NL-K)VIP (■) to inhibit MCF7 proliferation was determined. The mean value ± S.D. of 3 determinations is indicated; p < 0.01, **; p < 0.05; * using Student’s t-test. This experiment is representative of 3 others.
Figure 4.
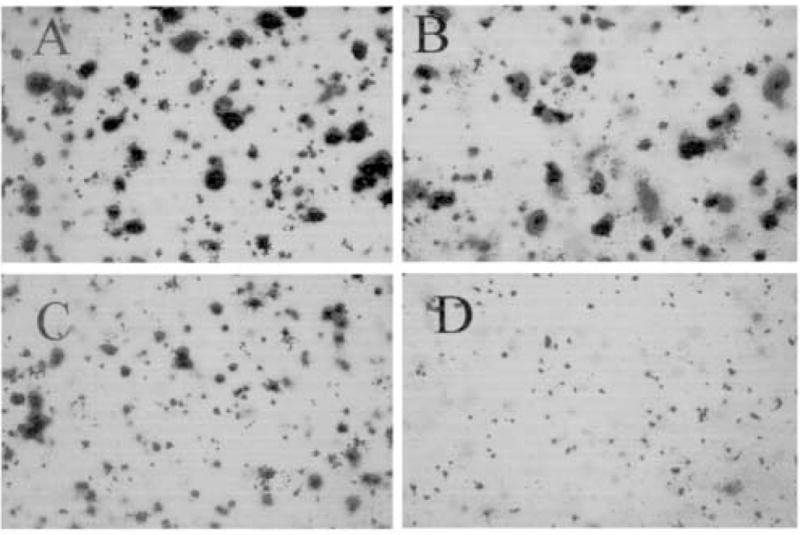
Effect of (A-NL-K) VIP-L2-CPT on MCF7 cells. The ability of 0 (A), 30 nM (B), 100 nM (C) and 300 nM (D) (A-NL-K) VIP-L2-CPT to inhibit MCF7 proliferation was determined. This experiment is representative of 3 others.
4. Discussion
VIP-like peptides are autocrine growth factors for some cancer cells [30]. Pre-pro-VIP and pre-pro-PACAP mRNA are present in lung cancer cells [16]. Also, VIP and pro-VIP immunoreactivity are present in lung cancer cells [25]. PACAP mRNA and immunoreactivity are present in breast cancer biopsy specimens. Pre-pro-PACAP but not PACAP-38 immunoreactivity was present in peri-tumoral and tumoral breast tissues as well as alveolar epithelial cells and leukocytes in connective tissue [12]. Fully processed VIP-like peptides and their precursors have biological activity [9].
Basic amino acids of VIP such as Arg14, Lys15 and Lys21 may interact electrostatically with acidic amino acids such as Glu36 on the VPAC1-R [36]. Substitution of Ala for VIP amino acids at positions 1, 6, 12, 14 or 23 reduced binding affinity to VPAC1-R by over 2-orders of magnitude [19,36]. Substitution of Ala for VIP amino acids at positions 3, 5, 7, 10, 15, 20 or 21 reduced binding affinity to VPAC1-R by over 1-order of magnitude [19,36]. The analog (Ala2,8,9,11,19,24,25,27,28)VIP is a potent VPAC1-R agonist that is resistant to degradation [20].
VIP receptors were detected in 68 of 68 breast cancer biopsy specimens, as determined using in vitro autoradiographic techniques [40]. Specific 125I-VIP binding was displaced by a selective VPAC1-R agonists but not Ro25-1553, a VPAC2-R agonist. Some arterial vessels in the smooth muscle also had VPAC1-R and VPAC2-R. By RT-PCR, mRNA for VPAC1-R was detected in 20 out of 20 breast cancer biopsy specimens whereas VPAC2-R mRNA was only present in 45% of the samples [29]. By immunocytochemistry, VPAC1-R and VPAC2-R were present in ductal and glandular epithelial cells both in the normal and malignant breast in 13 patients [13]. Immunoreactive VPAC1-R and VPAC2-R were present in carcinomatous tissue at the stromal level in leukocytes. Because 100 nM VIP, increases adenylyl cylase activity in breast membranes, the VIP receptors are biologically active.
Breast cancer VIP receptors bind radiolabeled VIP with high affinity [48]. Ala was substituted for VIP amino acids at non-essential positions 2,8,9,19,24,25,27, Nle was substituted at Met17 and Lys substituted at position 28 for conjugation to CPT. The resulting (A-NL-K)VIP bound with high affinity to VPAC1-R and VPAC2-R. (A-NL-K)VIP bound with higher affinity (Kd = 0.1 nM) to T47D cells than 3T3 cells transfected with VPAC1-R (Kd = 1.9 nM). This may result because T47D cells have fewer receptors (resulting in less ligand internalization and degradation) or have less guanine nucleotides (GTP impairs VIP receptor binding). Addition of L2 or L2-CPT to the Lys28 ε-amino group reduced the affinity of binding by approximately 1-order of magnitude to VPAC1-R and 2-orders of magnitude to VPAC2-R. Previously, we found that VIP and VIP-E analog had IC50 values of 10 and 100 nM using cells containing VPAC1-R [26]. The results indicate that position 28 of VIP or the C-terminal of VIP can be modified with retention of high affinity binding. Because addition of (A-NL-K)VIP, (A-NL-K)VIP-L2 or (A-NL-K)VIP-L2-CPT to MCF7 cells increased cAMP, each of the VIP analogs functioned as VPAC1-R agonists. 125I-(A-NL-K)VIP-L2-CPT was internalized by MCF7 cells at 37°C but not 4°C. In other cellular systems, peptide-CPT conjugates are rapidly internalized after binding to cell surface receptors [23,32].
VIP-CPT or VIP-E conjugates may be internalized after binding to VPAC1-R by receptor-mediated endocytosis. After targeting to lysosomes, VIP-LALA-E may be metabolized by endopeptidases, which degrade amide bonds. In contrast, (A-NL-K)VIP-L2-CPT is metabolized by P450 enzymes which metabolize carbamate bonds [4]. P450 enzymes are present in numerous human tumors including breast, colon, kidney, liver, lung and prostate, as well as to be up-regulated by human tumors [37]. A major difference, however, is that E targets topoisomerase II, whereas CPT targets topoisomerase I. Because (A-NL-K)VIP-L2-CPT is more resistant to degradation by proteases than is VIP-LALA-E, it is better at killing MCF7 cells.
VIP-CPT conjugates inhibited the proliferation of MCF7 cells. In the MTT assay, (A-NL-K)VIP-L2-CPT at 300 and 1000 nM significantly inhibited MCF7 proliferation, whereas (A-NL-K)VIP or (A-NL-K)VIP-L2 did not. In the clonogenic assay, (A-NL-K)VIP-L2-CPT significantly inhibited MCF7 proliferation at 100, 300 and 1000 nM, whereas (A-NL-K)VIP or (A-NL-K)VIP-L2 did not. The clonogenic assay may be more sensitive than the MTT assay, because MCF7 cells are exposed to VIP-CPT conjugates for 2 weeks as opposed to 3 days. Theses results indicate that CPT is essential for VIP-chemotherapeutic conjugates to kill breast cancer cells, however, it is also possible that VIP-chemotherapeutic conjugates will be cytotoxic for normal cells having high densities of VPAC1-R.
Prodrugs are being used to decrease drug associated side-effects and enhance tumor cytotoxicity [6]. A wide spectrum of prodrugs have been developed, however, the use of ligands for hormone/growth factor receptors that are overexpressed by tumors has received considerable attention [8]. Recent studies report that bombesin, luteinizing hormone-releasing hormone, somatostatin and VIP receptor ligands can be coupled to cytotoxic agents or radiolabels and have potent antitumor effects [2, 32, 35, 43]. The results of these studies with other peptide receptors, as well as the present study with VIP indicate that the chemotherapeutic cytotoxic analogs coupled to peptide receptor ligands can be developed that are biologically active. These analogs not only allow receptor-targeted delivery but hydrolyzed release of the cytotoxic agent within the cancer cell. The availability of peptide-chemotherapeutic analogs such as the VIP-CPT conjugates should allow a better assessment of this approach using in vivo models.
Acknowledgments
This research is supported by the Intramural Research Program of the NIH, NCI Center for Cancer Research, NIDDK, and Tulane University Peptide Research Fund.
Abbreviations
- VIP
vasoactive intestinal peptide
- CPT
camptothecin
- PACAP
pituitary adenylate cyclase activating polypeptide
- (A-NL-K)VIP
(Ala2,8.9.19.24.25.27, Nle17, Lys28) VIP
- Ki
inhibitory constant
- L2
N-(N-methyl-amino-ethyl)-glycine carbamate
- IP
inositol phosphate
- RPMI
Roswell Park Memorial Institute
- SIT medium
RPMI-1640 containing 3 × 10−8 M sodium selenite, 5 μg/ml bovine insulin and 10 μg/ml transferrin
- MTT
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide]
- VPAC1-R
VIP-1 receptor
- VPAC2-R
VIP-2 receptor
- PAC1-R
PACAP receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arimura A. Pituitary adenylate cyclase activating polypeptide (PACAP): Discovery and current status of research. Regul Peptides. 1992;37:287–303. [PubMed] [Google Scholar]
- 2.Buchholz S, Keller G, Schally AV, Halmos G, Hohla F, Heinrich E, Koester F, Baker B, Engel JB. Therapy of ovarian cancers with targeted cytotoxic analogs of bombesin, somatostatin, and luteinizing hormone-releasing hormone and their combinations. Proc Natl Acad Sci USA. 2006;103:10403–7. doi: 10.1073/pnas.0602971103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Casibang MC, Purdom S, Zia H, Jakowlew S, Neckers L, Ben-Av P, Hla T, You L, Jablons D, Moody TW. Prostaglandin E2 and VIP increase VEGF mRNAs in lung cancer cells. Lung Cancer. 2001;31:203–212. doi: 10.1016/s0169-5002(00)00168-9. [DOI] [PubMed] [Google Scholar]
- 4.Cha SW, Gu HK, Lee KP, Lee MH, Han SS, Jeong TC. Immunotoxicity of ethylcarbamate in female BALB/C mice: Role of esterase and cytochrome P450. Toxicol Lett. 2000;115:173–181. doi: 10.1016/s0378-4274(00)00176-4. [DOI] [PubMed] [Google Scholar]
- 5.Cheng Y, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 6.deGroot FM, Damen EW, Scheeren HW. Anticancer prodrugs for application in monotherapy: targeting hypoxia, tumor-associated enzymes, and receptors. Curr Med Chem. 2001;8:1093–1122. doi: 10.2174/0929867013372634. [DOI] [PubMed] [Google Scholar]
- 7.Dickson RB, Pestell RG, Lippman ME. Molecular biology of breast cancer. In: DeVita VT, Hellman S, Rosenberg SA, editors. Cancer: Principles and practice of oncology. Philadelphia: Lippincott, Williams and Wilkins; 2006. pp. 1399–1414. [Google Scholar]
- 8.Dubowchik GM, Walker MA. Receptor-mediated and enzyme-dependent targeting of cytotoxic anticancer drugs. Pharmacol Ther. 1999;83:67–123. doi: 10.1016/s0163-7258(99)00018-2. [DOI] [PubMed] [Google Scholar]
- 9.Fahrenkrug J. Glycine-extended processing intermediate of proVIP: A new bioactive form of VIP in the rat. Biomed Res. 1992;13(Sup2):19–23. doi: 10.1016/0006-291x(91)91795-e. [DOI] [PubMed] [Google Scholar]
- 10.Fisher B, Constantino JP, Wickerham DL, Cecchini WM, Cronin WM, Robidoux A, Bevers TB, Kavanah MT, Atkins JN, Margolese RG, Runowicz CS, James JM, Ford LG, Wolmark N. Tamoxifen for the prevention of breast cancer. Current status of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst. 2005;97:1652–1662. doi: 10.1093/jnci/dji372. [DOI] [PubMed] [Google Scholar]
- 11.Fuselier JA, Sun L, Woltering SN, Murphy WA, Vasilevich N, Coy DH. An adjustable release rate linking strategy for cytotoxin-peptide conjugates. Bioorg Med Chem Lett. 2003;13:799–803. doi: 10.1016/s0960-894x(03)00016-7. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Fernandez MO, Bodega G, Ruiz-Villaespesa A, Cortes J, Prieto JC, Carmena MJ. PACAP expression and distribution in human breast cancer and healthy tissue. Cancer Let. 2004;205:189–195. doi: 10.1016/j.canlet.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 13.Garcia-Fernandez MO, Collado B, Bodega G, Cortes J, Ruiz-Villaespesa A, Carmena MJ, Prieto JC. Pituitary adenylate cyclase-activating peptide/vasoactive intestinal peptide receptors in human normal mammary gland and breast cancer tissue. Gynecol Endocrinol. 2005;20:327–333. doi: 10.1080/09513590500098240. [DOI] [PubMed] [Google Scholar]
- 14.Gelber E, Granoth R, Fridkin M, Dreznik Z, Brenneman DE, Moody TW, Gozes I. A lipophilic vasoactive intestinal analog enhances the antiproliferative effect of chemotherapeutic agents on cancer cell lines. Cancer. 2001;92:2172–2180. doi: 10.1002/1097-0142(20011015)92:8<2172::aid-cncr1560>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 15.Gespach C, Bawab W, Decremox P, Calvo F. Pharmacology, molecular identification and functional characteristics of vasoactive intestinal peptide receptors in human breast cancer cells. Cancer Res. 1988;48:5079–5083. [PubMed] [Google Scholar]
- 16.Gozes I, Davidson A, Draoui M, Moody TW. The VIP gene is expressed in non small cell lung cancer cell lines. Biomed Res. 1993;13:37–40. [Google Scholar]
- 17.Gozes I, McCune SL, Jacobson L, Warren D, Moody T, Fridkin M, Brenneman DJ. A hybrid antagonist to vasoactive intestinal peptide. Effects on cellular function in the central nervous system. J Pharmacol Exp Ther. 1991;257:959–966. [PubMed] [Google Scholar]
- 18.Harmar T, Lutz EM. Multiple receptors for PACAP and VIP. Trends in Pharmacological Sciences. 1993;15:97–9. doi: 10.1016/0165-6147(94)90042-6. [DOI] [PubMed] [Google Scholar]
- 19.Igarishi H, Ito T, Hou W, Mantey SA, Pradhan TK, Ulrich HC, Hocart SJ, Coy DH, Jensen RT. Elucidation of vasoactive intestinal peptide pharmacophore for VPAC1 receptors in human, rat and guinea pig. J Pharmacol Expt Ther. 2002;301:37–50. doi: 10.1124/jpet.301.1.37. [DOI] [PubMed] [Google Scholar]
- 20.Igarashi H, Ito T, Mantey SA, Pradhan TK, Hou W, Coy DH, Jensen RT. Development of simplified vasoactive intestinal peptide analogs with receptor selectivity and stability for human vasoactive intestinal peptide/pituitary adenylate cyclase-activating polypeptide receptors. J Pharmacol Exp Ther. 2005;315:370–381. doi: 10.1124/jpet.105.088823. [DOI] [PubMed] [Google Scholar]
- 21.Igarashi H, Ito T, Pradhan TK, Mantey SA, Hou W, Coy DH, Jensen RT. Elucidation of the vasoactive intestinal peptide pharmacophore for VPAC2 receptors in human and rat, and comparison to the pharmacophore for VPAC1 receptors. J Pharmacol Exp Ther. 2002;302:445–460. doi: 10.1124/jpet.102.038075. [DOI] [PubMed] [Google Scholar]
- 22.Ishihara R, Shigemoto R, Mori K, Takahashi K, Nagata S. Functional expression and tissue distribution of a novel receptor for vasoactive intestinal polypeptide. Neuron. 1992;8:811–9. doi: 10.1016/0896-6273(92)90101-i. [DOI] [PubMed] [Google Scholar]
- 23.Laburthe M, Couvineau A, Marie JC. VPAC receptors for VIP and PACAP. Receptors Channels. 2002;8:137–153. [PubMed] [Google Scholar]
- 24.Mahmoud S, Staley J, Taylor J, Bogden A, Moreau JP, Coy D, Avis I, Cuttitta F, Mulshine JL, Moody TW. (Psi13,14)bombesin analogues inhibit the growth of small cell lung cancer in vitro and in vivo. Cancer Res. 1991;51:1298–1302. [PubMed] [Google Scholar]
- 25.Moody TW, Chan D, Fahreenkrug J, Jensen RT. Neuropeptides as autocrine growth factors in cancer cells. Current Pharmaceutical Design. 2003;9:495–509. doi: 10.2174/1381612033391621. [DOI] [PubMed] [Google Scholar]
- 26.Moody TW, Czerwinski G, Tarasova NI, Michejda CJ. VIP-ellipticine derivatives inhibit the growth of breast cancer cells. Life Sci. 2002;71:1005–1014. doi: 10.1016/s0024-3205(02)01741-1. [DOI] [PubMed] [Google Scholar]
- 27.Moody TW, Fuselier J, Coy DH, Mantey S, Pradhan T, Nakagawa T, Jensen RT. Camptothecin-somatostatin conjugates inhibit the growth of small cell lung cancer cells. Peptides. 205(26):1560–6. doi: 10.1016/j.peptides.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 28.Moody TW, Hill JM, Jensen RT. VIP as a trophic factor in the CNS and cancer cells. Peptides. 2003;24:163–177. doi: 10.1016/s0196-9781(02)00290-5. [DOI] [PubMed] [Google Scholar]
- 29.Moody TW, Jensen RT. Breast cancer VPAC1 receptors. Ann NY Acad Sci. 2006;1070:636–9. doi: 10.1196/annals.1317.058. [DOI] [PubMed] [Google Scholar]
- 30.Moody TW, Jensen RT. VIP and PACAP as autocrine growth factors in breast and lung cancer. In: Kastin A, editor. Handbook of Biologically Active Peptides. London: Elsevier Science; 2006. pp. 493–8. [Google Scholar]
- 31.Moody TW, Leyton J, Chan D, Brenneman DE, Fridkin M, Gelber E, Levy A, Gozes I. VIP receptor antagonists and chemotherapeutic drugs inhibit the growth of breast cancer cells. Breast Cancer Research and Treatment. 2001;68:55–64. doi: 10.1023/a:1017994722130. [DOI] [PubMed] [Google Scholar]
- 32.Moody TW, Sun LC, Mantey SA, Pradhan T, Mackey LV, Gonzales N, Fuselier JA, Coy DH, Jensen RT. In vitro and in vivo antitumor effects of cytotoxic camptothecin-bombesin conjugates are mediated by specific interaction with cellular bombesin receptors. J Pharmacol Exp Ther. 2006;318:1265–1272. doi: 10.1124/jpet.106.104141. [DOI] [PubMed] [Google Scholar]
- 33.Moody TW, Mantey SA, Pradhan TK, Schumann M, Nakagawa T, Martinez A, Fuselier J, Coy DH, Jensen RT. Development of high affinity camptothecin-bombesin conjugates which have targeted cytotoxicity for bombesin receptor-containing cells. J Biol Chem. 2004;279:23580–9. doi: 10.1074/jbc.M401938200. [DOI] [PubMed] [Google Scholar]
- 34.Moody TW, Zia F, Draoui M, Brenneman DE, Fridkin M, Davidson A, Gozes I. A novel VIP antagonist inhibits non-small cell lung cancer growth. Proc Natl Acad Sci USA. 1993;90:4345–9. doi: 10.1073/pnas.90.10.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagy A, Schally AV. Targeting cytotoxic conjugates of somatostatin, luteinizing hormone-releasing hormone and bombesin to cancers expressing their receptors: a “smarter” chemotherapy. Curr Pharm Des. 2005;11:1167–1180. doi: 10.2174/1381612053507594. [DOI] [PubMed] [Google Scholar]
- 36.Nicole P, Lins L, Rouyer-Fessard C, Bronuot C, Fulcrand P, Thomas A, Couvineau A, Martinez J, Brasseur R, Laburthe M. Identification of key residues for interaction of vasoactive intestinal peptide with human VPAC1 and VPAC2 receptors and development of highly selective VPAC1 receptor agonist. Alanine scanning and molecular modeling of the peptide. J Biol Chem. 2000;275:24003–24012. doi: 10.1074/jbc.M002325200. [DOI] [PubMed] [Google Scholar]
- 37.Patterson LH, McKeown SR, Robson T, Gallagher R, Raleigh SM, Orr S. Antitumor prodrug development using cytochrome P450 (CYP) mediated activation. Anticancer Drug Des. 1999;14:473–4. [PubMed] [Google Scholar]
- 38.Pisegna JR, Wank SA. Multiple cloning and functional expression of the pituitary adenylate cyclase activating polypeptide type I receptor. Proc Natl Acad Sci USA. 1993;90:6345–9. doi: 10.1073/pnas.90.13.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reubi JC. In vitro identification of vasoactive intestinal peptide receptors in human tumors: Implications for tumor imaging. J Nucl Med. 1995;36:1846–1853. [PubMed] [Google Scholar]
- 40.Reubi JC, Laderach U, Waser B, Gebbers JD, Robberecht P, Laissue JA. Vasoactive intestinal peptide/pituitary adenylate cyclase activating polypeptide receptor subtypes in human tumors and their tisssues of origin. Cancer Res. 2000;60:3105–3112. [PubMed] [Google Scholar]
- 41.Said SI, Mutt V. Polypeptide with broad biological activity: Isolation from the small intestine. Science. 1970;69:1217–8. doi: 10.1126/science.169.3951.1217. [DOI] [PubMed] [Google Scholar]
- 42.Spengler D, Waeber C, Pantaloni C, Holsboer F, Bockaert J, Seeberg PH, Journot L. Differential signal transduction by five splice variants of the PACAP receptor. Nature. 1993;365:170–5. doi: 10.1038/365170a0. [DOI] [PubMed] [Google Scholar]
- 43.Sun L, Fuselier JA, Coy DH. Effects of camptothecin conjugated to a somatostatin analog vector on growth of tumor cell lines in culture and related tumors in rodents. Drug Deliv. 2004;11:231–8. doi: 10.1080/10717540490446125. [DOI] [PubMed] [Google Scholar]
- 44.Tripathy D, Slamon DJ, Cobleigh M, Arnold A, Saleh M, Mortimer JE, Murphy M, Stewart SJ. Safety of treatment of metastatic breast cancer with trastuzumab beyond disease progression. J Clin Oncol. 2004;22:1063–1070. doi: 10.1200/JCO.2004.06.557. [DOI] [PubMed] [Google Scholar]
- 45.Waschek JA, Richards MI, Bravo DT. Differential expression of VIP/PACAP receptor genes in breast, intestinal and pancreatic cell lines. Cancer Res. 1995;92:143–9. doi: 10.1016/0304-3835(95)03768-r. [DOI] [PubMed] [Google Scholar]
- 46.Whitmarsh AJ, Davies RJ. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J Mol Med. 1996;74:589–607. doi: 10.1007/s001090050063. [DOI] [PubMed] [Google Scholar]
- 47.Wood WD, Muss HB, Solin LJ, Olopade O. Malignant tumors of the breast. In: DeVita VT, Hellman S, Rosenberg SA, editors. Cancer: Principles and practice of oncology. Philadelphia: Lippincott, Williams and Wilkins; 2006. pp. 1415–1477. [Google Scholar]
- 48.Zia H, Hida T, Jakowlew S, Birrer M, Gozes Y, Reubi JC, Fridkin M, Gozes I, Moody TW. Breast cancer growth is inhibited by VIPhybrid, a synthetic VIP receptor antagonist. Cancer Res. 1996;56:3486–9. [PubMed] [Google Scholar]